

ABSTRACT
Regressing tumors are usually associated with a large immune infiltrate, but the molecular and cellular interactions that govern a successful anti-tumor immunity remain elusive. Here, we have triggered type I Interferon (IFN) signaling in a breast tumor model (MMTV-PyMT) using 5,6-dimethylxanthenone-4-acetic acid (DMXAA), a ligand of the STimulator of Interferon Genes, STING. The 2 main events rapidly triggered by DMXAA in transplanted PyMT tumors are 1) the disruption of the tumor vasculature, followed by hypoxia and cell death; 2) the release of chemokines. Both events converged to trigger the recruitment of 2 waves of immune cells: a swift, massive recruitment of neutrophils, followed by a delayed rise in monocytes and CD8 T cells in the tumor mass. Depletion experiments in vivo revealed that myeloid cell subsets and T cells need to cooperate to achieve full-blown recruitment and activation at the tumor site and to induce effective secondary cell death leading to tumor regression (Illustration 1). Altogether, our study highlights that the tumor regression induced by the STING agonist DMXAA results from a cascade of events, with an initial vessel destruction followed by several infiltration waves of immune cells which have to cooperate to amplify and sustain the initial effect. We thus provide the first global and detailed kinetic analysis of the anti-tumoral effect of DMXAA and of its different articulated steps.
KEYWORDS: Cooperation, imaging, interferon, myeloid cells, STING, tumor regression, T lymphocytes
Abbreviations
- DMXAA
5,6-dimethylxanthenone-4-acetic acid
- IFN
interferon
- MMTV
Mouse mammary tumor virus
- PyMT
polyomavirus middle T antigen
- ROS
reactive oxygen species
- STING
Stimulator of interferon genes
- TAM
tumor-associated macrophages
- TIL
tumor-infiltrating T lymphocytes
- TNF
Tumor necrosis factor
- VDA
Vascular disrupting agents
Introduction
The cancer immunotherapies that appear most promising these days are those based either on the adoptive transfer of anti-tumor T cells with a chimeric antigen receptor1 or on checkpoints inhibitors like anti-PD1 or anti-CTLA-4.2 The latter treatments succeed in inducing long-lasting effects, but only in a limited percentage of the patients. It seems therefore important to devise other treatments that could be combined with these promising therapies. One of them could be the use of vascular disrupting agents (VDA), which damage specifically tumor vessels.
DMXAA is a VDA that has been reported to induce tumor regression following a single intra-peritoneal (i.p.) injection in multiple pre-clinical tumor models.3 Two different modes of action of DMXAA have been reported, i.e. damages of the tumor vasculature and induction of an anti-tumor immune response. This drug was initially shown to disrupt the existing vascular network, reducing the supply of blood and oxygen to the tumor.4,5 Soon after DMXAA injection, one can observe apoptosis of endothelial cells, the rupture of blood vessels, and an increase in tumor permeability.6 For ill-defined reasons, the endothelium of tumors is exquisitely sensitive to DMXAA, which allows a systemic drug administration with a specific effect.
Regarding the DMXAA-induced cytokines, some reports have underlined the importance of TNFα, whereas others have highlighted IFNβ. TNFα contributes to the recruitment of neutrophils and to the vascular damages upon DMXAA treatment.7 TNFα could be released by DMXAA-activated macrophages since DMXAA can trigger TNF-α release by macrophages in vitro, and clodronate liposomes reduce the DMXAA-induced necrotic hemorrhage in vivo.8,9 Thus, endothelial cell apoptosis could be induced directly by DMXAA or indirectly by TNFa -producing macrophages.10
DMXAA binds directly to murine STING, a master regulator of IFN genes.11 STING is a ubiquitous intracellular protein activated by cyclic dinucleotides that is generated from cytosolic DNA coming from DNA viruses or dying cells. The activation of STING leads mainly to the activation of TBK1/IRF3 and the downstream transcription of type I interferon genes. Therefore, IFNβ could be key to the DMXAA anti-tumor effect.12 Type I IFN production is a central sensor for the initiation of innate and adaptive T cell responses,13 and a type I IFN signature was found to be associated with tumor regression induced by various anti-tumor therapies.14 Thus, DMXAA is not only considered as a tumor VDA, but also as an adjuvant for immune responses, including anti-viral ones.15,16
Published data about DMXAA leave several unanswered questions: 1) DMXAA acts both as a VDA and as an immune response inducer. What is the precise link between these 2 effects? 2) Myeloid cells and CD8 T cells are recruited to the tumor site after DMXAA treatment. Do these cells act independently or cooperate to actively contribute to the DMXAA-induced anti-tumoral response?
These questions have been addressed in the present study by the analysis of the dynamic events taking place in the breast tumor model of transplanted PyMT tumors.
Results
Activation of STING by DMXAA triggers vascular damages and the regression of PyMT tumors
Human carcinomas display an architecture with tumor islets surrounded by a vascularized stroma populated by immune cells.17 We used mice transplanted with freshly dissociated MMTV-PyMT tumors that form tumors with a distinct tumor/stroma architecture (Fig S1). A single i.p. injection of DMXAA was sufficient to induce the regression of transplanted PyMT tumors with an average of 40% reduction in size, 7 days after injection (Fig. 1A). To visualize the sequential events taking place in the tumor during the DMXAA induced-regression, we first examined the damages caused to the tumor vasculature. Contrast-enhanced ultrasound was used to evaluate tumor perfusion: the contrast enhancement observed following i.v. bolus injection of the ultra-sound contrast agent is related to the level of perfusion in both main vessels and capillaries, allowing to visualize tumor microvessels (∼10 μm). This method showed that PyMT tumor vascularization was severely damaged 6 hours after DMXAA (Fig. 1B and movie 1 and 2). One week after DMXAA, the tumor vasculature was still dysfunctional (Fig. 1B). In untreated tumors, scarce regions that are distant from blood vessels show signs of hypoxia. Within 24h, the damage induced by DMXAA was followed by a significant increase in the extension of hypoxic regions (Fig S2A, B).
Figure 1.
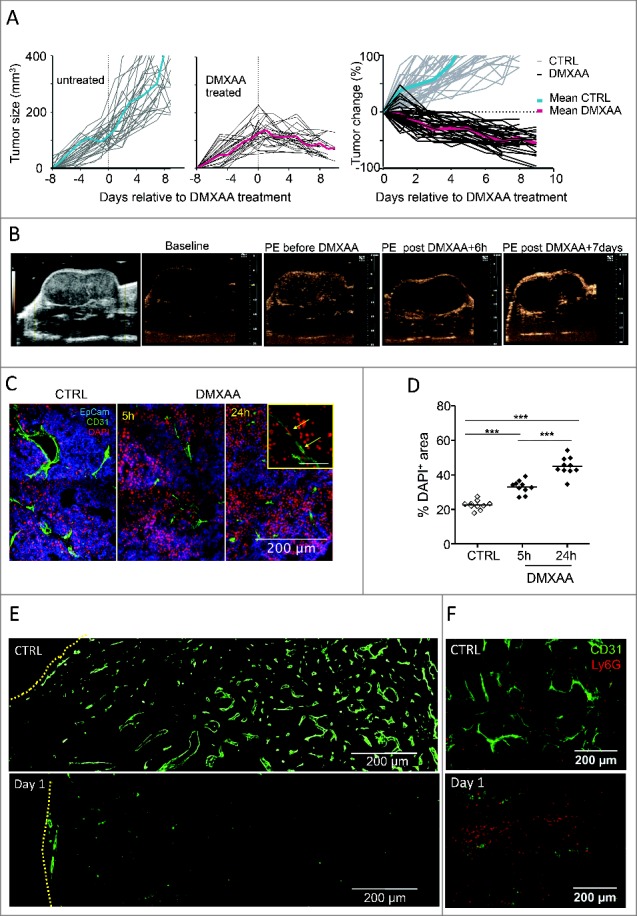
Anti-tumoral DMXAA effects begin with vascular damages and early cell death in transplanted PyMT tumors. A. Follow-up of transplanted PyMT tumor controls (left) and after one i.p. injection of DMXAA (23 mg/kg) (center). The right panel gives the relative tumor size changes after day 0, the day of treatment: [(size at day X)-(size at day 0)]/ (size at day 0). n = 20 tumors for each condition. The average curves are shown in color. B. Assessment of tumor perfusion using contrast ultrasound: from left to right: 1) acquisition in the largest diameter showing tumor anatomy; 2) baseline image in non-linear imaging mode before contrast enhancement of non-treated tumor; 3) Peak Enhancement (PE) image obtained after bolus injection of Ultrasound Contrast Agent (UCA) in the non-treated tumor; 4) UCA-PE image 6 hours after DMXAA injection; 5) UCA-PE image 7 d after DMXAA injection. Contrast enhancement observed in images after i.v. UCA injection illustrate that DMXAA treatment is associated with a strong alteration of vessel functionality as revealed by the absence of micro-bubbles circulation 6 h after DMXAA. Slight recovery of perfusion is observable at the tumor periphery after 7 d. C. DAPI staining shows an early DMXAA-induced cell death (DAPI+ cells) in both tumor and endothelial cells. D. Quantification of relative DAPI+ areas at different time points after DMXAA injection. Each point is a 20x image field, from n = 2 independent experiments. *** = p < 0.001 (t-test). E. As judged from CD31+ staining, 24 h after its i.p. injection, DMXAA has induced major vascular damages in the center of the tumor. The edge of the tumors is indicated with yellow lines. F. Ly6G+ neutrophils are much more abundant one day after DMXAA than in control conditions.
DMXAA-induced cell death was further analyzed by DAPI staining of fresh tumor slices, since only dead cells are expected to be labeled by DAPI. Dead DAPI+ CD31+ endothelial cells were clearly visible after DMXAA treatment, as well as dead cells at distance from the tumor vessels (Fig. 1C). The overall fraction of DAPI+ dead cell areas increased with time and covered about 50% of the tumor surface within 24 h of DMXAA treatment (Fig. 1D). In addition, strong alterations of tumor vessels were systematically found in the center of the tumors after DMXAA treatment as judged by CD31 labeling. Vessels were more scarce and fragmented in the tumor center than in its periphery. No such difference between periphery and center was observed in control PyMT tumor mice (Fig. 1E). We observed that Ly6G+ neutrophils infiltrate the tumor at this stage (Fig. 1F) and accumulate preferentially near disrupted vessels (Fig. S2C). These data demonstrate that DMXAA was able to initiate vascular damages and to induce regression of tumors in which immune cells surrounded tumor islets, as one can be found in human carcinoma. This extends the relevance of the conclusions of previous studies showing the therapeutic potential of DMXAA in more artificial tumors derived from cell lines.
DMXAA is an agonist of STING that drives the expression of type I IFN genes. Ifnα and Ifnβ gene expression was rapidly upregulated in the tumor microenvironment 3h after DMXAA injection and maintained at 24h (Fig. 2A). To investigate to what extent the expression of IFN genes is linked to the massive cell death and neutrophil recruitment in the tumor area, we conducted experiments in IFNα‐receptor deficient mice (Ifnr-1−/−) mice. As these mice are on the C57BL/6J background, the DMXAA efficacy in Ifnr-1−/− mice was tested in mice transplanted with the TC1 tumor cell line, isolated from the same background. In this system, type I IFN signaling is prevented in host cells but not in tumor cells. As in PyMT tumors, one DMXAA i.p. injection in TC1‐transplanted wild‐type mice induced a massive cell death and neutrophil recruitment in the tumor at 24h, as measured by flow cytometry (Fig. 2B). The 2 effects were blocked in Ifnr-1−/− mice. These results indicate that the DMXAA effects on cell death and early recruitment of some innate cells was indirect and required a type I IFN dependent activation of infiltrating immune and stromal cells.
Figure 2.

type (I)IFN activation in immune infiltrating cells is necessary for early DMXAA-induced cell death and immune cell recruitment. A. IFNβ and IFNα gene expression was upregulated early after DMXAA injection in PyMT tumor bearing mice. B. Ifnr-1 −/− mice (C57Bl6/J) transplanted with the TC1 tumor cell line and treated or not with DMXAA were used to determine the importance of IFN type I signaling in host cells in DMXAA‐induced tumor regression. In WT mice, a single injection of DMXAA is sufficient to induced cell death and neutrophil recruitment in TC1 tumors, whereas in Ifnr-1−/− mice, DMXAA fails to induce cell death and neutrophil recruitment. C. Upregulation of IFNβ and IFNα gene expression was abrogated in Ifnr-1 −/− mice treated with DMXAA, compared with wild type littermate. n n = 6 tumors, x = 2 independent experiments. One-way Anova was used for the statistics.
Immune infiltration induced by DMXAA treatment
DMXAA triggered a tumor infiltration of various immune cell types. After 24h, Ly6G+ neutrophils outnumbered the resident tumor-associated macrophages (TAM, defined as CD11b+ Ly6C− Ly6G− F4/80+) population and reached up to 40% of viable cells in the tumor as assessed by flow cytometry (Fig. 3A and Fig. S3). When the tumors started to shrink (around 48h after DMXAA treatment, see Fig. 1A), the immune infiltrate was mainly composed of neutrophils, whereas monocytes and CD8 T cells represented less than 5% of the living cells (Fig. 3A). In the following days, CD8+ T cells and monocytes kept on accumulating, and represented 20–30% and 5–10%,respectively, of living cells for up to 12 days.
Figure 3.
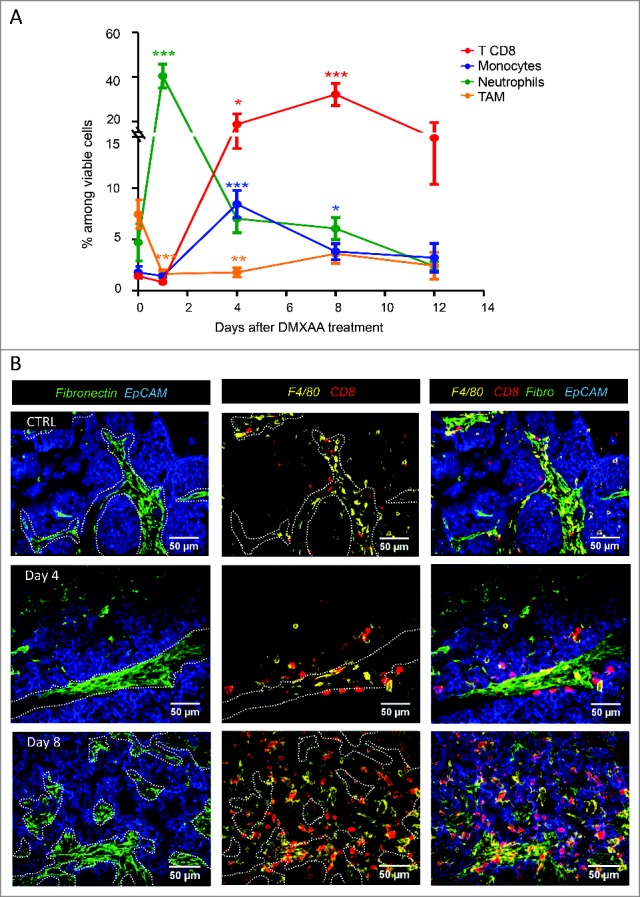
DMXAA injection induces a dynamic immune cell recruitment involving both innate and adaptive cells. A. Neutrophils are scarce in growing tumors (top), and abundant 24 hours after DMXAA injection (bottom). B. Kinetics of immune cell recruitment measured by flow cytometry reveals a high influx of neutrophils (CD11b+ Ly6C− Ly6G+ F4/80−) during the first 24h, followed by monocytes (CD11b+ Ly6C+ Ly6G− F4/80+) (day4) and CD8 T cell (day4–8) recruitment. DMXAA also induced a decrease of TAM (CD11b+ Ly6C− Ly6G− F4/80+). n = 10–20 tumors by time point and x = 4 independent experiments. Mann-Whitney test was used for * (p < 0.1), ** (p < 0.01) and *** (p < 0.001) in this and the following figures. C. Localization of immune cells in the tumor after DMXAA injection. Immunofluorescence staining was performed on tumor slices (350µm thick) at different time points after DMXAA injection (day 0 (ctrl), day 4 and day8). Before DMXAA injection (top), tumors showed a limited infiltrate of immune cells mostly located in stromal areas (fibronectin+) whereas after DMXAA injection (middle and bottom), immune cells also invaded EpCam+ tumor islets.
The localization of the immune infiltrate in tumors of control- and DMXAA-treated mice is shown in Fig. 3B. The stromal regions located between the EpCam+ tumor islets were labeled by fibronectin. In control tumors, F4/80+ and CD8 T cells were found in these stromal regions. Progressively after DMXAA treatment, the immune cells were no longer confined in the stromal zone but infiltrated the tumor islets on day 4 and day 7 post-DMXAA injection as illustrated in Fig. 3B. These data show that DMXAA induced a biphasic response with swift vascular damages followed by a drastic alteration of the composition and the localization of the immune cell infiltrate for at least 12 days following the treatment.
We next examined the local production of pro-inflammatory mediators known to be induced by DMXAA treatment. In addition to the production of IFNβ and IFNα (Fig. 2A), the levels of TNFα, IL-6 and reactive oxygen species (ROS) were increased during the first 24h and returned to baseline level after 4 days (Fig. 4A, B and Fig. S4). TAM constituted initially the main source of TNFα and IL-6, but they were rapidly reinforced at 24h by neutrophils and at 4 days by monocytes. In addition, the progressive accumulation of CD8 T cells in the tumor coincided with the production of CXCL9 by TAM, neutrophils and monocytes successively (Fig. 4C).
Figure 4.
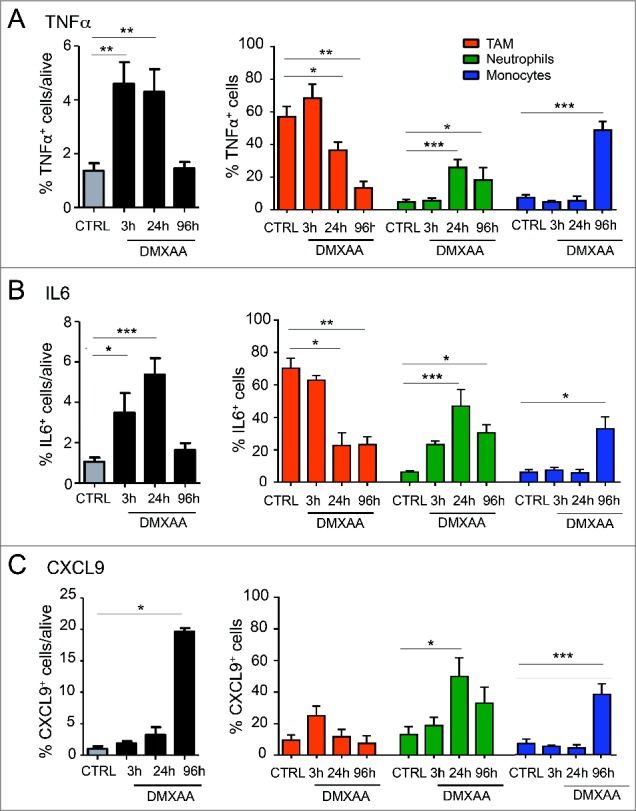
DMXAA induces TAM, neutrophils and monocytes to produce inflammatory cytokines and chemokines in the tumor microenvironment. Intracellular cytokines/ chemokines staining by flow cytometry indicates that innate immune cells (TAM, neutrophils and monocytes) participate to build the inflammatory tumor microenvironment (TNFα, IL-6 production) and allow the recruitment of other immune cells (CXCL9 production). n = 9 tumors per condition and x = 3 independent experiments. One-way Anova test was used for statistical test.
Immune cell subsets cooperate in regressing tumors
To determine to what extent the immune cell infiltrate contributes to the DMXAA-induced tumor regression, we selectively depleted neutrophils, macrophages and CD8 T cells. Removal of any of these immune cell subsets prevented the average regression induced by DMXAA and only a transient stabilization of the tumor growth was achieved after partial immune cell depletion (Fig. 5A). This shows that the 3 cell subsets contributed to the DMXAA effect.
Figure 5.
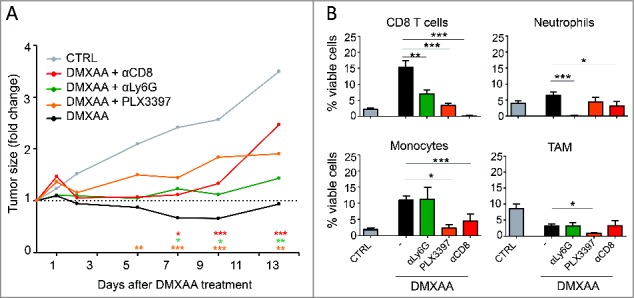
The clinical impact of DMXAA requires the contribution and the cooperation of several immune cell subsets. A. The follow up of tumor sizes after DMXAA injection associated with selective immune cells depletion reveals that all the immune cells tested are essential for an optimal tumor regression. Neutrophils (Ly6G+) and CD8 T cells were eliminated using depleting antibodies, macrophages were depleted with PL3397. n = 20 tumors minimum per condition and x≥ 3 independent experiments. For clarity, the statistical significance of all conditions compared with DMXAA alone is shown by the color coded stars above the x axis. B. Analysis of the tumor cell infiltrates 4 d after DMXAA treatment in depleted animals.
We further analyzed if the depletion of one immune cell subset affects the behavior of the other immune cells infiltrating the tumors after DMXAA. The composition of the infiltrates was analyzed by flow cytometry 4 days after DMXAA treatment in immune cell-depleted animals. At this time point, neutrophils, CD8 T cells and monocytes were all infiltrating DMXAA-treated tumors. Unexpectedly, the density of CD8 T cells was reduced in PyMT mice treated with DMXAA and PLX3397 or anti-Ly6G (Fig. 5B), indicating that TAM and neutrophils were necessary for an optimal infiltration of the tumor by CD8 T cells. Reciprocally, the densities of infiltrating neutrophils and monocytes following DMXAA treatment were reduced in mice depleted in CD8 cells. The depletion of TAM also reduced the monocyte infiltrate (Fig. 5B). On the other hand, the depletion of neutrophils or CD8 T cells did not impact the proportion of infiltrating TAM that was reduced rapidly after DMXAA treatment.
We next addressed whether the depletion of myeloid cells at the time of DMXAA treatment affected the function of CD8 T cells and vice versa. As shown in Fig. 6A, the capacity of CD8 T cells to secrete IFNγ was strongly reduced when DMXAA was injected in animals depleted in neutrophils and, to a lesser extent, after TAM depletion. On the other hand, the capacity of TAM and monocytes to produce CXCL9 or TNFα was reduced when DMXAA was injected in CD8 T cells depleted mice (Figs 6B and 5C). Altogether, these data indicate that a reciprocal cooperation takes place between activated CD8 T cells and myeloid cells for an optimal recruitment, accumulation and release of effector molecules at the tumor site 4 days after DMXAA injection.
Illustration 1.
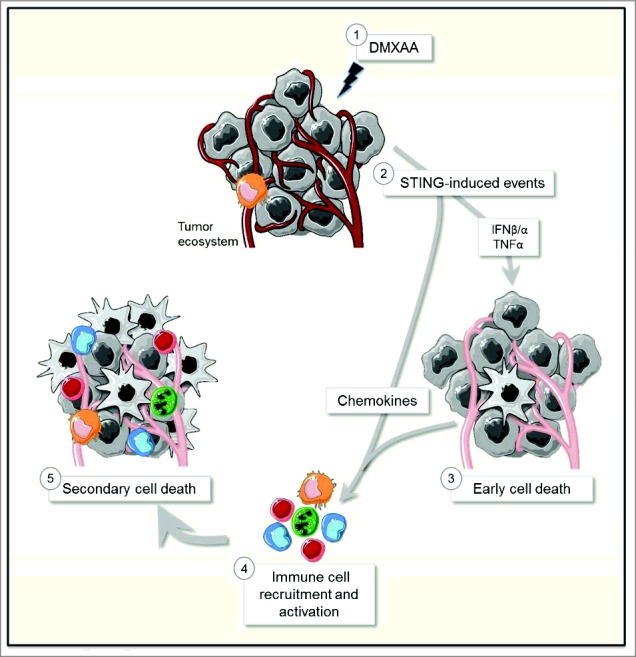
Schematic view of the anti-tumoral mode of action of DMXAA.
Figure 6.
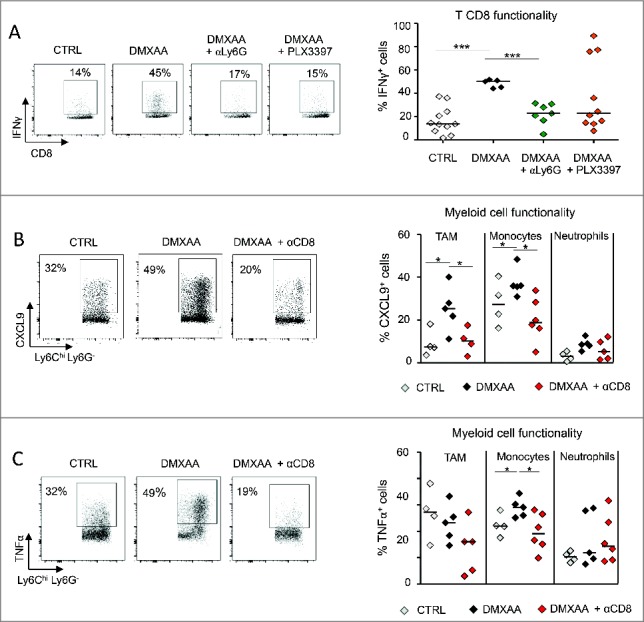
The functional state of DMXAA-induced immune cells is regulated in a cooperative way. In the 3 panels, the functional state of immune cells was measured 4 d after DMXAA tretament. A. The ability of CD8 T cells to produce IFNγ in response to PMA/ionomycine in vitro is affected in DMXAA treated animals that were depleted in neutrophils (anti-Ly6G) or monocytes/macrophages (PLX3397). B. Similarly, the capacity of myeloid cells to produce CXCL9 after LPS/IFNγ is inhibited in CD8-depleted DMXAA-treated mice. n = 6–9 tumors per condition and x = 3 independent experiments. C. Left: The capacity of myeloid cells to produce TNFα was increased by DMXAA. In CD8-depleted animals, this increase was completely prevented, even below the control level, n = 6–9 tumors per condition and x = 3 independent experiments. Right: this effect of CD8 depletion on TNFα production concerns TAM and monocytes, but not neutrophils.
Discussion
Interactions between myeloid cells and T cells in a tumor have long been considered to be detrimental for anti-tumor immunity according to most reports. However, a different point of view has emerged, which proposes that infiltrating myeloid cells may synergize with anti-tumor T cells to reject established tumors under appropriate stimuli (see e.g.18). Here, we took advantage of the acute inflammation induced by DMXAA in tumors after a single i.p. injection for providing a new example in which the coordinated activation of resident TAM, and successively recruited neutrophils, monocytes and T cells leads to tumor regression, by amplifying and sustaining the initial damages created by DMXAA to the tumor vasculature.
Kinetic analysis of the anti-tumoral effect of DMXAA
The literature describes DMXAA either as a tumor-specific VDA10 or as a molecule inducing an inflammatory state in tumors due to TNFα19 or to IFNβ.12 In the present study, we provide the first kinetic analysis of the anti-tumoral effect of DMXAA, taking into account the sequencing of a vascular disrupting action followed by the recruitment of the immune cells responsible for the release of inflammatory cytokines. We have shown that the effect of DMXAA on transplanted tumors is biphasic. First, in the 4–6 hours following DMXAA i.p. injection in PyMT-transplanted mice, we observed the collapse of tumor microvessels, enhanced tumor hypoxia and increased cell death of both endothelial and tumor cells. A second phase of the response to DMXAA occurred with the successive recruitment of several waves of immune cells, which replaced the resident TAM (CD11b+ F4/80+ Ly6C− Ly6G−). We show that the first infiltrating cells were neutrophils (CD11b+ Ly6Clow Ly6G+), which were recruited within less than one day. Some readers may wonder if these cells could be PMN-MDSC (polymorphonuclear myeloid-derived suppressor cells). Indeed, a fraction of myeloid-derived suppressor cells express Ly6G.20 However, Ly6G±-MDSC are not a separate cell lineage, they are a mere subset of neutrophils with suppressor properties. Thus, the Ly6G± cells massively recruited by DMXAA are clearly neutrophils. Moreover, our data with depleting Ly6G antibody indicate that these cells are not suppressive in this context. On the contrary, their presence appears necessary for the proper recruitment and activation of other anti-tumoral immune cells.
The use of Ifnr-1−/− mice allowed us to demonstrate that type I interferon signaling is mandatory for the early death events and neutrophils recruitment to the tumor. Concerning the innate cells recruited after DMXAA injection, it has been reported that the proportion of CD11b+ cells was increased in the tumor shortly after DMXAA treatment.21 However, the identity and the role of these myeloid cells have not been carefully analyzed so far. We show here that infiltrating neutrophils were responsible for prolonging the production of inflammatory cytokines (IL-6 and TNFα), initially produced by resident TAM. This first cellular wave was followed by a second one, consisting of monocytes and CD8 T cells. The size of both populations peaked at around 4 days after DMXAA injection. At that time, infiltrating CD8 T cells were about twice as abundant as monocytes, and persisted longer. These data are more complete and accurate, but compatible with those previously reported.7,21
Altogether, this kinetic analysis revealed a cascade of events initiated by type I IFN and TNFα release by resident cells. The initial vascular disruption and cell death could be due to inflammatory cytokines and/or to hypoxia, and was followed by the recruitment of 2 waves of immune cells. This recruitment presumably results initially from numerous cell death-associated signals for the recruitment of neutrophils, and from chemokines for the second wave of immune cells. This part of the sequence involves interactions between different cell subsets which will now be further discussed.
DMXAA-induced tumor regression requires cooperations between macrophages, neutrophils, monocytes and lymphocytes
We have shown that the successive activation and accumulation of neutrophils, monocytes and CD8 T cells at the tumor site in PyMT-mice involve a reciprocal cooperation between myeloid cells and CD8 T cells. CD8 T cell depletion prevents DMXAA-induced tumor regression (Fig. 5A) confirming that the establishment of an adaptive anti-tumor immune response is required for the efficacy of DMXAA-treatment.21 TAM and neutrophils were necessary for the accumulation of the anti-tumor CD8 T cells, since their depletion reduced the proportion of CD8 T cells in treated animals 4 days after DMXAA injection. In line with this, TAM and neutrophils are capable of producing large amounts of CXCL9 between 3 h–96 h after DMXAA-treatment. At later time points, when TAM and neutrophils have disappeared, monocytes recruited secondarily, may sustain the recruitment of CD8 T cells as they also produce the appropriate chemokines.
On the other hand, CD8 T cell depletion was associated with a decrease in the capacity of TAM and monocytes to produce CXCL9 after DMXAA treatment. The IFNγ produced by infiltrating CD8 T cells is a good candidate for stimulating CXCL9 production by myeloid cells. It is reasonable to propose that myeloid cells recruit CD8 T cells, and an IFNγ−dependent positive feedback loop helps recruiting more immune cells to the inflamed tumor.
TAM and neutrophils were not only necessary for the recruitment of CD8 T cells, but also for their optimal activation. CD8 T cells from neutrophil- and TAM-depleted-DMXAA-treated, animals produced smaller amounts of IFN-γ in vitro compared with those from non-depleted mice. Taken together, these data shed light on the communication between myeloid cell subsets (neutrophils, TAM and monocytes) and T cells for an optimal recruitment at the tumor site for DMXAA efficacy. Recent reports have also brought evidence for a cooperative activation and recruitment of myeloid and T cell in the context of tumors. For instance, a positive loop favoring T cell transmigration in tumors was observed following the administration of CpG or low dose irradiation.22 We have not addressed the direct effect of activated myeloid cells on the destruction of tumor cells but an earlier report proposed that the tumor hemorrage associated with DMXAA injection is mainly due to the TNFα released by resident TAM.23 Thus, i.p. injection of DMXAA, or peritumoral injection of type I IFN,18,24 may induce TAM to exert, directly and indirectly, an anti-tumor activity.
Spatial and temporal dimensions of the anti-tumoral immune cell cooperation
Part of the cooperation described here takes place in a specific microenvironment where several cell types coexist at a given time. For instance, before the treatment, rare CD8 T cells and resident TAM share the same stromal microenvironment where they may interact with each other. After DMXAA treatment, the space in which they may interact extends to the tumor islets that both of them infiltrate a few days after treatment. As we have previously shown in a different tumor model, the potential molecular basis of these interactions include local chemokines produced by myeloid cells and T cell-produced IFNγ18.
However, immune cells may cooperate without being simultaneously present at the same place. This is the temporal dimension. First, we have shown that, like in a relay race, the same molecule, CXCL9, may be produced first by one cell type (resident TAM), then by a second one (rapidly recruited neutrophils), before a third one (monocytes recruited a few days later). Another striking observation is that the recruitment of neutrophils affects that of T cells, even though neutrophils have completely disappeared when T cells arrive in the tumor. Everything happens as if T cells were following the footsteps of neutrophils. It has previously been described that, after a viral lung infection, CXCL12 left by neutrophils may form trails that guide influenza-specific CD8⁺ T cells in the airways.25 In a tumor microenvironment, it is unlikely that the situation is that simple, as several chemokines, including CXCR3 ligands, contribute to T cell recruitment.18
In conclusion, the effect of DMXAA was evaluated here in a tumor model presenting the common structure of human tumors. In this model, the anti-tumoral efficacy of DMXAA results from a whole cascade of events: initial vessel destruction followed by several waves of recruitment of immune cells which cooperate to the final effect. It will be important to examine if the same cascade may be elicited in spontaneous, rather than transplanted tumors. Indeed in transplanted tumors, the sudden appearance of a large number of tumor cells may lead to an immune and inflammatory response which has no counterpart in a spontaneous tumor occurring in mouse or human. This may modify the tumor susceptibility to subsequent treatments. Finally, given that DMXAA does not activate human STING, it will be necessary to evaluate if other STING agonists that may also target human STING can elicit similar immune cell cooperation and overall tumor regressions after systemic injection, in tumor models.
Experimental procedures
Animal studies
MMTV-PyMT transgenic mice were backcrossed with FvB/NCrl mice (Charles River laboratories). Ifnar−/− mice were kindly provided by A. Lebon, Institut Cochin, Paris, France. Mice were maintained at the Cochin Institute SPF animal facility. Animal care was performed by expert technicians in compliance with the Federation of European Laboratory Animal science association and under the approval of the animal experimentation ethics committee of Paris Descartes (CEEA 34, 16–063).
Mice and tumor cell suspensions
Tumor implantation was performed using primary tumors developed in donor mice. Donor mice with primary tumors of ∼10–15 mm were killed and tumors were pooled, dissociated and then re-injected orthopically in receiver mice. In practice, with one mouse bearing spontaneous tumors, one can transplant tumors in 10–20 recipient mice. Tumors were dissociated mechanistically and incubated for 30 minutes at 37°C with DNase I (100 µg/ml, Roche), collagenase (1mg/ml, Roche) and hyaluronidase (1 µg/ml, Sigma) in RPMI with 2% serum. After red blood cell lysis and filtration on a 70 µm sieve, cell suspension was rinsed 3 times in PBS and used for flow cytometry analysis or for tumor transplantation. For tumor transplantation, 106 cells isolated from tumors of MMTV-PyMT mice were injected in 100 µl of PBS in the mammary fatpad of 8 weeks-old FvB mice. The tumor cell suspension contained 10–15% viable cells that represent on average 75% of CD45− cells, 15% of CD11b+ myeloids cells and 10% of lymphoid cells. About 10 d after inoculation, transplanted PyMT tumors reached a diameter of ∼ 6mm.
The Ifnar−/− mice were inoculated in the flank with 105 TC1 cells. TC1 cell line was maintained in culture in complete RPMI, including 10% FCS (GE Healthcare), antibiotics (Penicillin 50U/ml, Streptomycin 50 μg/ml, GIBCO), L-Glutamine (4mM, GIBCO) and Sodium Pyruvate (1mM, GIBCO). TC1 tumors also reached a diameter of ∼ 6 mm after 10 days.
In vivo treatments
Mice with tumors of 6 mm in diameter received one i.p. injection of DMXAA (23mg/kg in DMSO, Sigma) or 100 µl of 50% DMSO in PBS as control. For immune cell depletion, 200 µg of anti-CD8 antibody (BioXcell, clone 53–6.72 or 300 µg of anti-Ly6G (BioXcell, clone 1A8) were injected every 2 days starting one day before DMXAA treatment. Macrophages were depleted with PLX3397 (obtained via an MTA with Plexxikon Inc.), a CSF1-R signaling inhibitor. Mice were fed with chow containing PLX3397, or control chow, starting 48h before DMXAA treatment.
Multicolor flow cytometry
Tumor cells suspension (4.106 cells) were stained in 96-wells round bottom plates with dead/live staining (blue fluorescent reactive dye, Invitrogen) during 20min at room temperature. Fc receptors were blocked with anti-FCR (anti-CD16 CD-32 at 5 µg/ml, BD PharMingen). After 2 washes in PBS 2% FCS, cells were stained with antibodies CD45-PeCy7, CD11b-ef450, Ly6C-APC, Ly6G-FITC, F4.80 PerCPCy5.5 (for myeloid staining), with CD45-PeCy7, TCRβ-BV605, CD4-BV711 and CD8-APCH7 antibodies (for lymphoid staining) or with CD45-PerCPCy5.5, CD11b-PeCy7, Ly6C-BV421, Ly6G-BV510, F4.80–605 antibodies, all purchased from BD PharMingen. After washing in PBS, cells were fixed in 1% PFA, stored at 4°C, and acquired the next day with LSR II flow cytometer (BD Bioscience) or LSR-Fortessa flow cytometer (BD Bioscience). For the detection of ROS production, cells were incubated 1h at 37°C with a ROS inhibitor NAC (1mM) or a ROS stimulator TBHP (200 µM) or nothing. The fluorescent probe CellROX® Deep Red (500 nM) was added for 1h at 37°C (CellROX® Reagent KIT Thermofisher). For intracellular cytokine staining, cell suspensions were incubated 4h with Golgi PLUG (1/1000), stimulated or not with LPS (100 ng/ml) and IFNγ (4.102 U/ml) or with PMA/Ionomycin (0.5 µg/ml each). After cell surface staining, cells were fixed/permeabilized with BD Cytofix/Cytoperm™. Cell suspensions were stained with anti-TNFα-AF700, anti-IL-6 PE, anti-IFNg-BV421, anti-MCP1-APC, anti-CXCL9-PE (from BD PharMingen), then fixed in PFA 1% and data acquired the following days.
Immunofluorescence
Tumor pieces were either used directly for live immunofluorescence or fixed overnight with Periodate-Lysine-Paraformaldehyde at 4°C. Immunofluorescence measurements in thick tumor slices (350 µm) were performed as described previously (Salmon et al., 2012). Immunostaining of surface markers was performed at room temperature for 1h with primary antibodies specific for CD8-PerCP, CD31-Biotin, EpCAM-BV421 (all from BD PharMingen), F4/80-biotin, Ly6G-biotin, EpCAM AF647, fibronectin (from Biolegend) and F4/80-PE (AbD Serotec) and rabbit anti-fibronectin (from Sigma). Immunodetection was performed using either secondary anti-rabbit fluorescent antibodies (BD PharMingen) or streptavidin-Alexa Fluor 647/488/561 (Invitrogen). Eventually, slices were counter-stained with DAPI for 5min at room temperature.
Images were obtained with a CSU X1 spinning disk microscope (Yokogawa, Roper Scientific, Lisses, France) equipped with an Orca Flash 4 LT camera (Hamamatsu) and a 25 x objective. All images were acquired with MetaMorph 7 imaging software (Molecular Devices) and analyzed with Image J.
Transcriptomic analysis
Tumor RNA was extracted using RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions and gene expression was analyzed by RT-qPCR with the LighCycler® 480 Real-Time PCR system. For cDNA synthesis, RNA was reverse transcribed using the Advantage® RT for PCR kit (Applied Clontech). Briefly, 1 µg of total RNA in a final volume of 12.5 µl DEPC-water was supplemented with 1 µl of Random Hexamers (20 pmol). The mix was heated at 70°C for 2min and cooled on ice. Then, 6,5 µl of master mix containing 5X Reaction buffer (50 mM Tris-HCl pH 8.3, 75 mM KCl, 3 mM MgCl2), MMLV Reverse Transcriptase (> 200U/µl RNA), dNTPs (10 µM each) and recombinant RNase inhibitor (1U/µl) was added. The reaction was performed in a total reaction volume of 20 µl, incubated at 42°C for 1h and heated at 94°C for 5min to stop the cDNA synthesis reaction and to shut off any DNase activity. The reaction was diluted to a final volume of 100 μl by adding 80 μl of DEPC-treated H2O. For each cDNA sample, 96 small RNAs were profiled using a gene maximization PCR plate setup in a 96-well plate. PCR amplification reactions were performed in a total volume of 10 µl. One part of q-PCR were realized using Syber Green and the other with TaqMan reagents. For Syber Green assay, a master mix, composed of Syber Green 2x, all the primers (10 µM) (supplementary table I) and H2O (LighCycler 480 Syber Green I Master kit. Roche) was prepared and put in the plate. Then, cDNA was added. Cycling conditions were as follows: 94°C for 2min followed by 40 cycles of 15s at 94°C and 1min at 60°C. For TaqMan assay, PCR amplification reaction was performed following the manufacturer recommendations (Thermo Fisher). GAPDH was used as a housekeeping gene to normalize mRNA expression. The ratio of gene of interest versus housekeeping gene was calculated according to the following formula: ratio = 2−ΔCt (ΔCT = mean Ct gene − mean Ct housekeeping). All the measures were performed in triplicate and validated when the difference in threshold cycle (Ct) between the 2 measures was <0.3. Raw Cq values were calculated using the lightcycler 480 v1.5.0 SP3 software. All PCR reactions were performed on the LightCycler® 480 Real-Time PCR system (Applied Roche).
Contrast-enhanced ultrasound
Mice were anesthetized by isoflurane in medical air (3% isoflurane for inducing the anethesia, 1.5% to maintain it) and were positioned in prone position on a heating pad (VEVO imaging station, Visualsonics, Canada and THM150, Indus Instruments, United States) allowing realtime physiologic monitoring of each animal (Temperature, ECG, Respiratory frequency). The skin at the tumor site was depilated using a depilatory cream (Veet). Ultrasound imaging was performed with a high resolution ultrasound system dedicated to small animals (VEVO2100, Visualsonics), using a high-resolution probe (MS550, 40 MHz center frequency) for measuring anatomic dimensions and a lower resolution probe (MS250, 18 MHz central frequency) allowing non-linear imaging of ultrasound contrast agent. Tail vein was first catheterized, the probe was positioned in sagittal view and then rotated along the central axis of the probe, to image the largest diameter of the tumor. Then acquisition of a 2D image sequence in nonlinear mode was triggered, and 50 µl of ultrasound contrast agent suspension (Micromarker, Visualsonics) containing 5.109 microbubbles was injected through the venous catheter, and 90s video loops showing contrast enhancement were stored.
Statistics
Unless otherwise indicated, results are expressed as means ± SEM of 3 to 6 mice. All experiments were repeated at least twice, yielding similar results. Data were analyzed with GraphPad Prism5 software. p values were calculated by the unpaired Student's t test or One-way ANOVA and Tukey test for multiple comparison. Values ≤ 0.05 were considered significant. * p < 0.05; ** p < 0.01; *** p < 0.001).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Plexxikon for providing the PLX3397 inhibitor. We would like to thank C. Peyssonnaux for providing the MMTV-PyMT mice and A. Le bon for providing the Ifnr-1−/− mice. We would like to thank also Emmanuel Donnadieu and Chahrazade Kantari-Mimoun for critical review of the manuscript, Georges Bismuth for his advice with ROS measurements, the staff of the IMAG'IC and CYBIO facilities of the Cochin Institute for their helpful advice along this study.
Funding
This work was granted by the Worldwide Cancer Research; “Le comité Ile-de-France de la” La Ligue Nationale Contre le Cancer; The cancer research personalized medicine (CARPEM-projet emergent); The CNRS, Inserm and University of Paris-Descartes. M. G. was granted by the Ministère de l’éducation Nationale, de l'enseignement supérieur et de la recherche. In vivo imaging was performed at the Life Imaging Facility of Paris Descartes University (Plateforme Imageries du Vivant - PIV), partly supported by France Life Imaging (grant ANR-11-INBS-0006), Région Ile-de-France and the GIS IBISA.
Authors contribution
J.M.W, M.V.G., N.B. and AT. designed, performed and analyzed the experiments. F.R, I.G-F, L.V., V.F, E.P and M.T. contributed to multicolor flow cytometry and immunostaining. G.R., A.T. and N.B. performed the contrast-enhanced ultrasound measurements. A.T. and N.B. designed the study and A.T, N.B. and M.V.G. wrote the manuscript and prepared the figures.
References
- 1.Maus MV, June CH. Making better chimeric antigen receptors for adoptive T-cell therapy. Clin Cancer Res Off J Am Assoc Cancer Res 2016; 22:1875-84; https://doi.org/ 10.1158/1078-0432.CCR-15-1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karaki S, Anson M, Tran T, Giusti D, Blanc C, Oudard S, Tartour E. Is there still room for cancer vaccines at the era of checkpoint inhibitors. Vaccines 2016; 4:37; PMID:27827885; `https://doi.org/ 10.3390/vaccines4040037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baguley BC, Siemann DW. Temporal Aspects of the Action of ASA404 (Vadimezan; DMXAA). Expert Opin Investig Drugs 2010; 19:1413-25; PMID:20964495; https://doi.org/ 10.1517/13543784.2010.529128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siemann DW. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by Tumor-vascular disrupting agents. Cancer Treat Rev 2011; 37:63-74; PMID:20570444; https://doi.org/ 10.1016/j.ctrv.2010.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao L, Ching LM, Kestell P, Kelland LR, Baguley BC. Mechanisms of tumor vascular shutdown induced by 5,6-dimethylxanthenone-4-acetic acid (DMXAA): Increased tumor vascular permeability. Int J Cancer 2005; 116:322-6; PMID:15800918; https://doi.org/ 10.1002/ijc.21005 [DOI] [PubMed] [Google Scholar]
- 6.Ching LM, Zwain S, Baguley BC. Relationship between tumour endothelial cell apoptosis and tumour blood flow shutdown following treatment with the antivascular agent DMXAA in mice. Br J Cancer 2004; 90:906-10; PMID:14970872; https://doi.org/ 10.1038/sj.bjc.6601606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L-CS, Thomsen L, Sutherland R, Reddy CB, Tijono SM, Chen C-JJ, Angel CE, Dunbar PR, Ching L-M. Neutrophil influx and chemokine production during the early phases of the antitumor response to the vascular disrupting agent DMXAA (ASA404). Neoplasia N Y N 2009; 11:793-803; https://doi.org/ 10.1593/neo.09506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fridlender ZG, Jassar A, Mishalian I, Wang L-C, Kapoor V, Cheng G, Sun J, Singhal S, Levy L, Albelda SM. Using macrophage activation to augment immunotherapy of established tumours. Br J Cancer 2013; 108:1288-97; PMID:23481183; https://doi.org/ 10.1038/bjc.2013.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao L, Ching L-M, Kestell P, Baguley BC. The antitumour activity of 5,6-dimethylxanthenone-4-acetic acid (DMXAA) in TNF receptor-1 knockout mice. Br J Cancer 2002; 87:465-70; PMID:12177785; https://doi.org/ 10.1038/sj.bjc.6600479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ching L-M, Cao Z, Kieda C, Zwain S, Jameson MB, Baguley BC. Induction of endothelial cell apoptosis by the antivascular agent 5,6-Dimethylxanthenone-4-acetic acid. Br J Cancer 2002; 86:1937-42; PMID:12085190; https://doi.org/ 10.1038/sj.bjc.6600368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prantner D, Perkins DJ, Lai W, Williams MS, Sharma S, Fitzgerald KA, Vogel SN. 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential. J Biol Chem 2012; 287:39776-88; PMID:23027866; https://doi.org/ 10.1074/jbc.M112.382986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roberts ZJ, Ching L-M, Vogel SN. IFN-beta-dependent inhibition of tumor growth by the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid (DMXAA). J Interferon Cytokine Res Off J Int Soc Interferon Cytokine Res 2008; 28:133-9; https://doi.org/ 10.1089/jir.2007.0992 [DOI] [PubMed] [Google Scholar]
- 13.Dunn GP, Bruce AT, Sheehan KCF, Shankaran V, Uppaluri R, Bui JD, Diamond MS, Koebel CM, Arthur C, White JM, et al.. A critical function for type I interferons in cancer immunoediting. Nat Immunol 2005; 6:722-9; PMID:15951814; https://doi.org/ 10.1038/ni1213 [DOI] [PubMed] [Google Scholar]
- 14.Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C, et al.. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med 2014; 20:1301-9; PMID:25344738; https://doi.org/ 10.1038/nm.3708 [DOI] [PubMed] [Google Scholar]
- 15.Shirey KA, Nhu QM, Yim KC, Roberts ZJ, Teijaro JR, Farber DL, Blanco JC, Vogel SN. The anti-tumor agent, 5,6-dimethylxanthenone-4-acetic acid (DMXAA), induces IFN-beta-mediated antiviral activity in vitro and in vivo. J Leukoc Biol 2011; 89:351-7; PMID:21084628; https://doi.org/ 10.1189/jlb.0410216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang CK, Aoshi T, Jounai N, Ito J, Ohata K, Kobiyama K, Dessailly BH, Kuroda E, Akira S, Mizuguchi K, et al.. The chemotherapeutic agent DMXAA as a unique IRF3-dependent type-2 vaccine adjuvant. PloS One 2013; 8:e60038; PMID:23555875; https://doi.org/ 10.1371/journal.pone.0060038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean M-C, Validire P, Trautmann A, Mami-Chouaib F, Donnadieu E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest 2012; 122:899-910; PMID:22293174; https://doi.org/ 10.1172/JCI45817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thoreau M, Penny HL, Tan K, Regnier F, Weiss JM, Lee B, Johannes L, Dransart E, Le Bon A, Abastado J-P, et al.. Vaccine-induced tumor regression requires a dynamic cooperation between T cells and myeloid cells at the tumor site. Oncotarget 2015; 6:27832-46; PMID:26337837; https://doi.org/ 10.18632/oncotarget.4940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joseph WR, Cao Z, Mountjoy KG, Marshall ES, Baguley BC, Ching LM. Stimulation of tumors to synthesize tumor necrosis factor-alpha in situ using 5,6-dimethylxanthenone-4-acetic acid: a novel approach to cancer therapy. Cancer Res 1999; 59:633-8; PMID:9973211 [PubMed] [Google Scholar]
- 20.Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest 2015; 125:3356-64; PMID:26168215; https://doi.org/ 10.1172/JCI80005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jassar AS, Suzuki E, Kapoor V, Sun J, Silverberg MB, Cheung L, Burdick MD, Strieter RM, Ching L-M, Kaiser LR, et al.. Activation of tumor-associated macrophages by the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid induces an effective CD8+ T-cell-mediated antitumor immune response in murine models of lung cancer and mesothelioma. Cancer Res 2005; 65:11752-61; PMID:16357188; https://doi.org/ 10.1158/0008-5472.CAN-05-1658 [DOI] [PubMed] [Google Scholar]
- 22.Sektioglu IM, Carretero R, Bender N, Bogdan C, Garbi N, Umansky V, Umansky L, Urban K, von Knebel-Döberitz M, Somasundaram V, et al.. Macrophage-derived nitric oxide initiates T-cell diapedesis and tumor rejection. Oncoimmunology 2016; 5:e1204506; PMID:27853636; https://doi.org/ 10.1080/2162402X.2016.1204506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Downey CM, Aghaei M, Schwendener RA, Jirik FR. DMXAA causes Tumor Site-Specific vascular disruption in Murine non-small cell lung cancer, and like the Endogenous Non-Canonical cyclic Dinucleotide STING Agonist, 2′3′-cGAMP, induces M2 Macrophage Repolarization. PLoS One [Internet] 2014. [cited 2016March15]; 9:e99988 Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4062468/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Long KB, Gladney WL, Tooker GM, Graham K, Fraietta JA, Beatty GL. IFNγ and CCL2 cooperate to redirect Tumor-Infiltrating Monocytes to degrade Fibrosis and enhance chemotherapy efficacy in Pancreatic Carcinoma. Cancer Discov 2016; 6:400-13; PMID:26896096; https://doi.org/ 10.1158/2159-8290.CD-15-1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim K, Hyun Y-M, Lambert-Emo K, Capece T, Bae S, Miller R, Topham DJ, Kim M. Neutrophil trails guide influenza-specific CD8+ T cells in the airways. Science 2015; 349:aaa4352; PMID:26339033; https://doi.org/ 10.1126/science.aaa4352 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.