

ABSTRACT
Myelofibrosis (MF) is a clonal neoplasia associated with chronic inflammation due to aberrant cytokine production. Mutations in Janus Kinase-2 (JAK2), calreticulin (CALR) and myeloproliferative leukemia protein (MPL) genes have been recently associated to MF and they all activate the JAK/STAT signaling pathway. Since this pathway is essential in shaping the immune response, we investigated the role of circulating immune subsets and cytokines in 38 patients (20 carrying JAK2(V617F),13 exon-9 CALR mutation and 5 triple negative). In comparison to healthy donors, patients presented a reduced amount of circulating dendritic cells (DCs) associated with a defective ability of monocytes in differentiating into DCs. In addition, we found a reduction in circulating T-helper (Th)1 and Th17 and hypo-functional innate lymphoid cells (ILC). Results analyzed according to the mutational status showed that patients carrying JAK2(V617F) mutation had a reduction in Th17, myeloid-DCs and effector Tregs as well as increased ILC1 and cytokine producing Tregs. The CALR mutated patients revealed high ILC3 levels, reduced Th1 and their monocytes had a reduced capacity to mature in vitro into fully committed DCs. Their Tregs were also less effective in inhibiting the proliferation of autologous effector T-cells due to an increased proliferative status induced by CALR mutation. Triple negative patients presented a reduced amount of total circulating CD3, effectors Tregs and Th1 with increased ILC1.
Overall, we have demonstrated that in MF different mutations lead to phenotypic and functional alterations in different immune subsets that may have a potential role in disease progression and susceptibility to infections.
KEYWORDS: Calreticulin, chronic inflammation, Immune dysregulation, JAK2(V617F), Myelofibrosis
Background
Myelofibrosis (MF) refers to the Philadelphia chromosome-negative myeloproliferative neoplasms (MPNs) originating in the multipotent haematopoietic stem cells. It is clinically characterized by progressive anemia, splenomegaly, debilitating constitutional symptoms and by an increased risk to evolve in acute leukemia.1 MF can develop de novo as primary MF (PMF) or secondary either from Polycythemia Vera (PPV-MF) or Essential Thrombocythaemia (PET-MF). Approximately 60% of MF patients carry a mutation in the Janus Kinase 2 (JAK2)2 gene, and an additional 10% in the myeloproliferative leukemia protein gene (MPL). Mutations in Calreticulin gene (CALR) have been reported in about 80% of JAK2 and MPL unmutated patients.3 Around 10% of patients have non-mutated JAK2, MPL and CALR genes (“triple negative”).
Of note, regardless the driver mutations, the JAK-STAT signaling pathway is hyper-activated in all the MPNs.4 Chronic inflammation, as result of aberrant cytokines production by mutated and unmutated cells, is considered the MF hallmark. In this scenario, infectious complications are the leading cause of morbidity and mortality constituting more than 10% of all patient deaths.5,6
To understand whether the atypical infectious events are caused by deficits in the innate or adaptive immune response, a comprehensive analysis of key immune cells is required.
To date, it is well established that in PMF, the monocytes composition is different with a reduction in the classical (CD14brightCD16−) compartment.7 Monocytes can differentiate, under inflammatory conditions, in dendritic cells (DCs); however, no data have been published so far about the ability of MF monocytes to differentiate into DCs. DCs are a heterogeneous group of professional antigen-presenting cells (APCs) including plasmacytoid (pDC) and myeloid (mDCs) DCs.8 Thus far, no data have been reported on the frequency of circulating DC subsets in MF.
A recent report studied the T helper (Th)1, Th2 and Th17 compartments in MPNs patients under treatment. Of note, no differences between healthy donors and patients were found in Th cells polarization at baseline level.9 Thymus derived regulatory T cells (Tregs) frequency has been already studied in MPNs, however conflicting results have been published.10-12
MPN, have reduced natural killer cell (NKs) compartment with impaired function.13,14 NKs are part of the recently described family of innate lymphoid cells (ILCs), which play a role in autoimmunity, inflammation15 and tumor immunosurveillance.16 Beside conventional NKs, 3 distinct ILCs subsets have been described based on their transcriptional regulation and cytokine profiles mirroring those of Th cells.17 We and others recently showed that acute myeloid leukemia patients present an impaired ILC compartment18,19 but no data are available in MF.
Based on this background and considering the essential role of the JAK/STAT pathways in shaping the immune response,20 we functionally evaluated key immune-cell subsets with the aim to investigate their putative role in immunosurveillance. We found that MF patients are characterized by a state of mutation-dependent immune alterations with key cellular components of the innate and adaptive immunity showing defective number and function.
Results
Patients characteristics
38 MF patients (20 JAK2(V617) mutated, 13 CALR mutated and 5 triple negative) were included in the study. Baseline features of the entire cohort are detailed in Table 1. Leukocytosis (leukocytes ≥ 25 × 109/L) was observed in 5 patients, while 7 patients had a low (≤ 4 × 109/L) leukocyte count; lymphopenia (lymphocytes ≤ 109/L) and monocytosis (monocytes ≥ 109/L) were present in 17 and 10 patients, respectively. We studied 18 patients at the diagnosis while 20 patients received previous treatment of MF (Hydroxyurea/Ruxolitinib), as detailed in Table 1. In all cases, therapies had been discontinued for at least 2 months before sample collection. Only 2 patients presented an autoimmune clinical history.
Table 1.
Clinical and laboratory features of MF patients according to mutational status
| Total MF (38 cases) | JAK2(V617F)+ (20 cases) | CALR+ (13 cases) | Triple negative (5 cases) | P 1 value | P 2 value | P 3 value | |
|---|---|---|---|---|---|---|---|
| Median age, years (range) | 71 (40–87) | 67,5 (40–82) | 73 (67–84) | 78 (73–87) | 0.02 | 0.01 | 0.6 |
| Males, no. (%) | 18 (47.4%) | 9 (45%) | 7 (54%) | 2 (40%) | 0.72 | 0.84 | 0.7 |
| Median Hemoglobin, g/dl; median (range) | 11 (7.5–15.1) | 12.6 (9.2–15.1) | 9.3 (7.7–14) | 10 (7.5–12.3) | 0.001 | 0.01 | 0.7 |
| Median Leukocytes, x 109/l; median (range) | 7.4 (2.3–48.3) | 9.3 (2.5–26.4) | 6.4 (2.3–48.3) | 7.4 (7.1–27.0) | 0.6 | 0.7 | 0.8 |
| Median Platelets, x 109/l; median (range) | 216 (38–618) | 248 (41–549) | 175 (90–419) | 259 (38–618) | 0.26 | 0.4 | 0.3 |
| Median Lymphocyte, x 109/l; median (range) | 1.5 (0.4–10.7) | 1.45 (0.5–2.7) | 1.6 (0.5–10.7) | 1.9 (0.4–5.0) | 0.16 | 0.5 | 0.24 |
| Median Monocyte, x 109/l; median (range) | 0.6 (0.1–7.6) | 0.58 (0.19–1.8) | 0.6 (0.1–7.6) | 0.5 (0.2–5.3) | 0.23 | 0.34 | 0.37 |
| BM fibrosis, no. of patients (%) | |||||||
| Grade 1 | 13(34%) | 11(55%) | 0 (0%) | 2 (40%) | 0.002 | 0.06 | 0.78 |
| Grade 2 | 18 (47%) | 6 (30%) | 9 (69%) | 3 (60%) | 0.03 | 0.05 | 0.78 |
| Grade 3 | 7 (21%) | 3(15%) | 4 (31%) | 0 (0%) | 0.39 | 0.78 | 0.07 |
| IPSS, Number of patients | |||||||
| Low | 3 (9%) | 3 (15%) | 0 (0%) | 0 (0%) | 0.53 | 0.68 | 1 |
| Intermediate-1 | 20 (53%) | 11 (55%) | 5 (38%) | 4 (80%) | 0.48 | 0.6 | 0.78 |
| Intermediate-2 | 4 (12%) | 3 (15%) | 1 (8%) | 0 (0%) | 1 | 0.78 | 0.85 |
| High | 11 (29%) | 3 (15%) | 7 (54%) | 1(20%) | 0.025 | 0.28 | 0.06 |
| Unfavorable Karyotype, no of patients (%) | 4 (10%) | 3 (15%) | 0 (0%) | 1 (20%) | 0.26 | 0.36 | 0.45 |
| Previous treatment, no of patients (%) | |||||||
| Hydroxyurea | 15 (39%) | 9 (45%) | 2 (20%) | 4 (80%) | 0.24 | 0.06 | 0.05 |
| Ruxolitinib | 4 (12%) | 2 (10%) | 2 (15%) | 0 (0%) | 1 | 0.79 | 0.64 |
| WHO Diagnosis | |||||||
| PMF | 26 (68%) | 14 (70%) | 8 (62%) | 4 (80%) | 0.71 | 0.64 | 0.72 |
| PPV-MF | 4 (12%) | 4 (20%) | 0 (0%) | 0 (0%) | 0.14 | 0.58 | 1 |
| PET-MF | 8 (21%) | 2 (10%) | 5 (38%) | 1 (20%) | 0.08 | 0.26 | 0.06 |
PMF: Primary Myelofibrosis; PPV-MF: Post Polycythemia Vera-Myelofibrosis; PET-MF: Post Essential Thrombocythemia-Myelofibrosis. The presence of 0, 1, 2 or 3 and >4 adverse factors defines low, intermediate-1, intermediate-2 and high-risk disease. IPSS, International Prognostic Scoring System; unfavorable karyotype (presence of one or 2 abnormalities including þ8, 7/7q-, i(17q), inv(3), 5/5q-, 12p- or 11q23 rearrangement). P1: P value between JAK2(V617F) and CALR mutated patients. P2: P value between JAK2(V617F) and triple negative mutated patients. P3: P value between CALR and triple negative mutated patients.
Dysregulated plasma levels of cytokines involved in differentiation/function of immune cells in MF patients
We first evaluated the plasma levels of cytokines involved in the differentiation and function of different immune cell types. In agreement with previous reports,21,22 we found reduced plasma levels of IL-4, -5 and IFN-γ with concomitant increased levels of IL-1β, -6, -10, -17, and TNF-α as compared with HD (Table S1). No correlation between allele burden and cytokine plasma levels was observed, with the notable exception of TNF-α which highly correlated (R = 0.63; p < 0.008) with JAK2(V617F) allele burden. Irrespective of mutational status, IL-12 and -13 plasma levels were negatively correlated with the IPSS score values (R = 0.47; p < 0.04; R = 0.49; p < 0.04, respectively). Conversely, we found a positive correlation between circulating IL-6 levels and splenomegaly/fibrosis (R = 0.46; p = 0.018 and R = 0.49; p = 0.003, respectively) (data not shown).
Reduced circulating mDCs in JAK2(V617F) mutated patients
Afterwards, we evaluated the number of circulating mDCs and pDCs in MF patients and controls (Fig. 1A). As shown in Fig. 1B, circulating mDCs were significantly reduced in MF compared with HD (7.8 ± 4.3 vs 12.7 ± 4 cells/μL, p ≤ 0.01). Interestingly, according to the mutational status, this reduction was significant only in JAK2(V617F) (12.7 ± 4 vs 6.2 ± 2.7 cells/μL, p ≤ 0.001), but not in CALR mutated patients. A significant reduction was also observed in the number of circulating pDCs (p ≤ 0.05; Fig. 1C).
Figure 1.
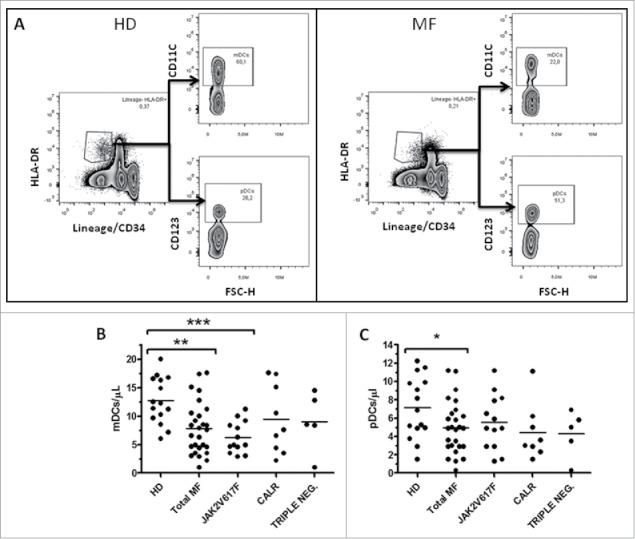
Reduced circulating mDCs in JAK2(V617F) mutated patients. A) Representative example of the gating strategy used to determine, in HD (left panels) and MF patients (right panels), the percentages used to calculate the circulating level of mDCs (identified as Lin−HLA-DR+CD11c+ cells) and pDCs (identified as Lin−HLA-DR+CD123+ cells). Circulating number of mDCs (B) and pDCs (C) in HD (n = 17), total MF (n = 27), JAK2(V617F) mutated (n = 13), CALR mutated (n = 9) and triple negative (n = 5) patients are shown; cell concentrations were calculated as follows: (percentage of positive cells) x (white blood cell count)/100. For all graphs one symbol represents one individual, and the height of the bar represents the mean (*p ≤ 0.05, **p ≤ 0.01, (***p ≤ 0.001).
Impaired DC differentiation capacity of monocytes from MF patients
Monocytes can differentiate into DCs in vivo mainly in infected or inflamed tissues, leading to the concept that monocytes are a precursor of inflammatory DCs. We thus studied the capacity of freshly isolated monocytes to differentiate into DCs in vitro. After 5 day culture, the phenotype of immature mo-DCs was evaluated by flow cytometry. As shown in Fig. 2A, monocytes from CALR and triple negative but not those from JAK2(V617F) mutated patients were not able to differentiate into immature DCs, as indicated by the persistence of CD14 expression. In addition, irrespective of mutational status, immature mo-DCs failed to up-regulate CD1a (85 ± 4.6% vs 73.5.7 ± 19.6%, p ≤ 0.01) and CD80 expression (70.6 ± 12.9% vs 38.8 ± 16.2%, p ≤ 0.01) as compared with the normal counterparts. Of note, although monocytes from triple negative patients were not able to fully differentiate in DCs they did not present defect in the upregulation of the CD1a (Fig. 2A).
Figure 2.
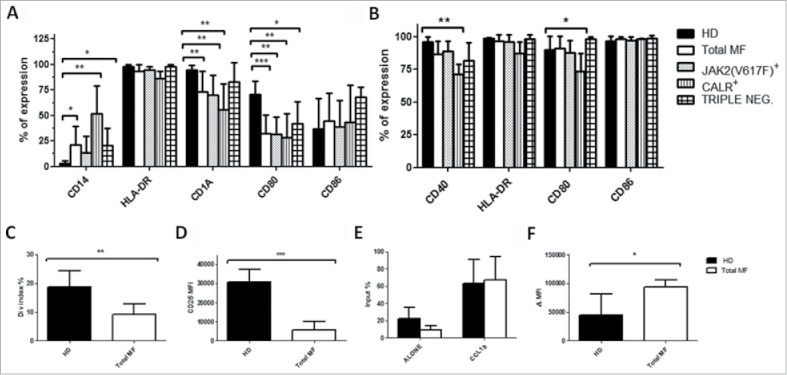
Impaired DCs differentiation capacity of monocytes from MF patients. Immature (A) and mature (B) mo-DCs phenotype from HD (n = 10) total MF (n = 16), JAK2(V617F) mutated (n = 7), CALR mutated (n = 5) and triple negative (n = 4) patients. The expression of HLA-DR, CD14, CD1a, CD40, CD80 and CD86 was evaluated by flow cytometry. Histograms represent the mean percentage of expression ± SD; C) ability of mo-DCs from HD (n = 8), total MF (n = 8) to prime allogeneic T-cell responses in vitro. mo-DCs were cultured with allogeneic Tresp (mo-DCs/Tresp ratio 1:10) labeled with CFSE. The assays were performed over a period of 5 days and T cell proliferation was evaluated by division index. Histograms represent the mean ± SD of the division index expressed as percentages; D) mo-DCs were cultured with allogeneic Tresp (mo-DCs/Tresp ratio 1:10) labeled with CFSE. The assays were performed over a period of 5 days and CD25 expression was evaluated by flow cytometry. Histograms represent the mean MFI ± SD of CD25 from HD (n = 8) and total MF (n = 8) to prime allogeneic T-cell responses in vitro; E) evaluation of spontaneous and toward CCL19 (400 ng/mL) mature mo-DCs migratory capacity in HD (n = 6) and MF patients (n = 8). 1 × 105 cells were seeded in a transwell chamber (diameter 6.5 mm, pore size 8 µm) for 4 hours. The amount of migrated cells is expressed as a percentage of the input: (number of migrated cells in the lower compartment/loaded cells in the upper compartment) X 100. Histograms represent the mean ± SD of the input; F) Immature mo-DCs dextran uptake in HD (n = 6) and MF patients (n = 8). Cells were incubated for 30 min at 37°C or on ice (used as a background control). After washing, fluorescence was analyzed by flow cytometry. Uptake of FITC-dextran was expressed as delta (Δ) mean fluorescence intensity (MFI): MFI (uptake at 37°C) – MFI (uptake on ice). Histograms represent the mean ± SD of dextran uptake (* p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001).
We then assessed the capacity of immature mo-DCs to mature in the presence of an inflammatory cocktail. Surprisingly, immature mo-DCs were able to respond and mature as the healthy counterpart (Fig. 2B). However, mature mo-DCs from CALR mutated patients continued to be defective in CD40 expression (63.4 ± 7.6% vs 93.7 ± 3.7%, p ≤ 0.01) and CD80 (73.7 ± 13% vs 98.8 ± 0.4%, p ≤ 0.01) as compared with the normal counterparts.
To investigate whether the impaired DCs phenotype was associated with altered function, we first assessed the ability of immature mo-DCs to prime allogeneic T-cell responses in vitro. Regardless of mutational status, patients’ derived mo-DCs were unable to stimulate T cell proliferation to the same extent as the HD counterpart (Fig. 2C). This data are supported by the defective CD25 upregulation in T cells (Fig. 2D). Migration toward the lymphonode and the capacity to capture antigens are essential for DCs function. For this reason, we performed migration and endocytosis assays. No significant differences were found in the migratory capacity of MF-derived mature mo-DCs, both spontaneous or in the presence of CCL19, a chemokine essential for lymphonode homing (Fig. 2E). However, MF-derived immature mo-DCs were more efficient in capturing the antigen than the control counterparts (Fig. 2F).
These results show an impaired MF-monocyte capacity to differentiate in vitro into mo-DCs associated with a defective priming ability.
Reduced Th1 compartment in CALR mutated patients
Th cells play critical roles in the development and progression of infections, autoimmune diseases and tumors. Here, we first analyzed the percentages of CD3+ cells and no significant differences between MF and HD were found (Fig. 3A). Nevertheless, triple negatve patients showed a significant reduction in CD3+ cells that was equally distributed between CD4 and CD8 (Fig. 3A). We then evaluated the Th1 and Th2 balance,23 showing a decrease in the Th1 (25.6 ± 8% vs 12.9 ± 9%, p ≤ 0.01) but not in the Th2 compartment (Fig. 3B and C). Interestingly, only triple negative and CALRmutated patients presented a significant reduction in Th1 (Fig. 3B).
Figure 3.
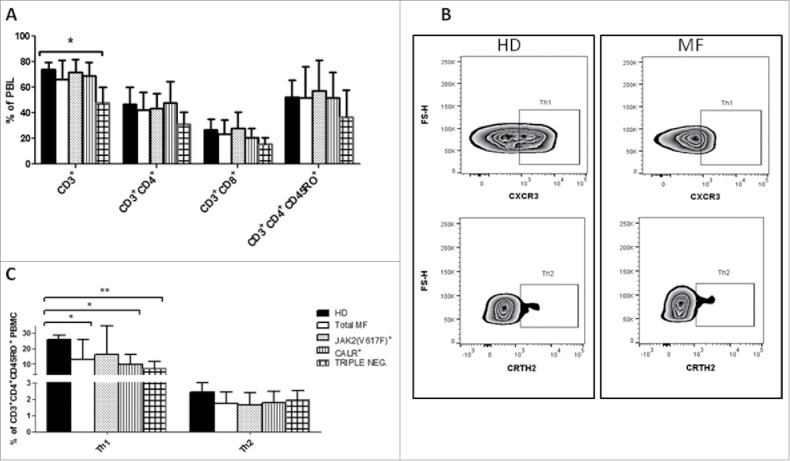
Reduced Th-1 compartment in CALR mutated patients A) Percentages of CD3+, CD3+CD4+, CD3+CD8+, CD3+CD4+ CD45RO+ cells on PBMC from HD (n = 14), total MF (n = 16), JAK2(V617F) mutated (n = 7), CALR mutated (n = 5) and triple negative (n = 4) patients evaluated by flow cytometry; B) Representative example of the gating strategy used to determine by flow cytometry, in HD (left panels) and MF patients (right panels), the percentages used to identify Th-1 and Th-2 (identified as CD3+CD4+CD45RO+CXCR3+CRTH2−CCR6− and CD3+CD4+CD45RO+CXCR3−CRTH2+ cells,respectively); C) Percentages of Th1 and Th2 cells on the CD3+CD4+CD45RO+ population from HD (n = 14), total MF (n = 16), JAK2(V617F) mutated (n = 7) and CALR mutated (n = 5) and triple negative (n = 4) patients. Histograms represent the mean percentages ± SD (* p ≤ 0.05, ** p ≤ 0.01).
Reduced Th17 compartment in JAK2(V617F) mutated patients
Th17 can promote anticancer immunity; however, these cells exhibit also tumor-promoting properties. This dichotomy in cancer may be related to the versatile nature of these cells.24
In MF the mean number of circulating Th17 (identified as CD3+CD4+CCR6+CD161+ cells) was reduced as compared with HD (24.73 ± 17.8 vs 38 ± 16.8 cells/μL, p ≤ 0.01) (Fig. 4A and B). However, when data were analyzed according to the mutational status, only JAK2(V617F) mutated patients showed a statistically significant reduction (18.16 ± 9.3 vs 38 ± 16.8 cells/μL, p < 0.01) (Fig. 4B). Subsequently, we focused our attention on the intermediate Th populations recently described based on the expression of chemokine receptor and their capacity to secrete IL-17/IFN-γ and IL17/IL22, namely Th17/Th1 and Th17/Th22 respectively.25,26 We found that MF patients showed a reduced percentage of circulating Th17/Th22 (p ≤ 0.001) and Th17/Th1 (p ≤ 0.001) cells (Fig. 4C). In particular the defect in Th17 was more prominent in the JAK2(V617F) mutated patients as the triple negative one showed a significant higher percentage of Th17/22 (Fig. 4C).
Figure 4.
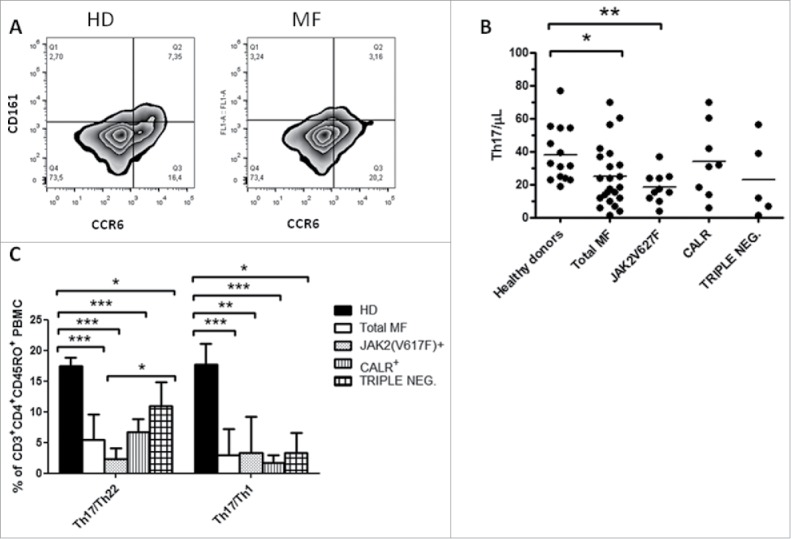
JAK2(V617F) mutated patients show a reduced Th17 compartment. A) Representative example of the gating strategy used to determine by flow cytometry, in HD (left panels) and MF patients (right panels), the percentages used to calculate the circulating level of Th17 (identified as CD3+CD4+CCR6+CD161+ cells); B) PB circulating number of Th17 in HD (n = 19), total MF (n = 23), JAK2(V617F) mutated (n = 10) CALR mutated (n = 8) and triple negative (n = 5) patients; cell concentrations were calculated as follows: (percentage of positive cells) x (Lymphocyte count)/100. Each symbol represents one individual and the height of the bar represents the mean; C) Percentages of Th17/Th1 and Th17/Th22 (identified as CD3+CD4+CD45RO+CXCR3+CRTH2−CCR6+ and CD3+CD4+CD45RO+CXCR3−CRTH2−CCR6+ cells, respectively) in HD (n = 14), total MF (n = 16), JAK2(V617F) mutated (n = 7) CALR mutated (n = 5) and triple negative (n = 4) patients. Histograms represent mean percentage expression on the CD3+CD4+CD45RO+ population ± SD (* p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
These results suggest that in MF the reduced number of circulating Th17 is associated with a defective plasticity of this compartment.
Tregs heterogeneity in periphery differs between MF patients
Circulating Treg numbers tended to be reduced in MF patients, however this reduction was significant only in the triple negative group. (Fig. 5A). A deep investigation considering the 3 Treg sub-populations described by Miyara et al. 27 on PBMCs showed a reduction of the effector Tregs compartment identified as CD3+CD4+CD45RA−CD25brightCD127low (Population II, p ≤ 0.05) (Fig. 5B and C). The analysis according to the mutational status revealed that triple negative and JAK2(V617F) but not CALR mutated patients, showed this reduction (Fig. 5C). Overall, Tregs heterogeneity was different between MF patients as triple negative were enriched in Population I and JAK2(V617F) in Population III; conversely CALR mutated patients Tregs resembled the healthy donors (Fig. 5C). Triple negative patients showed an imbalance between naive and memory cells and the ratio was significantly increased (p ≤ 0.05; Fig. S1).
Figure 5.
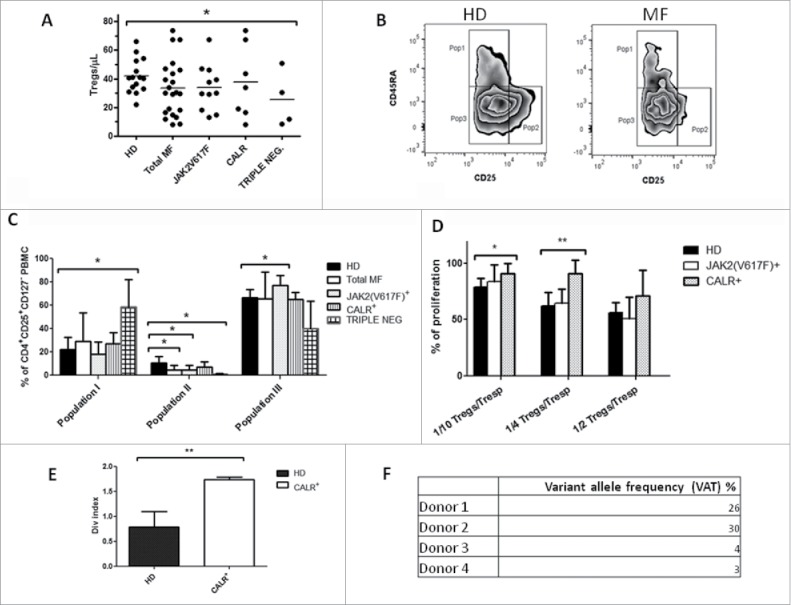
JAK2(V617F) mutated patients present a different Tregs heterogeneity in periphery. A) PB circulating number of Tregs (identified as CD3+CD4+CD25+CD127low cells) in HD (n = 17), total MF (n = 22), JAK2(V617F) mutated (n = 11), CALR mutated (n = 7) and triple negative (n = 4) patients; cell concentrations were calculated as follows: (percentage of positive cells) x (lymphocyte count)/100. Each symbol represents one individual and the height of the bar represents the mean; B) Representative example of the gating strategy used to determine by flow cytometry, in HD (left panels) and MF patients (right panels), the percentages of CD3+CD4+CD45RA+CD25+CD127low (population I), CD3+CD4+CD45RA−CD25brightCD127low (population II) and CD3+CD4+CD45RA−CD25+CD127− (population III) cells; C) Percentages of Population I, II and III in HD (n = 14), total MF (n = 16), JAK2(V617F) mutated (n = 7) CALR mutated (n = 5) and triple negative (n = 4) patients.. Histograms represent mean percentage expression on the CD3+CD4+CD25+CD127− population ± SD; D) Co-colture (5 days) of autologous Treg from HD (n = 9), JAK2(V617F) mutated (n = 4) and CALR mutated (n = 4) patients with autologous CD4+CD25− (Tresp) stimulated with anti-CD3 and anti-CD28 (5µg/mL) and labeled with CFSE. Percentage of proliferation was calculated as: (ratio between the” upper generation proliferation index” of Tresp cultured in the presence of increasing Treg ratios and the” upper generation proliferation index” of CTR culture, where no Treg were added)x100; histograms represent mean ± SD; E) Proliferation of CD4+CD25− from CALR mutated patients (n = 4) and HD (n = 4) stimulated with anti-CD3 and anti-CD28. Proliferation is calculated using the division index (average number of cell divisions that a cell in the original population has undergone); histograms represent mean ± SD; F) CALR exon 9 sequencing performed by Next Generation Sequencing to evaluate the variant allele frequency (VAT) expressed as percentages. Data indicate the percentages of mutated reads analyzed. (* p ≤ 0.05, **p ≤ 0.01).
To further understand the Treg role in MF we tested their suppressive ability in vitro and no significant differences were observed between patients and controls (data not shown). However, we found that Tregs from CALR, but not those from JAK2(V617F) mutated patients, do not show inhibition of T cell proliferation as effectively as the normal counterparts (Fig. 5D). In that regard, the effect of CALR mutation in T cell activation has been described.28 Specifically, Tregs from CALR deficient mice are functional but effectors T cells are less sensitive to suppression by their ability to produce pro inflammatory cytokines like IL-2. In line with this hypothesis, we compared the proliferative ability of CD4+CD25− T cells from CALR+ patients and HD. As predicted, CD4+CD25− T cells from patients showed increased proliferation as compared with HD counterparts (p ≤ 0.01) (Fig. 5E). Interestingly, the CD4+CD25− T cell population used in the assay carried the exon 9 CALR mutation (Fig. 5F).
In conclusion, we show that MF patients have a different Tregs heterogenity; moreover, the presence of CALR mutation in the effector T cells confers them a status of hyper-activation.
Selected subsets of ILC with reduced functional capacity are expanded in MF patients
ILC function is tightly regulated by cytokines, and uncontrolled activation and proliferation can contribute to severe inflammation.15,17 Due to the aberrant cytokine compartment in MF, we evaluated ILC frequencies, phenotype and function.
The total ILC frequency was similar between MF patients and healthy controls (data not shown). We therefore analyzed the relative frequency of the different ILC subsets (Fig. 6A). As shown in Fig. 6B, according to mutational status, ILC1 cells were significantly increased in triple negative and JAK2(V617F) patients (p ≤ 0.05) while CALR+ patients showed an increased ILC3 NCR+ compartment (p < 0.05). Interestingly, total ILC3 percentages were significantly higher in patients with intermediate-2/high IPSS score compared with those with low/intermediate-1 IPSS score (p < 0.05). Moreover, we found a positive correlation between the percentages of ILC3 NCR+ and the circulating levels of IL-6 (R = 0.51; p < 0.04)(data not shown). Finally, we tested the functionality of ILCs by evaluating their cytokine producing capacity. Following short-term ex vivo activation, ILCs from MF patients showed dramatically impaired production of IFN-γ, IL-4, -5 and -13 (Fig. 6C). In addition, ILC from triple negative patients were the only one defecting in TNF-α production, while preserving IL5 and IL13 production.
Figure 6.
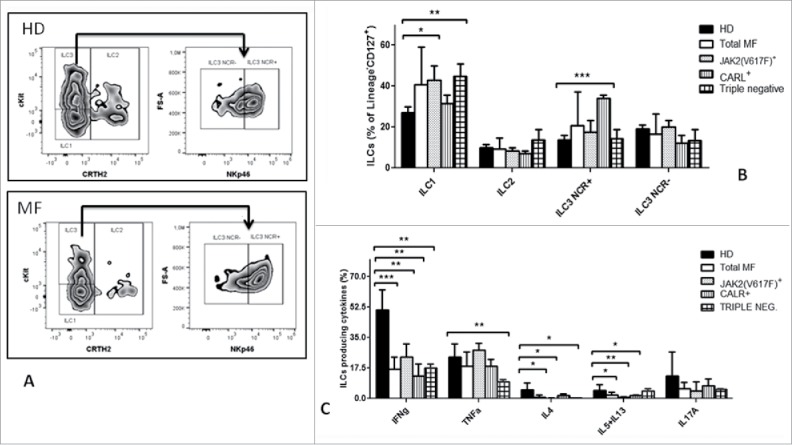
Selected subsets of ILCs with reduced functional capacity are expanded in MF patients. A) Representative example of the gating strategy used to determine by flow cytometry, in HD (upper panels) and MF patients (lower panels), the percentage of: ILC1 identified as Lin−CD127+CRTH2−cKit−CD56− cells; ILC2 identified as Lin−CD127+ CRTH2+cKit+/− cells; ILC3 identified as Lin−CD127+CRTH2− cKit+NKp46− cells that were further characterized by the expression of the natural cytotoxicity receptor(NCR). B) Histograms represent the percentages ± SD of selected ILC subpopulation in HD (n = 21), total MF (n = 23), JAK2(V617F) mutated (n = 12) and CALR mutated (n = 6) and triple negative (n = 5) patients. C) PBMCs were stimulated ex vivo for 3 hours, then an intracellular staining was performed. Histograms represent the mean ± SD of the percentage of ILCs (Lin−CD127+cells) producing IFN-γ, TNF-α, IL4, IL5 plus IL13, IL17A in HD (n = 21), total MF patients (n = 21), JAK2(V617F)+ (n = 12) CALR+ (n = 6) and triple negative patients (n = 4). (* p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
In conclusion, we demonstrated that in MF the ILC compartment is functionally dysregulated.
Discussion
MF is considered as “A Human Inflammation Model”29 where the uncontrolled myeloproliferation and cytokine secretion creates a pro-inflammatory milieu influencing the immune system. Here we have demonstrated that several subsets of the adaptive and innate immune response show quantitative and/or qualitative abnormalities. Our data demonstrate how circulating DCs, Th17, Th1, Tregs, ILCs and cytokine plasma levels are dysregulated in MF. Noteworthy, the absence or the presence of different mutations can affect this phenotype.
Specifically, Th17, mDCs and Treg Population II reduction, associated with an increase in Treg Population III and ILC1, was principally observed in the JAK2(V617F) mutated group. Patients carrying CALR mutation presented a dysregulated IFN-γ axis. In particular, reduced circulating levels of IFN-γ accompanied by Th1 reduction, hypofunctional ILC1 and mo-DCs. Lastly, CD3+ cells were reduced in triple negative patients; this reduction was equally distributed between CD4 and CD8. The analysis on CD4 revealed that Th1 and effector Tregs were the subpopulations significantly involved. ILC1 were increased but hypofunctional with defect in IFN-γ and TNF-α production. Notable, no association was found between allele burden and the number/phenotype/function of the studied cells. Furthermore, PET-MF and PV-MF did not show an immune pattern significantly different from PMF.
Along with a reduced amount of circulating myeloid and plasmacytoid DCs, we found an impaired ability of MF-derived monocytes to differentiate in vitro toward DCs when cultured in the presence of IL-4 and GM-CSF. On monocytes, IL-4 receptor signals through JAK1/330 while GM-CSF receptor through JAK2 and alternatively by IKK complex.31 In MF, JAK2 constitutive activation confers to monocytes an high sensitivity to GM-CSF (commonly used in vitro for macrophages differentiation32) that results in a reduced capacity toward DCs differentiation and an increased macrophage-like phenotype. In line with these observations, 5 day cultured monocytes failed to fully down-regulate CD14 and presented a reduced expression of CD1a and CD80 compared with the normal counterpart. As a consequence, MF mo-DCs showed a reduced capacity to stimulate T cell proliferation and an enhanced endocytosis ability. In addition, mature mo-DCs from CALR mutated patients continued to be defective in CD80 and CD40 molecules providing an incomplete co-stimulatory signal essential for T cells activation and differentiation.
Our results on DCs are in line with previous reports showing an increased level of myeloid derived suppressor cells in MPN33 confirming that the increased release of immature myeloid cells led to the reduction of differentiated myeloid subsets as DCs. All together, these findings can explain, at least in part, the high infection rates seen in MF patients that are further exacerbated by ruxolitinib,34 a JAK1/2 inhibitor affecting DCs differentiation and function in vitro.35
DCs have the unique capacity to direct T cell differentiation through the strength of TCR interaction and cytokines present in the microenvironment. Although no differences in CD3 were detected in the total population, the analysis executed according to the mutational status showed that triple negative patients were defective in CD3+ cells. Both CD4+ and CD8+ cells were influenced; a more detailed analysis on CD4 cells revealed a reduction in Th1 associated with a different Treg heterogeneity; in particular, they had a reduced percentages of Population II with an increase in Population I. The different Treg heterogeneity is due to an imbalance between naive and memory cells. Memory Tregs, especially Population II, are highly suppressive in vivo; however, they die by apoptosis after activation. In context of high inflammation, this might be the reason why the ratio between naive and memory cells is inverted. However these data need to be confirmed in a larger cohort of patients to better clarify if the reduction in CD3+ is mainly due to CD4 rather than CD8 cells and if the dysregulated Treg compartment may generate autoimmune phenomena.
The analysis of the different CD4 cell subsets in the other cohorts revealed a Th1 reduction in CALR mutated patients. IL-12 is a key factor for Th1 differentiation; consistently, mature mo-DCs from CALR mutated patients show a reduced expression of CD40, a marker linked with DCs IL-12 production ability and Th1 differentiation in vivo.36,37
Of note, Tregs from CALR mutated patients showed lower inhibition of autologous effector T cells proliferation than the normal counterpart. Specifically, responder T cells, used in the assay, carried exon 9 CALR mutation conferring them an higher proliferative capacity in vitro. To our knowledge, a mutation in the T cell compartment in MF has been reported in MPN patients carrying the JAK2(V617F) mutation, 38-40 with no data available on CALR mutated patients. The enhanced proliferative status do not correlate with the presence of autoimmune phenomena in vivo as none of the studied patients showed autoimmune clinical history, on the other hand they all presented an high IPSS risk indicating that the mutation in T cell may influence the prognosis and disease progression. To better understand the role and the incidence of CALR mutation in lymphoid cells specific studies need to be designed.
The JAK2(V617F) mutated group is characterized by Th17 reduction with an impaired context-dependent plasticity since the percentage of Th17/Th1 and Th17/22 populations were reduced compared with controls. In addition, effector Tregs were reduced as well. As explained for the triple negative patients, we can hypothesize that this deficiency may be the result of increased apoptosis or conversion in the context of chronic inflammation. In fact, we observed a negative correlation between effectors Tregs and IL-12 plasma levels, a cytokine increasing the outgrowth of non-Tregs in vivo.41
ILC rapidly respond to cytokines and microbial signals providing multiple pro-inflammatory and immuno-regulatory cytokines. Taking into account the aberrant cytokine production in MF we investigated the frequency of different ILC subsets. ILC1 were increased in triple negative and JAK2(V617F) mutated patients while ILC3 NCR+ in the CALR mutated group. Nevertheless, irrespective of the mutational status, ILCs were hypofunctional. The increase of ILC1 can be explained considering the high IL-12 detected in circulation. This cytokine is essential for ILC1 differentiation and ILC2 conversion into ILC1.42 Equally, the ILC3 NCR+ increase could be linked to the high circulating level of IL-1β and IL-23. Consistently, an ILC1 increase concomitantly with reduced functionality has been recently shown by us in patients with acute myeloid leukemia.18 Noteworthy, total ILC3 percentages were significantly higher in patients with intermediate-2/high IPSS score indicating a possible role in MF progression. However, because of the current limited understanding in ILC biology, additional work needs to be performed to explain how the chronic inflammation status and the cytokine milieu influence this compartment.
Although this study is based on a limited number of patients, this limitation can be easily addressed in multicentric studies aiming to monitor MF patients before and under treatment. Our data highlight the importance of investigating in larger cohorts of patients the role of the immune system in MF and other MPNs according to the mutational status.
Despite this limitation, this study gives an initial proof of concept that the immune landscape of MF vary among patients and that selected immune defects are principally associated with the presence/absence of the JAK2(V617F) or CALR mutation. Overall, these abnormalities might contribute to the development of an immune defecting status with the potential to promote immune evasion, cancer progression and increased susceptibility to infections. In addition, a better understanding of the immune biology in the setting of MF would be important for designing novel therapies for MF.
Methods
Patients samples and cell isolation
EDTA-anticoagulated peripheral blood (PB) was obtained from 30 healthy age-matched volunteers and 38 patients with MF. Patients were at diagnosis (18 cases) or untreated for at least 2 months. The diagnosis of MF was made according to the WHO 2008 criteria (Table 1). No patients were previously treated with Interferon-α. This study was approved by the medical Ethical Committee of the University Hospital of Bologna and was conducted in accordance with the Declaration of Helsinki. Patients/controls provided written informed consent for the study. PB mononuclear cells (PBMCs) were separated by Lympholyte 1.077 g/cm3 gradient (Cedarlane; CL5020) stratification. Subsequently, highly purified CD4+CD25+, CD4+CD25− and CD14+ cells were isolated using specific immunomagnetic cell isolation Kits (Miltenyi Biotech, 130–050–201) according to manufacturer's instructions.
Cell phenotype
The circulating immune cells were evaluated in PB from patients and controls by multiparametric flow cytometry. Th17, Tregs, mDCs and pDCs, were identified as listed in supplementary table S2. A minimum of 1 × 105 cells were acquired by flow cytometer BD Accuri C6 or FACSCanto (Becton Dickinson). Analysis was performed excluding cellular debris in a SSC/FSC dot plot. The percentage of positive cells was calculated subtracting the value of the appropriate isotype controls. Cell concentrations were calculated as follows: (percentage of positive cells) x (Lymphocyte count)/100 (Th17 and Tregs) or White Blood Cell counts/100 (mDCs and pDCs). PBMCs were used to assess the percentage of Th1, Th2, Th17/22, Th17/Th1, the 3 subpopulations of ILCs and Tregs. Gating strategy and antibodies used are listed in Table S2 (see Additional tables). The phenotype of circulating monocytes and monocyte-derived immature and mature DCs was also characterized (Table S1; see Additional tables). ILCs' cytokine production has been evaluated after PMA/Ionomycin stimulation by flow cytometry as described in Table S1 (see Additional tables).
Generation of monocyte-derived DCs
Monocyte-derived DCs (mo-DCs) were generated by a 5-day culture of CD14+ cells in complete RPMI 1640 medium (Gibco-Invitrogen, BE12–167F) supplemented with 50 ng/mL Granulocyte Macrophage-Colony Stimulating Factor (GM-CSF) and 800 U/mL IL-4 (all from Endogen, 14–8339–62; 14–8049–80), at 37°C in 5% CO2, as described previously by us.43 For maturation, day 5 mo-DCs were cultured for 48 hours in the presence of a pro-inflammatory cocktail: GM-CSF (50 ng/mL), IL4 (800 U/mL), IL6 (10 ng/mL; RIL6I), IL1β (10 ng/mL; RIL1BI), TNF-α (10 ng/mL; BMS301) and Prostaglandin (PGE)-2 (1 μg/mL; 14–8129–62) (all from Endogen).
Suppression assay
To assess the inhibitory capacity of freshly-isolated Tregs, we set-up a Mixed Leukocyte Reaction (MLR). Briefly, 105 CD4+CD25− (Tresp) were labeled with Carboxyfluorescein succinimidyl ester (CFSE; Invitrogen; C34554), 5μM, according to the manufacturer's instructions. Tresp were co-cultured, for 5 days, alone or with autologous and irradiated (3000 cGy) Tregs at different Tresp/Treg ratios. MLR was set-up in 96-well plates pre-coated with anti-CD3 monoclonal antibody (mAb; clone UCHT1; BioLegend,; 317301) in presence of soluble anti-CD28 mAb (clone CD28; BioLegend; 302901). CFSE dilution has been exploited to assess cell division by flow cytometry (BD FACSCanto™). The capacity of Treg to modulate Tresp proliferation was analyzed using ModFit LT™ 3.1 calculating the upper generation proliferation index.
Proliferation assay
Allogeneic purified CD4+CD25− cells from healthy donors (HD) were labeled with CFSE and stimulated to proliferate by using immature mo-DCs (Tresp/DCs ratio 1:10) from HD and patients. The assays were performed over a period of 5 days at 37°C and T cell proliferation was evaluated by flow-cytometry (BD FACSCanto™).
Endocytosis assay
Dextran uptake was measured by exposing 1 × 105 immature mo-DCs to fluorescein isothiocyanate (FITC)-conjugated dextran (0.5 mg/mL; Sigma Aldrich; 74817). Cells were incubated for 30 minutes at 37°C or on ice (used as a background control). After washing, fluorescence was analyzed by flow cytometry (BD FACSCanto™). Uptake of FITC-dextran was expressed as delta (Δ) mean fluorescence intensity (MFI):MFI (uptake at 37°C) – MFI (uptake on ice).
Migration assay
A total of 1 × 105 cells were seeded in a transwell chamber (diameter 6.5 mm, pore size 8 µm; Costar; Corning; CLS3464) in a 24-well plate and migration in response to CCL19 (400 ng/mL; Biolegend; 582104) was analyzed after 4 hours by Trypan Blue exclusion test. The amount of migrated cells was expressed as a percentage of the input: (number of migrated cells in the lower compartment/number of loaded cells in the upper compartment) x 100.
Plasma levels measurement of selected circulating cytokines
Selected cytokines plasma levels of patients/controls were measured by ELISA, according to the manufacturer's instructions. The IL-17 ELISA kit was provided by Boster Immunoleader (Boster Biological Technology Co.; EK0430). The Ciraplex™ immunoassay kit / Human 9-Plex Array (Aushon BioSystems, Cytokine 2 Array) was used for the measurement of various cytokines.
Mutation analysis
JAK2(V617F) allele-burden was assessed in granulocyte DNA with ipsogen JAK2 MutaQuant Kit (Qiagen, Marseille, France) on 7900 HT Fast Real Time PCR System (Applied Biosystem, Monza, Italy). CALR exon 9 sequencing was performed by Next Generation Sequencing (NGS) approach with GS Junior (Roche-454 platform; Roche Diagnostics, Monza, Italy); analysis was performed with AVA Software (GRCh38 as referenced). Rare CALR mutations identified by NGS were confirmed by Sanger sequencing. MPL mutations were investigated by ipsogen MPLW515K/L MutaScreen Kit (Qiagen) and by Sanger sequencing (for MPLS505N and other secondary exon 10 mutations).
Cytogenetic analysis
Chromosome banding analysis was performed on BM cells by standard banding techniques according to the International System for Human Cytogenetic Nomenclature. At least 20 metaphases were required. Unfavorable karyotype, defined according the Dynamic International Prognostic Score System-plus (DIPSS),44 included complex karyotype or single or 2 abnormalities including +8, -7/7q-, i(17q), -5%5q-, 12p-, inv(3) or 11q23 rearrangement.
Statistical analysis
Numerical variables have been summarized by their median and range, and categorical variables by count and relative frequency (%) of each category. All P values were considered significant when ≤.05 (2-tailed). Statistical analyses were performed with Graphpad (Graphpad Software Inc., La Jolla, USA) using unpaired t test.
Declarations
Ethics approval and consent to participate
This study was approved by the medical Ethical Committee of the University Hospital of Bologna (N° 96/2014/U/Tess) and was conducted in accordance with the Declaration of Helsinki. Patients/controls provided written informed consent for the study.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank L. Barber from the Hematology/Oncology Department of King's College for helpful advice in manuscript editing.
Funding
This research was funded by Associazione Italiana contro le Leucemie-Bologna section (BolognAIL) and by the University of Bologna (RFO 2014–2015 for L.C. and F.P.). D.S., M.P., S.L. and D.F. were supported by the University of Bologna. CJ an ST were supported by the Swiss National Science Foundation (Ambizione PZOOP3_161459 and Marie Heim Vögltin fellowship PMPDP3_164447, respectively).
Author contributions
M.R., D.S. and L.C. designed and performed research and wrote the paper; N.P, N.V. and F.P. provided samples and were involved in the clinical part; D.S., S.T., M.B., D.F. performed functional analysis and prepared figures; M.P. and S.L performed molecular and cytogenetic studies; N.V. and M.C. supervised the study; F.P., C.J. and L.C. gave intellectual input and corrected the manuscript.
References
- 1.Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, Harris NL, Le Beau MM, Hellström-Lindberg E, Tefferi A, et al.. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009; 114:937-51; https://doi.org/ 10.1182/blood-2009-03-209262 [DOI] [PubMed] [Google Scholar]
- 2.James C, Ugo V, Casadevall N, Constantinescu SN, Vainchenker W. A JAK2 mutation in myeloproliferative disorders: pathogenesis and therapeutic and scientific prospects. Trends Mol Med 2005; 11:546-54; https://doi.org/ 10.1016/j.molmed.2005.10.003 [DOI] [PubMed] [Google Scholar]
- 3.Lavi N. Calreticulin mutations in myeloproliferative neoplasms. Rambam Maimonides Med J 2014; 5:e0035; PMID:25386351; https://doi.org/ 10.5041/RMMJ.10169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rampal R, Al-Shahrour F, Abdel-Wahab O, Patel JP, Brunel JP, Mermel CH, Bass AJ, Pretz J, Ahn J, Hricik T, et al.. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014; 123:e123-33; PMID:24740812; https://doi.org/ 10.1182/blood-2014-02-554634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Passamonti F, Cervantes F, Vannucchi AM, Morra E, Rumi E, Pereira A, Guglielmelli P, Pungolino E, Caramella M, Maffioli M, et al.. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010; 115:1703-8; PMID:20008785; https://doi.org/ 10.1182/blood-2009-09-245837 [DOI] [PubMed] [Google Scholar]
- 6.Polverelli N, Breccia M, Benevolo G, Martino B, Tieghi A, Latagliata R, Sabattini E, Riminucci M, Godio L, Catani L, et al.. Risk factors for infections in myelofibrosis: role of disease status and treatment. A multicenter study of 507 patients. Am J Hematol 2016; 92:37-41; PMID:27701770; https://doi.org/ 10.1002/ajh.24572 [DOI] [PubMed] [Google Scholar]
- 7.Campanelli R, Rosti V, Fois G, Bonetti E, Barosi G, Massa M. CD14(bright)CD16(low) intermediate monocytes expressing Tie2 are increased in the peripheral blood of patients with primary myelofibrosis. Exp Hematol 2014; 42:244-6; PMID:24333662; https://doi.org/ 10.1016/j.exphem.2013.12.002 [DOI] [PubMed] [Google Scholar]
- 8.Satpathy AT, Wu X, Albring JC, Murphy KM. Re(de)fining the dendritic cell lineage. Nat Immunol 2012; 13:1145-54; PMID:23160217; https://doi.org/ 10.1038/ni.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keohane C, Kordasti S, Seidl T, Perez Abellan P, Thomas NS, Harrison CN, McLornan DP, Mufti GJ. JAK inhibition induces silencing of T Helper cytokine secretion and a profound reduction in T regulatory cells. Br J Haematol 2015; 171:60-73; PMID:26075866; https://doi.org/ 10.1111/bjh.13519 [DOI] [PubMed] [Google Scholar]
- 10.Massa M, Campanelli R, Fois G, Villani L, Bonetti E, Catarsi P, Poletto V, Viarengo G, De Amici M, Rosti V, et al.. Reduced frequency of circulating CD4+CD25brightCD127lowFOXP3+ regulatory T cells in primary myelofibrosis. Blood 2016; 128:1660-2; PMID:27531678; https://doi.org/ 10.1182/blood-2016-03-704577 [DOI] [PubMed] [Google Scholar]
- 11.Wang JC, Sindhu H, Chen C, Kundra A, Kafeel MI, Wong C, Lichter S. Immune derangements in patients with myelofibrosis: the role of Treg, Th17, and sIL2Rα. PLoS One 2015; 10:e0116723; PMID:25793623; https://doi.org/ 10.1371/journal.pone.0116723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riley CH, Jensen MK, Brimnes MK, Hasselbalch HC, Bjerrum OW, Straten PT, Svane IM. Increase in circulating CD4+CD25+Foxp3+ T cells in patients with Philadelphia-negative chronic myeloproliferative neoplasms during treatment with IFN-α. Blood 2011; 118:2170-3; PMID:21708889; https://doi.org/ 10.1182/blood-2011-03-340992 [DOI] [PubMed] [Google Scholar]
- 13.Froom P, Aghai E, Kinarty A, Lahat N. Decreased natural killer (NK) activity in patients with myeloproliferative disorders. Cancer 1989; 64:1038-40; PMID:2788028; https://doi.org/ 10.1002/1097-0142(19890901)64:5%3c1038::AID-CNCR2820640513%3e3.0.CO;2-W [DOI] [PubMed] [Google Scholar]
- 14.Briard D, Brouty-Boyé D, Giron-Michel J, Azzarone B, Jasmin C, Le Bousse-Kerdilès C. Impaired NK cell differentiation of blood-derived CD34+ progenitors from patients with myeloid metaplasia with myelofibrosis. Clin Immunol 2003; 106:201-12; PMID:12706407; https://doi.org/ 10.1016/S1521-6616(02)00046-3 [DOI] [PubMed] [Google Scholar]
- 15.Sonnenberg GF, Artis D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med 2015; 21:698-708; PMID:26121198; https://doi.org/ 10.1038/nm.3892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hazenberg MD, Spits H. Human innate lymphoid cells. Blood 2014; 124:700-9; PMID:24778151; https://doi.org/ 10.1182/blood-2013-11-427781 [DOI] [PubMed] [Google Scholar]
- 17.Artis D, Spits H. The biology of innate lymphoid cells. Nature 2015; 517:293-301; PMID:25592534; https://doi.org/ 10.1038/nature14189 [DOI] [PubMed] [Google Scholar]
- 18.Trabanelli S, Curti A, Lecciso M, Salomé B, Riether C, Ochsenbein A, Romero P, Jandus C. CD127+ innate lymphoid cells are dysregulated in treatment naïve acute myeloid leukemia patients at diagnosis. Haematologica 2015; 100:e257-60; PMID:25710455; https://doi.org/ 10.3324/haematol.2014.119602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munneke JM, Björklund AT, Mjösberg JM, Garming-Legert K, Bernink JH, Blom B, Huisman C, van Oers MH, Spits H, Malmberg KJ, et al.. Activated innate lymphoid cells are associated with a reduced susceptibility to graft-versus-host disease. Blood 2014; 124:812-21; PMID:24855210; https://doi.org/ 10.1182/blood-2013-11-536888 [DOI] [PubMed] [Google Scholar]
- 20.Shuai K, Liu B. Regulation of JAK–STAT signalling in the immune system. Nat Rev Immunol 2003; 3:900-11; PMID:14668806; https://doi.org/ 10.1038/nri1226 [DOI] [PubMed] [Google Scholar]
- 21.Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol 2011; 29:1356-63; PMID:21300928; https://doi.org/ 10.1200/JCO.2010.32.9490 [DOI] [PubMed] [Google Scholar]
- 22.Mondet J, Hussein K, Mossuz P. Circulating cytokine levels as markers of inflammation in philadelphia negative myeloproliferative neoplasms: Diagnostic and prognostic interest. Mediators Inflamm 2015; 2015:670580; PMID:26525644; https://doi.org/ 10.1155/2015/670580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brodie T, Brenna E, Sallusto F. OMIP-018: chemokine receptor expression on human T helper cells. Cytometry A 2013; 83:530-2; PMID:23504907; https://doi.org/ 10.1002/cyto.a.22278 [DOI] [PubMed] [Google Scholar]
- 24.Muranski P, Restifo NP. Essentials of Th17 cell commitment and plasticity. Blood 2013; 121:2402-14; PMID:23325835; https://doi.org/ 10.1182/blood-2012-09-378653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rutz S, Eidenschenk C, Ouyang W. IL-22, not simply a Th17 cytokine. Immunol Rev 2013; 252:116-32; PMID:23405899; https://doi.org/ 10.1111/imr.12027 [DOI] [PubMed] [Google Scholar]
- 26.Boniface K, Blumenschein WM, Brovont-Porth K, McGeachy MJ, Basham B, Desai B, Pierce R, McClanahan TK, Sadekova S, de Waal Malefyt R. Human Th17 cells comprise heterogeneous subsets including IFN-gamma-producing cells with distinct properties from the Th1 lineage. J Immunol 2010; 185:679-87; PMID:20511558; https://doi.org/ 10.4049/jimmunol.1000366 [DOI] [PubMed] [Google Scholar]
- 27.Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, Parizot C, Taflin C, Heike T, Valeyre D, et al.. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009; 30:899-911; PMID:19464196; https://doi.org/ 10.1016/j.immuni.2009.03.019 [DOI] [PubMed] [Google Scholar]
- 28.Porcellini S, Traggiai E, Schenk U, Ferrera D, Matteoli M, Lanzavecchia A, Michalak M, Grassi F. Regulation of peripheral T cell activation by calreticulin. J Exp Med 2006; 203:461-71; PMID:16492806; https://doi.org/ 10.1084/jem.20051519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hasselbalch HC. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leukemia Res 2013; 37:214-20; https://doi.org/ 10.1016/j.leukres.2012.10.020 [DOI] [PubMed] [Google Scholar]
- 30.Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. THE IL-4 RECEPTOR: Signaling Mechanisms and Biologic Functions. Annu Rev Immunol 1999; 17:701-38; PMID:10358772; https://doi.org/ 10.1146/annurev.immunol.17.1.701 [DOI] [PubMed] [Google Scholar]
- 31.van de Laar L, Coffer PJ, Woltman AM. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood 2012; 119:3383-93; PMID:22323450; https://doi.org/ 10.1182/blood-2011-11-370130 [DOI] [PubMed] [Google Scholar]
- 32.Lacey DC, Achuthan A, Fleetwood AJ, Dinh H, Roiniotis J, Scholz GM, Chang MW, Beckman SK, Cook AD, Hamilton JA. Defining GM-CSF– and Macrophage-CSF–Dependent macrophage responses by In Vitro models. J Immunol 2012; 188:5752-65; PMID:22547697; https://doi.org/ 10.4049/jimmunol.1103426 [DOI] [PubMed] [Google Scholar]
- 33.Wang JC, Kundra A, Andrei M, Baptiste S, Chen C, Wong C, Sindhu H. Myeloid-derived suppressor cells in patients with myeloproliferative neoplasm. Leuk Res 2016; 43:39-43; PMID:26943702; https://doi.org/ 10.1016/j.leukres.2016.02.004 [DOI] [PubMed] [Google Scholar]
- 34.Cervantes F, Vannucchi AM, Kiladjian JJ, Al-Ali HK, Sirulnik A, Stalbovskaya V, McQuitty M, Hunter DS, Levy RS, Passamonti F, et al.. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood 2013; 122:4047-53; PMID:24174625; https://doi.org/ 10.1182/blood-2013-02-485888 [DOI] [PubMed] [Google Scholar]
- 35.Heine A, Held SA, Daecke SN, Wallner S, Yajnanarayana SP, Kurts C, Wolf D, Brossart P. The JAK-inhibitor Ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood 2013;122: 1192-202; PMID:23770777; https://doi.org/ 10.1182/blood-2013-03-484642 [DOI] [PubMed] [Google Scholar]
- 36.Ma DY, Clark EA. The role of CD40 and CD154/CD40L in dendritic cells. Semin Immunol 2009; 21:265-72; PMID:19524453; https://doi.org/ 10.1016/j.smim.2009.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Magee CN, Boenisch O, Najafian N. The role of costimulatory molecules in directing the functional differentiation of alloreactive T helper cells. Am J Transplant 2012; 12:2588-600; PMID:22759274; https://doi.org/ 10.1111/j.1600-6143.2012.04180.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larsen TS, Christensen JH, Hasselbalch HC, Pallisgaard N. The JAK2 V617F mutation involves B- and T-lymphocyte lineages in a subgroup of patients with Philadelphia-chromosome negative chronic myeloproliferative disorders. Br J Haematol 2007; 136:745-51; PMID:17313377; https://doi.org/ 10.1111/j.1365-2141.2007.06497.x [DOI] [PubMed] [Google Scholar]
- 39.Bogani C, Guglielmelli P, Antonioli E, Pancrazzi A, Bosi A, Vannucchi AM. B-, T-, and NK-cell lineage involvement in JAK2V617F-positive patients with idiopathic myelofibrosis. Haematologica 2007; 92:258-9; PMID:17296581; https://doi.org/ 10.3324/haematol.10527 [DOI] [PubMed] [Google Scholar]
- 40.Delhommeau F, Dupont S, Tonetti C, Massé A, Godin I, Le Couedic JP, Debili N, Saulnier P, Casadevall N, Vainchenker W, et al.. Evidence that the JAK2 G1849T (V617F) mutation occurs in a lymphomyeloid progenitor in polycythemia vera and idiopathic myelofibrosis. Blood 2007; 109:71-7; PMID:16954506; https://doi.org/ 10.1182/blood-2006-03-007146 [DOI] [PubMed] [Google Scholar]
- 41.Zhao J, Zhao J, Perlman S. Differential effects of IL-12 on Tregs and non-Treg T cells: roles of IFN-γ, IL-2 and IL-2R. PLoS One 2012; 7:e46241; PMID:23029447; https://doi.org/ 10.1371/journal.pone.0046241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Belz GT. ILC2s masquerade as ILC1s to drive chronic disease. Nat Immunol 2016; 17:611-2; PMID:27196511; https://doi.org/ 10.1038/ni.3467 [DOI] [PubMed] [Google Scholar]
- 43.Trabanelli S, La Manna F, Romano M, Salvestrini V, Cavo M, Ciciarello M, Lemoli RM, Curti A. The human mesenchymal stromal cell-derived osteocyte capacity to modulate dendritic cell functions is strictly dependent on the culture system. J Immunol Res 2015; 2015:526195; PMID:26247040; https://doi.org/ 10.1155/2015/526195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gangat N, Caramazza D, Vaidya R, George G, Begna K, Schwager S, Van Dyke D, Hanson C, Wu W, Pardanani A, et al.. DIPSS plus: a refined dynamic international prognostic scoring system for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol 2011; 29:392-7; PMID:21149668; https://doi.org/ 10.1200/JCO.2010.32.2446 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.