

ABSTRACT
Checkpoint inhibitors, monoclonal antibodies that inhibit PD-1 or CTLA-4, have revolutionized the treatment of multiple cancers. Despite the enthusiasm for the clinical successes of checkpoint inhibitors, and immunotherapy, in general, only a minority of patients with specific tumor types actually benefit from treatment. Emerging evidence implicates epigenetic alterations as a mechanism of clinical resistance to immunotherapy. This review presents evidence for that association, summarizes the epi-based mechanisms by which tumors evade immunogenic cell death, discusses epigenetic modulation as a component of an integrated strategy to boost anticancer T cell effector function in relation to a tumor immunosuppression cycle and, finally, makes the case that the success of this no-patient-left-behind strategy critically depends on the toxicity profile of the epigenetic agent(s).
KEYWORDS: Immunotherapy, immunosuppression, immunogenic cell death, epigenetic modulation, checkpoint inhibitors, resistance
Introduction
Strictly speaking, the immunotherapy revolution began, not on the revolutionary date of March 25th 2011 with the approval of Ipilimumab (Yervoy) the first checkpoint inhibitor, but in September 18911 with Dr. William B. Coley's first published manuscript about the treatment of inoperable sarcomas with bacterial preparations—these preparations, a combination of gram-positive, heat-killed streptococci plus gram-negative, heat-killed Serratia marcescens, subsequently referred to as “Coley's toxins”2 or mixed bacterial vaccine (MBV), led to spontaneous tumor regressions in some patients due to activation of the immune system. Despite a reported cure rate of 10%,3 Coley's toxin was not widely accepted or used except as a last resort due to the anecdotal and unpredictable nature of the responses, nonspecificity to tumors and the dangerous side effects (e.g. high fever) that the treatment induced.2 The specific mechanism of action, unknown to Coley at the time, is probably related to a “massive induction of immunoregulatory cytokines,” specifically TNF-α, IFN-γ, and IL1-β, according to Karbach et al, 2012, who conducted a Phase 1 clinical trial with Coley's toxins.4
Almost one hundred years later, picking up where Dr. Coley left off, early non-specific immunotherapies such as human IFNα-2b for the treatment of multiple malignancies and interleukin-2 (IL-2) for metastatic renal cell carcinoma were developed to induce an immune response against the tumor.5 In addition to IL-2 and interferon therapy, a Calmette–Guérin (BCG) vaccine for tuberculosis was repurposed to drive cellular immunity in superficial bladder cancer.6 However, these early cytokine-based immunotherapies not only showed disappointing efficacy rates as monotherapies but were also in some cases severely toxic, especially IL-2, which limit their clinical use.7 Low efficacy is a function of multiple factors: non-selective targeting of tumors, the heterogeneity of host responses,8 and tumor immune escape5 notably from induction9 of tolerance via immunosuppressive cytokines such as TGF-β and IL-10 and upregulation of the negative checkpoint molecules, CTLA-4 and PDL-1/PD-1, to limit T cell responses.10
With regard to the latter, CTLA-4 and PDL-1/PD-1 checkpoint inhibitors have revolutionized the treatment landscape in advanced melanoma, lung cancer, RCC, bladder cancer, and Hodgkin lymphomas with the demonstration of durable clinical efficacy and improved overall survival. Nevertheless, despite the clear evidence of improved clinical outcomes, the fact remains that the majority of cancer patients do not respond to them. In an expansive clinical trial with the anti-PD-1 mAb, nivolumab, in NSCLC, melanoma, and renal cell cancer, objective responses occurred in 25% of patients,11 which is notable not only for the patients that did respond but also for the 75% that didn't, supporting a rationale for immunotherapy-based combination treatment to improve on this response rate.
Emerging data places the high rate of epigenetic modifications pathognomonic to cancer cells at the center or, perhaps more appropriately, the epicenter of tumor resistance to radiotherapy, chemotherapy and immunotherapy.12 The term epigenetics, originally coined in the middle of the twentieth century by Conrad Waddington, a British developmental biologist,13 is used herein to refer to changes in the pattern of gene expression and activity that are not the result of changes to the primary DNA sequence.14 Epigenetic modifications include alterations in DNA methylation and chromatin remodeling via acetylation and methylation. In cancer the accumulation of these epigenetic alterations leads to profound changes in the expression of proteins that contribute not only to the development of radio- and chemoresistance but also immunoresistance15-19 and immune escape through transcriptional silencing of immune-related genes.20
Epigenetic agents that increase the expression of these silenced immune-related genes have the potential to overcome cancer cell-induced T cell dysfunction not only in tumors with known susceptibility to checkpoint inhibitors, like melanoma or NSCLC, but also in tumors traditionally considered immune-resistant such as pancreatic or colorectal cancers. On the premise that a combination of nonoverlapping anticancer therapies is necessary to redress the suppressive mechanisms, discussed herein, by which tumors escape immune attack and immunogenic cell death, epigenetic inhibitors currently under clinical evaluation are highlighted in this review as an important part of an integrated strategy to boost anticancer B and T cell effector function and thereby improve therapeutic outcomes, so that fewer and fewer patients are “left behind.”
Epigenetic modifications
Epigenetic changes are a ubiquitous feature of carcinogenesis 21 and progression. Among the most intensively studied epigenetic aberrations are DNA methylation of CpG dinucleotides and post-translational histone modification,22 both of which work in concert to establish and maintain patterns of gene expression conducive to tumor proliferation.23 The intrinsic reversibility and plasticity of epigenetic alterations as compared with the relative immutability of genetic events renders cancer a suitable disease for treatment with epigenetic and chromatin modifiers. A brief summary of DNA methylation and histone modification is provided below. For more in-depth reviews on epigenetic alterations the reader is referred to other cited literature.24,25
DNA methylation
DNA methylation refers to the covalent attachment of a methyl group to the carbon-5 position of cytosine within clusters or islands of CpG dinucleotides26 (cytosine and guanine separated by a phosphate) found in or near promoter regions of genes where transcription is initiated27 via a reaction catalyzed by DNA methyltransferases (Fig. 1A).
Figure 1.
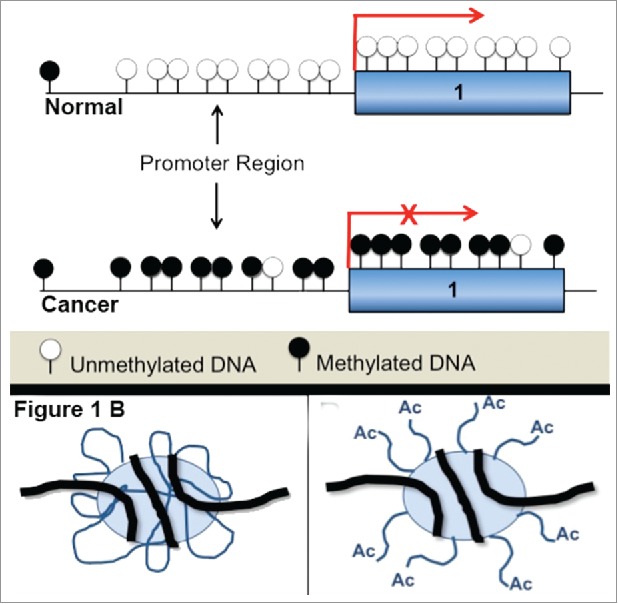
(A) Hypermethylation and transcriptional silencing. A hallmark of cancer is hypermethylation of DNA sequences in the promoter region leading to transcriptional silencing. (B) Histone acetylation leads to a switch between transcriptionally repressive and permissive chromatin. Histone acetylation induces a change from compacted to a more ‘open’ chromatin state and increases the accessibility of transcription complexes to genomic DNA. Acetylation is an important factor for the regulation of gene expression.
These methyl groups project into the major groove of DNA and sterically hinder the binding of RNA polymerase, which effectively silences transcription (Fig. 1A).28,29
Histone covalent modification
Space constraints in the micron-sized nucleus requires coiling of the 2 m30 long negatively-charged DNA molecule around an octamer of positively-charged histone proteins, which are, in turn, further compacted into chromatin fibers,31,32 rendering DNA largely inaccessible to transcriptional regulators. Acetylation, which occurs at the positively charged lysine residues on the amino-terminal histone tails, neutralizes the positive charge, decreasing the affinity of histones for DNA and thereby resulting in increased accessibility to the transcriptional machinery.33 Histone acetylation thus maintains chromatin in an open state conducive to active transcription.34 By contrast, histone deacetylases or HDACs deacetylate chromatin and reinstate the positive charge on the histone tails, resulting in high-affinity binding between histones and DNA,35 which, in turn, promotes a condensed chromatin state and transcriptional silencing (Fig. 1B and Fig. 2).27
Figure 2.
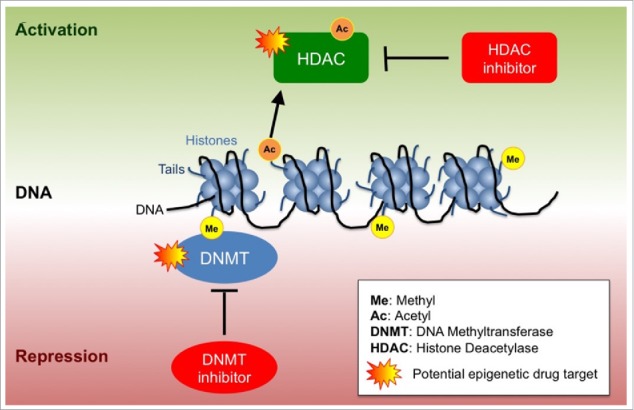
Epigenetic regulation of transcription. DNA is compressed in the nucleus as chromatin. The building block of chromatin is the nucleosome, which consists of 8 DNA wrapped histones.. Epigenetic regulation involves DNA methylation, histone acetylation and histone methylation that depending on the combination of modifications lead to transcriptional repression or transcriptional activation.
Epigenetic inhibitors as radio/chemosensitizers
Since DNA methyltransferases (DNMTs) and HDAC are multifunctional enzymes36 that impact a broad spectrum of pathways and cellular processes, including cell cycle regulation, DNA damage response, cell adhesion, angiogenesis, and immunity, it is not surprising that the gene expression changes wrought by epigenetic agents influence multiple facets of tumor progression and the tumor microenvironment.37
Epigenetic agents have demonstrated clinical efficacy not only in hematologic malignancies like acute myeloid leukemia and myelodysplastic syndrome where the DNMT inhibitors azacytidine (Vidaza, AZA, or 5-aza) and decitabine (Dacogen or 5AZA2) are approved, but also in solid tumors,38 most notably as radio- and chemosensitizers in patients with highly resistant tumors. In a Phase I/II study of 5-azacitidine and carboplatin 46% of patients with platinum-resistant or refractory ovarian cancer demonstrated durable responses and stable disease (median duration of therapy 7.5 months).39 In addition, an ongoing randomized Phase II trial (NCT02096354) acronymed ROCKET with the experimental systemically nontoxic epigenetic and tumor-associated myeloid repolarizing agent, RRx-001, followed by irinotecan rechallenge on progression of RRx-001 has resulted in tumor resensitization to irinotecan in multiple patients.15 Similar40 preliminary responses have been seen with RRx-001 in notoriously radioresistant melanoma brain metastases in combination with whole brain radiotherapy (WBRT) in a Phase I trial acronymed BRAINSTORM (NCT02215512).
One general mechanism of radio/chemoresensitization is related to epigenetic restoration of silenced tumor suppressor genes such as p53.25 Another mechanism of radio/chemoresensitization is through blood vessel normalization, which equilibrates oxygen levels within the tumor and enhances drug delivery.41 The endogenous vasodilator, nitric oxide (NO), has been reported as possessing epigenetic activity, modifying global histone methylation through inhibition of Jumonji C (JmjC) domain-containing demethylases.42 Consequently, NO-mediated radio- or chemo-sensitizing agents may exert their activity through a multiple mechanisms of action that include both vascular modification and NO or Reactive Oxygen Species and Reactive Nitrogen Species (ROS/RNS)-induced epigenetic modifications.41,43
A normalized tumor vasculature also makes it possible for immune cells to infiltrate the tumor and exert an anticancer effect, discussed below.
Epigenetic inhibitors as immunosensitizers
In addition to sensitization of chemotherapy and radiation, epigenetic agents (e.g., 5-AZA) have been used as part of a strategy44 to prime immunotherapy responses. In 5 patients with NSCLC who received 5-azacytidine and entinostat before treatment with either anti-PD-1 or anti-PD-L1 antibodies, durable objective responses were observed in 3 patients while stable disease for more than 6 months was observed in the other 2 patients.45 Based on these clinical responses, a study of azacitidine and entinostat or azacitidine alone before the PD-1 inhibitor nivolumab (NCT01928576) was initiated with the primary end point of overall response rate; however the inherent toxicity of nucleoside DNA methyltransferase inhibitors (DNMTs) such as azacitidine, which are poorly tolerated, is likely to significantly limit the efficacy of this strategy. While HDAC inhibitors such as entinostat are generally well tolerated, they, too, may induce dose-limiting cytopenias.46
In contrast to these myelosuppression-associated complications, preliminary results from the first cohort of patients treated with the combination of the experimental systemically non-toxic pan-epigenetic inhibitor and tumor-associated myeloid repolarizing agent, RRx-001, and the PD-1 inhibitor, nivolumab, in a Phase I dose escalation study called PRIMETIME (NCT02518958) indicate promising safety and activity.47 A low side effect profile is particularly important in the context of an immune priming strategy, due to the potential for a reverse ‘anti-priming’ effect in the event that bone marrow and other host toxicities from the epigenetic agent either reduce tolerance to or prevent subsequent treatment with immunotherapy.
Mechanisms of tumor-induced immune escape
In 1957 2 prominent immunologists, Burnett and Thomas,48 put forward the controversial immunosurveillance hypothesis, now largely accepted with revisions, that the ever-vigilant immune system constantly detects and eliminates malignant threats before they clinically present49 as full-blown tumors. However, immunosurveillance is not 100% effective even in immunocompetent patients; and, in fact, under the selective pressure of this active surveillance it is hypothesized that resistant tumor cells evolve a repertoire of epigenetically mediated escape mechanisms to resist immunogenic cell death (ICD) in a process called immunoediting.50
The term immunogenic cell death,51 refers to a multi-step cycle of immune activation initiated by the release of cell-associated antigens 52 and danger signals (step 1 in the cycle) from dying tumor cells. These released tumor antigens (TAs) are captured by antigen presenting cells (APCs) such as macrophages and DCs, which process and load TA-derived peptides on major histocompatibility complex (MHC) class I or class II (step 2); the loaded MHCs then activate MHC-restricted T cells (i.e., cytotoxic T-lymphocytes or helper T cells) in lymph nodes (step 3). In particular, cytotoxic T-lymphocytes traffic to (step 4) and infiltrate the tumor bed (step 5), specifically recognizing antigen on cancer cells through interactions between T-cell receptor (TCR) and MHC class I (step 6), and eliminate the targeted cells (Step 7), releasing additional antigens in the process that, in turn, further stimulate the immune response.53
This 7-step cycle is interrupted in tumors, which rely on stealth tactics and deploy multiple countermeasures including reduced expression of tumor associated antigens, deficient antigen presentation, reduced or absent costimulation, resistance to apoptosis and even counterattack to escape immune elimination. These countermeasures are, to a greater or lesser degree, regulated by epigenetic processes, and, therefore, potentially susceptible to reprogramming with epigenetic therapies.
The flip side or mirror image of the antitumor immunity cycle is an immunoescape paradigm, depicted schematically as a series of countermeasures to intercept immunosurveillance at each of the 7 steps. (Fig. 3). Each immunoescape countermeasure, described in more detail below, has been matched in boxes with a corresponding potential antitumor strategy.
Figure 3.
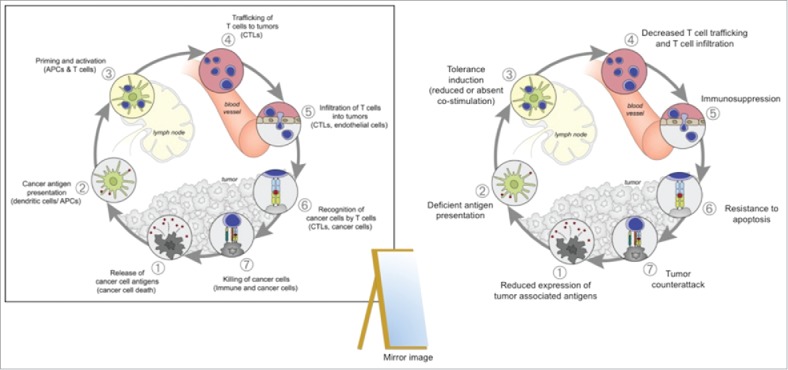
The Immunoescape Cycle. To the left, the Antitumor Immunity Cycle illustrating the 7 main steps to stimulate an effective antitumor response. In cancer this cycle is interrupted at one or more steps to prevent specific T-cell immunity. Adapted from Oncology Meets Immunology: The Cancer-Immunity Cycle Daniel S. Chen and Ira Mellman Immunity 39, July 25, 2013. In a mirror image of the beneficial immunity cycle pictured to the left, which is responsible for the elimination of tumor cells, the harmful Immunoescape Cycle pictured to the right illustrates 7 different mechanisms by which the tumor corrupts or evades the 7 steps of the immunity cycle.
Immunoescape mechanism 1: Reduced expression of tumor associated antigens
Despite the failure of effector cells to lyse tumors in patients with cancer, malignant cells are, in fact, immunogenic due, in part, to the variable expression of tumor associated antigens (TAAs). These immunogenic TAAs include cancer testis antigens (CTAs), such as MAGE-A3 and NY-ESO-1, normally restricted to the germ cells of the gonads and silenced in somatic cells, but aberrantly expressed in up to 40% of hematologic and solid tumor types.54
Increased CTA levels in both normal and tumor tissue are associated preclinically with epigenetic modification 55 and re-expression of these hypermethylated antigens following exposure to epigenetic agents has been demonstrated in clinical trials. For example, in a Phase II study (NCT01050790), post-transplant multiple myeloma patients receiving sequential azacitidine (Aza) and lenalidomide (Rev) demonstrated a significant increase in MAGEA4, MAGEA6, SPA17 and AKAP4 in bone marrow compared with pre-therapy samples. This induction was associated with a subsequent sustained T-cell-mediated immune response.56 Similarly, the phase I/II study (NCT02332889) in relapsed neuroblastoma and sarcoma tested the efficiency of decitabine to upregulate cancer testis antigen expression, followed by a dendritic cell MAGE-A1, MAGE-A3, and NY-ESO1 peptide vaccine. A complete response was documented in 1/10 patients and of the 2 patients who were disease-free at start of DC vaccine therapy one was disease-free 2 y post therapy.57
These results suggest that the mutational burden of tumors directly correlates with increased neoantigen presentation, immunogenicity and response to checkpoint inhibitors. Accordingly, mutagenic tumors like melanoma and NSCLC are more responsive to immunotherapy than AML or ALL, where the rate of mutations is correspondingly lower (Table 1).58
Table 1.
Somatic mutational burden per tumor type. In theory, tumor-specific neoantigen generation and presentation is directly proportional to mutational burden.
| Disease | Somatic mutation prevalence (average number mutations per megabase) | Formation of Neoantigens |
|---|---|---|
| Melanoma | ∼20 | Frequently |
| Lung Squamous | ∼9.5 | Regularly |
| Lung Adeno | ∼9.0 | Regularly |
| Stomach | ∼8.5 | Regularly |
| Esophagus | ∼8.0 | Regularly |
| Lung Small Cell | ∼8.0 | Regularly |
| Colorectal | ∼8.0 | Regularly |
| Bladder | ∼7.5 | Regularly |
| Uterus | ∼7.0 | Regularly |
| Cervix | ∼6.5 | Regularly |
| Liver | ∼6.0 | Regularly |
| Head and Neck | ∼5.0 | Regularly |
| Kidney Clear Cell | ∼2.0 | Regularly |
| Lymphoma B-Cell | ∼1.5 | Regularly |
| Kidney Papillary | ∼1.1 | Regularly |
| Breast | ∼1.0 | Occasionally |
| Pancreas | ∼1.0 | Occasionally |
| Myeloma | ∼1.0 | Occasionally |
| Ovary | ∼1.0 | Occasionally |
| Prostate | ∼1.0 | Occasionally |
| Glioblastoma | ∼0.9 | Occasionally |
| Glioma Low Grade | ∼0.85 | Occasionally |
| Neuroblastoma | ∼0.8 | Occasionally |
| Medulloblastoma | ∼0.7 | Occasionally |
| CLL | ∼0.65 | Occasionally |
| Kidney Chromophobe | ∼0.6 | Occasionally |
| Thyroid | ∼0.5 | Occasionally |
| ALL | ∼0.3 | Occasionally |
| AML | ∼0.2 | Occasionally |
| Pilocytic Astrocytoma | ∼0.05 | Occasionally |
Mismatch repair deficit and microsatellite instability
In similar way, tumors with mismatch-repair deficits and microsatellite instability status, which correlate with a higher mutational rate and neoantigen load, are more susceptible to treatment with PD-1 blockade.59 For example, patients with microsatellite instable (MSI) colorectal cancer (CRC) (approximately 15% of the CRC population) are candidates for PD-1 therapy while microsatellite stable (MSS) CRC patients, who comprise the remaining 85%, are not.60 Microsatellites are short sequences repeated throughout the genome that are vulnerable to mutation due to a defect in DNA mismatch repair.61 Despite the higher expression of neoantigens in MSI+ CRC tumors, the presence of commensal bacteria in the intestines may facilitate upregulation of checkpoint ligands, thereby preventing immune eradication.62
The premise that tumors with microsatellite instability that lead to a higher level of somatic mutations are more susceptible to immune therapy, once the immune checkpoint ‘brakes’ have been released is intriguing and suggests potential ways of extending the activity of immune therapy to tumors that are currently not susceptible to this therapeutic approach. It has been well know that exposing tumors to ionizing radiation induces cellular changes that result in the upregulation and cellular expression of neoantigens,63 often as a result ofROS-induced central necrosis,64 that can manifest as pseudoprogression.65 Indeed, ROS induced necrosis results in pro-inflammatory and immuno-stimulatory responses66 such that the effect may be more accurately described as immunogenic cell-death, mediated through important signaling to the immune system.67,68 HDAC and DNMT inhibitors both exert an immunostimulatory effect: DNMTs through the induction of an interferon response by viral mimicry,69,70 and HDAC inhibitors through induction of immunogenic cell death.71
ROS-mediated epigenetic agents, such as RRx-001, has shown effects on global methylation and histone acetylation,72 may therefore act through a dual mechanism of action – inducing an immune response through HDAC and DNMT inhibition, while amplifying that response by neoantigen presentation as a consequence of central necrosis. As a ROS-mediated epigenetic agent, extensive tumor central necrosis has been observed in the phase 1 clinical trial and subsequently in ongoing Phase 2 trials. Fig. 4 shows a typical PET-CT image of liver lesion with extensive tumor necrosis. Necrosis was surgically confirmed in both the central region and rim (> 90% necrotic) suggesting extensive lymphocyte recruitment to the tumor.73
Figure 4.
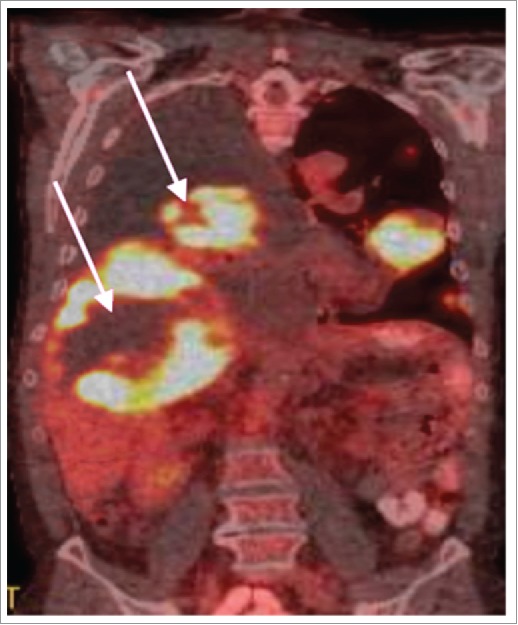
PET-CT showing tumor central necrosis and pseudoprogression in a patient recruited in the Phase 1 clinical trial. The pathology report indicated that the rim was >90% necrotic suggesting an influx of lymphocytes.
| Therapeutic strategy in MSI high tumors e.g., colon, gastric and endometrial tumors as well as patients with Lynch Syndrome: Combine an epigenetic agent with radiation, CAR-T, oncolytic viruses, bispecific T-cell engagers (BiTE) and DNA repair inhibitors to increase the complement of targetable T-cell antigens |
Immunoescape mechanism 2: Deficient antigen presentation
7The major histocompatibility complex (MHC) is a cluster of genes that function to bind and present antigens to the adaptive immune system. In humans, MHC, known as human leukocyte antigen (HLA), includes coding regions for different co-receptors on the T cells, that is, CD8 and CD4, hence allowing for identification of CD4+ T helper (Th) cells and CD8+ cytotoxic T lymphocytes (CTLs), which recognize antigen presented by MHC class I-derived molecules and execute anti-tumor responses74 MHC class I antigens also regulate the lytic activity of NK immune cells, which are programmed to kill any cells lacking MHC class I expression.74
As reviewed elsewhere,75 malignant transformation is associated with the epigenetic loss and/or downregulation of MHC class I antigens via hypermethylation of the HLA promoter, which impairs the recognition of tumor cells by CTLs, leading to escape from immune destruction; treatment with epigenetic agents76 restores expression of MHC class I and/or components of the class I antigen processing and presentation machinery (TAP1, TAP2, LMP7, Tapasin),77,78 which enhances tumor specific T-cell killing.79 MHC class I and antigen presentation machinery are also regulated by DNA methylation.80; likewise, treatment with demethylating agents restores their expression in cancer cells,81 which increases susceptibility to destruction by immune effector cells.
| Therapeutic strategy for tumors with low MHC class I expression: Combine an epigenetic agent with IFN-gamma, GM-CSF or high dose IL-2 to induce the upregulation of MHC class I expression. |
Immunoescape mechanism 3: Tolerance induction (Reduced or absent costimulation)
In accordance with the so-called “2 signal model,” T-cell activation requires at least a double signal.82 The first signal83 results from the interaction of the T-cell receptor (TCR) with its cognate antigen plus major histocompatibility complex on the surface of antigen-presenting cells (APCs) or target cells.84 The second set of antigen-independent signals termed costimulation are expressed on the surface of the APCs (Fig. 5). In the absence of this double signal, these anti-tumor T cells enter a state of unresponsiveness termed anergy.85 The major costimulatory signals implicated in T cell activation is the glycoprotein CD28, which interacts on the APC with CD80 and CD86 (also called B7.1 and B7.2) and the OX40-OX40 ligand interaction.86 The co-inhibitory molecules, CTLA-4, and PD-1, also interact with CD80 and CD86.87 The third signal is the production of cytokines (e.g., IL-2, which, in turn, induces IFN-γ production) required for differentiation of lytic CD8+ T cell lytic effectors.88 Epigenetic mechanisms are linked with the expression of costimulatory and coinhibitory molecules89 as90 well as the repression of several pro-inflammatory cytokines (IL-1, IL-2, IL-8, and IL-12).91 Chiappinelli et al70 recently demonstrated that 5-azacytidine directly induces the expression of interferon. The mechanism, which overlaps with the targeted inflammation induced during oncolytic virus therapy, is related to derepression of endogenous retroviral (ERV) and other viral gene sequences in the tumor, triggering a type I interferon response.
Figure 5.
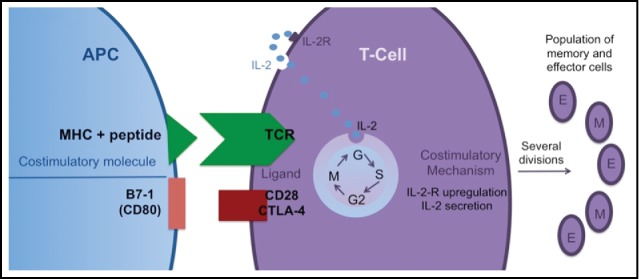
Naïve (T)cells require 2 signals to become activated: TCR/MHC and co-stimulation. The specific signal alone or the co-stimulatory signal alone leads to T cell anergy. The co-stimulatory signal is necessary for the synthesis and secretion of IL-2, which stimulates the T cell to divide and generate a larger population of memory and effector cells.
| Therapeutic strategy for tumors with low-to-intermediate expression of B7 and OX40 costimulatory molecules: Combine an epigenetic agent with a viral vector for transduction of IL-2 and B7.1 genes, antagonistic antibodies directed against the checkpoint inhibitors (e.g, CTLA-4, PD-1) and agonist antibodies such as OX40 to improve costimulatory signaling to T cells. |
Immunoescape mechanism 4: Decreased T-cell trafficking and T-cell infiltration
Tumor vessels differ from normal counterparts92 by virtue of their tortuosity, immaturity and hyperpermeability. In particular, the leakiness of the vessels due to the prevalence of proangiogenic signals results in raised interstitial pressure with compression of the vasculature, which further impedes blood flow and exacerbates pre-existing hypoxia.
Hypoxia not only increases resistance to chemotherapy and radiation but it also contributes to immune escape through different mechanisms including amplification of levels and activity of regulatory T cells (Tregs), myeloid suppressor cells (MDSC) and tumor associated macrophages (TAMs).93 In addition, hypoxia influences the cell adhesion molecule profile of the endothelium leading to preferential Treg adhesion, infiltration and immune suppression.94 The mechanisms of T cell adhesion and transmigration across the tumor endothelium have been covered in other reviews,95,96 and therefore due to space limitations will not be discussed here. Finally, hypoxia leads to global epigenetic changes and silencing of tumor suppressor genes like BRCA1 and DNA repair enzymes such as RAD51,97 which favors a proliferative phenotype. Examples of hypoxic tumor types include head and neck, cervix, sarcomas and glioblastomas.98 (Fig. 6)
Figure 6.
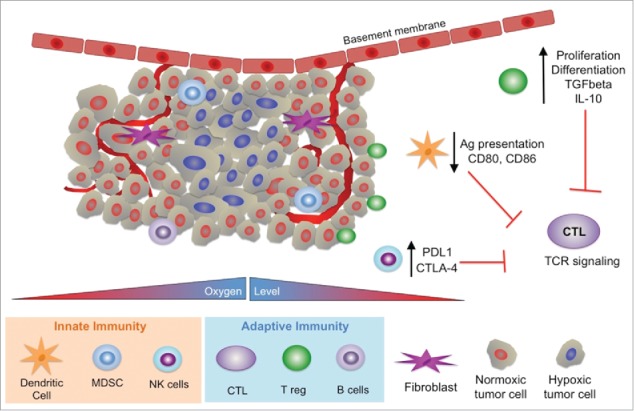
Influence of hypoxia in the tumor on the innate and adaptive immune response. In general hypoxia suppresses the response of the immune system. The adaptive immune system includes cytotoxic T lymphocytes (CTL), T regulatory cells (T reg) and B cells. The innate immune system includes dendritic cells, myeloid derived suppressor cells (MDSC) and natural killer cells (NK).
Treatment with anti-VEGF agents such as Bevacizumab or receptor tyrosine kinase (RTK) inhibitors like sunitinib prunes or disrupts the morphologically aberrant blood vessels that compromise blood flow in the tumor, leading to a more homogeneous distribution of functionally mature tumor vessels and a transient window of increased blood perfusion, assessable with magnetic resonance imaging, termed “vascular normalization”.99 Several preclinical studies have demonstrated that antiangiogenic therapy not only facilitates T-cell infiltration100 but also reverses the hypoxia-induced recruitment of immunosuppressive Tregs and the M2-like skewing of tumor associated macrophages,101 thereby reprogramming the microenvironment for tumor inhibition. Like bevacizumab and the RTK inhibitors, the experimental, pan-epigenetic and tumor-associated myeloid repolarizing agent, RRx-001102 has also been associated with normalization of the tumor vasculature, although, admittedly, this non-VEGF-related property may be attributable to the unique mechanism of RRx-001 rather than a general class effect of epigenetic agents.103 However, as a class, epigenetic therapies have the potential to reverse hypoxia-induced silencing of tumor suppressor and other beneficial genes with or without vascular normalizing agents.
| Therapeutic strategy for tumors with low blood flow and high vascular permeability as determined by DCE-MRI: Combine an epigenetic agent with a VEGF inhibitor to increase penetration of T-lymphocytes and amplify the response of the adaptive immune system. |
Immunoescape mechanism 5: Immunosuppression
Even if all the biochemical and physical barriers that might inhibit immune infiltration of tumors are overcome, it is by no means ensured that the TILs will manage to mount an effective anti-cancer response due to the induction of an immunosuppressive microenvironment. This section, which provides a basic—but far from exhaustive—overview of key immunosuppressive factors under epigenetic control, including secretion of cytokines, expression of negative regulatory checkpoint ligands that bind to cognate receptors on T cells, the induction of indoleamine 2,3-dioxygenase (IDO) and the recruitment of regulatory T cells.
Cytokines
The immunosuppressive cytokines, such as interleukin (IL)-10 and transforming growth factor-β (TGF-β), secreted from the malignant cells themselves as well as noncancerous cells present at the tumor site, such as immune, epithelial, or stromal cells inhibit the maturation and antigen-presenting function of dendritic cells (DC) and T cell effector responses through regulatory T cells.104 In addition to the epigenetically regulated expression of these tumor-derived cytokines,105 the small molecule prostaglandin E2 (PGE2), VEGF and macrophage-colony stimulating factor change the repertoire of immune cells and inhibit T cell proliferation, activation, and differentiation. Neutralizing antibodies that bind or “trap” these cytokines, in particular TGF-β, is a strategy to reverse local immunosuppression. As an example, oncolytic viruses may be armed with transgenes that express cytokine traps locally within the tumor microenvironment.106
Checkpoint inhibition
Checkpoint proteins such as cytotoxic T lymphocyte antigen-4 (CTLA-4) and programmed cell death 1 (PD-1) constitute a part of the normal suppressive mechanism, which serves in a physiologic setting to dampen inflammatory responses of autoimmune or infectious origin or in pathologic situations to downregulate anti-tumor responses107 (Fig. 7). PD-1, in particular, has 2 coinhibitory ligands: PD-L1 (also named B7-H1; CD274) and PD-L2 (B7-DC; CD273).108 PD-L1 is expressed in multiple tumor types especially NSCLC, melanoma, and renal cell carcinoma while PD-L2 expression is far less prevalent.109 The checkpoint inhibition pathways are co-opted and epigenetically upregulated by the tumor to direct the pattern of host immune responses in its own favor.44 Approved PD-1 inhibitors are nivolumab (Opdivo) and pembrolizumab (Keytruda).
Figure 7.
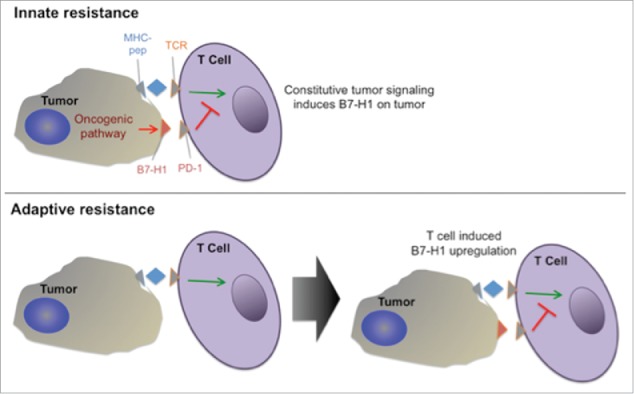
Innate vs adaptive resistance. PD-L1 can be constitutively expressed or induced adaptively if the right inflammatory cytokines are present in the immune environment.
Induction of indoleamine 2,3-dioxygenase (IDO)
In what has been termed immunosuppression by starvation,110 expression of IDO,111 a ubiquitous enzyme that converts tryptophan, the essential amino acid required by T cells for proliferation and survival, to the immunotoxic metabolite, kynurenine, mediates the immunosuppression of pregnancy. Moreover, as a component of the compensatory anti-inflammatory response syndrome (CARS), it is epigenetically inducible by pro-inflammatory cytokines,112,113 including interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α). Similar to the checkpoint pathway, cancer cells co-opt the physiologic mechanism of IDO-mediated tryptophan degradation to turn off antitumor responses.114 Inhibitors of IDO are currently under investigation in clinical trials.115
Recruitment of Regulatory T cells
One of the primary physiologic mechanisms to limit excessive and harmful immune responses is through the induction of a specialized subset of CD4+ T cells, CD4+CD25+ regulatory T cells (Tregs),116 which are conscripted to enhance tumor cell proliferation. As “major players” in the immune microenvironment,117 the presence of regulatory T cells, which correlates with poor survival in a variety of tumors, including prostatic, lung, hepatocellular and renal cell carcinomas,118 leads to the suppression of Thelper1 (Th1)/CD8+ T cell responses that mediate tumor cytotoxicity. Under the influence of cytokine CCL22,119 Tregs migrate to tumors where multiple factors including IL-2, TGF-β120 and, in particular, the epigenetically regulated transcription factor, Foxp3,121 are responsible for their expansion. Strategies to deplete Tregs include the administration of metronomic low dose therapy, cyclophosphamide and daclizumab, a US FDA-approved humanized anti-CD25 antibody. VEGF-A blockade may also reduce the numbers of tumor-infiltrating Tregs.122
| Therapeutic strategy to relieve immunosuppression: Combine an epigenetic agent with oncolytic viruses that encode cytokine traps, checkpoint inhibitors, the anti-CD25 antibody, daclizumab and a VEGF inhibitor to increase penetration of T-lymphocytes and amplify the response of the adaptive immune system. |
Immunoescape mechanism 6: Resistance to apoptosis
Resistance to apoptosis, a genetically and epigenetically regulated process of cell suicide,123 which underpins treatment failure, is a hallmark of cancer cells.124 Initiation of the intrinsic or the extrinsic apoptotic pathways induces the caspases, a family of cysteine proteases that act as common death effector molecules. Aberrant methylation in the promoters of genes coding for proteins implicated in the extrinsic and intrinsic pathways has been implicated as an important mechanism of apoptosis resistance in cancer cells.125 In addition, the epigenetic mechanism, which is arguably the best understood currently,126 involves the transcriptional silencing of a growing list of tumor suppressor genes, most notably the key guardian p53,127 that trigger apoptosis in the presence of cell abnormalities.
| Therapeutic strategy to induce apoptosis: In this context, an epigenetic agent may be combined with any number of different treatment options since, in general, most chemotherapies irrespective of the mechanism of action or molecular target have the potential to induce apoptosis. However, in particular, agents, which generate reactive oxygen species or deplete reduced glutathione and induce oxidative stress like the platinums, arsenic trioxide, motexafin gadolinium, nitroglycerin, and various flavonoids may activate p53 (and other tumor suppressors) and synergize with an epigenetic agent. |
Immunoescape mechanism 7: Tumor counterattack
In addition to the defensive strategies described above, a controversial hypothesis proposes that tumors also “go on the offensive” or on the “counterattack” through upregulation of Fas ligand (FasL/CD95L) expression. FasL is a transmembrane protein that belongs to the Tumor Necrosis Factor (TNF) family.128 The interaction of FasL with its cell surface receptor, Fas (CD95/APO-1) leads to apoptosis129 in Fas-positive cells, which is hypothesized to maintain the immune tolerance of the testis, hair follicle, placenta, eye and brain.130 Evidence suggests that tumors epigenetically131 upregulate the expression of Fas to ‘counterattack’ Fas-ligand-expressing tumor-infiltrating lymphocytes, which favors immune escape. If this controversial hypothesis is correct—the controversy relates to the fact that some studies support it,132,133 while others refute it134—then the tumor walks a fine line between protection and harm, since Fas expression also predisposes to apoptotic cancer cell death, and further upregulation of the protein may ultimately suppress tumor proliferation.
|
Therapeutic strategy to ‘counter the counterattack’: Further upregulate Fas expression in tumors with epigenetic therapies. In addition to epigenetic inhibition, other strategies include viral mediated delivery of Fas or fusion proteins connecting FasL to a single chain antibody fragment that specifically recognizes tumor cells or tumor stroma antigens is a promising field of research in therapeutics. Upon binding, the initially inactive protein would convert into a protein with membrane-bound-FasL-like activity. |
Summary and conclusions
According to the double-entendred Chinese proverb, “May you live in interesting times,” the modern era of immunotherapy has been both blessed and cursed. On the one hand, immunotherapy constitutes a revolution in cancer care with the promise of durable benefit and the potential of long-term survival in a subset (approximately 20-30%) of patients and tumor types.135 On the other hand, success has so far eluded the majority of patients (70-80%), for whom the promise of anti-tumor immune activity remains tantalizingly out of reach.
This review presents evidence for the centrality of epigenetic alterations i.e., histone modifications and DNA methylation in de novo and acquired immune resistance. Unlike genetic lesions, epigenetic changes are highly dynamic and potentially reversible with DNA-demethylators and HDAC inhibitors.136 By analogy with the United States No Child Left Behind Act of 2001 for disadvantaged students, the declarative premise in the title of this review, “No Patient Left Behind” is that the unacceptably high immunotherapy failure rate is conceptually as reversible as the methylation and acetylation modifications which underlie it: epigenetic reprogramming may reactivate silenced immune-related genes, including MHC class I, tumor-associated antigens, and accessory/costimulatory molecules, leading to enhanced immune recognition and tumor clearance, so that patients once stigmatized as “resistant to therapy” with an attendant poor prognosis will now have the opportunity to reap the same benefits as the minority of patients with intrinsically susceptible tumors.
Several approved (e.g., azacitidine and decitabine) and experimental (e.g., RRx-001) epigenetic therapies have demonstrated preclinical and preliminary clinical evidence of immune priming before or in combination with immune therapies in apparent contradiction to the “diminishing returns” usually associated with additional lines of treatment. However, whether or not the law of increasing or diminishing returns ultimately applies in this context depends on the side effect profile of the epigenetic agent(s): Grade 3-4 toxicities increase the risk and the likelihood that patients will not tolerate immunotherapy, which is the antithesis of a priming strategy. Immune checkpoint blockade, in particular, is associated with its own particular set of serious side effects termed immune-related adverse events or irAEs137; consequently, even if the overlap in toxicities between the PD-1 inhibitor and the epigenetic agent(s) is minimal the combination of toxicities may result in a dose and treatment limiting degradation of performance status and quality of life tantamount to an anti-priming effect. This is especially the case with decitabine,138 which is poorly tolerated as a result of hematologic side effects such as myelosuppression.
In addition, due to the broad spectrum of resistance and the multifactorial escape mechanisms as well as the multiplicity of targets, both known and unknown, in tumors, a therapeutic cocktail or combo, similar to treatment of the HIV virus, that includes an epigenetic agent may optimize immune priming responses, provided that the individual components combine advantageously i.e., that severe toxicities and/or antagonistic interactions do not interfere with activity. To that end, the specific strategies presented in this review are not mutually exclusive or incompatible but dovetail given the multifactorial nature of immune derangement in cancer.
Since the relative toxicities of the epigenetic agents in decreasing order are: DNMTs>HDACs>RRx-001, potential anticancer combinations might include RRx-001 as the epigenetic priming agent. Finally, as the strategy of immune priming with epigenetic agents gathers wider traction and momentum, the hope is that fewer and fewer patients will be left behind.
To paraphrase the Chinese proverb from the beginning of this Discussion section: May all cancer patients, especially those who were formerly resistant to the effects of immunotherapy, live in interesting prime times.
Disclosure of potential conflicts of interest
EpicentRx Inc. funds research of RRx-001. Authors B.O. and J.S. are employees of EpicentRx, Inc. The remaining authors have no conflicts of interest to declare.
EpicentRx Inc. funds research of RRx-001. Authors B.O. and J.S. are employees of EpicentRx, Inc. The remaining authors have no conflicts of interest to declare.
Acknowledgments
The authors acknowledge Dr. Harry Lybeck M.D., Ph.D., a pioneer in the field of endocrinology and an astute 98 y old physician with a wealth of information from over 7 decades of medical experience, for his invaluable insights during the writing of this review.
Author contributions
The authors declare that each author contributed to the writing of this manuscript.
References
- 1.Coley WB., II Contribution to the knowledge of sarcoma. Ann Surg 1891; 14:199-220; PMID:17859590; https://doi.org/ 10.1097/00000658-189112000-00015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parish CR. Cancer immunotherapy: The past, the present and the future. Immunol Cell Biol 2003; 81:106-13; PMID:12631233; https://doi.org/ 10.1046/j.0818-9641.2003.01151.x [DOI] [PubMed] [Google Scholar]
- 3.Wiemann B, Starnes CO. Coley's toxins, tumor necrosis factor and cancer research: A historical perspective. Pharmacol Ther 1994; 64:529-64; PMID:7724661; https://doi.org/ 10.1016/0163-7258(94)90023-X [DOI] [PubMed] [Google Scholar]
- 4.Karbach J, Neumann A, Brand K, Wahle C, Siegel E, Maeurer M, Ritter E, Tsuji T, Gnjatic S, Old LJ, et al.. Phase I clinical trial of mixed bacterial vaccine (Coley's toxins) in patients with NY-ESO-1 expressing cancers: Immunological effects and clinical activity. Clin Cancer Res 2012; 18:5449-59; PMID:22847809; https://doi.org/ 10.1158/1078-0432.CCR-12-1116 [DOI] [PubMed] [Google Scholar]
- 5.Firor AE, Jares A, Ma Y. From humble beginnings to success in the clinic: Chimeric antigen receptor-modified T-cells and implications for immunotherapy. Exp Biol Med (Maywood) 2015; 240:1087-98; PMID:25956686; https://doi.org/ 10.1177/1535370215584936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larson CL, Baker RE, Ushijima RN, Baker MB, Gillespie C. Immunotherapy of friend disease in mice employing viable BCG vaccine. Proc Soc Exp Biol Med 1972; 140:700-2; PMID:4556666; https://doi.org/ 10.3181/00379727-140-36534 [DOI] [PubMed] [Google Scholar]
- 7.Rosenberg SA. IL-2: The first effective immunotherapy for human cancer. J Immunol 2014; 192:5451-8; PMID:24907378; https://doi.org/ 10.4049/jimmunol.1490019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morikawa K, Fidler IJ. Heterogeneous response of human colon cancer cells to the cytostatic and cytotoxic effects of recombinant human cytokines: Interferon-alpha, interferon-gamma, tumor necrosis factor, and interleukin-1. J Biol Response Mod 1989; 8:206-18; PMID:2499665 [PubMed] [Google Scholar]
- 9.Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol 2006; 90:51-81; PMID:16730261; https://doi.org/ 10.1016/S0065-2776(06)90002-9 [DOI] [PubMed] [Google Scholar]
- 10.Rosenblatt J, Avigan D. Targetting the PD-L1/PD-1 axis holds promise in the treatment of malignancy. Transl Cancer Res 2012; 1:283-6. [Google Scholar]
- 11.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al.. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443-54; PMID:22658127; https://doi.org/ 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown R, Curry E, Magnani L, Wilhelm-Benartzi CS, Borley J. Poised epigenetic states and acquired drug resistance in cancer. Nat Rev Cancer 2014; 14:747-53; PMID:25253389; https://doi.org/ 10.1038/nrc3819 [DOI] [PubMed] [Google Scholar]
- 13.Waddington CH. The epigenotype. 1942. Int J Epidemiol 2012; 41:10-3; PMID:22186258; https://doi.org/ 10.1093/ije/dyr184 [DOI] [PubMed] [Google Scholar]
- 14.Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, Berdasco M, Fraga MF, O'Hanlon TP, Rider LG, et al.. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res 2010; 20:170-9; PMID:20028698; https://doi.org/ 10.1101/gr.100289.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ning S, Sekar TV, Scicinski J, Oronsky B, Peehl DM, Knox SJ, Paulmurugan R. Nrf2 activity as a potential biomarker for the pan-epigenetic anticancer agent, RRx-001. Oncotarget 2015; 6:21547-56; PMID:26280276; https://doi.org/ 10.18632/oncotarget.4249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cabrales P, Oronsky B, Scicinski J. Abstract 1420: RRx-001 inhibits glucose erythrocyte and tumor glucose 6-phosphate dehydrogenase. Cancer Res 2014; 74:1420; https://doi.org/ 10.1158/1538-7445.AM2014-1420 [DOI] [Google Scholar]
- 17.Cabrales P, Reid T, Oronsky B, Scicinski J, Chauchan D, Parker C, et al.. RRx-001 An EXO-based epigenetic anti-cancer agent in phase 2 clinical trials. ISEV International Society for Extracellular Vesicles 2014, Educational Event San Diego, Oct 26, 2014. [Google Scholar]
- 18.Juo YY, Johnston FM, Zhang DY, Juo HH, Wang H, Pappou EP, Yu T, Easwaran H, Baylin S, van Engeland M, et al.. Prognostic value of CpG island methylator phenotype among colorectal cancer patients: A systematic review and meta-analysis. Ann Oncol 2014; 25:2314-27; PMID:24718889; https://doi.org/ 10.1093/annonc/mdu149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell 2014; 54:716-27; PMID:24905005; https://doi.org/ 10.1016/j.molcel.2014.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, Diaz LA Jr, Papadopoulos N, Kinzler KW, Vogelstein B, et al.. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A 2014; 111:11774-9; PMID:25071169; https://doi.org/ 10.1073/pnas.1410626111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics 2009; 1:239-59; PMID:20495664; https://doi.org/ 10.2217/epi.09.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sigalotti L, Covre A, Fratta E, Parisi G, Colizzi F, Rizzo A, Danielli R, Nicolay HJ, Coral S, Maio M. Epigenetics of human cutaneous melanoma: setting the stage for new therapeutic strategies. J Transl Med 2010; 8:56; PMID:20540720; https://doi.org/ 10.1186/1479-5876-8-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hashimshony T, Zhang J, Keshet I, Bustin M, Cedar H. The role of DNA methylation in setting up chromatin structure during development. Nat Genet 2003; 34:187-92; PMID:12740577; https://doi.org/ 10.1038/ng1158 [DOI] [PubMed] [Google Scholar]
- 24.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3:415-28; PMID:12042769; https://doi.org/ 10.1038/nrg816 [DOI] [PubMed] [Google Scholar]
- 25.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis 2010; 31:27-36; PMID:19752007; https://doi.org/ 10.1093/carcin/bgp220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kulis M, Esteller M. DNA methylation and cancer. Adv Genet 2010; 70:27-56; PMID:20920744; https://doi.org/ 10.1016/B978-0-12-380866-0.60002-2 [DOI] [PubMed] [Google Scholar]
- 27.Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol 2005; 2(Suppl 1):S4-11; PMID:16341240; https://doi.org/ 10.1038/ncponc0354 [DOI] [PubMed] [Google Scholar]
- 28.Lund AH, van Lohuizen M. Epigenetics and cancer. Genes Dev 2004; 18:2315-35; PMID:15466484; https://doi.org/ 10.1101/gad.1232504 [DOI] [PubMed] [Google Scholar]
- 29.Golbabapour S, Abdulla MA, Hajrezaei M. A concise review on epigenetic regulation: Insight into molecular mechanisms. Int J Mol Sci 2011; 12:8661-94; PMID:22272098; https://doi.org/ 10.3390/ijms12128661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Josselyn S, Frankland PW. Another twist in the histone memory code. Cell Res 2015; 25:151-2; PMID:25342557; https://doi.org/ 10.1038/cr.2014.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muller WG, Rieder D, Karpova TS, John S, Trajanoski Z, McNally JG. Organization of chromatin and histone modifications at a transcription site. J Cell Biol 2007; 177:957-67; PMID:17576795; https://doi.org/ 10.1083/jcb.200703157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneider R, Grosschedl R. Dynamics and interplay of nuclear architecture, genome organization, and gene expression. Genes Dev 2007; 21:3027-43; PMID:18056419; https://doi.org/ 10.1101/gad.1604607 [DOI] [PubMed] [Google Scholar]
- 33.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev 1998; 12:599-606; PMID:9499396; https://doi.org/ 10.1101/gad.12.5.599 [DOI] [PubMed] [Google Scholar]
- 34.Luo RX, Dean DC. Chromatin remodeling and transcriptional regulation. J Natl Cancer Inst 1999; 91:1288-94; PMID:10433617; https://doi.org/ 10.1093/jnci/91.15.1288 [DOI] [PubMed] [Google Scholar]
- 35.Zupkovitz G, Tischler J, Posch M, Sadzak I, Ramsauer K, Egger G, Grausenburger R, Schweifer N, Chiocca S, Decker T, et al.. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol Cell Biol 2006; 26:7913-28; PMID:16940178; https://doi.org/ 10.1128/MCB.01220-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rajendran R, Garva R, Krstic-Demonacos M, Demonacos C. Sirtuins: Molecular traffic lights in the crossroad of oxidative stress, chromatin remodeling, and transcription. J Biomed Biotechnol 2011; 2011:368276; PMID:21912480; https://doi.org/ 10.1155/2011/368276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nebbioso A, Carafa V, Benedetti R, Altucci L. Trials with ‘epigenetic’ drugs: an update. Mol Oncol 2012; 6:657-82; PMID:23103179; https://doi.org/ 10.1016/j.molonc.2012.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wee S, Dhanak D, Li H, Armstrong SA, Copeland RA, Sims R, Baylin SB, Liu XS, Schweizer L. Targeting epigenetic regulators for cancer therapy. Ann N Y Acad Sci 2014; 1309:30-6; PMID:24571255; https://doi.org/ 10.1111/nyas.12356 [DOI] [PubMed] [Google Scholar]
- 39.Azad N, Zahnow CA, Rudin CM, Baylin SB. The future of epigenetic therapy in solid tumours–lessons from the past. Nat Rev Clin Oncol 2013; 10:256-66; PMID:23546521; https://doi.org/ 10.1038/nrclinonc.2013.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carter C, Reid T, Fisher G, Cho-Phan C, Kunz P, Kaiser H, Oronsky B, Fanger G, Caroen S, Parker C, et al.. O3.8Early Results: “ROCKET” a phase II Study of RRx-001, a novel triple epigenetic inhibitor, resensitization to irinotecan in colorectal cancer. Ann Oncol 2015; 26:ii4-5; https://doi.org/ 10.1093/annonc/mdv081.8 [DOI] [Google Scholar]
- 41.Oronsky B, Oronsky N, Lybeck M, Fanger G, Scicinski J. Targeting hyponitroxia in cancer therapy In: Bonavida B, ed. Nitric oxide and cancer: Pathogenesis and therapy: Springer, 2015:39-47. [Google Scholar]
- 42.Hickok JR, Vasudevan D, Antholine WE, Thomas DD. Nitric oxide modifies global histone methylation by inhibiting Jumonji C domain-containing demethylases. J Biol Chem 2013; 288:16004-15; PMID:23546878; https://doi.org/ 10.1074/jbc.M112.432294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scicinski J, Oronsky B, Ning S, Knox S, Peehl D, Kim MM, Langecker P, Fanger G. NO to cancer: The complex and multifaceted role of nitric oxide and the epigenetic nitric oxide donor, RRx-001. Redox Biol 2015; 6:1-8; PMID:26164533; https://doi.org/ 10.1016/j.redox.2015.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, Vancriekinge W, Demeyer T, Du Z, Parsana P, et al.. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget 2013; 4:2067-79; PMID:24162015; https://doi.org/ 10.18632/oncotarget.1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brahmer JR, Pardoll DM. Immune checkpoint inhibitors: Making immunotherapy a reality for the treatment of lung cancer. Cancer Immunol Res 2013; 1:85-91; PMID:24777499; https://doi.org/ 10.1158/2326-6066.CIR-13-0078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Juergens RA, Vendetti F, Coleman B, Sebree RS, Rudek MA, Belinsky SA. Phase I trial of 5-azacitidine (%AC) and SNDX-275 in advanced lung cancer (NSCLC). J Clin Onc 2008; 26:abstract no. 19036; https://doi.org/ 10.1200/jco.2008.26.15_suppl.19036 [DOI] [Google Scholar]
- 47.EpicentRx, unpublished data. [Google Scholar]
- 48.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: From immunosurveillance to tumor escape. Nat Immunol 2002; 3:991-8; PMID:12407406; https://doi.org/ 10.1038/ni1102-991 [DOI] [PubMed] [Google Scholar]
- 49.Dushyanthen S, Beavis PA, Savas P, Teo ZL, Zhou C, Mansour M, Darcy PK, Loi S. Relevance of tumor-infiltrating lymphocytes in breast cancer. BMC Med 2015; 13:202; PMID:26300242; https://doi.org/ 10.1186/s12916-015-0431-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gross E, Sunwoo JB, Bui JD. Cancer immunosurveillance and immunoediting by natural killer cells. Cancer J 2013; 19:483-9; PMID:24270347; https://doi.org/ 10.1097/PPO.0000000000000005 [DOI] [PubMed] [Google Scholar]
- 51.Krysko O, Love Aaes T, Bachert C, Vandenabeele P, Krysko DV. Many faces of DAMPs in cancer therapy. Cell Death Dis 2013; 4:e631; PMID:23681226; https://doi.org/ 10.1038/cddis.2013.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, Galluzzi L, Kepp O, Kroemer G. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol 2015; 6:187; PMID:25964783; https://doi.org/ 10.3389/fimmu.2015.00187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ishii H, Tanaka S, Masuyama K. Therapeutic strategy for cancer immunotherapy in head and neck cancer. Adv Cell Mol Otolaryngol 2015; 3:27690; https://doi.org/ 10.3402/acmo.v3.27690 [DOI] [Google Scholar]
- 54.Scanlan MJ, Simpson AJ, Old LJ. The cancer/testis genes: Review, standardization, and commentary. Cancer Immun 2004; 4:1; PMID:14738373 [PubMed] [Google Scholar]
- 55.Kim R, Kulkarni P, Hannenhalli S. Derepression of cancer/testis antigens in cancer is associated with distinct patterns of DNA hypomethylation. BMC Cancer 2013; 13:144; PMID:23522060; https://doi.org/ 10.1186/1471-2407-13-144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Toor AA, Payne KK, Chung HM, Sabo RT, Hazlett AF, Kmieciak M, Sanford K, Williams DC, Clark WB, Roberts CH, et al.. Epigenetic induction of adaptive immune response in multiple myeloma: Sequential azacitidine and lenalidomide generate cancer testis antigen-specific cellular immunity. Br J Haematol 2012; 158:700-11; PMID:22816680; https://doi.org/ 10.1111/j.1365-2141.2012.09225.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang M, Krishnadas D, Lucas K. Cellular and antibody based approaches for pediatric cancer immunotherapy. J Immunol Res 2015; 2015:675269.:Article ID 675269, 7 pages, 2015; PMID:26587548; https://doi.org/ 10.1155/2015/675269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348:69-74; PMID:25838375; https://doi.org/ 10.1126/science.aaa4971 [DOI] [PubMed] [Google Scholar]
- 59.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al.. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015; 372:2509-20; PMID:26028255; https://doi.org/ 10.1056/NEJMoa1500596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smyrk TC, Watson P, Kaul K, Lynch HT. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer 2001; 91:2417-22; PMID:11413533; https://doi.org/ 10.1002/1097-0142(20010615)91:12%3c2417::AID-CNCR1276%3e3.0.CO;2-U [DOI] [PubMed] [Google Scholar]
- 61.Richard GF, Kerrest A, Dujon B. Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol Mol Biol Rev 2008; 72:686-727; PMID:19052325; https://doi.org/ 10.1128/MMBR.00011-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold GL, et al.. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013; 14:207-15; PMID:23954159; https://doi.org/ 10.1016/j.chom.2013.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Corso CD, Ali AN, Diaz R. Radiation-induced tumor neoantigens: Imaging and therapeutic implications. Am J Cancer Res 2011; 1:390-412; PMID:21969260 [PMC free article] [PubMed] [Google Scholar]
- 64.Martens K, Meyners T, Rades D, Tronnier V, Bonsanto MM, Petersen D, Dunst J, Dellas K. The prognostic value of tumor necrosis in patients undergoing stereotactic radiosurgery of brain metastases. Radiat Oncol 2013; 8:162; PMID:23822663; https://doi.org/ 10.1186/1748-717X-8-162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parvez K, Parvez A, Zadeh G. The diagnosis and treatment of pseudoprogression, radiation necrosis and brain tumor recurrence. Int J Mol Sci 2014; 15:11832-46; PMID:24995696; https://doi.org/ 10.3390/ijms150711832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Festjens N, Vanden Berghe T, Vandenabeele P. Necrosis, a well-orchestrated form of cell demise: Signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta 2006; 1757:1371-87; PMID:16950166; https://doi.org/ 10.1016/j.bbabio.2006.06.014 [DOI] [PubMed] [Google Scholar]
- 67.Formenti SC, Demaria S. Systemic effects of local radiotherapy. Lancet Oncol 2009; 10:718-26; PMID:19573801; https://doi.org/ 10.1016/S1470-2045(09)70082-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, Kroemer G. Cell death modalities: Classification and pathophysiological implications. Cell Death Differ 2007; 14:1237-43; PMID:17431418; https://doi.org/ 10.1038/sj.cdd.4402148 [DOI] [PubMed] [Google Scholar]
- 69.Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al.. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 2015; 162:961-73; PMID:26317465; https://doi.org/ 10.1016/j.cell.2015.07.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al.. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 2015; 162:974-86; PMID:26317466; https://doi.org/ 10.1016/j.cell.2015.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maio M, Covre A, Fratta E, Di Giacomo AM, Taverna P, Natali PG, Coral S, Sigalotti L. Molecular pathways: At the crossroads of cancer epigenetics and immunotherapy. Clin Cancer Res 2015; 21:4040-7; PMID:26374074; https://doi.org/ 10.1158/1078-0432.CCR-14-2914 [DOI] [PubMed] [Google Scholar]
- 72.Zhao H, Ning S, Scicinski J, Oronsky B, Knox S, Peehl DM. Abstract 3515: RRx-001: A double action systemically non-toxic epigenetic agent for cancer therapy. Cancer Research 2015; 75:3515; https://doi.org/ 10.1158/1538-7445.AM2015-3515 [DOI] [Google Scholar]
- 73.Reid T, Infante JR, Paul A, Burris HA, Oronsky B, Scribner C, Knox S, Stephens J, Santini J, Scicinski J. Abstract LB-86: Preliminary results from an ongoing phase I trial of RRx-001, a tumor selective cytotoxic agent. Cancer Res 2013; 73:LB-86; https://doi.org/ 10.1158/1538-7445.AM2013-LB-86 [DOI] [Google Scholar]
- 74.Garcia-Lora A, Algarra I, Garrido F. MHC class I antigens, immune surveillance, and tumor immune escape. J Cell Physiol 2003; 195:346-55; PMID:12704644; https://doi.org/ 10.1002/jcp.10290 [DOI] [PubMed] [Google Scholar]
- 75.Chang CC, Ferrone S. Immune selective pressure and HLA class I antigen defects in malignant lesions. Cancer Immunol Immunother 2007; 56:227-36; PMID:16783578; https://doi.org/ 10.1007/s00262-006-0183-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Campoli M, Ferrone S. HLA antigen changes in malignant cells: Epigenetic mechanisms and biologic significance. Oncogene 2008; 27:5869-85; PMID:18836468; https://doi.org/ 10.1038/onc.2008.273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Khan AN, Magner WJ, Tomasi TB. An epigenetically altered tumor cell vaccine. Cancer Immunol Immunother 2004; 53:748-54; PMID:14997346; https://doi.org/ 10.1007/s00262-004-0513-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Magner WJ, Kazim AL, Stewart C, Romano MA, Catalano G, Grande C, Keiser N, Santaniello F, Tomasi TB. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol 2000; 165:7017-24; PMID:11120829; https://doi.org/ 10.4049/jimmunol.165.12.7017 [DOI] [PubMed] [Google Scholar]
- 79.Tomasi TB, Magner WJ, Khan AN. Epigenetic regulation of immune escape genes in cancer. Cancer Immunol Immunother 2006; 55:1159-84; PMID:16680460; https://doi.org/ 10.1007/s00262-006-0164-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Coral S, Sigalotti L, Gasparollo A, Cattarossi I, Visintin A, Cattelan A, Altomonte M, Maio M. Prolonged upregulation of the expression of HLA class I antigens and costimulatory molecules on melanoma cells treated with 5-aza-2′-deoxycytidine (5-AZA-CdR). J Immunother 1999; 22:16-24; PMID:9924695; https://doi.org/ 10.1097/00002371-199901000-00003 [DOI] [PubMed] [Google Scholar]
- 81.Fonsatti E, Nicolay HJ, Sigalotti L, Calabro L, Pezzani L, Colizzi F, Altomonte M, Guidoboni M, Marincola FM, Maio M. Functional up-regulation of human leukocyte antigen class I antigens expression by 5-aza-2′-deoxycytidine in cutaneous melanoma: immunotherapeutic implications. Clin Cancer Res 2007; 13:3333-8; PMID:17545540; https://doi.org/ 10.1158/1078-0432.CCR-06-3091 [DOI] [PubMed] [Google Scholar]
- 82.Capece D, Verzella D, Fischietti M, Zazzeroni F, Alesse E. Targeting costimulatory molecules to improve antitumor immunity. J Biomed Biotechnol 2012; 2012:926321; PMID:22500111; https://doi.org/ 10.1155/2012/926321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Garcia KC, Teyton L, Wilson IA. Structural basis of T cell recognition. Annu Rev Immunol 1999; 17:369-97; PMID:10358763; https://doi.org/ 10.1146/annurev.immunol.17.1.369 [DOI] [PubMed] [Google Scholar]
- 84.Aleksic M, Dushek O, Zhang H, Shenderov E, Chen JL, Cerundolo V, Coombs D, van der Merwe PA. Dependence of T cell antigen recognition on T cell receptor-peptide MHC confinement time. Immunity 2010; 32:163-74; PMID:20137987; https://doi.org/ 10.1016/j.immuni.2009.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med 1987; 165:302-19; PMID:3029267; https://doi.org/ 10.1084/jem.165.2.302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ishii N, Takahashi T, Soroosh P, Sugamura K. OX40-OX40 ligand interaction in T-cell-mediated immunity and immunopathology. Adv Immunol 2010; 105:63-98; PMID:20510730; https://doi.org/ 10.1016/S0065-2776(10)05003-0 [DOI] [PubMed] [Google Scholar]
- 87.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 2007; 27:111-22; PMID:17629517; https://doi.org/ 10.1016/j.immuni.2007.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK, Mescher MF. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J Immunol 1999; 162:3256-62; PMID:10092777 [PubMed] [Google Scholar]
- 89.Wang LX, Mei ZY, Zhou JH, Yao YS, Li YH, Xu YH, Li JX, Gao XN, Zhou MH, Jiang MM, et al.. Low dose decitabine treatment induces CD80 expression in cancer cells and stimulates tumor specific cytotoxic T lymphocyte responses. PLoS One 2013; 8:e62924; PMID:23671644; https://doi.org/ 10.1371/journal.pone.0062924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, Fang Z, Nguyen M, Pierce S, Wei Y, et al.. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014; 28:1280-8; PMID:24270737; https://doi.org/ 10.1038/leu.2013.355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Villagra A, Sotomayor EM, Seto E. Histone deacetylases and the immunological network: Implications in cancer and inflammation. Oncogene 2010; 29:157-73; PMID:19855430; https://doi.org/ 10.1038/onc.2009.334 [DOI] [PubMed] [Google Scholar]
- 92.Nagy JA, Chang SH, Dvorak AM, Dvorak HF. Why are tumour blood vessels abnormal and why is it important to know? Br J Cancer 2009; 100:865-9; PMID:19240721; https://doi.org/ 10.1038/sj.bjc.6604929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barsoum IB, Koti M, Siemens DR, Graham CH. Mechanisms of hypoxia-mediated immune escape in cancer. Cancer Res 2014; 74:7185-90; PMID:25344227; https://doi.org/ 10.1158/0008-5472.CAN-14-2598 [DOI] [PubMed] [Google Scholar]
- 94.Mauge L, Terme M, Tartour E, Helley D. Control of the adaptive immune response by tumor vasculature. Front Oncol 2014; 4:61; PMID:24734218; https://doi.org/ 10.3389/fonc.2014.00061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Muller WA. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol 2003; 24:327-34; PMID:12810109; https://doi.org/ 10.1016/S1471-4906(03)00117-0 [DOI] [PubMed] [Google Scholar]
- 96.Muller WA. Mechanisms of transendothelial migration of leukocytes. Circ Res 2009; 105:223-30; PMID:19644057; https://doi.org/ 10.1161/CIRCRESAHA.109.200717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lu Y, Chu A, Turker MS, Glazer PM. Hypoxia-induced epigenetic regulation and silencing of the BRCA1 promoter. Mol Cell Biol 2011; 31:3339-50; PMID:21670155; https://doi.org/ 10.1128/MCB.01121-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yeh JJ, Kim WY. Targeting tumor hypoxia with hypoxia-activated prodrugs. J Clin Oncol 2015; 33:1505-8; PMID:25800764; https://doi.org/ 10.1200/JCO.2014.60.0759 [DOI] [PubMed] [Google Scholar]
- 99.Jain RK. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005; 307:58-62; PMID:15637262; https://doi.org/ 10.1126/science.1104819 [DOI] [PubMed] [Google Scholar]
- 100.Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res 2010; 70:6171-80; PMID:20631075; https://doi.org/ 10.1158/0008-5472.CAN-10-0153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tripathi C, Tewari BN, Kanchan RK, Baghel KS, Nautiyal N, Shrivastava R, Kaur H, Bhatt ML, Bhadauria S. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget 2014; 5:5350-68; PMID:25051364; https://doi.org/ 10.18632/oncotarget.2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Paulmurugan R, Oronsky B, Brouse CF, Reid T, Knox S, Scicinski J. Real time dynamic imaging and current targeted therapies in the war on cancer: A new paradigm. Theranostics 2013; 3:437-47; PMID:23781290; https://doi.org/ 10.7150/thno.5658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kim M, Parmar H, Cao Y, Pramanik P, Schipper M, Hayman J, Junck L, Mammoser A, Heth J, Carter CA, et al.. Whole brain radiotherapy and RRx-001: A case report of two partial responses in radioresistant melanoma brain metastases. Transl Oncol 2016; 9:108-13; PMID:27084426; https://doi.org/ 10.1016/j.tranon.2015.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kim R, Emi M, Tanabe K, Arihiro K. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res 2006; 66:5527-36; PMID:16740684; https://doi.org/ 10.1158/0008-5472.CAN-05-4128 [DOI] [PubMed] [Google Scholar]
- 105.Yasmin R, Siraj S, Hassan A, Khan AR, Abbasi R, Ahmad N. Epigenetic regulation of inflammatory cytokines and associated genes in human malignancies. Mediators Inflamm 2015; 2015:201703; PMID:25814785; https://doi.org/ 10.1155/2015/201703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hedjran F, Shantanu K, Tony R. Deletion analysis of Ad5 E1a transcriptional control region: Impact on tumor-selective expression of E1a and E1b. Cancer Gene Ther 2011; 18:717-23; PMID:21818136; https://doi.org/ 10.1038/cgt.2011.41 [DOI] [PubMed] [Google Scholar]
- 107.Oelkrug C, Ramage JM. Enhancement of T cell recruitment and infiltration into tumours. Clin Exp Immunol 2014; 178:1-8; PMID:24828133; https://doi.org/ 10.1111/cei.12382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.He J, Hu Y, Hu M, Li B. Development of PD-1/PD-L1 pathway in tumor immune microenvironment and treatment for non-small cell lung cancer. Sci Rep 2015; 5:13110; PMID:26279307; https://doi.org/ 10.1038/srep13110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al.. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med 2002; 8:793-800; PMID:12091876; https://doi.org/ 10.1038/nm0902-1039c [DOI] [PubMed] [Google Scholar]
- 110.Mellor AL, Munn DH. Tryptophan catabolism and T-cell tolerance: Immunosuppression by starvation? Immunol Today 1999; 20:469-73; PMID:10500295; https://doi.org/ 10.1016/S0167-5699(99)01520-0 [DOI] [PubMed] [Google Scholar]
- 111.Lob S, Konigsrainer A, Rammensee HG, Opelz G, Terness P. Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: Can we see the wood for the trees? Nat Rev Cancer 2009; 9:445-52; PMID:19461669; https://doi.org/ 10.1038/nrc2639 [DOI] [PubMed] [Google Scholar]
- 112.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest 2007; 117:1147-54; PMID:17476344; https://doi.org/ 10.1172/JCI31178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xue ZT, Sjogren HO, Salford LG, Widegren B. An epigenetic mechanism for high, synergistic expression of indoleamine 2,3-dioxygenase 1 (IDO1) by combined treatment with zebularine and IFN-gamma: potential therapeutic use in autoimmune diseases. Mol Immunol 2012; 51:101-11; PMID:22424783; https://doi.org/ 10.1016/j.molimm.2012.01.006 [DOI] [PubMed] [Google Scholar]
- 114.Opitz CA, Litzenburger UM, Opitz U, Sahm F, Ochs K, Lutz C, Wick W, Platten M. The indoleamine-2,3-dioxygenase (IDO) inhibitor 1-methyl-D-tryptophan upregulates IDO1 in human cancer cells. PLoS One 2011; 6:e19823; PMID:21625531; https://doi.org/ 10.1371/journal.pone.0019823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Beatty G, O'Dwyer P, Clark J, Shi J, Newton R, Schaub R, et al.. Phase I study of the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of the oral inhibitor of indoleamine 2,3-dioxygenase (IDO1) INCB024360 in patients (pts) with advanced malignancies. J Clin Oncol 2013; 31:Abstract 3025; PMID:2377595423775954 [Google Scholar]
- 116.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol 2006; 6:295-307; PMID:16557261; https://doi.org/ 10.1038/nri1806 [DOI] [PubMed] [Google Scholar]
- 117.Beyer M, Schultze JL. Regulatory T cells: major players in the tumor microenvironment. Curr Pharm Des 2009; 15:1879-92; PMID:19519430; https://doi.org/ 10.2174/138161209788453211 [DOI] [PubMed] [Google Scholar]
- 118.deLeeuw RJ, Kost SE, Kakal JA, Nelson BH. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: A critical review of the literature. Clin Cancer Res 2012; 18:3022-9; PMID:22510350; https://doi.org/ 10.1158/1078-0432.CCR-11-3216 [DOI] [PubMed] [Google Scholar]
- 119.Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer 2010; 127:759-67; PMID:20518016; https://doi.org/ 10.1002/ijc.25429 [DOI] [PubMed] [Google Scholar]
- 120.Andersson J, Tran DQ, Pesu M, Davidson TS, Ramsey H, O'Shea JJ, Shevach EM. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J Exp Med 2008; 205:1975-81; PMID:18710931; https://doi.org/ 10.1084/jem.20080308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, et al.. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol 2007; 5:e38; PMID:17298177; https://doi.org/ 10.1371/journal.pbio.0050038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Morita R, Hirohashi Y, Sato N. Depletion of tregs in vivo: A promising approach to enhance antitumor immunity without autoimmunity. Immunotherapy 2012; 4:1103-5; PMID:23194360; https://doi.org/ 10.2217/imt.12.116 [DOI] [PubMed] [Google Scholar]
- 123.Paschall AV, Liu K. Epigenetic regulation of apoptosis and cell cycle regulatory genes in human colon carcinoma cells. Genom Data 2015; 5:189-91; PMID:26309812; https://doi.org/ 10.1016/j.gdata.2015.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fulda S. Tumor resistance to apoptosis. Int J Cancer 2009; 124:511-5; PMID:19003982; https://doi.org/ 10.1002/ijc.24064 [DOI] [PubMed] [Google Scholar]
- 125.Hervouet E, Cheray M, Vallette FM, Cartron PF. DNA methylation and apoptosis resistance in cancer cells. Cells 2013; 2:545-73; PMID:24709797; https://doi.org/ 10.3390/cells2030545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tallen G, Riabowol K. Keep-ING balance: Tumor suppression by epigenetic regulation. FEBS Lett 2014; 588:2728-42; PMID:24632289; https://doi.org/ 10.1016/j.febslet.2014.03.011 [DOI] [PubMed] [Google Scholar]
- 127.Zuckerman V, Wolyniec K, Sionov RV, Haupt S, Haupt Y. Tumour suppression by p53: The importance of apoptosis and cellular senescence. J Pathol 2009; 219:3-15; PMID:19562738; https://doi.org/ 10.1002/path.2584 [DOI] [PubMed] [Google Scholar]
- 128.Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell 1993; 75:1169-78; PMID:7505205; https://doi.org/ 10.1016/0092-8674(93)90326-L [DOI] [PubMed] [Google Scholar]
- 129.Nagata S, Golstein P. The Fas death factor. Science 1995; 267:1449-56; PMID:7533326; https://doi.org/ 10.1126/science.7533326 [DOI] [PubMed] [Google Scholar]
- 130.Green DR, Ferguson TA. The role of Fas ligand in immune privilege. Nat Rev Mol Cell Biol 2001; 2:917-24; PMID:11733771; https://doi.org/ 10.1038/35103104 [DOI] [PubMed] [Google Scholar]
- 131.Maecker HL, Yun Z, Maecker HT, Giaccia AJ. Epigenetic changes in tumor Fas levels determine immune escape and response to therapy. Cancer Cell 2002; 2:139-48; PMID:12204534; https://doi.org/ 10.1016/S1535-6108(02)00095-8 [DOI] [PubMed] [Google Scholar]
- 132.Hahne M, Rimoldi D, Schroter M, Romero P, Schreier M, French LE, Schneider P, Bornand T, Fontana A, Lienard D, et al.. Melanoma cell expression of Fas(Apo-1/CD95) ligand: Implications for tumor immune escape. Science 1996; 274:1363-6; PMID:8910274; https://doi.org/ 10.1126/science.274.5291.1363 [DOI] [PubMed] [Google Scholar]
- 133.O'Connell J, O'Sullivan GC, Collins JK, Shanahan F. The Fas counterattack: Fas-mediated T cell killing by colon cancer cells expressing Fas ligand. J Exp Med 1996; 184:1075-82; PMID:9064324; https://doi.org/ 10.1084/jem.184.3.1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Favre-Felix N, Fromentin A, Hammann A, Solary E, Martin F, Bonnotte B. Cutting edge: The tumor counterattack hypothesis revisited: Colon cancer cells do not induce T cell apoptosis via the Fas (CD95, APO-1) pathway. J Immunol 2000; 164:5023-7; PMID:10799856; https://doi.org/ 10.4049/jimmunol.164.10.5023 [DOI] [PubMed] [Google Scholar]
- 135.McDermott D, Lebbe C, Hodi FS, Maio M, Weber JS, Wolchok JD, Thompson JA, Balch CM. Durable benefit and the potential for long-term survival with immunotherapy in advanced melanoma. Cancer Treat Rev 2014; 40:1056-64; PMID:25060490; https://doi.org/ 10.1016/j.ctrv.2014.06.012 [DOI] [PubMed] [Google Scholar]
- 136.Mack GS. Epigenetic cancer therapy makes headway. J Natl Cancer Inst 2006; 98:1443-4; PMID:17047192; https://doi.org/ 10.1093/jnci/djj447 [DOI] [PubMed] [Google Scholar]
- 137.Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol 2015; 33:1974-82; PMID:25605845; https://doi.org/ 10.1200/JCO.2014.59.4358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Garcia JS, Jain N, Godley LA. An update on the safety and efficacy of decitabine in the treatment of myelodysplastic syndromes. Onco Targets Ther 2010; 3:1-13; PMID:20616953 [DOI] [PMC free article] [PubMed] [Google Scholar]