

Abstract
In this work, a novel effective heterogeneous catalyst metal-organic framework MIL-53(Fe) has been synthesized for the purpose of activating persulfate (PS). Catalytic performance of MIL-53(Fe) activated under different vacuum conditions was investigated; stability and reusability of the catalyst were evaluated, and the activation mechanism was also investigated. The results indicated that vacuum activation could cause variation of the FeII/FeIII relative amount ratio of the catalyst, and thus would change the catalytic activities of MIL-53(Fe), because FeII or FeIII CUS (coordinative unsaturated metal site) are alternative active sites. It was found that MIL-53(Fe)-2 exhibits good performance for PS activation and could be used for multiple cycles. A removal rate of 98% for Orange G was obtained within 120min (95.7% mineralization efficiency), and 94.3% was attained in the fifth cycle. The mechanism of the activation of PS by MIL-53(Fe) was also suggested, which involves a predominant heterogeneous reaction and an auxiliary homogeneous reaction. The findings of this study provide new insight into the application of the reactive metal-organic frameworks in activating persulfate for the degradation of environmental contaminants.
1. Introduction
Advanced oxidation processes (AOPs) have been developed to successfully degrade various recalcitrant organics. Activated persulfate (PS) oxidation is an emerging AOP for organic pollutant degradation. PS is a strong oxidant with a higher reduction potential (E0 = 2.01 V) than hydrogen peroxide (E0 = 1.76 V); its activation results in generation of sulfate free radical (SO4−·, E0 = 2.5~3.1 V), a powerful oxidant with high redox potential, even higher than hydroxyl free radical (OH·, E0= 1.9~2.7 V) under certain pH conditions [1]. During the past several years, SO4−· has attracted great interest as a substitution to OH· for the degradation of recalcitrant pollutants. SO4−· possesses certain advantages over OH· such as longer half-life period (T SO4-·≈4s, yet T OH·≈10−9s) [2], higher stability, lower reactivity with background natural organic matter [3, 4], and thus it may have higher efficiency under certain conditions. And above all, since SO4−·tends to react with contaminants via electron transfer [5, 6], it is efficient in the degradation of aromatic contaminants containing a benzene ring. However, persulfate has to be effectively activated to generate SO4−· under certain conditions.
The activation method of persulfate directly determines the generation rate of SO4−·and its utilization efficiency in degrading contaminants. Zero-valent iron (ZVI), ferrous iron (Fe2+), and ferric iron (Fe3+) are the most commonly used activators [7~9]. When using ferrous ion for activating persulfate, a large amount of soluble Fe2+ will instantly react with persulfate to rapidly release SO4−·. However, excess quantities of Fe2+ will react with SO4−·to generate its oxidation product Fe3+ [10]. Consequently, SO4−· cannot be fully utilized, thus the degradation efficiency of the ferrous iron-persulfate process decreases. Besides, this activation system is significantly affected by the pH value since Fe2+ will convert to Fe3+ or its (oxo) hydroxide. At neutral and alkaline pH, Fe3+ will precipitate as iron oxide resulting in reduced activity. In order to regulate and control the concentration of soluble Fe2+ in the solution, various complexing agents such as ethylene diamine tetraacetic acid (EDTA), ethylene diamine disuccinic acid (EDDS), oxalic acid, and citric acid have been used to chelate ferrous iron [11, 12]. Although the addition of a complexing agent can successfully complex iron, the complexing agent itself would react with SO4−· [13], which results in scavenging a certain fraction of the free radicals [14]. Concerning zero valent iron activated persulfate, the catalytic efficiency is still influenced greatly by the initial pH value, though ZVI successfully serves as a slow-release source of dissolved Fe2+ and slows down the generation rate of SO4−· [15]. In addition, the surface of ZVI would easily corrode to generate species such as FeOOH and Fe2O3 [16] which cover the ZVI core and thus inhibit the release of Fe2+ needed to activate persulfate and degrade the target contaminant.
Metal Organic Frameworks (MOFs) are interesting materials that have attracted more and more attention in recent years from both academia and industry. MOFs have been successfully applied in various fields such as gas storage, separation, light and electricity, sensors and drug-delivery, especially in catalysis such as chemical, photo and biomimetic applications [17~19]. MOFs are a class of multifunctional inorganic-organic hybrid materials which usually possess a three-dimensional well-defined periodic infinite network structure [20]. MOFs can self-assemble via coordination or covalent interaction by metal ions or clusters linked with organic ligands [21]. So far, much of the research interest on MOFs was centred on their composition, structure type, tunability, and the accessible, coordinative unsaturated metal sites (CUS) [22]. The existence of CUS can significantly facilitate the interaction between MOFs and guest molecules (gases or liquids). Therefore CUS can promote the redox reactions of MOFs with other substances and thus this topic is of particular importance in catalytic processes. Previous studies have shown that MOFs exhibit semiconducting behaviour that can activate H2O2 or PS via capture of the photo-generated electrons in the conduction band under visible light irradiation [23, 24]. Though one function of MOFs is as good photocatalysts, another important function is that they can participate in AOPs through Fenton-like reaction pathways via CUS acting as alternative active sites [25]. Hence, MOFs can be promising heterogeneous catalysts for the degradation of pollutants by AOPs. For example, MIL-88A was successfully used as heterogeneous catalyst to activate persulfate for the decolorization of Rhodamine B dye [26]. MIL-100(Fe), [Cu2(btec)(btx)1.5]n was determined to have high catalytic activity towards H2O2 [27, 28] as a Fenton catalyst, and cobalt-based MOF, like ZIF-67 and Co3(BTC)2·12H2O were all proved to be good heterogeneous peroxymonosulfate(PMS) catalyst [29, 30]. MIL-53(Fe) is an iron-based MOF with appreciable stability. It is one type of low-toxicity MOF and can be synthesized through facile hydrothermal methods. Although it is an effective photocatalyst, it has been proved to have intrinsic peroxidase-like activity towards peroxides like H2O2 [31, 32] due to the existence of Fe(II) and Fe(III) in its framework which was generated via coordination between iron cations and DMF molecules [33, 34]. But up to the present, its activity towards persulfate is undeveloped and reports using MIL-53(Fe) activated persulfate to degrade contaminants are very limited. Therefore, it is of great interest to explore the feasibility of using MIL-53(Fe) as a persulfate catalyst and investigate its activation performance as well as mechanisms.
Orange G was chosen as the target contaminant of this study because it is often present in many industry wastewaters, such as textile, pulp and paper, printing, cosmetic and others [35]. It is toxic, potential carcinogenic, difficult to degrade and mineralize by traditional physical and biological treatment methods due to its complex structure and stability [36]. In this work, MIL-53(Fe) was prepared by a modified mild solvothermal method for the activation of persulfate to degrade organic contaminant. The objectives of the present study were to (i) investigate the catalytic performance of MIL-53(Fe) post-processed under different vacuum activation conditions; (ii) analyse the reason that causes the differences in catalytic capacity between MIL-53(Fe) with different structures; (iii) evaluate the stability and reusability of MIL-53(Fe) and (iv) illustrate the mechanism for activating persulfate by MIL-53(Fe).
2. Experimental
2.1 Reagents and materials
Water used in this study for the preparation of all solutions was purified using a Millipore reverse osmosis (RO) system. Chemicals used were purchased from the following sources: Orange G (OG, 80.0%), persulfate (PS, Na2S2O8, 98.0%) and p-Phthalic acid (1,4-BDC, C8H6O4, 99.0%) were purchased from the Aladdin chemistry Co., Ltd (Shanghai, China). Methyl alcohol (MeOH, CH4O, 99.5%), ethanol (C2H6O, 99.7%), N, N-dimethylformamide (DMF, HCON(CH3)2, 99.5%), ferric chloride (FeCl3·6H2O, 99.0%) and sodium bicarbonate (NaHCO3, 99.5%) were purchased from Sinopharm Chemical Reagent Co., Ltd (Beijing, China). Potassium iodate (KI, 99.0%) were purchased from Shanghai Yindian Chemical Co., Ltd (Shanghai, China). 1, 10-phenanthroline (C12H8N2·H2O, 99.0%), ammonium acetate (CH3COONH4, 98.0%), hydroxylamine hydrochloride (HONH3Cl, 98.5%), hydrochloric acid (HCl, 36%~38%) and acetic acid (C2H4O2, 99.5%) were obtained from Guangzhou Chemical Reagent Factory (Guangzhou, China). All the above reagents were of AR and purchased directly for use without further purification.
2.2 Preparation of catalysts
MIL-53(Fe) was prepared according to the method as previously reported [37] with certain after-treatment modifications. Specifically, FeCl3·6H2O (5mmol, 1.35g), p-Phthalic acid (1, 4-BDC) (5mmol, 0.83g) and N,N-dimethylformamide (DMF) (25mL) were mixed and stirred for 20min at first. The mixture was then poured into a 100mL Teflon-lined steel autoclave, placed in a fan oven preheated to 150°C and maintain for 5h. After that, the autoclave was removed from the oven and allowed to cool naturally to room temperature, and the products were collected by filtration. To remove the solvent molecule DMF, the obtained power was washed sequentially with 150mL MeOH and an equivalent amount of deionized water. The power was suspended into clean water next, followed by stirring for a whole night. Finally, the powder was dried in a vacuum oven at a required temperature (120°C, 170°C or 220°C) for a certain time length (12h, 24h or 48h) to remove H2O molecule and transform the valence state of ferrum. The resulting powder was stored at room temperature in a covered glass container until needed. The prepared MIL-53(Fe) catalysts were marked as MIL-53(Fe)-1/2/3/4/5 where 1, 2, 3, 4, 5 represents different vacuum chemical activation conditions (120°C&12h, 170°C&12h, 220°C &12h, 170°C&24h and 170°C&48h.) of synthesis.
2.3 Catalyst characterization
Crystallite structures were analyzed with a Bruker D8 Advance Powder X-ray diffraction (PXRD) using Cu Kα radiations (λ=0.15418nm). The elemental composition of the samples and valence state of each element were characterized by X-ray photoelectron spectroscopy (XPS, Kratos Axis Ultra DLD) with Al Kα radiation was operated at 1486.6eV, 10mA×15KV, 700×300 μm. Binding energies were calibrated versus the carbon signal at 284.6eV. The morphology was observed with a Merlin Compact scanning electron microscope (SEM). The Brunauer-Emmett-Teller (BET) surface area and porous structure were measured using a surface area and porosity analyzer (ASAP 2020, micromeritics). All the samples were degassed in vacuum at 393K for 12h prior to the measurement, the nitrogen adsorption-desorption isotherms were measured at 77K. Zeta potential of MIL-53(Fe) in ultrapure water was measured with Malvern Zetasizer Nano instrument (Malvern, UK). The mineralization of OG solution was established on the basis of total organic carbon (TOC) content, performed by using a TOC analyzer (GE Sievers 800) after the sample was quenched by 2M sodium thiosulfate pentahydrate.
2.4 Analytical methods
OG concentration was detected using UV-vis spectrophotometry at the wavelength of 478 nm. The concentration of persulfate anions was determined by iodometric titration also using UV-vis spectrophotometry at the wavelength of 352 nm. Leachable amount of iron ion (including total iron and ferrous iron) during the oxidative process was measured by 1, 10-phenanthroline spectrophotometry at the wavelength of 510nm. EPR spectra were recorded at liquid nitrogen temperature with a Bruker A300 spectrometer (Germany), with a resonance frequency of 9.875 GHz, microwave power of 18.44 mW, modulation frequency of 100 kHz, modulation amplitude of 2.0 G, sweep width of 500 G, time constant of 81.92 ms, sweep time of 40.96 s, and receiver gain of 1.00 × 103. 5, 5-Dimethyl-1-pyrroline N-oxide (DMPO) was used as the radical spin trap.
2.5 Experimental procedures
All experiments were conducted in a constant temperature shaker shaken at 180rpm, 25°C in dark environment. In a typical procedure, the solid catalysts (MIL-53(Fe)) were added into a series of 250 ml conical flasks filled with desired concentration of OG and persulfate solution. The reaction was timed as soon as MIL-53(Fe) was added. These conical flasks were rotated for the desired time length (2h, 3h or 5h) in the shaker throughout the experimental period. The initial pH value of the solutions was adjusted with 0.05M sulfuric acid (H2SO4) or sodium hydroxide (NaOH) when needed.
At given reaction time intervals, a certain amount of the sample were taken out from the flask, and injected into a colorimetric tube filled with ethanol solution to quench the reaction immediately. The mixed solution was then reserved for the detection of the residual concentration of OG, persulfate or iron. All samples were filtrated through 0.22μm filter membrane prior to analysis. Selected samples were analyzed in duplicate within accepted analytical error (±5%), for which the average was used.
2.6 EPR measurements
Electron Paramagnetic Resonance (EPR) experiments were conducted as follows: Reactions were performed with 9 mL Na2S2O8 (32mM) solution, 1 mL 5, 5-dimethyl-1-pyrroline N-oxide (DMPO, 88mM) and 0.01 g MIL-53(Fe) in a 10mL glass sample vial with PTEE-lined cap. The final concentration of DMPO was 8.8mM in the reaction solutions. PS and DMPO solutions were prepared with ultrapure water. At each sampling event, the sample was taken out using a glass capillary and recorded immediately by EPR.
3. Results and discussion
3.1 Synthesis and characterization of MIL-53(Fe) activated under different vacuum conditions
Since it is the iron CUS of MIL-53(Fe) that act as the main active site in persulfate activation and promote the decomposition of aqueous persulfate to generate SO4−·, seeking effective methods to change the relative content of iron CUS is essential for improving the catalytic activity of MIL-53(Fe). Preparation of mixed valence MOFs has been widely reported. The location of the ions with different oxidation states of such mixed valence MOFs may be exchangeable without change of the structure [34]. One of the studies have shown that part of the H2O molecules on the terminal of iron octahedral of MIL-100(Fe) could be removed from the framework through heating above 100°C under vacuum or in an inert gas flow, leading to the exposure of a large number of FeIII CUS acting as Lewis acid sites in the pore. And FeII CUS could be created above 150°C not due to the direct reduction of FeIII CUS but rather to the departure of anionic ligands (F− or OH−). This important feature indicated that the amount of FeIII CUS and FeII CUS may be changed by thermal activation under vacuum whilst keeping the structure intact [38, 39]. Other studies have also shown that iron-based trimeric MOFs such as MIL-88(Fe) also possess this kind of controlled reducibility property of Fe(III) and Fe(II) CUS [40] through vacuum or inert atmosphere treatment under certain conditions. Although MIL-53(Fe) is one type of iron-based dimeric MOF and possess different secondary building unit (SBU) with MIL-100(Fe) or MIL-88(Fe) [41], this controlled reducibility may still be applicable. This is because MIL-53(Fe) has a similar primary building unit (PBU) to MIL-100(Fe) and MIL-88(Fe): all of the central iron of these MOFs adopt six-fold coordination pattern [42], where four of the oxygen atoms originate from the ligand that occupies the equatorial positions while the other two disordered atoms originate from the unbound hydroxyl or H2O molecules. Together they form an octahedral geometry backbone [43]. Thus, during the vacuum activation process, with the removal of H2O and OH−, Fe(II) and (III) would finally form. For this purpose, MIL-53(Fe) was post-processed using vacuum chemical activation under different time and temperature to obtain different Fe CUS state and catalytic capacities.
As mentioned above, In order to verify the above assumption about the change of iron CUS during vacuum activation, X-ray photoelectron spectroscopy (XPS) was used to analyze the chemical states of Fe. XPS is a surface characterization technique that provides qualitative and quantitative information from the outermost few atomic layers [44]. It is extremely sensitive to Fe2+ and Fe3+ and is often used to study the compositions of mixed-valence iron oxides. Primarily, XPS results proved that the as-pre pared MIL-53(Fe) only contained C, Fe and O elements, the relative content of N was too low to quantify, which means that DMF was successfully removed from MIL-53(Fe) during the elution process. Since iron cations would coordinate with DMF during the crystallization process, the complete removal of DMF molecule via ligand dissociation would cause the exposure of iron coordinative unsaturated metal center [45], and hence could provide the feasible site for activating persulfate. Therefore, this suggested that iron CUS were successfully formed under the conditions employed. Secondly, Fe2p spectra were fitted to three components as illustrated (Fig. 1). Peak parameters used are presented in Table 1.
Fig. 1.

Fe2p XPS spectra of MIL-53(Fe) synthesized by different vacuum activation conditions: (a) MIL-53(Fe)-3/2/1 and (b) MIL-53(Fe)-2/4/5.
Table 1.
Chemical states of Fe elements and there corresponding parameters used in this study.
| Conditions | Sub level (photo electron core level) | Assignment | Peak position | FWHM | Area (%) | FeII/FeIII ratio |
|---|---|---|---|---|---|---|
| MIL-53(Fe)-3 (220°C, 12h) | Fe 2p3/2 | FeII | 709.3 | 2.70 | 47.8% | 1.95 |
| FeIII | 711.5 | 2.70 | 33.9% | |||
| FeII | 714.2 | 2.70 | 18.2% | |||
| MIL-53(Fe)-1 (120°C, 12h) | Fe 2p3/2 | FeII | 709.6 | 2.48 | 37.6% | 1.07 |
| FeIII | 712.0 | 2.70 | 48.2% | |||
| FeII | 714.7 | 2.70 | 14.2% | |||
| MIL-53(Fe)-2 (170°C, 12h) | Fe 2p3/2 | FeII | 709.4 | 2.10 | 39.8% | 1.76 |
| FeIII | 711.3 | 2.18 | 36.1% | |||
| FeII | 713.9 | 2.70 | 24.1% | |||
| MIL-53(Fe)-4 (170°C, 24h) | Fe 2p3/2 | FeII | 709.3 | 2.22 | 39.5% | 1.53 |
| FeIII | 711.1 | 2.02 | 37.8% | |||
| FeII | 712.7 | 2.43 | 22.7% | |||
| MIL-53(Fe)-5 (170°C, 48h) | Fe 2p3/2 | FeII | 709.5 | 2.58 | 35.3% | 1.24 |
| FeIII | 711.4 | 1.78 | 44.7% | |||
| FeII | 712.5 | 3.10 | 20.0% |
As can be seen, the relative content ratio of FeII/FeIII of MILs activated under different vacuum activation conditions differs from each other. FeII/FeIII ratio of MIL-53(Fe) activated under 220°C, 170°C and 120°C were 1.95, 1.76 and 1.07, respectively, which revealed that high temperature was effective to promote the generation of FeII CUS. However, prolonging heating time could not further increase the relative amount of FeII CUS. Conversely, FeII/FeIII ratios were decreased to 1.53 and 1.24 when the activation time was 24h and 48h. This finding was also in accordance with a previous report [34] that used bond valence calculations.
The catalysts prepared under different vacuum conditions were also analysed by XRD. As depicted in Fig. 2, there are conspicuous differences between the XRD patterns of different catalysts. As for MIL-53(Fe)-1/2/3, the peak at 12.7° assigned to the important characteristic pattern of MIL-53(Fe) greatly decreases with the increasing of the vacuum temperature, indicating that the crystalline lattice of MIL-53(Fe) was deformed when dehydrated at high temperature [46]. Likewise, the intensities of the characteristic peaks gradually abated with prolonged heating time, which means that long time vacuum activation could also destroy the crystal structure of MIL-53(Fe). The pattern of MIL-53(Fe)-2 match well with those reported in previous studies [47], the characteristic peaks of were at 2θ of 9.3°, 12.7°, 17.6°, 18.5°, 25.5° and 27.3°, thus demonstrating that pure MIL-53(Fe) was prepared successfully in this condition.
Fig. 2.

XRD patterns of MIL-53(Fe) activated under different vacuum conditions.
3.2 Reactivity of MIL-53(Fe) activated under different vacuum conditions as PS activator
Theoretically, the increase of relative amount of FeII CUS would afford more alternative activation sites for PS, and thus could enhance the catalytic activity of MIL-53(Fe). This hypothesis was verified by the degradation of OG in different MIL-53(Fe) activated PS systems. As shown in Fig. 3, persulfate alone exhibited a relatively low reactivity towards OG, at the reaction time of 120min, the removal rate of OG were only about 1% in PS alone, while the presence of MIL-53(Fe) could obviously promote the degradation of OG. And vacuum activation did have an influence on catalytic activity of MIL-53(Fe). After 60min treatment, OG removal rate reached 96.3%, 83.7% and 64% in MIL-53(Fe)-3/PS, MIL-53(Fe)-2/PS and MIL-53(Fe)-1/PS system, respectively. While it changed to 77.2% and 70.6% in MIL-53(Fe)-4/PS and MIL-53(Fe)-5 systems, respectively. This implies that higher vacuum activation temperature could increase the catalytic activity of MIL-53(Fe), whereas prolonging the activation time had an inhibitory effect. It can be seen from the above analysis that the variation between catalytic activities of the as-prepared MIL-53(Fe) was in accordance with the changing regulation of relative content ratio of FeII/FeIII. This implies that vacuum activation could change the relative content of iron CUS and hence would change the catalytic activity of MIL-53(Fe). It is worth noting that the adsorption of OG by MIL-53(Fe)-2 alone only contributed around 2% removal, and similar weak adsorption behaviour was also observed in the case of MIL-53(Fe)-1/3/4/5 (data not shown in this paper).
Fig. 3.

Effect of vacuum activation conditions on catalytic activity. Experimental conditions: [OG]=0.2mM, [PS]=32mM; Catalyst dose=1g/L, T=25°C, ambient pH.
In order to better understand the weak adsorption ability of MIL-53(Fe), Zeta potential was measured to analyse the electrostatic interactions between MIL-53(Fe) and OG. Fig. S1 shows that the isoelectric point of MIL-53(Fe)-2 was at 10.4, and the quantity of electric charge of MIL-53(Fe)-2 is determined to be below 10mV, hence there is a weak electrostatic interaction between positively charged MIL-53(Fe)-2 and OG anions. Although theoretically, the electrostatic interaction was helpful for the adsorption, MIL-53(Fe) showed low adsorption ability towards OG. This is likely due to the large amount of SO42− anions in the solution (brought by PS) blockingthe electrostatic interactions [48]. Also, since H2BTC molecule has a short branched-chain, the pore size and BET surface area of MIL-53(Fe) is too small (the data was given later in Table 2) to accept OG for entering the inner space of its pore, and the interaction of π-π stacking between the aromatic rings of OG and MIL-53(Fe) is not strong enough for significant adsorption [49]. Therefore, it is clear that the enhanced removal of OG in MIL-53(Fe)/PS system was mainly due to catalytic persulfate oxidation rather than adsorption. Also, pHZPC of MIL-53(Fe) before vacuum activation was determined to be 10.7 (also see Fig. S1), and was around 10.2 after vacuum activation. This suggest that vacuum activation had little effect on the charged surface of MIL-53(Fe) in aqueous solution, and thus did not change the adsorption ability of MIL-53(Fe).
Table 2.
Structural properties of MIL-53(Fe) prepared using different vacuum activation conditions
| Catalysts | SBET (m2/g) | Total pore volume (cm3/g×10−2) | Pore size (nm) |
|---|---|---|---|
| MIL-53(Fe)-1 | 77.63 | 9.29 | 5.96 |
| MIL-53(Fe)-2 | 88.64 | 12.06 | 3.03 |
| MIL-53(Fe)-3 | 89.69 | 12.71 | 3.01 |
| MIL-53(Fe)-4 | 85.34 | 10.71 | 3.50 |
| MIL-53(Fe)-5 | 80.75 | 9.98 | 5.11 |
In addition, BET surface area and total pore volume of the mentioned MIL-53(Fe) were determined, and the results are recorded in Table. 2. It can be seen from the results that BET surface area and total pore volume of MILs material are very small, hence resulted in the weak adsorption ability towards OG. Also, changes of surface area and pore volume of different MIL-53(Fe) were relatively small, the SBET and Total pore volume of MIL-53(Fe)-1/2/3/4/5 were 77.63, 88.64, 89.69, 85.34, 80.75 m2/g and 0.0929, 0.1206, 0.1271, 0.1071, 0.0998 cm3/g, respectively. It can be seen from the above data that BET surface area and total pore volume of the as-prepared samples rises with the increase of vacuum activation temperature, while it reduces with the increase of vacuum activation time. And the variation between BET was also in agreement with the change of catalytic activities shown in Fig. 3, which suggests that the change of BET did have some influence on the catalytic activities of MIL-53(Fe).
Therefore, it can be concluded that the catalyst MIL-53(Fe)-2 prepared under 170°C for 12h maintained higher catalytic activities as well as intact structures. Furthermore, the oxidation efficiency of MIL-53(Fe)-2/PS for OG was monitored by total organic carbon (TOC). TOC removal rate was determined to be 95.7% in 120 min without pH adjustment. Overall, this indicates that MIL-53(Fe) is an effective PS catalyst for degrading organic pollutants.
3.3 Reusability and stability of MIL-53(Fe)-2
The recyclability of the catalyst is crucial to the catalytic application of novel catalysts. Herein, the reusability of MIL-53(Fe) was checked by five circulating runs in the catalytic degradation of OG with MIL-53(Fe)-2/PS system without additional treatment on the spent MIL-53(Fe). Initially, the catalyst was isolated via filtration after each cycle and was then collected for use in the next cycle. But then it was noticed that a large portion of the catalyst was washed away during this filtration process, which seemed to have an influence on the removal efficiency of the next cycle (Fig. 4(a)). Therefore, another experiment was conducted to test the reusability of the catalyst without filtration. Specifically, additional OG solution was added into the system at intervals of 120min and added a total of 5 times, each time the initial concentration of OG was adjusted to 0.2mM (the remaining concentration of OG from the last cycle in the solution was assumed to be vanishingly small and thus negligible). During this process, the catalyst or PS was consumed and there was none remaining. The results are shown in Fig. 4(b). Contrary to Fig. 4(a), it was found that under this situation OG removal rate at the terminal of each cycle were 97.9%, 96.5%, 95.1%, 95.0% and 94.3%, respectively. This indicates that even after 5 cycles, the MIL-53(Fe)-2/PS system still exhibited a stable and effective activity to oxidize OG. This illustrates that the loss of the catalyst in filtration process did have an obvious influence on the oxidation of OG, because MIL-53(Fe) exists as nanoparticles which could pass through the filterable membrane and wash away with the leachate. Moreover, in comparison with the 1st cycle, the removal rate of OG of the 2nd cycle had significantly enlarged, which may due to the change of generation rate of sulfate radical in this oxidative system. But it tends to decrease slightly in the following 3rd, 4th and 5th cycles, which can be ascribed to the minor loss of FeII active catalytic sites of MIL-53(Fe).
Fig. 4.

The recycling runs of the degradation of OG for the MIL-53(Fe)-2/PS system. (a) The catalyst was filtered after each cycle and (b) without filtration. Experimental conditions: [OG]=0.2mM, [PS]=32mM, Catalyst dosage=1g/L, T=25°, ambient pH
This assumption was also verified by XPS results, after 5 cycles, the FeII/FeIII ratio of MIL-53(Fe)-2 changed from 1.76 to 0.96 (Fig. S2). And the reason for maintaining high OG removal efficiency for an extended period can be attributed to the conversion between FeIII CUS and FeII CUS due to electron transfer within the framework. The same phenomenon was also observed in other Fe-MOFs/AOPs systems by previous studies [50]. This finding suggested that MIL-53(Fe) could be continuously used for multiple cycles with its catalytic activity remaining almost the same. This feature enables MIL-53(Fe) to be a durable and effective catalyst for the persulfate activation process.
Another reason for the loss of FeII CUS can be explained by leachable iron in the solutions. It can be seen from Fig. 5(a) that the concentration of total iron gradually increased during the reaction, and attained 32mg·L−1after reaction for 120min. This means that a small fraction of MIL-53(Fe)-2 was dissolved into the solution and iron leached from the catalyst caused the decrease of active catalytic sites provided by FeII CUS or FeIII CUS on the framework. The amount of dissolved ferrous iron marginally decreased after reaction, while that of ferric iron rose continuously, indicating that a large part of dissolved Fe2+ also participated in the reaction and transferred into dissolved Fe3+, thus the amount of dissolved Fe3+ apparently grew as reaction time passed by. Furthermore, in order to clarify why the iron leaching was so high, the as-prepared catalyst was dissolved in deionized water with the initial pH adjusted to different values. The solution was vibrated using a shaker for 3 hours. During this time, water samples were taken from both groups at different time points for detecting the concentration of iron in the solutions. The results as presented in Fig. 5(b) indicated that MIL-53(Fe) was not so stable in acidic conditions, iron leaching amount was about 11 to 30 mg/L when solution pH was 2.5, and was about 6.7 to 14 mg/L when solution pH was 3.0. But the amount was low when solution pH value was above 5.0, which means that the catalyst can be stable in near-neutral and alkaline condition. Hence MIL-53(Fe) must be corrodible in activated persulfate system. The initial solution pH before activation was 2.5 (PS initial concentration was 32mM), and it was detected to be 2.3 after MIL-53(Fe)-2 activated PS for 2h, this may explain why iron leaching amount was high after reaction.
Fig. 5.

(a) Total iron leaching during reaction process (b) total iron leaching when MIL-53(Fe)-2 was dissolved in deionized water with the initial pH adjusted to different value. The values of an entire bar in figure (a) represent the total iron amount.
Based on the high concentration of iron leaching, it is crucial to decide whether the reaction is completely heterogeneous or there exists homogeneous process in this system. Hence, the percolate from the above experiment (Fig. 5(b)) was collected to do the homogeneous catalytic reaction after MIL-53(Fe) was filtered out. As can be seen from Fig. 6, OG can be partly degraded when percolate pH was 2.5, after 90 min, the removal rate achieved 29.3%, and was of only 3%–5% (even after 180min) in other pH conditions (from 3 to 11). Compared to the heterogeneous reaction shown in the first column, it is apparent that the degradation was mainly due to heterogeneous reaction, with homogeneous reaction providing a minor contribution.
Fig. 6.

Degradation of OG when using the percolate of MIL-53(Fe)-2 (after 3h’s vibration) as PS activator. The initial percolate pH value was also adjusted to 2.5, 3, 5, 7, 9 and 11. Experimental conditions: [OG]=0.2mM, [PS]=32mM, T=25°C, ambient pH
Examining the surface morphology and the crystal structure before and after reaction are necessary to evaluate the stability of MIL-53(Fe). As seen in Fig. 7, there were obvious distinctions between as-prepared and used MIL-53(Fe)-2 after activating PS for 5 cycles in the overview of the catalyst. The raw catalyst MIL-53(Fe)-2 was composed of collapsed octahedron crystals with a non-uniform diameter from 100–500 nm, which is consistent with the previous report [31], whereas the image of the used catalyst was consisted of thin rod-like particles, each with a length of 100–200nm and about 25nm in diameter.
Fig. 7.

SEM image of (a) as-prepared MIL-53(Fe)-2 and (b) MIL-53(Fe)-2 after activate PS for 5 cycles.
Furthermore, the catalyst was analyzed by XRD after reaction. As shown in Fig. 8, the XRD patterns of MIL-53(Fe)-2 after activating PS for a single cycle and five cycles are quite different from that of the freshly prepared catalyst. In particular, the intensity of the characteristic peaks at 2θ of 9.3°, 12.7° and 18.5° of the catalyst after activating PS for 1 cycle decreased sharply, and the peaks finally disappeared after 5 cycles. The intensity of peaks at 25.5° decreased apparently, and so did that of 17.6° (a marginally decrease). And a new peak at 28.0° emerged after reaction. The results indicate that the crystal structure as well as morphology of the used catalyst had some changes after activating PS, which supports that MIL-53(Fe) could activate persulfate to decompose to free radicals due to catalytic reaction.
Fig. 8.

XRD pattern of MIL-53(Fe)-2 before and the after activated persulfate.
3.4 Identification of predominant free radical species
In order to gain insight into the predominant free radicals, electron paramagnetic resonance (EPR) spectroscopy is a good choice to detect and identify free radicals. The hyperfine splitting constants of radicals-DMPO adduct can be analyzed from the obtained data. As shown in Fig. 9(c), mixing of 32mM PS and 8.8mM DMPO could form the typical six split lines of DMPO-SO4 and four split lines of DMPO-OH signals based on their hyperfine splitting constants (DMPO-OH: aN = aH = 14.2G; DMPO-SO4: aN = 13.1G, aH = 10.2G, ). The formation of DMPO-OH and DMPO-SO4 signal in MIL/PS was significantly higher than that in PS alone (see Fig. 9(a)), indicating that the concentration of SO4−· and OH· was higher than that in PS alone. According to previous reports [51], this signal is characteristic for the stable adduct with SO4−·. This demonstrated that SO4−· and OH· were produced during PS decomposition, and both of them contributed to the oxidative degradation of OG. Compared with EPR spectra conducted in the situation of complete darkness, the peak intensities of DMPO-OH and DMPO-SO4 of Fig. 9(b) were slightly lower, which indicated that MIL-53(Fe) might activate persulfate to produce free radicals through Fenton-like reaction approach without light radiation. Meanwhile, the production of free radicals was also influenced marginally by faint light, because as mentioned, MIL-53(Fe) is also one kind of photosensitizer. The effect of organic pollutants on the intensity of EPR spectra was also tested by adding OG into the mixture under identical conditions. Fig. 9(d) shows that the intensity of DMPO-OH and DMPO-SO4 signals decreased slightly when OG was added, indicating that some of the free radicals were captured and consumed by OG.
Fig. 9.

EPR spectra of DMPO mixture in different systems (a) PS+DMPO (b) MIL-53(Fe)-2+PS+DMPO under dark conditions (c) MIL-53(Fe)-2+PS+DMPO (visible light conditions) and (d) MIL-53(Fe)-2+PS+DMPO+OG. The
 symbol represented the DMPO-SO4−· adduct, and
symbol represented the DMPO-SO4−· adduct, and
 represented the DMPO-OH· adduct. All the initial pH value were without adjusted.
represented the DMPO-OH· adduct. All the initial pH value were without adjusted.
3.5 Mechanism for activation of PS by MIL-53(Fe)
The prior result strongly indicates that heterogeneous reaction was the predominant reaction involved in this system, which is also supported by the excellent sustained catalytic activity and recycling capability of MIL-53(Fe)-2. Thus, according to the above experiments, the activation mechanism is assumed as follows (Fig. 11): FeII CUS which acted as the active site undergoes a one-electron transition reaction and activates PS to generate SO4−·, meanwhile, FeII CUS returned to the trivalent state (Eq. (1)). At the same time, FeIII CUS in the framework was also able to initiate the decomposition of PS to generate oxidative persulfate radical (S2O8−·) as FeIII CUS was reduced to FeII CUS (Eq. (2)), which could be supported by several previous studies [52, 53, 54]. Under this circumstance, FeII CUS together with FeIII CUS embedded in the framework transferred to each other continuously. Simultaneously, SO4−· could be directly converted into OH· via hydrolysis reaction with water (at all pHs) (Eq. (3)) [54], this could explain why OH· existed in this system (solution pH was acidic). Besides, SO4−· and OH· could react with persulfate anion to generate additional S2O8−· (Eq. (4) and (5)) [53, 55]. In addition, iron leaching from MIL-53(Fe) into the solution played a minor role in the oxidative processes. Homogeneous reaction caused by trace amount of dissolved Fe2+ and Fe3+ also activated PS to generate a small number of free radicals (Eq. (6) and (7)). But overall, the activation reaction initiated by FeII CUS and dissolved Fe2+ were more predominant than that of FeIII CUS and dissolved Fe3+, as a result of which the amount proportion of FeII/FeIII on the node of the framework decreased and the content of dissolved Fe3+ in the solution surged after reaction. And OG was oxidized by SO4−·, S2O8−· or OH· mainly through heterogeneous reactions.
Fig. 11.
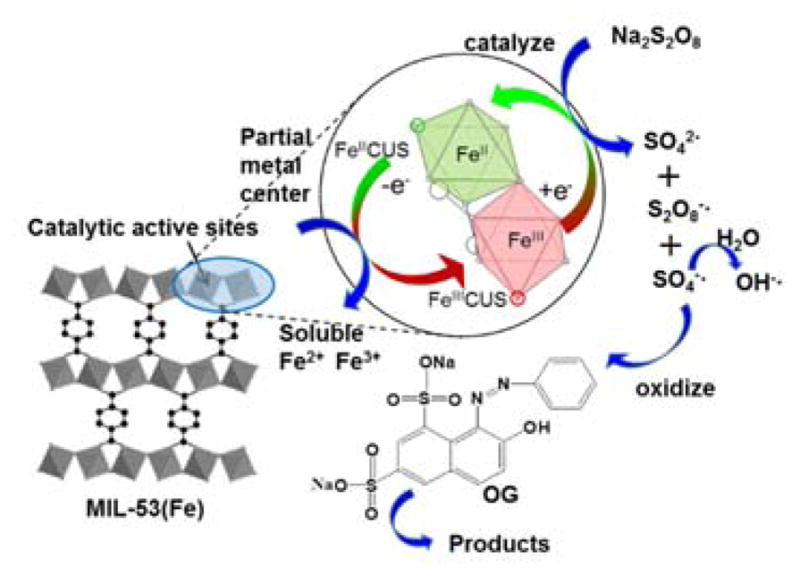
Proposed activation mechanism in MIL-53(Fe)/PS systems in ambient pH conditions.
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
4. Conclusions
In summary, this study represents the first attempt to use an iron-based MOF: MIL-53(Fe) as a heterogeneous catalyst to activate PS, which could become a promising water remediation method. MIL-53(Fe) was activated under different vacuum conditions and characterized by various techniques. The results showed that vacuum activation time or temperature did have apparent impact on crystal structure and catalytic activity of MIL-53(Fe). But this effect was not only due to the differences between BET surface areas, but also resulted from the diversity of FeII/FeIII relative amount ratio of each catalyst. Either increasing the vacuum activation temperature or reducing the vacuum activation time could increase the amount of FeII CUS, thus enhancing the catalytic activity of MIL-53(Fe). MIL-53(Fe)-2 showed best performance in maintaining high catalytic activity as well as intact structures after vacuum activation. MIL-53(Fe)-2 exhibits good reactivity as a PS activator and could be used for multiple cycles. 98% OG removal efficiency was achieved within 120min and the MIL-53 (Fe)-2/PS system exhibited a stable, durable and effective activity to oxidize OG even after 5 cycles, but the crystal structure and morphology of the used catalyst had some obvious changes. Furthermore, a possible mechanism for activation of PS by MIL-53(Fe) was proposed, involving a predominant heterogeneous reaction on the surface of the catalyst and an auxiliary homogeneous reaction in the solution. FeII together with FeIII embedded in the framework of MIL-53(Fe) could transfer to each other continuously and participate in the activation processes. This is because both FeII CUS and FeIII CUS acted as active catalytic sites and could activate PS to generate SO4−· or S2O8−·. And the existence of OH· was due to the hydrolysis reaction of SO4−·. In addition, PS could react with SO4−· and OH· to generate additional S2O8−·.
Fig. 10.

Fe2p XPS spectra of used MIL-53(Fe) samples activated under various vacuum conditions (after activating PS for 5 cycles).
Acknowledgments
This study was funded by National Natural Science Foundation of China (Grant No. 31570568, 31670585), Guangdong High level talent project (No. 201339), Science and Technology Planning Project of Guangzhou City, China (No. 20160701007, 201607020007), State key laboratory of Pulp and Paper Engineering in China (No. 201535) and SCUT Doctoral Student Short-Term Overseas Visiting Study Funding Program. The contribution of Mark Brusseau was supported by the NIEHS SRP (P42 ES04940).
References
- 1.Johnson Richard, Tratnyek Paul, Johnson Reid Obrien. Persulfate Persistence under Thermal Activation Conditions. Environmental Science & Technology. 2008;42:9350–9356. doi: 10.1021/es8019462. [DOI] [PubMed] [Google Scholar]
- 2.Liang Chenju, Huang Chiu-Fen, Chen Yan-Jyun. Potential for activated persulfate degradation of BTEX contamination. Water Research. 2008;42:4091–4100. doi: 10.1016/j.watres.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 3.Anipsitakis George P, Dionysiou Dionysios D. Degradation of Chlorinated Aromatics with Sulfate Radicals Generated by the Conjunction of Peroxymonosulfate with Cobalt. J Environmental Science and Technology. 2003;37:4790–4797. doi: 10.1021/es0263792. [DOI] [PubMed] [Google Scholar]
- 4.Furman Olha S, Teel Amy L, Watts Richard J. Mechanism of Base Activation of Persulfate. J Environmental Science & Technology. 2010;44:6423–6428. doi: 10.1021/es1013714. [DOI] [PubMed] [Google Scholar]
- 5.Fang Guodong, Liu Cun, Gao Juan, Dionysiou Dionysios D, Zhou Dongmei. Manipulation of Persistent Free Radicals in Biochar To Activate Persulfate for Contaminant Degradation. J Environmental Science & Technology. 2015;49:5645–5653. doi: 10.1021/es5061512. [DOI] [PubMed] [Google Scholar]
- 6.Monteagudo JM, Durán A, González R, Expósito AJ. In situ chemical oxidation of carbamazepine solutions using persulfate simultaneously activated by heat energy, UV light, Fe2+ ions, and H2O2. J Applied Catalysis B: Environmental. 2015;176–177:120–129. [Google Scholar]
- 7.Oh Seok-Young, Kim Hyeong-Woo, Park Jun-Mo, Park Hung-Suck, Yoon Chohee. Oxidation of polyvinyl alcohol by persulfate activated with heat, Fe2+, and zero-valent iron. J Journal of Hazardous Materials. 2009;168:346–351. doi: 10.1016/j.jhazmat.2009.02.065. [DOI] [PubMed] [Google Scholar]
- 8.Rastogi Aditya, Al-Abed Souhail R, Dionysiou Dionysios D. Sulfate Radicals Based Ferrous-Peroxymonosulfate Oxidative System for PCBs Degradation in Aqueous and Sediment Systems. J Applied Catalysis B: Environmental. 2009;85 (1):171–179. [Google Scholar]
- 9.Bolobajev Juri, Trapido Marina, Goi Anna. Improvement in iron activation ability of alachlor Fenton-like oxidation by ascorbic acid. J Chemical Engineering Journal. 2015;281:566–574. [Google Scholar]
- 10.Marchesi Massimo, Aravena Ramon, Sra Kanwartej S, Thomson Neil R, Otero Neus, Soler Albert, Mancini Silvia. Carbon isotope fractionation of chlorinated ethenes during oxidation by Fe2+ activated persulfate. J Science of the Total Environment. 2012;433:318–322. doi: 10.1016/j.scitotenv.2012.06.051. [DOI] [PubMed] [Google Scholar]
- 11.Yan Dickson YS, Lo Irene MC. Removal effectiveness and mechanisms of naphthalene and heavy metals from artificially contaminated soil by iron chelate-activated persulfate. J Environmental Pollution. 2013;178:15–22. doi: 10.1016/j.envpol.2013.02.030. [DOI] [PubMed] [Google Scholar]
- 12.Han Donghui, Wan Jinquan, Ma Yongwen, Wang Yan, Li Ying, Li Dongya, Guan Zeyu. New insights into the role of organic chelating agents in Fe(II) activated persulfate processes. J Chemical Engineering Journal. 2015;269:425–433. [Google Scholar]
- 13.Wu Xiaoliang, Gu Xiaogang, Lu Shuguang, Xu Minhui, Zang Xueke, Miao Zhouwei, Qiu Zhaofu, Sui Qian. Degradation of trichloroethylene in aqueous solution by persulfate activated with citric acid chelated ferrous ion. J Chemical Engineering Journal. 2014;255:585–592. [Google Scholar]
- 14.Kwan CY, Chu W. The role of organic ligands in ferrous-induced photochemical degradation of 2,4-dichlorophenoxyacetic acid. J Chemosphere. 2007;67:1601–1611. doi: 10.1016/j.chemosphere.2006.11.052. [DOI] [PubMed] [Google Scholar]
- 15.Liang Chenju, Lai Ming-Chun. Trichloroethylene degradation by zero valent iron activated persulfate oxidation. J Environmental Engineering Science. 2008;25(7):1071–1077. [Google Scholar]
- 16.Li Huanxuan, Wan Jinquan, Ma Yongwen, Huang Mingzhi, Wang Yan, Chen Yangmei. New insights into the role of zero-valent iron surface oxidation layers in persulfate oxidation of dibutyl phthalate solutions. J Chemical Engineering Journal. 2014;250:137–147. [Google Scholar]
- 17.Ferrando-Soria Jesús, Khajavi Hossein, Serra-Crespo Pablo, Gascon Jorge, Kapteijn Freek, Julve Miguel, Lloret Francesc, Pasán Jorge, Ruiz-Pérez Catalina, Journaux Yves, Pard Emilio. Highly Selective Chemical Sensing in a Luminescent Nanoporous Magnet. J Advanced Materials. 2012;24:5625–5629. doi: 10.1002/adma.201201846. [DOI] [PubMed] [Google Scholar]
- 18.Tanh Jeazet Harold B, Staudt Claudia, Janiak Christoph. Metal-organic frameworks in mixed-matrix membranes for gas separation. J Dalton Transactions. 2012;41:14003–14027. doi: 10.1039/c2dt31550e. [DOI] [PubMed] [Google Scholar]
- 19.Farrusseng David, Aguado Sonia, Pinel Catherine. Metal-Organic Frameworks: Opportunities for Catalysis. J Angewandte Chemie-international Edition. 2009;48:7502–7513. doi: 10.1002/anie.200806063. [DOI] [PubMed] [Google Scholar]
- 20.Kundu Tanay, Mitra Shouvik, Patra Prasun, Goswami Arunava, Daz David Daz, Banerjee Rahul. Mechanical Downsizing of a Gadolinium (III)-based Metal-Organic Framework for Anticancer Drug Delivery. J Chemistry A European Journal. 2014;20:10514–10518. doi: 10.1002/chem.201402244. [DOI] [PubMed] [Google Scholar]
- 21.Wang Jin-Liang, Wang Cheng, Lin Wenbin. Metal-Organic Frameworks for Light Harvesting and Photocatalysis. J ACS Catalysis. 2012;2(12):2630–2640. [Google Scholar]
- 22.Huo Shu-Hui, Yan Xiu-Ping. Facile magnetization of metal–organic framework MIL-101 for magnetic solid-phase extraction of polycyclic aromatic hydrocarbons in environmental water samples. J Analyst. 2012;137:3445–3451. doi: 10.1039/c2an35429b. [DOI] [PubMed] [Google Scholar]
- 23.Xu Wen-Tao, Ma Lin, Ke Fei, Peng Fu-Min, Xu Geng-Sheng, Shen Yu-Hua, Zhu Jun-Fa, Qiua Ling-Guang, Yuan Yu-Peng. Metal-organic frameworks MIL-88A hexagonal microrods as a new photocatalyst for efficient decolorization of methylene blue dye. J Dalton Transactions. 2014;43:3792–3798. doi: 10.1039/c3dt52574k. [DOI] [PubMed] [Google Scholar]
- 24.Li Xiyi, Pi Yunhong, Xia Qibin, Li Zhong, Xiao Jing. TiO2 encapsulated in Salicylaldehyde-NH2-MIL-101(Cr) for enhanced visible light-driven photodegradation of MB. J Appiled Catalysis B: Environmental. 2016;191:192–201. [Google Scholar]
- 25.Ai Lunhong, Zhang Caihong, Li Lili, Jiang Jing. Iron terephthalate metal-organic framework: Revealing the effective activation of hydrogen peroxide for the degradation of organic dye under visible light irradiation. J Applied Catalysis B: Environmental. 2014;148–149:191–200. [Google Scholar]
- 26.Lin Kun-Yi Andrew, Chang Hsuan-Ang, Hsu Chung-Jun. Iron-based metal organic framework, MIL-88A, as a heterogeneous persulfate catalyst for decolorization of Rhodamine B in water. J RSC Advances. 2015;5:32520–32530. [Google Scholar]
- 27.Lv Huanli, Zhao Hongying, Cao Tongcheng, Qian Lin, Wang Yanbin, Zhao Guohua. Efficient degradation of high concentration azo-dye wastewater by heterogeneous Fenton process with iron-based metal-organic framework. J Journal of Molecular Catalysis A: Chemical. 2015;400:81–89. [Google Scholar]
- 28.Cui Guang-Hua, He Cui-Hong, Jiao Cui-Huan, Geng Jian-Chen, Blatov Vladislav A. Two metal-organic frameworks with unique high-connected binodal network topologies: synthesis, structures, and catalytic properties. J CrystEngComm. 2012;14:4210–4216. [Google Scholar]
- 29.Lin Kun-Yi Andrew, Chang Hsuan-Ang. Zeolitic Imidazole Framework-67 (ZIF-67) as a heterogeneous catalyst to activate peroxymonosulfate for degradation of Rhodamine B in water. J Journal of the Taiwan Institute of Chemical Engineers. 2015;53:40–45. [Google Scholar]
- 30.Li Huanxuan, Wan Jinquan, Ma Yongwen, Wang Yan, Chen Xi, Guan Zeyu. Degradation of refractory dibutyl phthalate by peroxymonosulfate activated with novel catalysts cobalt metal-organic frameworks: Mechanism, performance, and stability. J Journal of Hazardous Materials. 2016;318:154–163. doi: 10.1016/j.jhazmat.2016.06.058. [DOI] [PubMed] [Google Scholar]
- 31.Dong Wenfei, Liu Xidong, Shi Wenbing, Huang Yuming. Metal–organic framework MIL-53(Fe): facile microwave-assisted synthesis and use as a highly active peroxidase mimetic for glucose biosensing. J RSC Advances. 2015;5(23):17451–17457. [Google Scholar]
- 32.Ai Lunhong, Li Lili, Zhang Caihong, Fu Jian, Jiang Jing. MIL-53(Fe): A Metal–Organic Framework with Intrinsic Peroxidase-Like Catalytic Activity for Colorimetric Biosensing. J Chemistry -A European Journal. 2013;19(45):15105–15108. doi: 10.1002/chem.201303051. [DOI] [PubMed] [Google Scholar]
- 33.Sun Qiao, Liu Min, Li Keyan, Zuo Yi, Han Yitong, Wang Junhu, Song Chunshan, Zhang Guoliang, Guo Xinwen. Facile synthesis of Fe-containing metal-organic frameworks as highly efficient catalysts for degradation of phenol at neutral pH and ambient temperature. J CrystEngComm. 2015;17:7160–7168. [Google Scholar]
- 34.Medina Manuela Eloísa, Dumont Yves, Grenèche Jean-Marc, Millange Franck. FeIII/FeII regular charge order in metal-organic framework. J Chemical Communications. 2010;46:7987–7989. doi: 10.1039/c0cc02161j. [DOI] [PubMed] [Google Scholar]
- 35.Stylidi Maria, Kondarides Dimitris I, Verykios Xenophon E. Pathways of solar light-induced photocatalytic degradation of azo dyes in aqueous TiO2 suspensions. J Applied Catalyst B: Environmental. 2003;40:271–286. [Google Scholar]
- 36.Suna Jianhui, Qiao Liping, Sun Shengpeng, Wang Guoliang. Photocatalytic degradation of Orange G on nitrogen-doped TiO2 catalysts under visible light and sunlight irradiation. J Journal of Hazardous Materials. 2008;155:312–319. doi: 10.1016/j.jhazmat.2007.11.062. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Caihong, Ai Lunhong, Jiang Jing. Solvothermal synthesis of MIL-53(Fe) hybrid magnetic composites for photoelectrochemical water oxidation and organic pollutant photodegradation under visible light. J Journal of Materials Chemistry A. 2015;3:3074–3081. [Google Scholar]
- 38.Yoon Ji Woong, Seo You-Kyong, Hwang Young Kyu, Chang Jong-San, et al. Controlled Reducibility of a Metal-Organic Framework with Coordinatively Unsaturated Sites for Preferential Gas Sorption. J Angewandte Chemie-International Edition. 2010;49:5949–5952. doi: 10.1002/anie.201001230. [DOI] [PubMed] [Google Scholar]
- 39.Wuttke Stefan, Bazin Philippe, Vimont Alexandre, Serre Christian, Seo You-Kyong, Hwang Young Kyu, Chang Jong-San, Férey Gérard, Daturi Marco. Discovering the Active Sites for C3 Separation in MIL-100(Fe) by Using Operando IR Spectroscopy. J Chemistry - A European Journal. 2012;18:11959–11967. doi: 10.1002/chem.201201006. [DOI] [PubMed] [Google Scholar]
- 40.McKinlay AC, Eubank JF, Wuttke S, Xiao B, Wheatley PS, Bazin P, Lavalley J-C, Daturi M, Vimont A, De Weireld G, Horcajada P, Serre C, Morris RE. Nitric Oxide Adsorption and Delivery in Flexible MIL-88(Fe) Metal-Organic Frameworks. J Chemistry of Materials. 2013;25:1592–1599. [Google Scholar]
- 41.Tranchemontagne David J, Mendoza-Cortés José L, O’Keeffe Michael, Yaghi Omar M. Secondary building units, nets and bonding in the chemistry of metal–organic frameworks. J Chemical Society Reviews. 2009;38:1257–1283. doi: 10.1039/b817735j. [DOI] [PubMed] [Google Scholar]
- 42.Monge Ángeles, Gándara Felipe, Gutiérrez-Puebla Enrique, Snejko Natalia. Lanthanide, Y and Sc MOFs: where amazing crystal structures meet outstanding material properties. J CrystEngComm. 2011;13:5031–5044. [Google Scholar]
- 43.Combelles C, Ben Yahia M, Pedesseau L, Doublet M-L. FeII/FeIII mixed-valence state induced by Li-insertion into the metal-organic-framework Mil53(Fe): A DFT+U study. J Journal of Power Sources. 2011;196:3426–3432. [Google Scholar]
- 44.Mielczarski Jerzy A, Atenas Gonzalo Montes, Mielczarski Ela. Role of iron surface oxidation layers in decomposition of azo-dye water pollutants in weak acidic solutions. J Applied Catalysis B: Environmental. 2005;56:289–303. [Google Scholar]
- 45.Zhou Yinxi, Song Jinliang, Liang Shuguang, Hu Suqin, Liu Huizhen, Jiang Tao, Han Buxing. Metal-organic frameworks as an acid catalyst for the synthesis of ethyl methyl carbonate via transesterification. J Journal of Molecular Catalysis A: Chemical. 2009;308(1–2):68–72. [Google Scholar]
- 46.Bezverkhyy Igor, Weber Guy, Bellat Jean-Pierre. Degradation of fluoride-free MIL-100(Fe) and MIL-53(Fe) in water: Effect of temperature and pH. J Microporous and Mesoporous Materials. 2016;219:117–124. [Google Scholar]
- 47.Du Jing-Jing, Yuan Yu-Peng, Sun Jia-Xin, Peng Fu-Min, Jiang Xia, Qiu Ling-Guang, Xie An-Jian, Shen Yu-Hua, Zhu Jun-Fa. New photocatalysts based on MIL-53 metal-organic frameworks for the decolorization of methylene blue dye. J Journal of Hazardous Materials. 2011;190:945–951. doi: 10.1016/j.jhazmat.2011.04.029. [DOI] [PubMed] [Google Scholar]
- 48.Yokoi Toshiyuki, Tatsumi Takashi, Yoshitake Hideaki. Fe3+ coordinated to amino-functionalized MCM-41: an adsorbent for the toxic oxyanions with high capacity, resistibility to inhibiting anions, and reusability after a simple treatment. J Journal of Colloid and Interface Science. 2004;274(2):451–457. doi: 10.1016/j.jcis.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 49.Yi Xiao, Fang Chen, Xixi Zhu, Hongling Qin, Hongmei Huang, Youyu Zhang, Dulin Yin, Xiaoxiao He, Kemin Wang. Ionic Liquid-assisted Formation of Lanthanide Metal-organic Framework Nano/Microrods for Superefficient Removal of Congo Red. J Chemical Research in Chinese Universities. 2015;31(6):899–903. [Google Scholar]
- 50.Lin Kun-Yi Andrew, Hsu Fu-Kong. Magnetic iron/carbon nanorods derived from a metal organic framework as an efficient heterogeneous catalyst for the chemical oxidation process in water. J RSC Advances. 2015;5:50790–50800. [Google Scholar]
- 51.Ranguelova Kalina, Rice Annette B, Khajo Abdelahad, Triquigneaux Mathilde, Garantziotis Stavros, Magliozzo Richard S, Mason Ronald P. Formation of reactive sulfite-derived free radicals by the activation of human neutrophils: An ESR study. J Free Radical Biology & Medicine. 2012;52:1264–1271. doi: 10.1016/j.freeradbiomed.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Haizhou, Bruton Thomas A, Doyle Fiona M, Sedlak David L. In Situ Chemical Oxidation of Contaminated Groundwater by Persulfate: Decomposition by Fe(III) and Mn(IV)-Containing Oxides and Aquifer Materials. J Environmental Science and Technology. 2014;48:10330–10336. doi: 10.1021/es502056d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu Xiao-Ying, Bao Zhen-Chuan, Barker John R. Free Radical Reactions Involving Cl·, Cl2−·, and SO4−· in the 248nm Photolysis of Aqueous Solutions Containing S2O82− and Cl−. J Journal of Physical Chemistry A. 2004;108:295–308. [Google Scholar]
- 54.Liu Haizhou, Bruton Thomas A, Li Wei, Van Buren Jean, Prasse Carsten, Doyle Fiona M, Sedlak David L. Oxidation of Benzene by Persulfate in the Presence of Fe(III)- and Mn(IV)-Containing Oxides: Stoichiometric Efficiency and Transformation Products. J Environmental Science & Technology. 2016;50:890–898. doi: 10.1021/acs.est.5b04815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McElroy WJ, Waygood SJ. Kinetics of the reactions of the SO4− radical with SO4−, S2O82−, H2O and Fe2+ J Journal of the Chemical Society, Faraday Transactions. 1990;86(14):2557–2564. [Google Scholar]