

ABSTRACT
Immunotherapy with anti-GD2 antibody (Ab) ch14.18/CHO is effective for treatment of high-risk neuroblastoma (NB) patients and is mainly based on GD2-specific Ab-dependent cellular cytotoxicity (ADCC). Strategies to further enhance the efficacy are important and currently explored in prospective clinical trials randomizing ch14.18/CHO ± IL-2. Recently, expression of programmed death 1 (PD-1) inhibitory receptor by effector cells and its ligand (PD-L1) by tumor cells has been shown. Here, we report for the first time effects of PD-1 blockade on ch14.18/CHO-based immunotherapy and mechanisms involved.
Expression of PD-1 and PD-L1 on NB and effector cells was analyzed by RT-PCR and flow cytometry in the presence of ch14.18/CHO and/or IL-2. The effect of PD-1 blockade on ch14.18/CHO-mediated anti-NB immune response was evaluated using anti-PD-1 Ab both in vitro (Nivolumab) and in a syngeneic PD-L1+/GD2+ NB mouse model (anti-mouse PD-1).
Culture of NB cells LA-N-1 (low PD-L1 baseline expression) with leukocytes and subtherapeutic ch14.18/CHO concentrations for 24 h induced strong upregulation of PD-L1, which was further increased by IL-2 resulting in complete inhibition of ch14.18/CHO-mediated ADCC. Importantly, blockade with Nivolumab reversed the PD-L1-dependent inhibition of ADCC. Similarly, co-incubation with anti-CD11b Ab abrogated the PD-L1 upregulation and restored ADCC. Mice treated with ch14.18/CHO in combination with PD-1 blockade showed a strong reduction of tumor growth, prolonged survival and the highest cytotoxicity against NB cells.
In conclusion, ch14.18/CHO-mediated effects upregulate the inhibitory immune checkpoint PD-1/PD-L1, and combination of ch14.18/CHO with PD-1 blockade results in synergistic treatment effects in mice representing a new effective treatment strategy against GD2-positive cancers.
KEYWORDS: ch14.18/CHO, immune checkpoint blockade, neuroblastoma, programmed cell death 1 (PD-1) ligand, PD-1
Abbreviations
- AM
acetoxymethyl ester
- ADCC
antibody-dependent cell-mediated cytotoxicity
- anti-Id
anti-idiotype
- CHO
Chinese Hamster Ovary
- CDC
complement dependent cytotoxicity
- ELISA
Enzyme-linked Immunosorbent Assay
- E:T
effector:target
- EFS
event-free survival
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- IL-2
interleukin-2
- mAb
monoclonal antibody
- NB
neuroblastoma
- OS
overall survival
- PD-1
programmed cell death 1
- PD-L1
programmed cell death ligand 1
- RT
room temperature
- SD
standard deviation
- SEM
standard error of the mean
- TH
tyrosine hydroxylase
- COG
Children's Oncology Group
- NK
natural killer
Introduction
One of the major challenges in pediatric oncology is the effective treatment of high-risk neuroblastoma (NB) patients. The 5-year event-free survival (EFS) rate following standard therapy still remains less than 50%.1 The Children's Oncology Group (COG) reported that treatment of high-risk NB patients with the human/mouse chimeric Ab ch14.18 produced in SP2/0 cells in combination with cytokines (GM-CSF and IL-2) improved 2-year event-free survival (EFS) and overall survival (OS) by 20% and 11%, respectively.2 This Ab/cytokine combination therapy was approved by the American Food and Drug Administration for the treatment of NB. In Europe, ch14.18/CHO was remanufactured in Chinese Hamster Ovary (CHO) cells and applied with and without IL-2 for the treatment of NB showing similar clinical activity and pharmacokinetics compared with the ch14.18 produced in SP2/0 cells,3 and ch14.18/CHO was approved by the European Medicines Agency for the treatment of NB.
The primary mechanism of action of ch14.18/CHO is the induction of antibody-dependent cell-mediated cytotoxicity (ADCC)4,5 that requires immune effector cells such as natural killer (NK) cells, monocytes, neutrophils and macrophages.6 The cytotoxic activity of these cells is mediated by immunoglobulin receptors including FCGR3A (CD16) expressed primarily on NK cells and FCGR2A (CD32) expressed on monocytes, macrophages and neutrophils.6 Both receptors recognize the Fc fragment of ch14.18/CHO bound to NB cells and induce cytotoxic effector functions.
One major side effect associated with anti-GD2 therapies is the induction of neuropathic pain.2,3 These side effects could be improved by changing the application method from short-term infusions (STI; 4–5 days; 8 h/day; cumulative dose 100 mg/m2) to a long-term infusion (LTI; 10 days; 24 h/day, cumulative dose 100 mg/m2).7
However, the survival rates of high-risk NB patients treated with anti-GD2 Ab are still far behind other malignant diseases in pediatric oncology (i.e. acute lymphoblastic leukemia, nephroblastoma), therefore new approaches are urgently needed.
In the context of strategies built on directing the patient´s immune system against cancer cells, additional blockade of inhibitory pathways is a potential step forward. Application of immunomodulatory Abs that block immunologic immune checkpoints was clinically effective in some cancers even when used as a monotherapy.8 Under physiologic conditions, immune checkpoints maintain self-tolerance during immune responses; however, in cancer they can negatively regulate anti-tumor immunity thereby reducing the treatment efficacy. Programmed Death 1 (PD-1) is one key immune checkpoint receptor expressed mainly by activated T cells and, as recently has been reported, also by NK cells.9 PD-1 mediates immunosuppression after binding to its ligands PD-L1, expressed by activated haematopoietic cells and epithelial cells, and PD-L2, expressed by dendritic cells and macrophages.8 Importantly, upregulation of both PD-1 ligands has been found in many human cancers with major expression of PD-L1 in solid tumors and PD-L2 in B cell lymphomas.8 Several studies revealed that the ligand expression, especially PD-L1, was associated with aggressive disease and adverse clinical outcome.10 Although the inhibitory role of the PD-1/PD-L1 pathway has been identified in different cancers, little is known about the mechanisms of the regulation of this immune checkpoint. Recently, involvement of CD11b receptor and p38 MAPK signaling pathway has been shown to be critical for the expression of PD-L1 by tumor cells.11
In NB, both a constitutive and inducible PD-L1 expression was recently reported in different cell lines.12,13 In NB cells with low PD-L1 levels inflammatory cytokines, such as interferon gamma, induced PD-L1 expression. Moreover, an active role of PD-L1 in immunosuppression of CD4+ and CD8+ T cells was demonstrated in vitro after blocking of PD-L1 expressed by NB cells.12 Importantly, expression of PD-1 receptor by αβ- and γδ T cells as well as by NK cells has emphasized PD-1/PD-L1-mediated negative regulation of the induced anti-NB immune response.13 PDL-1 expression was also evaluated by immunohistochemistry in a series of pediatric malignancies and reported to be positive in 72% for high-risk NB patients.14 Taken together, NB cells express ligand of the immune checkpoint receptor PD-1 and thus inhibit anti-tumor immune response. Based on these considerations, we addressed the question whether ch14.18/CHO-mediated anti-tumor effects could be augmented by the blockade of the immune checkpoint pathway PD-1/PD-L1.
We first analyzed PD-L1 and PD-1 expression by NB cells and leukocyte populations as well as ch14.18/CHO-mediated effects and involvement of CD11b in the regulation of this immune checkpoint. We then determined anti-tumor effects of ch14.18/CHO-based immunotherapy in combination with PD-1 blockade in vivo.
Our results demonstrate that ch14.18/CHO-mediated ADCC induces the inhibitory PD1/PD-L1 checkpoint on NB- and effector cells (lymphocytes, monocytes and granulocytes), a process that is CD11b-dependent. Importantly, a synergistic anti-NB effect of the combinatorial ch14.18/CHO and anti-PD-1 immunotherapy was observed, emphasizing its clinical potential as an immunotherapy strategy.
Results
PD-1 ligand expression by human neuroblastoma cell lines
We first analyzed baseline expression of PD-1 ligand mRNA in well-known human NB cell lines CHLA-20, LA-N-1, LA-N-6 and SK-N-SH as well as in primary cell lines HGW-1, HGW-2, HGW-3 and HGW-5 originated from high-risk NB patients15 using semi-quantitative RT-PCR analysis (Fig. 1A-B). All NB cell lines analyzed were found to be PD-L1-positive (Fig. 1A) with varying baseline expression levels (Fig. 1B). Since LA-N-1 cells showed the lowest baseline expression level from this panel (Fig. 1B), we selected this cell line to evaluate strategies to induce PD-L1 expression. We then confirmed weak baseline PD-L1 protein expression by LA-N-1 using flow cytometry (Fig. 1C-D). For calculation of baseline expression levels we used the mean fluorescence intensity relative to isotype control as described in Materials and Methods. Additionally, we analyzed PD-L1 baseline expression by 3 leukocyte populations (granulocytes, monocytes and lymphocytes) using flow cytometry and detected higher levels compared with LA-N-1 (Fig. 1C-D). Comparison of leukocyte populations revealed a 2-fold weaker baseline expression level of PD-L1 by lymphocytes compared with granulocytes and monocytes (Fig. 1D).
Figure 1.
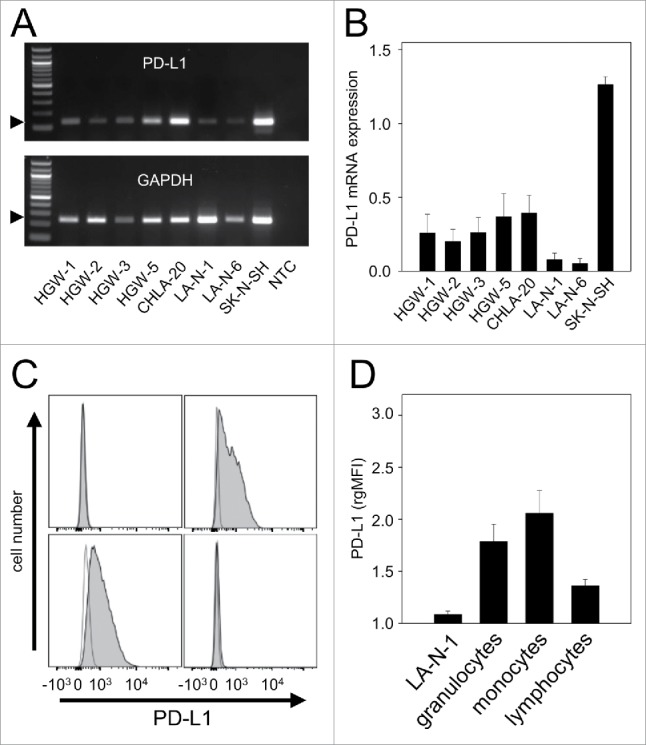
PD-L1 gene expression by NB- and effector cells. (A-B) Representative RT-PCR (A) and densitometric analysis of PD-L1 mRNA (B) by human NB cell lines CHLA-20, LA-N-1, LA-N-6 and SK-N-SH as well as by primary cell lines HGW-1, HGW-2, HGW-3, and HGW-5. PD-L1 mRNA Expression (A, upper panel) was evaluated relative to GAPDH (A, lower panel) which served as internal control according to the formula: PD-L1 signal intensity/GAPDH signal intensity. (B) Values are given as means ± SEM of 5 independent experiments. (C) Representative histograms and (D) flow cytometry analysis of PD-L1 baseline expression by NB cells LA-N-1 (C, upper left panel) and leukocytes (granulocytes (C, upper right panel), monocytes (C, lower left panel) and lymphocytes (C, lower right panel)). Cells were stained with PE-labeled anti-mouse PD-L1 Ab (filled black curve). Anti-mouse IgG2b, κ-PE was used as isotype control (open gray curve). The PD-L1 expression level was quantified using relative geometric mean fluorescence intensity (rgMFI) according to the formula: MFI of PD-L1 stained sample/MFI of isotype control. When the control histogram (isotype control) is not visible it is covered by the experimental histogram (PD-L1 staining). Data are shown as mean values ± SEM of at least 3 independent experiments.
Impact of ch14.18/CHO-mediated ADCC and IL-2 on PD-1 ligand expression
The current standard of care for NB maintenance therapy is the application of anti-GD2 Abs in combination with cytokines2 and in Europe this treatment consists of ch14.18/CHO combined with IL-2. Therefore, we investigated by flow cytometry effects of ch14.18/CHO-mediated ADCC and IL-2 on PD-L1 expression by LA-N-1 NB cells (GD2+/CD45−/PD-L1+) and 3 leukocyte populations: granulocytes, monocytes and leukocytes (GD2−/CD45+/PD-L1+). To distinguish between the leukocyte populations, a gating strategy based on cell granularity and intensity of the CD45 fluorescence of GD2-negative cells was used. To calculate changes of the PD-L1 expression level, we used mean fluorescence intensity relative to the respective untreated cell population as described in Materials and Methods.
To analyze PD-L1 expression on NB cells, GD2-positive LA-N-1 cells were co-cultured for 24 h with further components to induce ADCC i.e., leukocytes (E/T 10:1) (granulocytes, lymphocytes and monocytes), and subtherapeutic levels of anti-GD2 mAb ch14.18/CHO (10 ng/ml). A strong increase of the PD-L1 expression compared with controls was observed (Fig. 2A-B and D). Importantly, ch14.18/CHO (10 ng/ml) combined with IL-2 (100 IU/ml) further increased these effects showing significantly higher PD-L1 expression level compared with ch14.18/CHO without IL-2 (Fig. 2D). This effect was GD2-specific since co-treatment of the culture containing LA-N-1, ch14.18/CHO and leukocytes with an anti-idiotype Ab to the ch14.18/CHO (ganglidiomab)16 resulted in complete abrogation of PD-L1 induction (Fig. 2C and D). Incubation of NB cells with only one component of the ADCC reaction such as only ch14.18/CHO or exchanging ch14.18/CHO with an irrelevant Ab rituximab or with only effector cells did not affect PD-L1 expression, indicating that all 3 components (target cell, Ab and effector cells) are required. Moreover, IL-2 alone did not affect PD-L1 expression by NB cells (Fig. 2D).
Figure 2.

Effects of ADCC culture conditions on PD-L1 expression by NB cells. PD-L1 expression was analyzed on LA-N-1 cells by flow cytometry and data are shown by representative histograms (A-C) and rgMFI values (D) as described in Materials and Methods. Results show PD-L1 expression after 24 h incubation with ch14.18/CHO (A), ch14.18/CHO in combination with leukocytes (ADCC conditions, B) and ch14.18/CHO with leukocytes and anti-idiotype ganglidiomab (anti-Id) (specificity control, C). When the control histogram (isotype control) is not visible it is covered by the experimental histogram (PD-L1 staining). The effect of ADCC conditions on PD-L1 expression was also analyzed in the presence of IL-2 (D). Cells were stained with PE-labeled anti-human PD-L1 Ab. To distinguish between PD-L1-positive NB cells (PD-L1+/GD2+/CD45−) and leukocytes (PD-L1+/GD2−/CD45+), Alexa647-labeled anti-GD2 and PE/Cy7-labeled anti-CD45 mAb were used. The PD-L1 expression level was quantified using relative geometric mean fluorescence intensity (rgMFI) according to the formula: MFI of PD-L1 by treated cells - MFI of PD-L1 by the respective untreated control. Ch14.18/CHO and rituximab served as controls. Data represent mean values ± SEM of at least 3 independent experiments. t-test, ##P < 0.01 vs. ch14.18/CHO, ###P < 0.001 vs. leukocytes, §§§P <0.001 vs. ADCC without IL-2, *P < 0.05 vs. ADCC without IL-2, **P < 0.01 vs. leukocytes, ***P < 0.001 vs. LA-N-1 incubated with IL-2.
We also evaluated effects of ch14.18/CHO-mediated ADCC with and without IL-2 on PD-L1 expression by 3 leukocyte populations (granulocytes, monocytes and lymphocytes) (Fig. 3). Similar to PD-L1 expression by NB cells, culture conditions with all components involved in ADCC, i.e., NB cells, ch14.18/CHO Ab and effector cells, mediated the highest increase of PD-L1 on granulocytes and monocytes, respectively (Fig. 3A and B) and unchanged PD-L1 expression on lymphocytes (Fig. 3C). In contrast to NB cells, incubation of leukocytes with IL-2 in absence of NB cells or ch14.18/CHO resulted in significant increase of PD-L1 expression by granulocytes, monocytes and lymphocytes (Fig. 3A-C). Interestingly, we observed the highest level of PD-L1 expression by granulocytes, monocytes and in particular also lymphocytes when ADCC cultures contained IL-2 (Fig. 3A-C). Again, this effect was GD2-specific since the addition of ganglidiomab (anti-idiotype Ab of ch14.18/CHO)16 or exchanging ch14.18/CHO with an irrelevant mAb (rituximab) resulted in complete abrogation of PD-L1 induction. Finally, removal of one critical component of the ADCC reaction, such as ch14.18/CHO or effector cells abrogated PD-L1 induction, indicating that all 3 components (target cell, Ab and effector cells) are required (Fig. 3A-C).
Figure 3.

Effects of ADCC culture conditions on PD-L1 expression by 3 leukocytes populations: granulocytes (A), monocytes (B) and lymphocytes (C). Leukocytes were cultured for 24 h before analysis with ch14.18/CHO (10 ng/ml) and LA-N-1 NB cells (E:T 10:1) in the absence or presence of IL-2 (100 IU/ml). PD-L1 expression was analyzed by flow cytometry using PE-labeled anti-human PD-L1. Alexa647-labeled anti-GD2 and PE/Cy7-labeled anti-CD45 mAb were used to distinguish between NB cells (PD-L1+/GD2+/CD45−) and leukocytes (PD-L1+/GD2−/CD45+). PD-L1 expression level was quantified using the relative geometric mean fluorescence intensity (rgMFI) according to the formula: MFI of PD-L1 by treated cells - MFI of PD-L1 by the respective untreated control. Leukocytes incubated only with ch14.18/CHO, NB cells or IL-2 served as controls. To show GD2-specificity, ch14.18/CHO was replaced by rituximab or bound by ganglidiomab (anti-Id. of ch14.18/CHO) that was added to the ADCC culture condition. Data are shown as mean values ± SEM of at least 3 independent experiments. (A) t-test, ***P < 0.001 vs. ch14.18/CHO, §§P < 0.01 vs. ADDC, ##P < 0.01 vs. ch14.18/CHO and NB cells, ###P < 0.001 vs. rituximab, $$$P < 0.001 vs. granulocytes with IL-2 and NB cells with IL-2. (B) t-test, *P < 0.05 vs. ch14.18/CHO, ***P < 0.001 vs. NB cells and rituximab, §§P < 0.01 vs. ADCC, #P < 0.05 vs. ch14.18/CHO, ##P < 0.01 vs. NB cells, ###P < 0.001 vs. rituximab, $P < 0.05 vs. NB cells with IL-2, $$P < 0.01 vs. monocytes with IL-2. (C) t-test, *P < 0.05 vs. ch14.18/CHO, ***P < 0.001 vs. rituximab, ##P < 0.01 vs. ch14.18/CHO, NB cells and ADCC, ###P < 0.001 vs. rituximab and ADCC + anti-Id.
In summary, these data demonstrate strong upregulation of the PD-L1 under GD2-specific ADCC culture conditions (ch14.18/CHO, leukocytes, NB cells), that is enhanced by additional IL-2.
Effect of ch14.18/CHO-mediated ADCC with and without IL-2 on PD-1 expression
Similar to the analysis of PD-1 ligand, we evaluated expression of PD-1 receptor on NB cells (Fig. 4A) and on the 3 leukocyte populations (granulocytes (Fig. 4B), monocytes (Fig. 4C) and lymphocytes (Fig. 4D)). We did not detect PD-1 expression on LA-N-1 cells even under ADCC conditions irrespective of IL-2 co-treatment (Fig. 4A). This is in contrast to the findings on leukocytes (Fig. 4B-D). The strongest stimulatory effect on PD-1 expression was observed after induction of ch14.18/CHO-mediated ADCC in all effector cell populations analyzed (Fig. 4B-D); however the observed differences for granulocytes were not statistically significant. Co-incubation with IL-2 further increased PD-1 expression with significant effects in the lymphocyte population (Fig. 4D). Similar to the results of the PD-L1 analysis, incubation of the ADCC components with the anti-idiotype Ab ganglidiomab or replacement of ch14.18/CHO with irrelevant Ab rituximab abrogated the induction of PD-1 expression by all effector cells populations analyzed (Fig. 4B-D).
Figure 4.

Effects of ADCC culture conditions on PD-1 expression by LA-N-1 NB cells (A) and 3 leukocytes populations: granulocytes (B), monocytes (C) and lymphocytes (D). ADCC was induced by 24 h incubation of LA-N-1 with ch14.18/CHO and leukocytes in the presence or absence of IL-2. PD-1 expression was analyzed by flow cytometry with PE-labeled anti-human PD-1 Ab. Alexa647-labeled anti-GD2 and PE/Cy7-labeled anti-CD45 mAb were used to distinguish between NB cells (GD2+/CD45−) and leukocytes (GD2−/CD45+). PD-1 expression level was determined using relative geometric mean fluorescence intensity (rgMFI) according to the formula: MFI of PD-1 by treated cells - MFI of PD-1 by the respective untreated control. Ch14.18/CHO, IL-2, leukocytes and NB cells, respectively, served as controls. To show GD2-specificity of ADCC, ch14.18/CHO was replaced by rituximab or bound by ganglidiomab (anti-Id of ch14.18/CHO) that was added to the ADCC culture condition. Data are shown as mean values ± SEM of at least 3 independent experiments. (C) *P <0.05 vs. ch14.18/CHO, §§P <0.01 vs. NB cells, ***P < 0.001 vs. ch14.18/CHO, #P < 0.05 vs. monocytes with IL-2, ###P < 0.001 vs. monocytes with IL-2. (D) *P < 0.05 vs. ch14.18/CHO, **P < 0.01 vs. ch14.18/CHO, #P < 0.05 vs. lymphocytes with IL-2, ##P < 0.01 vs. ch14.18/CHO.
These data show strong upregulation of the PD-1 receptor on leukocytes under the culture conditions of ADCC (ch14.18/CHO, leukocytes, NB cells), which is GD2-specific. These effects were further enhanced by IL-2 in all 3 leukocyte populations analyzed.
PD-1/PD-L1 checkpoint blockade and ADCC
Based on our data of PD-1/PD-L1 checkpoint upregulation under ADCC conditions, we investigated whether this checkpoint inhibits the cytotoxic activity of effector cells and evaluated the effect of PD-1 blockade.
This was tested with untreated LA-N-1 cells that show low PD-L1 expression (Fig. 1) compared with LA-N-1 cells with high PD-L1 expression induced following 24 h culture under ADCC conditions (co-incubation with leukocytes (E:T 10:1) and subtherapeutic ch14.18/CHO concentration (10 ng/ml) for 24 h) (Fig. 2B and D). We then used both types of LA-N-1 cells with high and low PD-L1 expression as target cells for evaluation of ch14.18/CHO-dependent ADCC in a subsequent 4 h ADCC assay performed with ch14.18/CHO (10 µg/ml) and leukocytes of a healthy donor pre-treated with IL-2 (100 IU/ml) for 24 h. We detected a strong lysis (56%) of NB cells expressing low level of PD-L1 (Fig. 5). In contrast, ADCC against high PD-L1 expressing LA-N-1 cells was significantly inhibited (16%, Fig. 5), which was similar to the negative control rituximab (14%, Fig. 5). These data clearly show the inhibition of ADCC by the PD-1/PD-L1 pathway.
Figure 5.
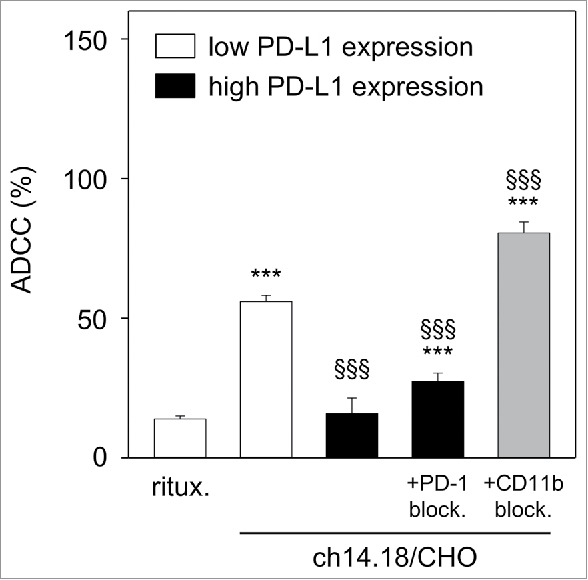
Effect of PD-L1 on ch14.18/CHO-dependent cellular cytotoxicity (ADCC). To show the impact of PD-L1 on ADCC, a 2-step assay was used: first, LA-N-1 NB cells were cultured under ADCC conditions in the presence of subtherapeutic concentrations of ch14.18/CHO (10 ng/ml, 24 h) and effector cells (E:T 10:1) for induction of high level of PD-L1 on NB cells (black columns). After incubation, NB cells were harvested, labeled with calcein-AM as described in Materials and Methods and used for the second step: cytotoxicity assay with therapeutic ch14.18/CHO concentrations (10 µg/ml, 4 h) and effector cells (E:T 40:1). LA-N-1 expressing low level of PD-L1 (white columns) served as positive control and rituximab served as negative control. To show effects of PD-1- (gray column) and CD11b- (right black column) blockade, anti-PD-1 was added to leukocytes and anti-CD11b mAb was added to ADCC culture, respectively. The cytotoxicity assay (step 2) was performed after labeling of LA-N-1 cells with calcein-AM and performed in the absence of anti-CD11b Ab. Data are shown as mean values ± SEM of at least 3 independent experiments. ***P < 0.001 vs. rituximab, §§§P <0.001 vs. low expressing PD-L1 LA-N-1.
Next, we induced ADCC against high PD-L1 expressing LA-N-1 in the presence of anti-PD-1 mAb. Importantly, blockade of PD-1 reversed the inhibitory effect of PD-1/PD-L1 pathway resulting in significant increase (27%) of anti-tumor cytotoxicity by effector cells (Fig. 5).
Our results show positive effects of blockade of PD-1/PD-L1 immune checkpoint on cytotoxic activity of effector cells.
Based on the fact that CD11b has been shown to be involved in the induction of PD-L1,11 we analyzed the impact of CD11b blockade on PD-L1 expression by tumor and effector cells. Analysis of LA-N-1 NB cells cultured under ADCC conditions (leukocytes, E/T 10:1; 10 ng/ml ch14.18/CHO) in the presence of anti-CD11b mAb showed reduced upregulation of PD-L1 (Fig. 6A and B). Similar effects were observed in granulocytes (Fig. 6C and E) and monocytes (Fig. 6D and E) compared with controls. Blockade of CD11b did not affect expression of PD-L1 by lymphocytes (Fig. 6E).
Figure 6.

Effect of CD11b-blockade on PD-L1 expression by NB cells LA-N-1 and 3 leukocytes populations (granulocytes, monocytes and lymphocytes). PD-L1 expression was induced by ADCC culture conditions (24 h incubation of LA-N-1 with subtherapeutic concentration of ch14.18/CHO (10 ng/ml) and leukocytes (E:T 10:1)) with or without addition of anti-CD11b Ab. PD-L1 expression was analyzed by flow cytometry with PE-labeled anti-human PD-L1 Ab. Alexa647-labeled anti-GD2 and PE/Cy7-labeled anti-CD45 mAb were used to distinguish between NB cells (PD-L1+/GD2+/CD45−) and leukocytes (PD-L1+/GD2−/CD45+) (A, C-D) Representative histograms of PD-L1 expression by NB cells LA-N-1 (A), granulocytes (C) and monocytes (D) (since ADCC did not affect PD-L1 expression by lymphocytes, the representative histogram for these effector cells is not shown). PD-L1 expression was determined under ADCC conditions in the presence (dashed black line) and absence of anti-CD11b Ab (solid black line) relative to untreated controls (filled gray line). PD-L1 expression level on NB cells LA-N-1 (B) and leukocytes (E) was determined using relative geometric mean fluorescence intensity (rgMFI) according to the formula: MFI of PD-L1 by treated cells - MFI of PD-L1 by the respective untreated control. Data are shown as mean values ± SEM of at least 3 independent experiments. t-test, **P < 0.01 vs. ADCC + anti-CD11b.
To evaluate whether the observed effects of CD11b blockade on PD-L1 have a functional effect, we analyzed the ADCC response against LA-N-1 NB cells cultured under ADCC conditions (leukocytes E/T 10:1; 10 ng/ml ch14.18/CHO) in the presence of anti-CD11b mAb (Fig. 5). LA-N-1 cells treated with anti-CD11b were found to be significantly more sensitive to the subsequent ADCC assay (81%) compared with the control (27%). Interestingly, ADCC levels were higher following CD11b blockade during ADCC culture conditions compared with untreated LA-N-1 cells characterized by low PD-L1 expression (81% vs 56%, respectively) (Fig. 5).
These results show that CD11b blockade reverse PD-L1 expression and inhibition of ADCC by the PD-L1/PD-1 checkpoint and suggest a possible therapeutic potential of CD11b blockade.
Effect of combined immunotherapy with ch14.18/CHO and anti-PD-1 Ab on tumor growth and survival
For evaluation of anti-tumor effects of the combinatorial immunotherapy, we used the murine NB cell line NXS2-HGW generated from NXS2 cells by in vivo passage in A/J mice to generate a syngeneic model. Immunohistochemical analysis of these NB cells showed a homogeneous GD2 expression in cell culture (Fig. 7A-B) and in primary tumor tissue after inoculation of NXS2-HGW cells in vivo (Fig. 7C). We observed a stable expression over time showing unchanged high GD2 levels even after 29 passages (Fig. 7F).
Figure 7.

Characterization of the murine NB cell line NXS2-HGW. (A-C) Representative immunohistochemical images of GD2 expression (left) and respective negative controls (right). NXS2-HGW cells cultivated onto chamber slides (A-B) and primary tumors obtained from A/J mice inoculated subcutaneously with NXS2-HGW cells (C) were analyzed for GD2-expression (magnification of 100x (A), 200x (B) and 400x (C)). (D) PD-L1 expression analysis by the parental NXS2 cells (left histogram) in comparison with NXS2-derived NXS2-HGW cells (right histogram) by flow cytometry. Cells were stained with PE-labeled anti-mouse PD-L1 Ab (filled black curve) relative to isotype control (open gray curve). Results are shown as representative histograms from at least 5 independent experiments. (E) RT-PCR analysis of MYCN- (product size: 248 bp), TH- (187 bp) and PD-L1 (180 bp) mRNA in NXS2-HGW cells. GAPDH (223 bp) served as internal control. NTC - no template control. (F) GD2 expression by the parental NXS2 cells in comparison with NXS2-derived NXS2-HGW cells (passage 49 for NXS2 and passage 1, 12 and 29 for NXS2-HGW) by flow cytometry. Cells were stained with chimeric ch14.18/CHO and PE-labeled anti-human IgG served as primary and secondary antibody, respectively (filled black curve). Rituximab served as isotype control (open gray curve). Results are shown as representative histograms from independent experiments.
Additionally, we examined 2 NB-specific markers MYCN and tyrosine hydroxylase (TH) and observed a strong mRNA expression of both genes (Fig. 7E) confirming NB-typical characteristics of NXS2-HGW cells.
Next, we investigated PD-L1 expression by NXS2-HGW cells using RT-PCR and flow cytometry (Fig. 7D-E). We could clearly detect PD-L1 expression with both methods underlining the applicability of the model. Subcutaneous injection of these cells into syngeneic A/J mice induced primary tumor growth and spontaneous metastasis to liver, lymph nodes, lung and adrenal glands (data not shown).
We then evaluated the efficacy of ch14.18/CHO and anti-PD-1 on primary tumor growth using in this model. Immunotherapy with either ch14.18/CHO or anti-PD-1 mAb resulted in delay of tumor growth beginning on day 13 after tumor cell inoculation (Fig. 8 A-B). Importantly, we observed the strongest anti-NB effect in mice treated with a combination of ch14.18/CHO and anti-PD-1 Ab.
Figure 8.
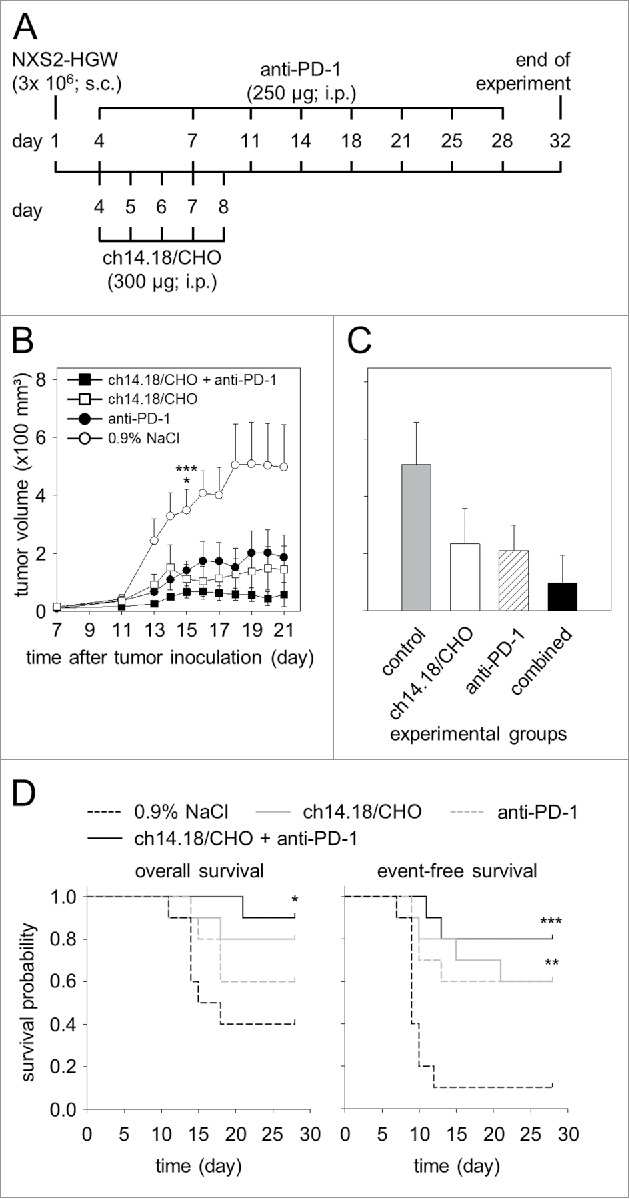
Anti-tumor effects of the combinatorial treatment with ch14.18/CHO and anti-PD-1 antibodies in vivo. (A) Schematic overview of the treatment schedule. The GD2- and PD-L1 expressing murine NB cells NXS2-HGW were injected subcutaneously into A/J mice followed by treatment with ch14.18/CHO in combination with PD-1 blockade. Four days after the last anti-PD-1 application blood samples were collected and mice were killed followed by isolation of splenocytes for evaluation of anti-tumor cytotoxicity. (B) Analysis of tumor growth (closed squares for combinatorial treatment; open squares and closed circles for treatment with ch14.18/CHO and anti-PD-1, respectively). Controls received 0.9% NaCl (open circles). In the case of appearance of having tumor burden, mice were killed ahead of schedule and the data of the last tumor volume measurement were included into subsequent calculation for the respective treatment group. Data are shown as mean values ± SEM. Kruskal-Wallis test: *P < 0.05 vs. ch14.18/CHO group, ***P < 0.001 vs. combined group. (C) Comparison of end point tumor volume was performed at the time point when mice were killed either ahead of schedule or at the end of the experiment (day 32). Mice were treated with ch14.18/CHO in combination with PD-1 blockade (black column), ch14.18/CHO (white column) or anti-PD-1 (white and striped column). Control mice received 0.9% NaCl (gray column). Data are shown as mean values ± SEM. Kruskal-Wallis test, differences between the groups were not significant. (D) Comparison of overall survival (left diagram) and event-free survival probabilities (right diagram) of mice treated with ch14.18/CHO in combination with PD-1 blockade (black solid line), ch14.18/CHO (gray solid line), anti-PD-1 (gray dashed line) or control mice (0.9% NaCl, black dashed line). For overall and event-free survival, death ahead of schedule and a tumor volume of 150 mm3 were defined as event, respectively. Statistical analysis was performed using LogRank test. For overall survival: *P < 0.05 vs. control group. For event-free survival: ***P < 0.001 and **P = 0.01 vs. control.
When mice developed tumors of a critical volume of 1000 mm3 or showed appearance of metastatic tumor burden as described in the “Material and Method” section, they were removed from the experiment. This resulted in reduction of the number of mice per group from day 15 after tumor cell inoculation. Therefore, statistical analysis of the primary tumor volume between groups was performed at this time point.
Analysis of tumors on day 15 in mice treated with ch14.18/CHO showed clearly significant tumor reduction compared with controls (Fig. 8B). Similarly, we observed a reduced tumor volume in mice treated with anti-PD-1; however this difference was not statistically significant (Fig. 8B). Importantly, mice treated with ch14.18/CHO in combination with anti-PD-1 Ab the statistical analysis revealed an extremely significant reduction of tumor volume (Fig. 8B). These data clearly show beneficial effects of the combined immunotherapy based on application of anti-GD2 Ab ch14.18/CHO and anti-PD-1 Ab.
These data were confirmed evaluating tumor volume determined at the time point when mice were killed either ahead of schedule or at the end of the experiment (day 32) (Fig. 8C). We observed the strongest anti-tumor effects in mice of the combined immunotherapy group showing the lowest tumor volume compared with control groups (Fig. 8C).
We also evaluated the percentage of mice that were killed ahead of schedule due to tumor burden as described in “Material and Methods.” 60% of mice (6/10) of the control group receiving 0.9% NaCl were removed from the experiment ahead of schedule compared with 20% (2/10) and 40% (4/10) of mice treated with ch14.18/CHO and anti-PD-1 monotherapy, respectively. In the group of mice treated with ch14.18/CHO and anti-PD-1, only one mouse (1/10; 10%) developed a critical tumor volume ahead of schedule. These data support synergistic anti-tumor effects of the combinatorial treatment with both Ab.
This finding is supported by OS and EFS analyses. We found a significant difference between the combinatorial treatment group and controls for OS (Fig. 8D, left diagram). Ch.14.18/CHO treated mice showed also improved survival rates compared with controls. The difference in EFS was statistically significant. Also, treatment with anti-PD-1 Ab alone resulted in only slightly improved OS compared with the control group. Similar results were observed with EFS (event = tumor volume of ≥ 150 mm3) (Fig. 8D, right diagram). The EFS difference between the anti-PD-1 and control groups (Fig. 8D, right diagram) was significant. Importantly, the differences between the combinatorial group and controls in EFS probabilities were found to be extremely significant. These data clearly indicate that ch14.18/CHO-mediated anti-tumor effects were augmented by PD-1/PD-L1 checkpoint blockade.
Immune response following combined immunotherapy with ch14.18/CHO and anti-PD-1
The cytotoxic activity of effector cells and serum of experimental mice was analyzed at the end of the experiment (32 d after the tumor cell inoculation) in vitro against the murine GD2- and PD-L1-positive NB target cells NXS2-HGW (Fig. 9). The levels of anti-tumor cytotoxicity of mice treated with ch14.18/CHO at that late time point was 7% compared with untreated controls (3%) and mice that received anti-PD-1 Ab (6%). Importantly, the anti-tumor response in the combined group was 18% (Fig. 9). These data are in line with effects on primary tumor growth and survival.
Figure 9.
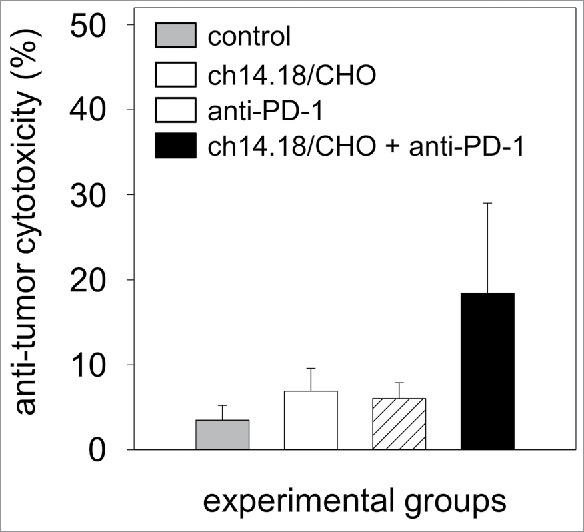
Anti-tumor effects of the combinatorial treatment with ch14.18/CHO and anti-PD-1 antibodies in vitro. Anti-tumor cytotoxicity mediated by effector cells (E:T ratio of 100:1) and serum (12.5% final concentration) of experimental mice was analyzed against murine GD2- and PD-L1-positive NB cells NXS2-HGW using calcein-AM-based cytotoxicity assay. Data are shown as mean values ± SEM of experiments performed at least in triplicate. Kruskal-Wallis test, differences between the groups were not significant.
In this experiment it is important to consider remaining ch14.18/CHO drug level on day 32 which is 24 d after the last ch14.18/CHO application. Therefore, we analyzed ch14.18/CHO levels as described previously.17,18 As expected, we did not detect ch14.18/CHO in the serum of ch14.18/CHO treated mice at this time point (Table 2). These results suggest that the anti-NB cytotoxicity observed on day 32 was not mediated by ch14.18/CHO and my therefore point toward the induction of an adaptive immune response.
Table 2.
Serum level of ch14.18/CHO. Concentration of anti-GD2 antibody ch14.18/CHO in serum of treated mice was evaluated 24 d after the last Ab injection ELISA.
| Experimental group | ch14.18/CHO concentration (µg/ml) |
|---|---|
| ch14.18/CHO + anti-PD-1 | 0.00 ± 0.01 |
| ch14.18/CHO | 0.01 ± 0.01 |
| anti-PD-1 | 0.00 ± 0.01 |
| control | 0.00 ± 0.01 |
Discussion
Although the application of anti-GD2 Ab ch14.18/CHO has demonstrated improved survival rates, not all NB patients benefit from the treatment. Thus, new ways to further optimize this approach are highly relevant. NB is a poorly immunogenic tumor characterized by low MHC class I expression and by low expression of the PD-1/PD-L1 inhibitory immune checkpoint (Fig. 1, 2).13 However, the combination of PD-L1 and MHC-I tumor cell density has been shown to be a prognostic biomarker for predicting OS in NB patients19 and NB patients with high PD-L1 expression had significantly better survival when high numbers of CD8-positive tumor-infiltrating lymphocytes were present.14 Therefore, anti-PD1/PD-L1 directed treatment strategies are reasonable approaches in NB, but based on the low expression less likely to succeed when used as monotherapy. The primary mechanism of action of ch14.18/CHO is the induction of ADCC, which is a process associated with the release of inflammatory cytokines.20 Inflammatory cytokines induce PD-L1 expression in NB21 and thereby, upregulate the PD-1/PD-L1 immune checkpoint in NB. In this context, interferon gamma has been shown to increase PD-L1 expression by NB and effector cells.12 Therefore, we hypothesized that PD-1/PD-L1 checkpoint inhibition may be synergistic to ch14.18/CHO immunotherapy.
In fact, we could demonstrate that ch14.18/CHO-mediated ADCC conditions are associated with upregulation of the PD-1/PD-L1 checkpoint in NB (Figs. 2–4), and that this upregulation was further augmented in the presence of IL-2. Based on these findings, upregulation of the PD-1/PD-L1 checkpoint can be anticipated in NB patients treated with ch14.18 and IL-2, which is the recommended treatment regimen.2 The clinical impact of PD-1/PD-L1 upregulation induced by this therapy is not clear at the moment, but is of concern, as we could demonstrate that the PD-1/PD-L1 checkpoint inhibited ch14.18/CHO-mediated ADCC (Fig. 5). However, we could also show that this inhibitory effect was reversed by anti-PD-1 therapy (Fig. 5). This observation was transferred to a syngeneic NB mouse model and revealed for the first time a synergistic effect combining ch14.18/CHO with anti-PD-1 (Figs. 8, 9).
Until now, the regulation mechanisms of tumor surface PD-L1 expression are not clearly understood. Recently, MYC- and MYCN-dependent effects on PD-L1 expression have been shown in NB both in vitro and in vivo.19 ADCC leads to a local inflammatory response including cytokine release that is a likely factor contributing to the PD-1/PD-L1 immune checkpoint upregulation in NB. However, a direct influence of CD11b-positive immune cells has been reported recently to be critical for PD-L1 expression by tumor cells.11 In murine melanoma, incubation of tumor cells with bone marrow cells resulted in induction of PD-L1 expression by tumor cells through CD11b-dependent cell-cell contact.11 Here we expand these findings and show for the first time that PD-L1 upregulation under ch14.18/CHO-mediated ADCC conditions is also CD11b-dependent (Fig. 6) and the inhibitory effect of the PD-1/PD-L1 immune checkpoint on ADCC is reversed in the presence of anti-CD11b Ab (Fig. 5)).
Moreover, anti-CD11b Ab also increased the ADCC response against low PD-L1 expressing LA-N-1 cells, suggesting an inhibitory effect of CD11b independently on the PD1/PD-L1 checkpoint. Interestingly, myeloid-derived suppressor cells (MDSCs) with immunosuppressive ability are characterized by expression of CD11b and have been shown to inhibit effector activity of NK- and T-cells.22 Moreover, MDSCs with immunosuppressive activity were found in cancer patients emphasizing their adverse role in tumor immune surveillance. Thus, CD11b-positive MDSCs represent potential candidates responsible for effects observed in our experiments. Beyond that, our present data suggest that CD11b may be a promising target in combinatorial immunotherapy not only by preventing immunotherapy-dependent induction of PD-1/PD-L1 immune checkpoints but also by reducing direct inhibitory effects delivered by CD11b-positive immunosuppressive cells.
Another interesting observation is the cytotoxic activity observed in mice treated with the ch14.18/CHO and anti-PD1 combination at a time point where ch14.18/CHO was not detectable in the serum of treated mice (Fig. 9). It is not clear at the moment if this result points toward the induction of a long lasting adaptive immunity.
In summary, we could demonstrate for the first time that a ch14.18/CHO-based immunotherapy upregulates the PD-1/PD-L1 immunosuppressive checkpoint in NB that is further augmented by IL-2. Importantly, this CD11b-dependent effect was reversed by co-administration of anti-PD-1 Ab, which translated to an increased anti-NB immune response in vitro and in vivo. Based on these data, the combination of ch14.18/CHO with anti-PD1 Ab is a promising strategy for clinical evaluation in NB and other GD2-positive malignancies.
Materials and methods
Ethic statement
All procedures involving human participants were in accordance with the ethical standards of the institutional and national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants. All procedures involving animal experiments were approved by the animal welfare committee (Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei Mecklenburg-Vorpommern, LALLF M-V/7221.3–1–049/15) and approved and supervised by the commissioner for animal welfare at the University Medicine Greifswald representing the Institutional Animal Care and Use Committee (IACUC).
Cell lines
The murine NB cell lines NXS223 and NXS2-HGW were cultured in DMEM (Capricorn Scientific GmbH, DMEM-HPSTA) supplemented with 4.5 g/l glucose, 2 mM stable glutamine, 10% FCS (Capricorn Scientific GmbH, FBS-12A) and 100 U/ml penicillin and 0.1 mg/ml streptomycin (1x P/S; PAN BIOTECH, P06–07100). Human GD2-positive NB cell lines CHLA-20, LA-N-1 and LA-N-6 were cultured in RPMI (PAN BIOTECH, P04–016520) supplemented with 4.5 g/l glucose, 2 mM stable glutamine, 10% FCS and 1x P/S. Human primary GD2-positive NB cell lines from patients with relapsed NB HGW-1, HGW-2, HGW-3 and HGW-5 15 were cultured in IMDM (PAN BIOTECH, P04–20250) supplemented with 4 mM stable glutamine, 20% FCS, 1x ITS (BD Biosciences, 3220669) and 1x P/S. Human GD2-negative NB cell line SK-N-SH was cultured in DMEM supplemented with 4.5 g/l glucose, 2 mM stable glutamine, 10% FCS and 1x P/S.
Prior to cultivation, mycoplasma contamination analysis was performed for every cell line using MYCOALERT detection Kit (Lonza Cologne GmbH, LT07–318). Only mycoplasma negative cell lines were used for experiments. All cell lines were passaged not more than 30 times. To exclude a cell cross-contamination, short tandem repeat (STR) analysis was performed regularly for each cell line used.
Mice
Analysis of anti-tumor effects of the combinatorial immunotherapy with ch14.18/CHO (Polymun Scientific) and anti-PD-1 mAb (InVivoMab, Bio X Cell, BE0146) was performed using lethal syngeneic mouse NB model established in our laboratory.23 The murine MYCN- and TH-positive NB cells NXS2-HGW expressing GD2 and PD-L1 were used for tumor inoculation (subcutaneously, 3 × 106 cells, ≥ 95% viability) in female A/J mice (10 weeks of age; Charles River Laboratories, Sulzfeld, Germany). Mice were housed in standard animal laboratories (12 h light/dark cycle) with free access to water and standard laboratory chow ad libitum. Mice were treated intraperitoneally either with 300 µg of ch14.18/CHO per day beginning on day 4 for 5 consecutive days or every fourth day with 250 µg of anti-PD-1 mAb per day beginning on day 7 after tumor inoculation (Fig. 8A). The combined group received ch14.18/CHO in combination with PD-1 blockade as described for the respective single treatment group. Rituximab and rat IgG2a (InVivoMab, Bio X Cell, BE0089) served as isotype controls. Controls received equivalent volume of 0.9% NaCl. To determine tumor volume and clinical signs of tumor burden, mice were examined daily 4 d after tumor cell inoculation. Primary tumor volume was calculated according to the formula: (length × width × height)/2. To show anti-tumor effects, tumor growth and tumor volume at the end of the experiment (day 32) for each group were analyzed. Tumor volume of mice killed ahead of schedule was also included using the data of the last measurement before removal. In such a lethal model we defined further parameters that define the well-being of mice: changes in body weight, behavior and physical appearance and appearance of metastases. These signs and symptoms were used to create a score (0 for none, 1 - low, 2 - medium and 3 - high) to assess mice in the experiment as a requirement of our internal review board supervising animal experiments. Mice were removed from the experiment with a score >7 or when tumors exceeded a volume of 1000 mm3. An additional parameter for evaluation of anti-tumor effects is the time to development of a primary tumor of 150 mm3. This tumor volume correlates with suffering and exponential tumor growth in experimental mice. Therefore, we defined development of tumor volume of 150 mm3 as event as described previously.24 Finally, 4 d after the last anti-PD-1 application (day 32) blood samples were collected and mice were killed followed by isolation of splenocytes for evaluation of anti-tumor cytotoxicity as described below.
PD-L1, MYCN- and TH mRNA expression by NB cells
To investigate expression of PD-L1 by human NB cells (CHLA-20, LA-N-1, LA-N-6, SK-N-H, HGW-1, -2, -3 and -5) PD-L1, MYCN and TH by the murine NB cells NXS2-HGW, we used gene-specific primers (Table 1) that were designed with the online primer design tool Primer3 or recently published.25 Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as internal control.
Table 1.
Primer sequences. Gene-specific primer sequences for murine MYCN (mMYCN), murine tyrosine hydroxylase (mTH), murine and human PD-L1 (mPD-L1 and hPD-L1, respectively). Murine and human GAPDH served as internal control, respectively.
| Gene | Primer | Sequence (5′ - 3′) | Product size (bp) |
|---|---|---|---|
| mMYCN | forward | GCTGCGGTCACTAGTGTGTC | 248 |
| reverse | CGCACAGTGATCGTGAAAGT | ||
| mTH | forward | GCCGTCTCAGAGCAGGATAC | 175 |
| reverse | CGAATACCACAGCCTCCAAT | ||
| mPD-L1 | forward | CTGCCAAAGGACCAGCTTTT | 180 |
| reverse | GGCTGGATCCACGGAAATTC | ||
| mGAPDH | forward | AACTTTGGCATTGTGGAAGG | 223 |
| reverse | ACACATTGGGGGTAGGAACA | ||
| hPD-L1 nested | forward | CAAGGCCGAAGTCATCTGGA | 456 |
| reverse | CCCGATGAACCCCTAAACCA | ||
| hPD-L125 | forward | TGGTGTAGCACTGACATTCA | 133 |
| reverse | TCCAATGCTGGATTACGTCT | ||
| hGAPDH | forward | GAGTCAACGGATTTGGTCGT | 238 |
| reverse | TTGATTTTGGAGGGATCTCG |
First, total RNA was isolated from 5 × 106 NB cells using the Nucleo Spin ® RNA II Kit (Macherey Nagel, 740955.250) and concentration was determined spectrophotometrically (Bio-Photometer plus, Eppendorf, Hamburg, Germany). Next, 2 μg of total RNA were used for cDNA synthesis with qScript™ cDNA Synthesis Kit (Quanta Biosciences™ Beverly, 95047) according to the manufacturer's guidelines.
MYCN was amplified by 28 cycles consisting of +94°C (15 s) for denaturation, +55°C (15 s) for primer-specific annealing and +72°C (30 s) for elongation. The PCR profile for TH and GAPDH was: +94°C for 15 s (denaturation), +56°C for 15 s (annealing) and +72°C for 15 s (elongation) for 25 and 22 cycles, respectively. For amplification of PD-L1, 35 PCR cycles of +94°C for 15 s (denaturation), +60°C for 15 s (annealing) and +72°C for 15 s (elongation) were performed. To analyze expression of PD-L1 in human NB cells, we developed a gene-specific nested PCR. In the first reaction, a set of external primers (hPD-L1 nested, Table 1) was used for amplification of a 456 bp DNA fragment. The PCR profile was: 35 cycles of +94°C (15 s) for denaturation, +39°C (15 s) for primer-specific annealing and +72°C (30 s) for elongation. In the second reaction, 1 µl of the 1:200 diluted product of the first PCR served as template to amplify a 133 bp DNA fragment using a set of internal primers25 (hPD-L1, Table 1) by 20 cycles of +94°C for 15 s (denaturation), +40°C for 15 s (annealing) and +72°C for 30 s (elongation). Finally, PCR products were analyzed by agarose gel electrophoresis and mRNA expression was quantified relative to GAPDH using densitometric analysis (ImageJ 1.48v).
Flow cytometry
GD2 expression by NB cells. For flow cytometric analysis of GD2 surface expression by human and murine NB cell lines, cells were harvested, cell viability was determined and then 1 × 106 cells were washed with 500 µl wash buffer (1x PBS, 1% BSA, 0.1% NaN3, 0.1% EDTA, pH 7.4) (300x g, 5 min, RT). After supernatant was discarded, cells were resuspended in 50 µl wash buffer followed by incubation with 1 µl FcR blocking reagent for 10 min at +4°C. Cells were then stained with 1 μg of ch14.18/CHO mAb in a total volume of 100 µl for 20 min at +4°C. The anti-CD20 chimeric Ab rituximab was used as isotype control. After washing once with 1 ml wash buffer, cells were incubated with 0.1 µg PE-labeled anti-human IgG Ab (Biolegend, 409304) in a total volume of 100 µl for 20 min at 4°C in the dark, washed again and resuspended in 500 µl wash buffer. To exclude dead cells from the analysis, 4 µl of a 0.1 mg/ml 4′,6-diamino-2-phenylindole (DAPI) solution (Sigma-Aldrich, D9542) were added 5 min before acquisition. For each sample, 20,000 live cells were analyzed using a BD CANTO II cytometer and FACS Diva software (BD Biosciences, Heidelberg, Germany). Data were analyzed with FlowJo V10 software (Ashland, OR, USA) using geometric mean fluorescence intensity (MFI). The maximum scaling of cell number in histograms was achieved by normalizing to the mode of the distribution.
PD-L1 expression by murine NB cells
Analysis of PD-L1 expression by the murine NB cells was performed in analogy to the GD2-detection using anti-murine PE-labeled anti-PD-L1 Ab (1:80) (Biolegend, 124308) and the respective isotype control (rat IgG2b, κ-PE (1:80) (Biolegend, 400607). The comparison of PD-L1 expression levels was performed using relative geometric mean fluorescence intensity (rgMFI) according to the formula: MFI of PD-L1/MFI of isotype control.
PD-L1 expression by human NB- and effector cells
Analysis of PD-L1 expression by the human NB cells LA-N-1 and 3 leukocyte populations (granulocytes, monocytes and lymphocytes) was performed similar to the analysis of PD-L1 expression by the murine NB cells using anti-human PE-labeled anti-PD-L1 Ab (1:80) (Biolegend, 329706). To clearly distinguish between PD-L1-positive NB cells (PD-L1+/GD2+/CD45−) and leukocytes (PD-L1+/GD2−/CD45+), Alexa 647-labeled anti-GD2 Ab 14G2a (1:790) (Biolegend) and anti-human PE/Cy7-labeled anti-CD45 mAb (1:30) (Beckman Coulter, IM3548) were used, respectively. To distinguish between granulocytes, monocytes and lymphocytes, a gating strategy based on cell granularity and intensity of the CD45 fluorescence of GD2-negative cells was used. As isotype controls served IgG2b, κ-PE (1:80) (Biolegend, 400312), IgG2a-Alexa Fluor 647 (1:160) and IgG1-PE/Cy7 (1:30) (Biolegend, 400126). The evaluation of PD-L1 baseline expression or comparison of PD-L1 levels after induction of ADCC as well as treatment with IL-2, PD-1 and CD11b was performed using geometric mean fluorescence intensity (rgMFI) relative to either isotype control or untreated cells, according to the formula: MFI of PD-L1/MFI of isotype control for baseline expression level and MFI of PD-L1 by treated cells - MFI of PD-L1 by the respective untreated control for evaluation of ADCC-mediated effects.
PD-1 expression by human NB- and effector cells
Expression of PD-1 by human NB- and effector cells was performed similar to the PD-L1 analysis using anti-human PE-labeled anti-PD-1 mAb (1:20; Biolegend, 329906). To distinguish between PD-1-positive NB- (PD-1+/GD2+/CD45−) and effector cells (PD-1+/GD2−/CD45+), Alexa Fluor 647-labeled anti-GD2 Ab 14G2a and anti-human PE/Cy7-labeled anti-CD45 Ab (1:300) (Beckman Coulter, IM3548), respectively were additionally used. Mouse IgG1, κ-PE (1:80) (Biolegend, 400112) served as isotype control for anti-PD-1 Ab. The evaluation of PD-1 baseline expression or comparison of PD-1 levels after induction of ADCC was performed using geometric mean fluorescence intensity (rgMFI) relative to either isotype control or untreated cells, according to the formula: MFI of PD-1/MFI of isotype control for baseline expression level and MFI of PD-1 by treated cells - MFI of PD-1 by the respective untreated control for evaluation of ADCC-mediated effects.
Effects of ADCC conditions on PD-1 and PD-L1 expression by neuroblastoma cells and leukocytes
To investigate impact of ch14.18/CHO-mediated ADCC on PD-L1 expression, the GD2-positive NB cells LA-N-1 were incubated with effector cells and ch14.18/CHO for 24 h. Effector cells were first isolated from sodium-heparin blood samples of a healthy donor using erythrocyte lysis buffer (10 min, RT, 0.15 M ammonium chloride, 10 mM potassium bicarbonate, 0.1 mM ethylenediaminetetraacetic acid, pH 7.4) and washed twice (1x PBS, 5 min, 300x g, RT), followed by cell counting and evaluation of cell viability. Then, 0.5 × 106 LA-N-1 cells were harvested and incubated with effector cells at effector-to-target (E:T) ratio of 10:1 for 24 h in the presence of ch14.18/CHO at the final concentration of 10 ng/ml. We used rituximab instead of ch14.18/CHO as negative control. Moreover, we evaluated impact of IL-2 using cell culture medium supplemented with 100 IU/ml of IL-2 (Novartis). Additionally, the effect of ch14.18/CHO on NB- and effector cells as well as effects of NB cells on effector cells and vice versa of effector cells on NB cells were investigated. Finally, to confirm ch14.18/CHO-mediated impact of ADCC, incubation with the anti-idiotype ganglidiomab16 was additionally performed.
By analogy with analysis of PD-L1, ADCC-mediated effects on PD-1 expression were investigated using NB- and effector cells incubated with 10 ng/ml ch14.18/CHO for 24 h with and without IL-2 as well as the respective controls as described for PD-L1.
Prior to flow cytometry analysis, NB- and effector cells were harvested, washed twice with 1 × PBS (pH 7.4, 300 × g, 5 min, RT) and stained as described above.
Analysis of impact of CD11b blockade on PD-L1 expression
Based on the fact that induction of PD-L1 expression by tumor cells has been shown to implicate CD11b-positive effector cells,11 we addressed whether blockade of CD11b can negatively affect ADCC-mediated induction of PD-L1 expression. For this, human NB cells LA-N-1 and leukocytes of a healthy donor (10:1 E:T ratio) were incubated for 24 h with ch14.18/CHO (10 ng/ml final concentration) and anti-CD11b (2 µg/ml final concentration) (Biolegend, 301312) followed by flow cytometry evaluation of PD-L1 expression and subsequent analysis of ADCC as described below. Experiments without blockade with anti-CD11b Ab served as controls.
PD-1/PD-L1 checkpoint blockade and role of CD11b in ADCC
To investigate whether blockade of PD-1 could result in decreased inhibitory impact of PD-1/PD-L1 pathway on effector cell cytotoxicity and, thus augment ADCC, we compared ch14.18/CHO-dependent cytotoxic activity of effector cells against NB cells expressing high levels of PD-L1 with and without blockade of PD-1 (10 µg/ml final concentration) (Nivolumab, OPDIVI®, Bristol-Myers Squibb). To induce high levels of PD-L1, LA-N-1 cells were first cultivated for 24 h under ADCC conditions (co-incubation with leukocytes at E:T ratio of 10:1 and subtherapeutic ch14.18/CHO concentration of 10 ng/ml). Then, LA-N-1 were harvested and washed twice (1x PBS, 5 min, 300x g, RT), followed by cell counting and analysis of cell viability. After cells were incubated with a calcein-acetoxymethyl ester as described previously,15,16 ADCC was evaluated 4 h after co-incubation of PD-L1 high expressing LA-N-1 with ch14.18/CHO (final concentration: 10 µg/ml) and leukocytes (E:T ratio of 40:1) that were cultivated in RPMI supplemented with IL-2 for 24 h. Additionally, untreated LA-N-1 expressing low levels of PD-L1 and rituximab served as controls.
Finally, we investigated whether blockade of CD11b could result in decreased PD-L1 expression by tumor cells and, thus augment ADCC. For this, human NB cells LA-N-1 and leukocytes of a healthy donor (10:1 E:T ratio) were incubated for 24 h with ch14.18/CHO (10 ng/ml final concentration) and anti-CD11b mAb (2 µg/ml final concentration) (Biolegend, 301312) followed by analysis of ADCC as described below. Untreated LA-N-1 and rituximab served as controls.
Determination of ch14.18/CHO concentration in serum
Specific detection of ch14.18/CHO in serum samples collected from experimental mice was performed by Enzyme-linked Immunosorbent Assay (ELISA) as described previously.17,18 Anti-idiotype Ab ganglidiomab16 (BioGenes GmbH) was used as a capture and the peroxidase-conjugated anti-human IgG (Fc-specific) as a secondary Ab (Sigma Aldrich, A0170). Absorption was analyzed at 450 nm in a plate reader (BioTek Instruments GmbH, SIAFRTD).
Development of the NXS2 HGW cell line
Prior to inoculation, the previously established A/J mice syngeneic NB cell line NXS223 was repeatedly in vivo passaged. For this, NXS2 cells were harvested and washed twice with 1x PBS (pH 7.4, PAN BIOTECH, P04–36500). Then, 3 × 106 NXS2 cells were inoculated subcutaneously into left flank of A/J mice. The mice were examined daily and the resulting tumors were surgically removed when they reached a maximum volume of 1000 mm3. The tumors were then mechanically dispersed using 70 µm cell strainer (BD Biosciences, Heidelberg, Germany) and washed twice with 10 ml 1x PBS supplemented with 0.5% BSA (Sigma Aldrich, A9647) and 2 mM EDTA (Sigma Aldrich, CN06.1) (pH 7.4, 5 min, 300x g, RT). After 2 wash steps (1x PBS/0.5% BSA/2 mM EDTA buffer; pH 7.4; 5 min, 300x g, RT) cell pellets were resuspended in 10 ml 1x PBS and layered over 10 ml of Lymphocytes Separation Media (Capricorn Scientific GmbH, LSM-A). After centrifugation (30 min, 300x g, RT without brake) the upper layer was discarded, a layer of primary cells was carefully transferred into a new tube and the remaining Lymphocyte Separation Medium was washed off with 1x PBS/0.5% BSA/2 mM EDTA buffer (pH 7.4; 10 ml, 5 min, 300x g, RT, brake on). Thereafter, the supernatant was carefully removed and the cell pellet was resuspended with 10 ml 1x PBS/0.5% BSA/2 mM EDTA buffer (pH 7.4) followed by cell counting and evaluation of cell viability. Finally, 1 × 107 vital cells were used for isolation of GD2-positive cell subset with MACS technique.
For this purpose, 1 × 107 primary cells were resuspended in 200 µl 1x PBS/0.5% BSA/2 mM EDTA buffer (pH 7.4) and then incubated on ice with 20 µl FcR-blocking reagent (Miltenyi Biotec, 120–003–855) for 5 min. Thereafter, the murine anti-GD2 Ab 14G2a (1.0 mg) was added followed by incubation for 10 min on ice. After washing with 1x PBS/0.5% BSA/2 mM EDTA buffer (15 ml, pH 7.4, 5 min, 300x g, +4°C) cell pellet was resuspended in 100 µl 1x PBS/0.5% BSA/2 mM EDTA buffer and 20 µl magnetic microbeads conjugated with anti-mouse IgG (Miltenyi Biotec, 130–048–401) were added followed by incubation on ice for 15 min. Next, the cells were washed again by adding 15 ml 1x PBS/0.5% BSA/2 mM EDTA buffer (pH 7.4) and centrifuged at 300x g for 5 min at +4°C. 1 ml of 1x PBS/0.5% BSA/2 mM EDTA buffer was used to resuspend the cell pellet and magnetic separation of GD2-positive cells was proceeded in MACS-separator (Miltenyi Biotec, Teterow, Germany) by applying the cell suspension onto the LS-separation column (Miltenyi Biotec, Teterow, Germany). The column was washed 3 times (3 ml, 1x PBS/0.5% BSA/2 mM EDTA buffer, pH 7.4) and then removed from the MACS-separator and placed into a collection tube. Cell fraction containing magnetically labeled GD2-positive cells were immediately flushed out by firmly applying the plunger supplied with the column (Miltenyi Biotec, Teterow, Germany). Finally, primary cells were washed with 1x PBS (pH 7.4, 5 min, 300x g, RT), the cell pellet was resuspended in NXS2 cell culture medium as described above and the cells were cultivated for 6 weeks. Prior to the next step of the in vivo passage procedure, GD2-expression was confirmed using flow cytometry as described below. We repeated the in vivo passage procedure 3 times using for the last step metastasis-derived cells instead of primarily tumor cells. The NXS2-derived cell line was called “NXS2-HGW." Finally, analysis of typical NB marker GD2, MYCN and TH was performed. Furthermore, PD-L1 expression by NXS2-HGW cells was additionally evaluated.
Immunohistochemical analysis of GD2-expression by NXS2-HGW cells.
Prior to immunohistochemical evaluation of GD2 expression by NXS2-HGW cells, we first characterized morphology of this cell line. For this, NXS2-HGW cells were cultivated in chamber slide flasks (Nunc, Roskilde, Denmark) for 5 d. After the cell layer confluence of 90% was achieved, cell culture medium was removed, cells were washed once with 1x PBS (pH 7.4) and 4,5% formaldehyde (pH 7, 20 min; RT) was used for cell fixation followed by 3 wash steps with 1x PBS (pH 7.4, 3 min). For immunohistochemically staining of GD2, samples were incubated with 3% H2O2 (Roth, 8070.2) for 20 min at RT, washed 3 times and treated with 3% BSA/PBS for 30 min at RT. Next, samples were again washed 3 times and incubated with 50 µg/ml 14G2a for 1 h at RT followed by another 3 wash steps. Then, peroxidase-conjugated secondary Ab goat anti-mouse IgG (1:400) (Sigma Aldrich, A0170) was incubated for 10 min at RT and afterwards removed by 3 wash steps. 3,3' diaminobenzidine (DAB) Substrate Kit (BD Biosciences, 550880) was used as chromogen. The samples were finally counterstained with Mayer's hemalum solution (Merck Chemicals GmbH, T865.3) for 2 min and examined by light microscopy (BX53 System Microscope, Olympus, Hamburg, Germany). Rituximab (MabThera®, Roche) served as isotype control.
Additionally, we evaluated GD2 expression in NXS2-HGW-derived primary tumor tissue. For this, NXS2-HGW cells were subcutaneously injected into A/J mice. 14 d after tumor inoculation, primary tumors were surgically rejected, tumor samples were frozen in liquid nitrogen using O.C.T. compound medium (VWR, 361603E) and stored at −80°C until used. Frozen tumor tissue samples were then cut (5–6 µm) with the cryomicrotome at −20°C, collected on SuperFrost®Plus microscope slides (R. Langenbrinck, Emmendingen, Germany) and fixed using 4,5% formaldehyde (pH 7, 20 min; RT). The tissue sections were then immediately used or stored frozen until needed at −20°C. The analysis of GD2 expression was performed as described above for the cell cultivation in chamber slide flask.
Determination of anti-tumor cytotoxicity in mice
To evaluate anti-NB cytotoxicity, serum and lymphocytes of immunized mice were incubated with murine PD-L1- and GD2-positive NB cells NXS2-HGW that were used as target cells. Serum and lymphocytes were prepared from whole blood and spleen samples, respectively, collected at the end of the in vivo experiment. For serum, whole blood was collected in BD Vacutainer plastic serum tubes followed by centrifugation at 1,700x g for 10 min at RT. For lymphocytes, spleens were mechanically dispersed using 70 µm cell strainer (BD Biosciences, Heidelberg, Germany) and washed twice with 10 ml 1x PBS (pH 7.4, 5 min, 300x g, RT). Cell pellets were then resuspended in 10 ml 1x PBS and layered over 10 ml of Lymphocytes Separation Media. After centrifugation (30 min, 300x g, RT without brake) the upper layer was discarded, a layer of primary cells was carefully transferred into a new tube and the remaining Lymphocyte Separation Medium was washed off with 1x PBS (pH 7.4; 10 ml, 5 min, 300x g, RT, brake on). Thereafter, the supernatant was carefully removed and the cell pellet was resuspended with culture medium (RPMI supplemented with 4.5 g/l glucose, 2 mM stable glutamine, 10% FCS, 1x P/S, 50 µM 2-mercaptoethanol (Sigma Aldrich, M7522) and 100 IU/ml rIL-2) followed by cell counting, evaluation of cell viability and cultivation for 72 h. Finally, effector cells were harvested and used for evaluation of anti-NB cytotoxicity with acetoxymethyl ester of calcein (calcein-AM) (Sigma Aldrich, 17783) assay as described previously.16 Briefly, calcein labeling of target cells (NXS2-HGW, 1 × 106/ml) was accomplished by incubation with 10 µM calcein-AM (30 min, +37°C, 100 rpm). After 5 wash steps (+37°C), cells were added to a 96-well plate and incubated for 4 h at +37°C with 12.5% mouse serum and effector cells (E:T ratio of 100:1). Next, supernatants (50 µl) of each well were transferred to a black 96-well plate (VWR, Darmstadt, Germany) for determination of fluorescence at 495 nm excitation and 515 nm emission wavelengths by a Synergy HT multimode microplate reader (BioTek Germany, Bad Friedrichshall, Germany). Experiments were analyzed in triplicates using at least 6 replicate wells for spontaneous (target cells only) and maximum release (target cells treated for 20 s with ultrasonic homogenizer) (Hielscher Ultrasonics, Teltow, Germany). Cytotoxicity in percent was calculated according to the formula: (test release - spontaneous release)/(maximum release - spontaneous release) × 100%.
Statistic
Statistical analysis was performed using Sigma Plot software (Jandel Scientific Software, San Rafael, CA, USA). After testing for normality, differences between independent groups were assessed using unpaired t-test or the non-parametric Mann-Whitney U test. For more than 2 groups, the non-parametric Kruskal-Wallis test was used followed by appropriate post hoc comparison. A P value of < 0.05 was considered significant (*), P < 0.01 very significant (**) and P < 0.001 extremely significant (***). All data are presented as mean ± SEM (standard error of the mean). Survival probabilities were estimated using Kaplan Meier analysis and compared using LogRank statistics (Mantel-Cox). For OS and EFS, death ahead of schedule and a tumor volume of 150 mm3 representing a well-established tumor were defined as event, respectively.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Maria Asmus, Manuela Brüser, Christin Eger and Theodor Koepp (University Medicine Greifswald, Pediatric Hematology and Oncology, Greifswald, Germany) for excellent technical assistance. Financial support was provided by the University Medicine Greifswald, Deutsche Forschungsgemeinschaft (DFG), Germany, H.W. & J. Hector Stiftung, Germany, Apeiron Biologics, Vienna, Austria, and Deutsche Kinderkrebsstiftung, Germany.
Funding
Financial support was provided by the University Medicine Greifswald, Deutsche Forschungsgemeinschaft (DFG), Germany under Grant SI-2147/1–1, H.W. & J. Hector Stiftung, Germany under Grant M57, Apeiron Biologics, Vienna, Austria under Grant APN, and Deutsche Kinderkrebsstiftung, Germany under Grant DKS 2014.05 A/B.
References
- 1.Matthay KK, George RE, Yu AL. Promising therapeutic targets in neuroblastoma. Clin Cancer Res 2012; 18:2740-53; PMID:22589483; https://doi.org/ 10.1158/1078-0432.CCR-11-1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med 2010; 363:1324-34; PMID:20879881; https://doi.org/ 10.1056/NEJMoa0911123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ladenstein R, Weixler S, Baykan B, Bleeke M, Kunert R, Katinger D, Pribill I, Glander P, Bauer S, Pistoia V, et al. Ch14.18 antibody produced in CHO cells in relapsed or refractory Stage 4 neuroblastoma patients: A SIOPEN Phase 1 study. MAbs 2013; 5:801-9; PMID:23924804; https://doi.org/ 10.4161/mabs.25215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siebert N, Jensen C, Troschke-Meurer S, Zumpe M, Juttner M, Ehlert K, Kietz S, Müller I, Lode HN. Neuroblastoma patients with high-affinity FCGR2A, -3A and stimulatory KIR 2DS2 treated by long-term infusion of anti-GD2 antibody ch14.18/CHO show higher ADCC levels and improved event-free survival. Oncoimmunology 2016; 5:e1235108; PMID:27999754; https://doi.org/ 10.1080/2162402X.2016.1235108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeng Y, Fest S, Kunert R, Katinger H, Pistoia V, Michon J, Lewis G, Ladenstein R, Lode HN. Anti-neuroblastoma effect of ch14.18 antibody produced in CHO cells is mediated by NK-cells in mice. Mol Immunol 2005; 42:1311-9; PMID:15950727; https://doi.org/ 10.1016/j.molimm.2004.12.018 [DOI] [PubMed] [Google Scholar]
- 6.Koehn TA, Trimble LL, Alderson KL, Erbe AK, McDowell KA, Grzywacz B, Hank JA, Sondel PM. Increasing the clinical efficacy of NK and antibody-mediated cancer immunotherapy: potential predictors of successful clinical outcome based on observations in high-risk neuroblastoma. Front Pharmacol 2012; 3:91; PMID:22623917; https://doi.org/ 10.3389/fphar.2012.00091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lode HN, Jensen C, Endres S, Pill L, Siebert N, Kietz S, et al. Immune activation and clinical responses following long-term infusion of anti-GD2 antibody ch14.18/CHO in combination with interleukin-2 in high-risk neuroblastoma patients. J clin oncol 2015; 32:5. [Google Scholar]
- 8.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015; 27:450-61; PMID:25858804; https://doi.org/ 10.1016/j.ccell.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Dam LS, de Zwart VM, Meyer-Wentrup FA. The role of programmed cell death-1 (PD-1) and its ligands in pediatric cancer. Pediatr Blood Cancer 2015; 62:190-7; PMID:25327979; https://doi.org/ 10.1002/pbc.25284 [DOI] [PubMed] [Google Scholar]
- 10.Wang X, Teng F, Kong L, Yu J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther 2016; 9:5023-39; PMID:27574444; https://doi.org/ 10.2147/OTT.S105862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noh H, Hu J, Wang X, Xia X, Satelli A, Li S. Immune checkpoint regulator PD-L1 expression on tumor cells by contacting CD11b positive bone marrow derived stromal cells. Cell Commun Signal 2015; 13:14; PMID:25889536; https://doi.org/ 10.1186/s12964-015-0093-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boes M, Meyer-Wentrup F. TLR3 triggering regulates PD-L1 (CD274) expression in human neuroblastoma cells. Cancer Lett 2015; 361:49-56; PMID:25697485; https://doi.org/ 10.1016/j.canlet.2015.02.027 [DOI] [PubMed] [Google Scholar]
- 13.Dondero A, Pastorino F, Della CM, Corrias MV, Morandi F, Pistoia V, Olive D, Bellora F, Locatelli F, Castellano A, et al. PD-L1 expression in metastatic neuroblastoma as an additional mechanism for limiting immune surveillance. Oncoimmunology 2016; 5:e1064578; PMID:26942080; https://doi.org/ 10.1080/2162402X.2015.1064578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chowdhury F, Dunn S, Mitchell S, Mellows T, Ashton-Key M, Gray JC. PD-L1 and CD8+PD1+ lymphocytes exist as targets in the pediatric tumor microenvironment for immunomodulatory therapy. OncoImmunology 2015; 4:e1029701; https://doi.org/ 10.1080/2162402X.2015.1029701 [DOI] [Google Scholar]
- 15.Siebert N, Seidel D, Eger C, Juttner M, Lode HN. Functional bioassays for immune monitoring of high-risk neuroblastoma patients treated with ch14.18/CHO anti-GD2 antibody. PLoS One 2014; 9:e107692; PMID:25226154; https://doi.org/ 10.1371/journal.pone.0107692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lode HN, Schmidt M, Seidel D, Huebener N, Brackrock D, Bleeke M, Reker D, Brandt S, Mueller HP, Helm C, et al. Vaccination with anti-idiotype antibody ganglidiomab mediates a GD(2)-specific anti-neuroblastoma immune response. Cancer Immunol Immunother 2013; 62:999-1010; PMID:23591980; https://doi.org/ 10.1007/s00262-013-1413-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siebert N, Seidel D, Eger C, Brackrock D, Reker D, Schmidt M, Lode HN. Validated detection of anti-GD2 antibody ch14.18/CHO in serum of neuroblastoma patients using anti-idiotype antibody ganglidiomab. J Immunol Methods 2013; 398-399:51-9; PMID:24055592; https://doi.org/ 10.1016/j.jim.2013.09.008 [DOI] [PubMed] [Google Scholar]
- 18.Siebert N, Eger C, Seidel D, Juttner M, Zumpe M, Wegner D, Kietz S, Ehlert K, Veal GJ, Siegmund W, et al. Pharmacokinetics and pharmacodynamics of ch14.18/CHO in relapsed/refractory high-risk neuroblastoma patients treated by long-term infusion in combination with IL-2. MAbs 2016; 8:604-16; PMID:26785755; https://doi.org/ 10.1080/19420862.2015.1130196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melaiu O, Mina M, Chierici M, Boldrini R, Jurman G, Romania P, D'Alicandro V, Benedetti MC, Castellano A, Liu T, et al. PD-L1 Is a therapeutic target of the bromodomain inhibitor JQ1 and, combined with HLA Class I, a promising prognostic biomarker in neuroblastoma. Clin Cancer Res 2017; PMID:28270499; https://doi.org/ 10.1158/1078-0432.CCR-16-2601 [DOI] [PubMed] [Google Scholar]
- 20.Kircheis R, Halanek N, Koller I, Jost W, Schuster M, Gorr G, Hajszan K, Nechansky A. Correlation of ADCC activity with cytokine release induced by the stably expressed, glyco-engineered humanized Lewis Y-specific monoclonal antibody MB314. MAbs 2012; 4:532-41; PMID:22665069; https://doi.org/ 10.4161/mabs.20577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagner LM, Adams VR. Targeting the PD-1 pathway in pediatric solid tumors and brain tumors. Onco Targets Ther 2017; 10:2097-106; PMID:28442918; https://doi.org/ 10.2147/OTT.S124008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Y, Wu T, Shao S, Shi B, Zhao Y. Phenotype, development, and biological function of myeloid-derived suppressor cells. Oncoimmunology 2016; 5:e1004983; PMID:27057424; https://doi.org/ 10.1080/2162402X.2015.1004983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lode HN, Xiang R, Varki NM, Dolman CS, Gillies SD, Reisfeld RA. Targeted interleukin-2 therapy for spontaneous neuroblastoma metastases to bone marrow. J Natl Cancer Inst 1997; 89:1586-94; PMID:9362156; https://doi.org/ 10.1093/jnci/89.21.1586 [DOI] [PubMed] [Google Scholar]
- 24.Seidel D, Shibina A, Siebert N, Wels WS, Reynolds CP, Huebener N, Lode HN. Disialoganglioside-specific human natural killer cells are effective against drug-resistant neuroblastoma. Cancer Immunol Immunother 2015; 64:621-34; PMID:25711293; https://doi.org/ 10.1007/s00262-015-1669-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang M, Meng Q, Guo H, Wang L, Zhao Z, Zhang L, Kuang J, Cui Y, Mai L, Zhu J. Programmed Death 1 (PD-1) is involved in the development of proliferative diabetic retinopathy by mediating activation-induced apoptosis. Mol Vis 2015; 21:901-10; PMID:26321864 [PMC free article] [PubMed] [Google Scholar]