

ABSTRACT
Growth suppression mediated by connexin 37 (Cx37; also known as GJA4) requires interaction between its C-terminus and functional pore-forming domain. Using rat insulinoma cells, we show that Cx37 induces cell death and cell cycle arrest, and slowed cell cycling. Whether differential phosphorylation might regulate intramolecular interactions, and consequently the growth-suppressive phenotype, is unknown. Protein kinase C inhibition increased the open state probability of low-conductance gap junction channels (GJChs) and reduced GJCh closed state probability. Substituting alanine at serine residues 275, 302 and 328 eliminated Cx37-induced cell death, supported proliferation and reduced the GJCh closed state probability. With additional alanine for serine substitutions at residues 285, 319, 321 and 325, Cx37-induced cell death was eliminated and the growth arrest period prolonged, and GJCh closed state probability was restored. With aspartate substitution at these seven sites, apoptosis was induced and the open state probability of large conductance GJChs (and hemichannels) was increased. These data suggest that differential phosphorylation of the C-terminus regulates channel conformation and, thereby, cell cycle progression and cell survival.
KEY WORDS: Gap junction, Connexin, Cell cycle, Apoptosis, Gating, Phosphorylation
Summary: Connexin 37 may serve as a molecular switch that contributes to cell fate decisions, between cell cycle arrest, cell cycle progression and cell death, in a phosphorylation- and channel-dependent manner.
INTRODUCTION
Regulated intercellular exchange of ‘inorganic ions, metabolites, and high energy phosphates’ through what we now know as gap junction channels (GJChs) has been recognized since the early 1960s as essential to proper growth, differentiation and tissue homeostasis in multicellular organisms (Loewenstein, 1973; Loewenstein and Azarnia, 1990). The proteins comprising these GJChs, the connexins, form a family with 21 members, each with a unique tissue expression profile and able to form conductive intercellular channels. In the past decade or so, it has become clear that connexins facilitate tissue growth responses through channel-dependent and -independent mechanisms, the latter involving connexin-specific interactions with intracellular growth regulatory proteins (Goodenough and Paul, 2003; Haefliger et al., 2004; Kardami et al., 2007). Which combination of mechanisms – intercellular channels, transmembrane channels (hemichannels, HChs) or intracellular protein–protein interactions – mediates growth regulation appears to be both connexin- and tissue-specific.
Some studies of Cx26 (also known as GJB2; Hirschi et al., 1996), Cx32 (GJB1; Eghbali et al., 1991; Omori et al., 2001) and Cx43 (GJA1; Zhu et al., 1991) have shown that the extent of gap junction-mediated intercellular communication and cell proliferation are inversely related, suggesting GJCh function may be essential to growth suppression. However, other studies, particularly those focused on growth regulation by Cx43, suggest that the C-terminus (CT), independent of the channel-forming domain, is sufficient to mediate the growth regulatory effects of Cx43 (Dang et al., 2006). These results led to an examination of the residues in the CT of Cx43 required for growth regulation. The Cx43 CT includes multiple serine and tyrosine residues that are known (or putative) targets for phosphorylation by growth-factor-activated kinases including: PKC, MAPK, Src, CK1, CDC and others (Bao et al., 2004; Solan and Lampe, 2008). Multiple studies have shown that site-specific substitution of kinase-targeted residues with aspartate or glutamate can result in a protein that functionally mimics many of the effects of kinase activation (Grosely et al., 2013; Remo et al., 2011), whereas substitution of the targeted residues with alanine (or phenylalanine for tyrosine) results in a protein that prevents the effects of kinase activation but retains other functional properties. For Cx43, this approach has revealed that many of the functions of Cx43, including growth effects and channel regulation, are regulated through differential phosphorylation of the CT (Ek Vitorín et al., 2016; Moreno et al., 2002; Nakamura and Shibata, 1999; Shin et al., 2001; Sohl and Willecke, 2004; Solan and Lampe, 2009, 2017).
Cx43 is the most extensively studied member of the connexin protein family; less is known about the mechanisms underlying growth regulation by other connexins. We and others have shown that Cx37, a vascular connexin, regulates vascular development (Kanady et al., 2011; Simon and McWhorter, 2002, 2003) and, in adult animals, regulates the angiogenic and arteriogenic responses to injury (Fang et al., 2011; McKinnon et al., 2009). Cx37 is a known phosphoprotein, with evidence for phosphorylation on serines, threonines and tyrosines; it is also known to form GJChs of multiple conductances in several cell types (Larson et al., 2000; Morel et al., 2010; Traub et al., 1998). We have used metastatic, connexin-deficient rat insulinoma (Rin) cells to explore the unique channel and growth regulatory properties of Cx43, Cx40 (GJA5) and Cx37. Unlike Cx43 and Cx40, Cx37 expression slows proliferation of Rin cells and, in a serum-sensitive manner, results in G1 accumulation (Burt et al., 2008). Decreases in cell number occurring within 24-48 h of induced Cx37 expression suggest that Cx37 may also trigger cell death in some cells. These ‘growth’ effects of Cx37 require both the CT and channel-forming domains of the protein; neither of these domains without the other is sufficient (Good et al., 2012, 2014, 2011; Nelson et al., 2013). In addition, the Cx37 CT must be able to interact with its own channel-forming domain for Cx37 to regulate growth of Rin cells (Nelson et al., 2013).
In the current study we test the hypothesis that the many Cx37 channel conformations and ‘growth’ phenotypes triggered by wild-type (WT) Cx37 expression result from differential phosphorylation of the CT. We show that: (1) Cx37 GJCh function is regulated by PKC in a manner distinct from its regulation of Cx43 GJCh function; (2) preventing (with alanine substitution) or simulating (with aspartate substitution) phosphorylation of Cx37 at serine sites with high predicted probability of phosphorylation by growth factor activated kinases alters GJCh and HCh function in a manner predictive of altered ‘growth’ phenotype; and (3) Cx37 HChs are active in normal extracellular Ca2+ concentrations, regulated by differential phosphorylation and contribute to cell phenotype. Our data suggest that whereas the Cx37 GJCh must be able to function for Cx37 to exert a growth suppressive effect, it is the closed conformations of the GJCh and HCh that support growth arrest and specific open conformations that support cell growth, with some HCh conformations promoting cell death. We conclude that Cx37 may serve as a molecular switch (for example, in the endothelium of the vasculature) that contributes to cell fate decisions (cell cycle arrest versus cell cycle progression and cell death) in a phosphorylation- and channel-dependent manner.
RESULTS
PKC-dependent regulation of Cx37-WT function
Primary mouse skeletal muscle endothelial cells (Fig. S1) and iRin cells that express Cx37-WT are growth suppressed, have a prolonged cell cycle time and accumulate in G1 with serum deprivation (Burt et al., 2008). These effects of Cx37 rely on the protein being able to form functional GJChs regulated by interaction of the Cx37-CT with its own pore-forming domain (Good et al., 2012, 2014, 2011; Nelson et al., 2013). Cx37-WT, as expressed in iRin cells, forms GJChs of multiple conductances (40-390 pS), indicative of multiple channel conformations; however, it is not known which of these GJCh conformations is required for growth-suppressive function. Together, these observations, along with its known phosphoprotein status, suggest that growth-factor-activated kinases may phosphorylate the CT of Cx37, altering its interactions with the pore-forming domain (and possibly other proteins) and thereby the growth-suppressive properties of Cx37.
Because most growth factor receptors activate PKC, we began our exploration of this possibility by asking whether PKC activators and inhibitors regulate Cx37 GJCh function. The conductance of gap junctions comprising Cx43 and Cx40 is stable over time during dual whole-cell voltage clamp recordings; we show in Fig. 1A that this is not the case for Cx37-WT gap junctions. For other channel types, such a decline in current over time indicates the loss of ongoing channel regulation by kinases or phosphatases, as a result of the recording conditions, and is termed ‘channel run-down’ (Yamaoka and Kameyama, 2003). We show in Fig. 1A that the run-down of junctional conductance (Gj) typical of Cx37-WT gap junctions was prevented by pretreating the cells with the specific PKC antagonist, bisindolylmaleimide (BIM) (Roberts et al., 2004). Examination of the single-channel conductance profiles of untreated iRin37-WT cells and cells treated with BIM or 12-T-tetradecanoylphorbol-13-acetate (TPA, non-specific PKC agonist; Solan and Lampe, 2017) revealed a broad range of GJCh conductances in untreated cells (see also Burt et al., 2008; Nelson et al., 2013) that was shifted to favor smaller conductance states upon BIM treatment and larger conductance states with TPA treatment (Fig. 1B and inset).
Fig. 1.

Growth-factor-activated kinases regulate conductance and open probability of Cx37-WT GJChs. (A) The time-dependent decrease in junctional conductance typical of Cx37-WT gap junctions is eliminated by pretreatment of the cells with the specific PKC antagonist BIM (WT-BIM, N=4; WT-Untx, N=5). (B) Untreated Cx37-WT GJChs display transitions of multiple amplitudes. Pretreatment with BIM induces a shift to smaller transition amplitudes whereas pretreatment with TPA induces a small increase in the frequency of larger-amplitude transitions. Vj=25 mV. Inset shows difference plot wherein relative frequency of transitions in each bin for the TPA-treated iRin37-WT cells was subtracted from the relative frequency of transitions in each bin for BIM-treated iRin37-WT cells, thereby highlighting the larger transitions favored in the ‘phosphorylated’ state and smaller transitions favored in the ‘dephosphorylated’ state (Untx, N=17 cell pairs, n=1917 events; BIM, N=6, n=440; TPA, N=7, n=1064). (C) Combined relative frequency of amplitude transition and Po plots show predominant behavior of Cx37-WT BIM-treated (top plot) versus untreated (inverted plot) GJChs (if identical they would appear as mirror images). Cx37-WT GJChs spend the bulk of their time closed (63%) but are able to transition from the closed state to and between multiple open states. After PKC inactivation (BIM treatment), Cx37-WT GJChs resided predominantly in the 30-90 pS conductance state (77%), with frequent but brief transitions to the closed state. Asterisk indicates significant difference in closed state probability. All events as in B. Po: WT-BIM, N=2, 4.55′ data; WT-Untx, N=10, 10.6′ data. Po fit: WT-Untx (R2=0.99938), 4 peaks [center (in pS); relative area (%)]: closed, 55%; 35 pS, 34%; 141 pS, 8.9%; 254 pS, 1.77%; WT-BIM (R2=0.9788), 2 peaks: 65 pS, 77%; 130 pS, 23% (closed state was not fit as a distinct peak). All values are mean±s.e.m.
Despite the absence of obvious peaks in the transition amplitude histogram of untreated Cx37-WT cells, open probability analysis showed a preference of the channel to reside in the closed (55%) or 30-90 pS (34%) conductance state (Fig. 1C), suggesting that the observed large-amplitude transitions represent brief openings from the closed or 30-90 pS states to these larger-conductance states where the dwell time is short and open state probability (Po) low. Following BIM treatment, not only does the transition amplitude histogram show increased numbers of small conductance events, the open state probability analysis shows a clear preference of the channel to reside in these low conductance states (77%), with fewer transitions to short-lived closed or larger-conductance states (Fig. 1C).
That the PKC-specific antagonist had a larger net effect on channel behavior than the non-specific PKC agonist suggests that Cx37 may be PKC phosphorylated in untreated iRin37-WT cells maintained in the presence of serum. Together, these data strongly suggest that PKC-phosphorylated Cx37 GJChs reside predominantly in the closed state and transition to multiple open state configurations whereas the PKC-dephosphorylated channel resides predominantly in a 30-90 pS conductance state and transitions most commonly to the closed state, but also to larger conductance states. These data further suggest that PKC-phosphorylated Cx37 and the closed state configuration may be crucial for Cx37-induced growth arrest.
Like many phosphorylated proteins, Cx37 migrates as multiple bands on standard SDS-PAGE. Total protein isolated from untreated iRin37-WT cells reveals two bands (Fig. 2A). Previous studies on Cx43 demonstrated that PKC-phosphorylated protein can appear in multiple bands (Solan et al., 2003), with large mobility shifts more commonly due to phosphorylation at proline-flanked sites (Smith et al., 1989). The intensity of the slower-migrating form is augmented by TPA treatment and is absent when the sample is treated with alkaline phosphatase prior to loading, suggesting that the more slowly migrating band in untreated samples is indeed a phosphorylated form of Cx37 present in untreated and TPA-treated iRin37-WT cells.
Fig. 2.

Identification of putative phosphorylation sites in the CT that may regulate Cx37 channel function and proliferative phenotype. (A) Standard Tris-glycine SDS-PAGE western blot shows a double banding pattern for Cx37-WT. The intensity of the more slowly migrating band is augmented by TPA pretreatment and the band is eliminated following alkaline phosphatase (AP) treatment. MM, mass marker. (B) Phosphorylation consensus site prediction programs [NetPhos 2.0 (www.cbs.dtu.dk/services/NetPhos/), KinasePhos (kinasephos.mbc.nctu.edu.tw/) and GPS analysis (gps.biocuckoo.org/online.php)] collectively regarded seven serine residues in the region of the Cx37-CT required for Cx37-mediated growth suppression as probable targets of growth-factor-activated kinases. For each site, the kinase family and kinase with highest probability are indicated. (C) Primary sequence alignment of mCx37 and mCx43 revealed predicted Cx37 phosphorylation sites that align with known MAPK, CK1 or PKC sites in Cx43 (green). Four additional sites (red) of high probability were identified in Cx37. Underlined residues are those shown by mass spectrometry to be phosphorylated following in vitro PKC-mediated phosphorylation.
Owing to off-target effects, long-term (21 days) exposure of iRin37-WT cells to PKC agonists or antagonists to assess growth effects of phosphorylated or dephosphorylated Cx37 is not practical; therefore, we sought to convert high-probability serine targets of PKC and other growth factor activated kinases to alanine, to prevent their phosphorylation, or to aspartate, to mimic their phosphorylation. Since residues 274-333 of the Cx37-CT are required for Cx37 to exert growth suppressive effects in iRin cells (Nelson et al., 2013), we used phosphorylation consensus site prediction programs to identify high-probability serine targets in this region. Seven serine residues with >90% probability of phosphorylation (Fig. 2B; NetPhos2) by growth-factor-activated kinases were identified, three of which align with known targets for phosphorylation in Cx43 (Fig. 2C): S275 with S282 of Cx43 (ERK1/2 site; Kanemitsu and Lau, 1993; Solan and Lampe, 2008; Warn-Cramer et al., 1996); S302 with S325, 328, 330 of Cx43 (CK1 sites; Cooper and Lampe, 2002; Lampe et al., 2006); and S328 with S368 of Cx43 (PKC site; Lampe et al., 2000). Two of these sites, S302 and S328, were targeted by PKC in in vitro phosphorylation/mass spectrometry assays (see underlined residues in Fig. 2C). The data in Figs 1 and 2 led us to examine the function of three mutants of Cx37: first, an alanine for serine substitution at residues 275, 302, 328 (Cx37-S3A3); second, an alanine for serine substitution at all seven (275, 285, 302, 319, 321, 325, 328) high-probability target serine residues (Cx37-S7A7); and third, an aspartate for serine substitution at those same seven high-probability sites (Cx37-S7D7). Since the results in Fig. 1 suggested that Cx37 may be phosphorylated by PKC during the initial period of profound growth suppression, we hypothesized that: (1) channel activity in one or both serine to alanine mutants would resemble that of BIM-treated Cx37-WT-expressing cells, and (2) one or both serine to alanine mutants would fail to suppress proliferation of iRin cells. In contrast, channel activity in the serine to aspartate mutant was predicted to resemble that observed in untreated (or TPA-treated) Cx37-WT-expressing cells and this mutant was predicted to suppress iRin cell proliferation.
Cx37-S3A3 channel function and proliferation
We transfected iRin cells with the Cx37-S3A3 sequence and selected multiple antibiotic-resistant clonal cell lines (iRin37-S3A3) for further study. The iRin37-S3A3 1D2 clone expressed 7.96 pmol Cx37-S3A3 per mg total protein after 24 h in doxycycline (dox) (Fig. S2A), a level well within the range demonstrated effective for growth suppression by Cx37-WT (0.423-8.21 pmol/mg total protein; Burt et al., 2008). iRin37-S3A3 cells formed functional GJChs and Gj was comparable in iRin37-S3A3 and -WT cells (iRin37-S3A3: 4.6±1.7 nS, N=15; iRin37-WT: 2.68±1.7 nS, N=8).
Single-channel studies (Fig. 3A,B) showed that Cx37-S3A3 GJCh behavior differed from both untreated and BIM-treated iRin37-WT cells. Cx37-S3A3 channels rarely visited the closed state and spent little time in this state. Instead, Cx37-S3A3 channels transitioned between multiple open states and the 30-90 pS state, where they spent the bulk of their time (66%). The differences between Cx37-S3A3 and BIM-treated Cx37-WT GJChs (Fig. 3B) suggested that Cx37 is either a target of PKC-mediated phosphorylation at sites distinct from those altered in the Cx37-S3A3 mutant or a target for phosphorylation by other TPA-activated kinases (Solan and Lampe, 2017). Consistent with this possibility, Cx37-S3A3 channels continued to respond to PKC activation and inhibition. BIM did not prevent run-down of Cx37-S3A3 Gj (Fig. 3C) as it did for Cx37-WT, suggesting involvement of the S3A3 sites in PKC regulation of GJCh function; however, the conformations adopted by Cx37-S3A3 channels remained sensitive to TPA stimulation and PKC inhibition (Fig. 3D,E), suggesting the involvement of additional sites.
Fig. 3.

Cx37-S3A3 gap junction channel behavior differs from both untreated and BIM-treated Cx37-WT channels and remains PKC sensitive. (A) Single-channel traces and corresponding all-points histograms of Cx37-WT and Cx37-S3A3; Cx37-S3A3 channels transitioned between the fully open state and the 30-90 pS state. Vj=25 mV. Scale bars: 1 s and 2 pA. (B) Combined plots of the relative event and Po frequencies highlight the preference of Cx37-S3A3 channels (green) to transition between the 30-90 pS and larger conductance states, whereas BIM-treated Cx37-WT channels (blue) prefer to transition between the 30-90 pS and closed states. Cx37-S3A3 Po data (n=4; 5.7 min of data) were best fit (R2=0.994) by three peaks: 48 pS, 66%; 209 pS, 31%; 332 pS, 6% (closed state was not fit as a distinct peak); WT-BIM Po fit as in Fig. 1. (C) In iRin37-S3A3 cells, BIM no longer blocks run down of junctional conductance. BIM, n=6; Untx, n=8 (through 14′) and 7 (through 18.5′). (D,E) As with Cx37-WT, PKC inhibition increases the relative frequency of smaller-amplitude events whereas TPA stimulation increases the relative frequency of larger-amplitude events in Cx37-S3A3-expressing cells. Subtracting bin frequency in TPA stimulated (phosphorylated) from PKC inhibited (BIM; dephosphorylated) highlights the effect of PKC-dependent phosphorylation in channel behavior. Vj=25 mV. All values are mean±s.e.m.
We next determined whether the evident differences in Cx37-S3A3 and Cx37-WT GJCh behavior were associated with alleviation of the growth-suppression and cell cycle effects of Cx37-WT. Proliferation was assessed over a 21 day period (Fig. 4A; dox+, serum+ throughout, initial low plating density). As shown in Fig. 4B (top) the S3A3 mutations appeared to produce an intermediate growth-suppressed state. However, Fig. 4B (bottom) and 4C show that these mutations prevented the early cell death typically observed in iRin37-WT cells, reduced the duration of any Cx37-induced growth arrest period, and thereafter supported proliferation at comparable rates to WT and non-induced cells. Cell cycle consequences of serum deprivation were also examined for iRin37-WT and iRin37-S3A3 cells (see Fig. 4A for experimental paradigm; intermediate plating density). A significant increase in the percentage of iRin37-WT cells in G1 was evident after only 24 h of induced expression and was sustained for the duration of the serum-deprivation period (Fig. 4D, top). Throughout this serum-deprivation period, cell number increased more slowly compared with levels measured in the presence of serum (Fig. 4D, bottom), consistent with a prolonged cell cycle time in the serum-deprived cells (Burt et al., 2008). In contrast, the percentage of iRin37-S3A3 cells in G1 was initially significantly decreased, rather than increased, by serum deprivation and only after extended exposure did accumulation in G1 occur. Since Cx37-S3A3 cell number did not change between 24 and 120 h, this accumulation represented completion of the cell cycle by cells in S and G2 phases (Fig. 4D; Fig. S3). These data suggest that phosphorylation at these serine sites contributes to the death and growth-arrest phenotypes of Cx37-WT-expressing cells. The results in Figs 3 and 4 show that preventing phosphorylation at serine residues 275, 302 and 328 resulted in GJChs that were rarely closed, instead favoring the 30-90 pS conductance state that supported proliferation. In addition, continued responsiveness of iRin37-S3A3 cells to serum deprivation indicated that additional sites must be involved in determining the channel and growth-suppressive properties of Cx37-WT.
Fig. 4.

Cx37-induced cell death and growth arrest are eliminated, and G1 accumulation is slowed upon serum starvation of Cx37-S3A3 cells. (A) Experimental paradigms for 21 day proliferation assay and serum-deprivation assay are illustrated. Initial cell density for 21 day protocol was 3125 cells/cm2 and for serum-deprivation protocol 12,732 cells/cm2. (B,C) Growth curves for iRin37-WT and iRin37-S3A3 cells induced (dox+) or not (dox−) to express Cx37 (B, top). Induced expression of Cx37-WT (blue) led initially to cell death (days 1-3), followed by a period of growth arrest (days 3-9; B, bottom) and finally restored proliferative capacity (C; regression lines for periods of exponential growth are not different; cell cycle time for 12-21 days was 2.97 days for WT-expressing versus 1.82 days for non-expressing cells). In contrast, Cx37-S3A3 (green) expression did not induce cell death nor growth arrest (B, bottom); instead, these cells proliferate from day 3 onwards at a rate comparable to non-expressing cells (C; cell cycle time 2.13 days versus 2.16 days for non-expressing cells). n=4 in triplicate for each. *P<0.05 compared with dox+ of same genotype. (D) Serum deprivation leads to accumulation of Cx37-WT cells in G1 after only 24 h of induced expression (top); since the cells were proliferating slowly between 24 and 120 h (bottom), this increase in G1 percentage suggests an increase in time in G1. In contrast, the percentage of G1 Cx37-S3A3 cells initially decreases upon serum deprivation, but ultimately increases to the level seen in Cx37-WT cells; however, since Cx37-S3A3 cell number does not change between 24 and 120 h (bottom), this accumulation represents completion of the cell cycle by cells in S and G2 phases (Cx37-WT, n=5; Cx37-S3A3, n=4). *P<0.05 versus 0 h of same serum condition; #P<0.05 versus serum+ of same genotype. All values are mean±s.e.m.
Cx37- S7A7 channel function and proliferation
Since both channel properties and serum-sensitive growth behavior of iRin37-S3A3 suggested that additional sites in Cx37-WT must be targets for phosphorylation, we next mutated to alanine the remaining four serine residues identified as high-probability phosphorylation targets (see Fig. 2; residues 285, 319, 321 and 325) to create the Cx37-S7A7 mutant. This mutant was stably transfected into iRin cells and iRin37-S7A7 clones characterized for Cx37 expression. A clone expressing Cx37-S7A7 at a level comparable to that in iRin37-WT cells (6.56 pmol/mg total protein; Fig. S2C), was selected for further study. The iRin37-S7A7 cells formed functional GJChs with coupling levels comparable to those observed in iRin37-WT cells 24 h after plating with dox induction (Cx37-S7A7, 5±2 nS; N=21).
Contrary to our hypothesis (that channel activity in one or both serine to alanine mutants would resemble that of BIM-treated Cx37-WT-expressing cells) the conductance and Po behaviors of Cx37-S7A7 and untreated Cx37-WT GJChs were nearly identical. Single-channel recordings (Fig. 5A) revealed channels that appeared to favor the 30-90 pS and 260 pS conductance states. The transition-amplitude histogram for Cx37-S7A7 further supported the prevalence of 270 pS events. However, Cx37-S7A7 Po data were, like Cx37-WT, best fit with four peaks where the closed state and 30-90 pS conductance states predominated (Fig. 5B). This similarity between Cx37-S7A7 and Cx37-WT in Po and conductance behaviors suggested that this mutant might be growth suppressive rather than growth permissive as hypothesized. We investigated this possibility using the 21 day proliferation and serum-deprivation assays described in Fig. 4A (with low and intermediate plating densities, respectively). Proliferation assay data (Fig. 5C and inset) showed that Cx37-S7A7-expressing cells did not die early in the proliferation assay and were, instead, growth arrested for an extended period (12 days); indeed, cell cycle time (doubling time) could not be calculated for the iRin37-S7A7 cells over the 21 day experiment as cell number did not double. At higher plating densities (in the presence of serum) the growth-suppressive effect of Cx37-S7A7 was attenuated and progression through G1 was slowed, leading to an increased percentage of cells in G1 (Fig. 5D,E). As seen in Cx37-S3A3-expressing cells, serum deprivation of Cx37-S7A7-expressing cells led to an early decrease in the percentage of cells in G1 that slowly increased over the ensuing 120 h as cells in S and G2 phases completed the cell cycle (Fig. S3). The density dependence of growth suppression by Cx37-S7A7 suggests possible involvement of HChs (see results below) versus GJChs in growth suppression, for at the low plating densities used in proliferation assays and channel studies, cell–cell contact, and consequently GJCh formation, is rare. Collectively, the data in Fig. 5 suggest that preventing phosphorylation at these seven sites resulted in GJChs that favored the closed state and growth arrest.
Fig. 5.

The closed state of GJChs and growth-arrested phenotype are favored in Cx37-S7A7-expressing cells. (A) Representative single-channel current trace and corresponding all-points histogram for Cx37-S7A7 GJChs. Vj=25 mV. Scale bars: 1 s and 2 pA. (B) Combined relative event and Po frequency plots for Cx37-S7A7 (red) and Cx37-WT (blue); note the similarity (mirror image between genotypes) of channel behavior. Cx37-S7A7 data (n=7; 4.65 min of data) best fit (R2=0.9994) by four peaks with centers and areas: closed, 63%; 36 pS, 28%; 140 pS 8%; and 255 pS, 1.5%. (C) Proliferation assays reveal that Cx37-S7A7 expression does not induce cell death; instead, a prolonged period of growth arrest is evident in Cx37-S7A7-expressing cells (n=4 in triplicate for both isoforms). At day 21, there were 2.18×105 Cx37-S7A7 cells and 6.51×105 Cx37-WT cells. *P<0.05 versus dox+ of same genotype. Inset shows results of 6 day proliferation assays, emphasizing the early proliferative phenotypes of Cx37-WT and Cx37-S7A7. (D) As for Cx37-S3A3 cells, the percentage of G1 Cx37-S7A7 cells initially decreases upon serum deprivation, but slowly increases over the 24-120 h induction period. Since Cx37-S7A7 cell number did not change significantly between 24 and 120 h (E), this accumulation represents arrest in G1 with completion of the cell cycle by cells in S and G2 phases. For D and E: Cx37-WT, n=5; Cx37-S7A7, n=4. *P<0.05 versus 0 h of same serum condition; #P<0.05 versus serum+ of same genotype. All values are mean±s.e.m.
Cx37-S7D7 channel function and growth phenotype
The Cx37-S7A7 and Cx37-S3A3 data suggested that the closed state of the GJCh (and possibly HCh, see below) was a configuration that supported growth arrest, whereas the 30-90 pS conductance state was a configuration that supported proliferation. That both mutants largely eliminated Cx37-induced cell death, suggested that one or more phosphorylation-induced configurations might induce cell death. To test this possibility, the seven high-probability serine residues (see Fig. 2) were mutated to aspartates and the resulting sequence stably transfected into iRin cells. A clone that expressed Cx37-S7D7 at 12.4 pmol/mg total protein (Fig. S2C) was identified for further study. As expected, the iRin37-S7D7 cells formed functional GJChs that supported a Gj of 9±1.5 nS (N=28). Fully open (∼370 pS) GJChs transitioning to an 80 pS conductance state were evident in Cx37-S7D7 records (Fig. 6A) and the relative frequency amplitude histograms revealed the prevalence of these transitions (Fig. 6B). Despite a similar range of event amplitudes, the Po profile of Cx37-S7D7 differed substantially from that of Cx37-WT (Fig. 6B). Notably, the mutant spent very little time in the closed state or 30-90 pS conductance state, instead, the Cx37-S7D7 GJChs spent the bulk of their time open in intermediate conductance states.
Fig. 6.

Large-conductance GJChs are favored by Cx37-S7D7 GJChs and likely contribute to induced (apoptotic) cell death. (A) Representative single-channel current trace of Cx37-S7D7 GJChs and corresponding all-points histogram. Vj=25 mV. Scale bars: 1 s and 2 pA. (B) Combined transition and Po relative frequency plots for Cx37-S7D7 (black) and Cx37-WT (blue) GJChs accentuate the considerable differences between these isoforms. Note the low probability of the closed and 60-90 pS states and high open state probability (56%) of an intermediate conductance state in the Cx37-S7D7 cells. Cx37-S7D7 Po data (n=3; 4.22 min of data) best fit (R2=0.995) by four peaks: 27 pS, 12%; 84 pS, 8%; 161 pS, 23% and 218 pS 56%. (C) Cx37-S7D7 expressing cells did not proliferate (n=4 in triplicate for each isoform). *P<0.05 versus dox+ of same genotype. Instead, Cx37-S7D7 induces cell death (D), evident (by FACS analysis of propidium iodide-labeled cells) to a small extent at 24 h (4.21%) and large extent at 48 h (70.2%) of induced expression. Arrowheads on x-axis denote G1 and G2 peak centers as defined by ModFit LT program. (E) The presence of cleaved caspase-3, a marker of apoptosis, in non-adherent cells expressing Cx37-S7D7 for 72 h suggested that death of at least some cells occurred via an apoptotic mechanism. Lane content: MM, mass markers; +, positive control for caspase activation (note ∼15 kDa band); A, adherent; NA, non-adherent; induced (dox+) or not (dox−) to express Cx37-S7D7.
We next determined the impact of Cx37-S7D7 expression on proliferation and cell cycle progression of the iRin cells. In proliferation assays, Cx37-S7D7 expression appeared to suppress proliferation of iRin cells (Fig. 6C). However, visual inspection of the cells suggested that rather than being growth arrested, the iRin37-S7D7 cells were detaching from the substrate and dying. FACS analysis of cell cycle position for iRin37-S7D7 cells in the presence of serum revealed that expression of Cx37-S7D7 induced cell death in a time-dependent manner (Fig. 6D), as indicated by an increasing number of dead cells (debris) (24 h, 4.21%; 48 h, 70.2% of analyzed events). Both adherent and non-adherent cells were tested for activated caspase-3 (Fig. 6E); non-adherent cells were positive whereas adherent cells were not. These results suggested that at least some of the iRin37-S7D7 cells died following the apoptotic pathway.
Hemi-channel activity
The Cx37-S7A7 and Cx37-S7D7 data both suggested that HChs might be involved in the ‘growth’-suppressive effects of Cx37 – in particular, the closed state for growth arrest and the fully open state for cell death. Consequently, we next characterized HCh behavior for each Cx37 mutant as well as for Cx37-WT. Fig. 7A shows examples of the HChs observed in standard external solution (with 1.0 mM Ca2+) in single isolated iRin cells expressing Cx37-WT, -S3A3, -S7A7 or -S7D7. Despite comparable levels of connexin expression, the number of Cx37-S7D7 cells with detectable HCh activity was nearly double that observed in iRin37-WT, -S3A3 and -S7A7 cells, which were comparable to each other (Fig. 7B). The zero-adjusted current was not different between the isoforms (current to clamp at +25 mV, WT 23±2.5 pA, N=8; S3A3, 23±3.1 pA, N=7; S7A7, 24±2.7 pA, N=6; S7D7, 21±2.5 pA, N=6), indicating membrane integrity was comparable across genotypes. The Po frequency graphs for the four Cx37 isoforms revealed that the closed state of Cx37-S7D7 HChs was less frequent than for Cx37-WT HChs, and the Po of all open states (100-300, 300-500, 500-800 pS) was greater than in Cx37-WT (Fig. 7C). Cx37-S3A3 HCh Po did not differ from the Cx37-WT for any closed or open state. In contrast, Cx37-S7A7 spent less time in the 100-300 pS sub-state and more time in the 300-500 pS sub-state than Cx37-WT, and Cx37-S7D7 occupied all open states more frequently than Cx37-WT HChs. Interestingly, the probability of multiple channels being open (>800 pS) was greater in all mutants than in Cx37-WT. Relative frequency difference of Po for Cx37 phospho-mutants is summarized in Fig. 7D. These plots suggest that the proliferative phenotype results from a preference for the 100-300 pS conductance state, the growth-arrested phenotype results from a preference for the closed state and 300-500 pS conductance state, and the death phenotype results from a preference for all open states, including fully open. That multiple open channels occur with all three mutants as well as the Cx37-WT suggests that HCh activity, per se, is not death inducing.
Fig. 7.

Cx37 HCh activity could contribute to phenotypic consequences of expression, especially for Cx37-S7D7. (A) HCh activity is detected in the presence of 1 mM [Ca2+]o for each Cx37 isoform. Vm=+25 mV for all traces. (B) As a percentage of studied cells, HCh activity is most common in iRin37-S7D7 cells (Cx37-WT, 41%, n=27; Cx37-S3A3, 50%, n=16; Cx37-S7A7, 44%, n=27; Cx37-S7D7, 73%, n=26); *P<0.05. (C) Relative frequency Po for each isoform, with comparisons against WT of closed state, 100-300, 300-500, 500-800 and >800 pS conductance states. The closed state probability is significantly less for Cx37-S7D7 (N=6) compared with Cx37-WT (N=8) and significantly greater than Cx37-WT for all open states. The Po of the 100-300 pS state is less in Cx37-S7A7 (N=6) than Cx37-WT, but greater than WT in the 300-500 pS range, with no differences in the closed state or >500 pS states. No differences were detected between Cx37-S3A3 (N=7) and WT. *P<0.05, Cx37-S7D7 versus Cx37-WT; #P<0.05, Cx37-S7A7 versus Cx37-WT. (D) Po relative frequency difference plots highlighting differences between mutants. ANOVA with Tukey's post hoc test, *P<0.05 between genotypes.
DISCUSSION
Cx37 exerts a growth suppressive effect in vivo and in vitro; it suppresses vasculogenesis and angiogenesis (Allagnat et al., 2017; Fang et al., 2011) and is a potent suppressor of cancer cell proliferation (Burt et al., 2008; Morel et al., 2010). Available data (Good et al., 2012, 2014, 2011; Nelson et al., 2013) suggest: (1) interactions between the CT and a functional GJCh pore-forming domain are necessary for Cx37-mediated regulation of growth suppression; (2) Cx37 is a target for phosphorylation (Larson et al., 2000; Morel et al., 2010; Traub et al., 1998), and (3) phosphorylation of proteins alters their conformation and thereby their functions, including their intra- and inter-molecular interactions (Groban et al., 2006; Grosely et al., 2013). The growth-suppressive efficacy of Cx37 in cancer cells lessens with continuous expression over weeks (Burt et al., 2008) and months (our unpublished observations) and is augmented by serum deprivation (Burt et al., 2008). This suggests that growth-factor-activated kinases target Cx37 to regulate both channel function and growth-suppressive effects. In the current study, using alanine and aspartate substitutions at putative targets of growth-factor-activated kinases, we explored the necessity and sufficiency of phosphorylation at select serine residues in the region of the Cx37-CT required for the growth-suppressive function. We show, for the first time, that Cx37 could serve a ‘gate keeper’ function in the response of Cx37-expressing cells to growth factors and cytokines, regulating the growth phenotype in a phosphorylation-dependent manner and thereby the balance between cell cycle arrest, proliferation and cell death (Fig. 8).
Fig. 8.
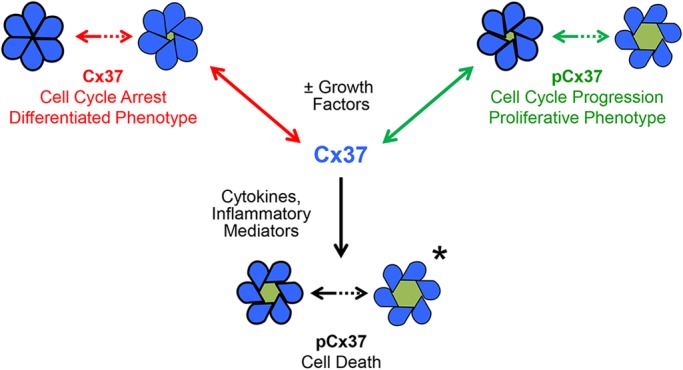
Cx37 serves as a molecular switch between arrest, proliferation and death in a phosphorylation- and channel-conformation-dependent manner. Our data suggest that the closed state (favored by dephosphorylation at seven high-probability serine residues) of the GJCh and HCh support conformations that arrest cell cycling. Phosphorylation states that support GJChs and HChs that reside predominantly in the lowest conductance state (60-90 pS for GJCh; 100-300 pS for HCh) and briefly transition between this state and larger conductance states are in a conformation that supports proliferation. Phosphorylation states that support significant activity of fully open HChs and GJChs and a low closed state probability induce cell death. Channel conformations in bold denote the preferred GJCh conductance state. Asterisk indicates possible causality.
Cx37 is prominently expressed in the endothelium of the arterial vasculature (Haefliger et al., 2004). In normal, unstressed settings, these cells are quiescent (non-cycling); however, vascular disease, injury, or even altered shear stress, induce changes in gene expression that support an increased endothelial cell turnover rate (Langille and O'Donnell, 1986). Such changes include the decreased expression of Cx37 and increased expression of Cx43 in affected vessels; Cx40 expression remains largely unchanged (Gabriels and Paul, 1998). Opposite regulation of Cx37 and Cx43 expression is also observed in other settings of stimulated growth (Cai et al., 2001; Kwak et al., 2002; Larson et al., 1997; Li et al., 2007). Along with the demonstrated growth-suppressive effect of Cx37 in vivo, our in vitro observations led us to question how Cx37 and Cx43 differ such that one supports while the other suppresses cell proliferation and whether channel properties might also be dichotomous. In Rin cells, Cx43 expression has no effect on cell proliferation, whereas Cx37 profoundly suppresses proliferation (Burt et al., 2008), making this system ideal for study of the mechanistic basis for growth suppression. As shown herein, the profound growth suppression mediated by Cx37 in Rin cells actually involved an initial period of cell death, followed by a period of growth arrest and finally partial recovery of proliferative potential (Fig. 4).
Since cell proliferation and response to injury are typically activated by growth-factor-activated kinases (e.g. PKC), we determined here whether these kinases altered the function of Cx37 gap junctions. Gap junction conductance, Gj, is defined as the product of channel number, unitary conductance and open probability. We show here that the conductance of junctions formed by Cx37-WT cells declined over time in a PKC-dependent manner. At the channel level, kinase activation favored transitions of fully open or partially open GJChs to the closed or 30-90 pS conductance state. In contrast, PKC inhibition favored transitions of GJChs between the 30-90 pS conductance and closed states, with a simultaneous reduction in closed state probability and increased probability of the channel residing in the 30-90 pS conductance state (see Fig. 1). This response of Cx37 channels to PKC activation/inhibition is opposite to that observed for Cx43 GJChs. PKC activation causes a loss Cx43 junctional conductance and this acute loss in Gj can be accounted for by reduced conductance of individual Cx43 channels (Lampe et al., 2000, and others). The corollary here is that PKC (and/or kinases regulated by PKC) targets Cx37 in GJChs, altering GJCh conductance and open state probability.
Long-term exposure to inhibitors or agonists of PKC can have off-target (Cx37) effects that make data interpretation challenging. Therefore, to understand the cell cycle and cell death consequences of kinase-dependent regulation of Cx37, we substituted alanine for serine at likely phosphorylation targets in Cx37, expecting to alter early consequences of Cx37 expression in proliferation and serum deprivation experiments and Cx37 channel properties in electrophysiology experiments. In settings where Cx43 is growth permissive (including Rin cells), phosphorylation-mimicking mutations favor proliferation while phosphorylation-preventing substitutions tend to limit proliferation (Johnstone et al., 2012; Dyce et al., 2012). In contrast, for Cx37, preventing phosphorylation at S275, S302 and S328 (Cx37-S3A3) largely eliminated the cell death and growth arrest specific to Cx37-WT expression; instead Cx37-S3A3 supported proliferation with cell cycle times comparable to non-expressing iRin cells. Interestingly, the iRin37-S3A3 cells remained serum sensitive, although the time course for their accumulation in G1 when serum deprived was prolonged compared with iRin37-WT cells. Since cell number did not increase when serum deprived, the accumulation of iRin37-S3A3 cells in G1 likely represented cells in S and/or G2 completing the cell cycle and arresting in G1. Recognizing that Cx37-mediated growth suppression relies on interaction of the Cx37-CT with a Cx37 pore-forming domain able to form a functional GJCh, the significant reduction in closed state probability of the proliferative iRin37-S3A3 cells suggests that the closed state may be necessary for Cx37-induced growth arrest. Perhaps the closed state conformation allows for interaction of Cx37 with, as yet unidentified, cell cycle regulators. In contrast, phospho-isoforms that support a high Po of the 30-90 pS GJCh conductance state, frequent transitions from this state to intermediate or fully open conductance states, and 100-300 pS conductance state of the Cx37 HCh may indicate a proliferative form of the protein.
Results from the iRin37-S7A7 and iRin37-S7D7 cells reinforce and expand on these conclusions. The iRin37-S7A7 cells were growth arrested for a prolonged period (∼12 days) when plated at low cell densities, but proliferated when plated at higher cell densities. At low cell densities, when GJChs were unable to form, Cx37-S7A7 HChs were either closed or active in the 300-500 pS conductance state; this state of the HCh and the closed state of both the HCh and GJCh supported growth arrest. As cell density increased and the incidence of GJCh formation increased, the GJCh conductance state favored by proliferative Cx37-S3A3 cells (30-90 pS) was available to support proliferation of the iRin37-S7A7 cells. The capacity of the Cx37-S7A7 GJChs to favor these two states may explain, or at least contribute to, the proliferative phenotype of these cells when plated at higher densities. In contrast to both the iRin37-S3A3 and -S7A7 cells, cells at both low and high plating density induced to express Cx37-S7D7 died (at least some by an apoptotic mechanism) with little evidence of growth arrest or proliferation prior to death. This HCh behavior, previously unreported, could explain reported apoptosis of endothelial cells induced to express Cx37 (Seul et al., 2004). The GJChs and HChs formed by Cx37-S7D7 had a very low closed state probability, instead displaying a high Po for a ∼180 pS GJCh conductance state and fully open HChs. These behaviors of the Cx37-S7D7 GJChs and HChs were unique amongst the isoforms studied, as was the death phenotype, suggesting a causative relationship.
Previous data indicate that HCh function is not sufficient for Cx37-mediated growth suppression (Good et al., 2014). Indeed, data from the current study further imply that it is not simply the ability for the HCh to function, but rather the unique conformation/conductance state of HChs that contribute to cell phenotype. It has been previously reported that phosphorylation can regulate HCh conductance, in addition to the well-documented effects of phosphorylation on GJCh conductance (Saez et al., 2005) and permselective properties (Ek-Vitorin et al., 2006). However, because of differences in proliferation of iRin37-S7A7 cells between low- and higher-density conditions (and therefore differing amounts of HChs versus GJChs), it is possible that there is differential phosphorylation of Cx37 in HChs versus GJChs that influences the net growth effect of Cx37 phospho-isoform expression.
The data presented herein indicate that phosphorylation-dependent regulation at one or more sites within the Cx37-CT modifies Cx37 conformation, as evidenced by altered channel function, and thereby growth phenotype. The specific sites dictating the growth arrest, proliferation and death phenotypes remain to be determined. It is likely that specific phosphorylation events are short lived (hence the run-down of Gj and the diversity of channel amplitudes), such that, on average, only a small percentage of the total Cx37 present is phosphorylated in the combinations necessary to produce the death, arrest and proliferative phenotypes. If correct, this would suggest that differential Cx37 phosphorylation and associated protein conformation and protein–protein interactions represent a potent regulator of cell phenotype – one that could potentially be exploited to prevent tumor angiogenesis but also to enhance vascular remodeling in other settings. For example, in the endothelium of the adult arterial vasculature, Cx37 could facilitate the typical growth-arrested state as well as the response to injury and disease with apoptosis or proliferation, as appropriate, to support vascular repair and remodeling. As the specific sites dictating these phenotypes are identified, their involvement in vascular repair and remodeling should be investigated. Relative to identifying the specific sites, channel behavior appears to predict phenotype. Specifically, phosphorylation states that support increased probability of the closed state of the GJCh (and HCh) predict growth arrest. Phosphorylation states that reduce the closed state probability but increase the GJCh Po of the 30-90 pS state with transient openings to larger conductance states, predict cell cycling and proliferation. Phosphorylation states that produce HCh and GJCh with low closed state probability and significant open state probability for fully open and intermediate conductance states predict (apoptotic) cell death.
Because aspartate substitution does not always completely mimic the effects of phosphorylation (Groban et al., 2006), future studies should determine the specific sites targeted by PKC and other growth-factor-activated kinases. It will also be important to determine the impact of differential phosphorylation on the selective permeability of the GJChs and HChs formed by Cx37. For example, if the ∼180 pS GJCh and corresponding open state of HChs (∼360 pS) formed by Cx37-S7D7 were more permeable to Ca2+, the resulting Ca2+ influx could be the stimulus for induced apoptosis. It also remains to be determined whether a unique combination of the explored sites, additional sites, or the timing of differential phosphorylation might contribute to the Cx37-induced growth phenotypes. Finally, it will be important to determine whether and how phosphorylation alters the interaction of the CT with the pore-forming domain and/or cell cycle mediators. Even without answers to these questions, however, the data herein demonstrate that Cx37-mediated growth suppression requires a GJCh-competent pore-forming domain that is able to interact with phosphorylation-dependent conformations of the CT that likely enable site-specific interactions of the Cx37-CT with cell cycle mediators to regulate cell cycle progression and cell survival.
MATERIALS AND METHODS
Cell culture
iRin cells (Burt et al., 2008; rat insulinoma cells with TET-ON expression system) were maintained in RPMI 1640 medium (Sigma Aldrich, St. Louis, MO) supplemented with 10% Fetal Plex serum (FP; Gemini Bio Products, West Sacramento, CA), G418 (300 µg/ml; Life Technologies, Grand Island, NY), penicillin (300 µg/ml; Sigma Aldrich), and streptomycin (500 µg/ml; Sigma Aldrich). Cells were passed weekly using 0.25% trypsin in divalent-cation-free saline and were maintained at 37°C in a humidified, 5% CO2 incubator. iRin cells transfected to inducibly express an isoform of mCx37, for example iRin37-WT (Burt et al., 2008), were maintained in hygromycin (100 µg/ml; Life Technologies) and Cx37 expression initiated with dox.
Mutant plasmid generation, transfection and clone isolation
Using the QuikChange Site-Directed Mutagenesis or QuikChange Multi Site-Directed Mutagenesis Kits (Agilent Technologies, La Jolla, CA) and appropriate oligonucleotide primers (Operon Biotechnologies, Huntsville, AL; Fig. S4), multi-site mutations were introduced sequentially into the pTRE2h-mCx37 plasmid (Burt et al., 2008). All sequences were confirmed at the University of Arizona Genomics Core Sequencing Facility. The mutant plasmids were transfected into iRin cells using lipofectamine (Life Technologies) according to the manufacturer's instructions. Cells resistant to hygromycin were isolated and dilution cloned. For further study, the following clones were selected: Cx37-S3A3 1D2 and 1C8; Cx37-S7A7 F5; Cx37-S7D7 1C2. Maximal induction of protein expression was initiated by the addition of 2 µg/ml dox for iRin37-WT and iRin37-S3A3, and 4 μg/ml dox for iRin37-S7A7 and iRin37-S7D7.
Immunoblotting
Whole-cell protein was isolated, the protein content assessed using the BCA Assay (Pierce), and 20 or 50 µg of sample protein loaded onto precast 12% SDS-PAGE gels (Bio-Rad). Alkaline phosphatase treatment of samples was performed as previously described (Solan et al., 2003). After transfer to nitrocellulose, Cx37 content of samples was quantified (Nelson et al., 2013) for each clonal cell line by comparison of sample band intensity to a standard curve generated with GST-Cx37 fusion protein (residues 233-333). Blots were blocked with 5% non-fat dried milk and Cx37 detected with anti-Cx37-18264 (a gift from Dr Alexander Simon, University of Arizona, Tucson, AZ, USA; Simon et al., 2006) used at 1:5000 dilution. Primary antibody was visualized using enhanced chemiluminescence strategies with SuperSignal West Dura or Femto Systems (mixed at a 1:1 ratio; Thermo Scientific, Waltham, MA) and anti-rabbit HRP-conjugated secondary antibody (GE Healthcare Life Sciences, Pittsburgh, PA) and visualized on a Kodak Image Station 2000.
Electrophysiology
To characterize GJCh and HCh activity, cells were plated at low density on glass coverslips and dox added to induce maximal protein expression. After 24 h (or 48 h if no functional channels were detected at 24 h), coverslips were mounted in a custom-made chamber and recordings made on attached, healthy appearing cells. Cells were immersed in an external solution containing: 142.5 mM NaCl, 4 mM KCl, 1 mM MgCl2, 5 mM glucose, 2 mM Na Pyruvate, 10 mM HEPES, 15 mM CsCl, 10 mM TEA-Cl, 1 mM CaCl2, pH adjusted to 7.2 (315 mOsM). Patch pipets (3-10 MΩ) contained: 124 mM KCl, 14 mM CsCl, 9 mM HEPES, 9 mM EGTA, 0.5 mM CaCl2, 5 mM glucose, 9 mM TEA-Cl, 3 mM MgCl2, 5 mM Na2ATP, pH 7.2, 315 mOsM.
GJCh electrophysiology
Gj was determined as previously described (Burt et al., 2008; Kurjiaka et al., 1998) using dual whole-cell voltage-clamp techniques with Axopatch1D amplifiers and pClamp software (Molecular Devices, Sunnyvale, CA). Gj was assessed with 10 mV transjunctional pulses delivered every 30 s; electrode seal integrity was monitored every 5 min over long recording periods. Single-channel activity was recorded using a transjunctional voltage of 25 mV; halothane was used, as necessary, to reduce Gj below 1 nS, but never in experiments assessing channel Po. Current records were digitized at 2.5 kHz, filtered at 50 Hz, and the amplitudes of current transitions (events, ‘n’) in the record, where current levels before and after the transition were stable for greater than 50 ms, were measured. Event amplitudes were converted to conductances (pA/25 mV), events grouped into 10 pS bins and the number of events in each bin relative to total events for each cell pair calculated. Mean data for each bin from multiple cell pairs (‘N’) collected on multiple days was then determined and plotted as a relative frequency histogram. Displayed current traces include the corresponding all-digitized-points histograms for the trace.
Po was assessed after decimating the unfiltered record (500 Hz sampling frequency) and compiling digitized points from the entire record into 10 pS bins (total number of minutes and cell pairs, ‘N’, reported), calculating the relative frequency of digitized points in each bin, and plotting the mean for each bin across multiple cell pairs as frequency. These data were then fit with 1, 2, 3 or 4 peaks using Origin software with no imposed limitations for peak centers, widths or areas in the fitting process. The best fit was selected by comparing the R2 values for the data fit with one, two, three and four peaks; the relative areas of those peaks were used to define Po for each identified open and closed state. In some cases, the closed state was assigned a negative conductance value, likely reflecting noise and some amount of drift in the zero GJCh current level over the duration of the recording. Regardless, the relative area of this peak is reported as the closed state. For combined Po and event frequency plots, the y-axis displays positive values in both directions as a convenient method of comparison between treatment groups or between genotypes (if there were no differences, these plots would appear as mirror images of each other).
In some GJCh experiments, kinase activity was manipulated prior to assessment of junctional and channel conductances. BIM, a specific PKC inhibitor, was applied (0.2 μM, from DMSO stock) for 60 min at 37°C. 12-O-Tetradecanoylphorbol-13-acetate (TPA), a non-specific PKC activator, was applied (50 ng/ml from ethanol stock) for 30 min at room temperature. Vehicle controls revealed no effect of ethanol or DMSO at the low final concentrations used for drug delivery (data not shown).
HCh electrophysiology
HCh activity was assessed using the same general approach as for GJChs with +25 mV transmembrane voltage pulses applied to single isolated cells. Event amplitudes were measured and plotted as described for GJChs. Po was determined from current records in which the pulse protocol included an initial ∼30 s pulse to +25 mV followed by ∼2 s at 0 mV and a second pulse to 25 mV of ∼4.5 min duration. 240 s of the decimated, filtered, second pulse were compiled into 0.25 pA bins (from −4 to +100 pA), the relative frequency of digitized points (each 0.002 s) calculated for each bin, and frequency versus current plotted. The current level associated with the center of the first peak (determined visually or following peak fitting with Origin software) was assumed to represent the closed state current, which was subtracted from all current bins, thereby defining the 0 pA (closed state) bin. Data from multiple records were aligned at the closed state bin, and the average relative frequency for each current bin across cells (N) determined. The average relative frequencies for each Cx37 isoform were plotted as a function of current as were differences between mutant isoforms. Isoforms were analyzed for differences (ANOVA and Tukey's multiple comparisons test using Prism software) in frequency (Po) in the −2.5-2.5, 2.5-7.5, 7.5-12.5 and >12.5 pA, which correspond to the closed state, 100-300, 300-500 and >500 pS conductance levels.
In vitro kinase phosphorylation
Mouse Cx37CT233-333 in a pGEXKT vector was expressed in the E.coli BL21 (DE3) strain. Briefly, bacterial cells grown in LB medium were induced with IPTG (1 mM) at a cell density of 0.6 (OD at 600 nm). Cells were grown for an additional 4 h. To purify the GST-tagged Cx37CT, cells were pelleted, and lysed in 1× phosphate-buffered saline (PBS) lysis buffer using an EmulsiFlex-C3 homogenizer. Cell debris was discarded and the supernatant was incubated with glutathione resin. Non-specific binding proteins were removed by sequential washes with 2× PBS and 1× PBS in presence of 2 mM DTT, at pH 6.0. The GST-tag was cleaved overnight at 4°C by incubation with thrombin (7 units/liter of culture). A 3 kDa Amicon® Ultracentrifugal filter was used to concentrate the protein to 0.35 mM. To perform the in vitro kinase assay, 300 µg Cx37-CT was buffer exchanged to the reaction buffer (MOPS 5 mM pH 7.2, 2.5 mM β-glycerol-phosphate, 5 mM MgCl2, 1 mM EGTA, 0.4 mM EDTA and 0.05 mM DTT) by using Zeba™ desalt spin column (Pierce). Then, Cx37-CT was incubated with each kinase (PKCα, ERK1 or CDK2) and 2 mM ATP at 30°C for 15 h. The reaction was stopped on ice; samples were run on a SDS-PAGE gel and stained with Coomassie Blue. The Cx37-CT band was cut and sent to the Beth Israel Deaconess Medical Center Mass Spectrometry Facility for analysis (Breitkopf and Asara, 2012).
Proliferation
As described previously (Burt et al., 2008), cells were seeded at low density (3×104 cells in six-well plates; 3125 cells/cm2) in triplicate. At 24 h after plating, dox was added to three of the six wells allocated for each time point. For 21 days, medium was refreshed every 48 h, with or without dox, as appropriate. Every 3 days, the cells in every well of the plate designated for a time point were harvested using trypsin. The number of cells per well was determined using a Cellometer (Nexcelom, Lawrence, MA). Six-day proliferation assays were plated at the same low density (5.5×105 cells/145 mm plate) and dox added 24 h later to induce protein expression. Thereafter, medium (with or without dox, as appropriate) was refreshed every 48 h and cells were counted every day for 6 days.
Serum-sensitive cell cycle analysis
Cells were plated at 106 cells/100 mm plate (12,732 cells/cm2). After 24 h, plates were divided into two groups: those maintained in medium containing 10% serum or switched to medium without serum. For both groups, 72 h after plating, dox was introduced to induce maximal protein expression (this is the zero time point on cell cycle analysis graphs). Adherent and non-adherent cells were counted, to assess extent of proliferation, and analyzed for cell cycle position (using propidium iodide staining as previously described; Burt et al., 2008). Fluorescence-activated cell sorting (FACS) was performed at the UACC/ARL Cytometry Core Facility. Data were collected using a FACScan or FACSCanto II (BD Biosciences, San Jose, CA) and cell cycle distribution was analyzed using ModFit LT software with doublet discrimination. Debris and dead cells were classified as events that contained less than a full complement of DNA.
Caspase-3 detection and immunoblotting
Protein expression was induced for 72 h and total protein was isolated from adherent and non-adherent cells, separately, as previously described (Burt et al., 2008). Caspase-3 primary (Cell Signaling, #9662) and horseradish peroxidase-conjugated secondary antibodies were visualized with the SuperSignal West Dura Chemiluminescence system (Thermo Scientific) and a Kodak ImageStation 2000.
Primary endothelial cell isolation and culture
In this study, male and female 1- to 3-month-old wild-type and Cx37−/− C57Bl/6 mice were used (Simon et al., 1997). Animals were permitted access to food and water ad libitum and were housed together when possible. All procedures were approved and conducted in compliance with the Institutional Animal Care and Use Committee of the University of Arizona. For each isolation, animals were euthanized by cervical dislocation and all major muscles from each hindlimb of one mouse were removed. Tissue was washed in ice-cold wash solution [1× PBS with 1% bovine serum albumin (BSA) and 1% antibiotic/mycotic] before mincing with fine scissors. The minced tissue was transferred to warm isolation solution [divalent-cation-free PBS with 10% (v/v) DMEM, 20 μg/ml elastase, 1.25 mg/ml collagenase type I, 1% BSA and 1% antibiotic/mycotic], and incubated at 37°C for 40 min. Every 10 min during the digestion, the sample was mixed (vigorous shaking) to encourage additional dissociation. The tissue homogenate was then strained over 100 μm and 40 μm sieves to remove any undigested components. The heterogeneous cell mixture was centrifuged, resuspended in staining buffer (PBS with 0.3% BSA), and cells were counted with a Cellometer. Fluorescent antibody (FITC-PECAM-1 or PE-VE-Cadherin) was used at 500 ng per 1×106 cells to stain cells for 45 min on ice. The cell mixture was sorted using a FACSAria IIu cell sorter (FITC-PECAM-1: 488 nm laser, 530/30 nm filter; PE-VE-Cadherin: 488 nm laser, 576/26 nm band-pass filter). Viable cells were gated based on forward and side-scatter profiles to exclude damaged cells; positive cells were selected by those exceeding a fluorescence intensity threshold determined by unstained control cells. Cell yield was comparable between genotypes (∼106 cells). All positive cells were collected in staining buffer, centrifuged, and resuspended in medium for culture. Cells were cultured in endothelial cell culture medium (LG-DMEM containing 10% fetal bovine serum, 20 μg/ml EC growth supplement, 40 μg/ml heparin, 1% heparin, 1% glutamine and 1% sodium pyruvate) on 1% gelatin coated six-well plates. Medium was refreshed every 2 days. After allowing 2 days for adherence to the gelatin matrix and recovery from live-cell sorting, the growth of cells was monitored by light microscopy and counting the number of cells in the field of view, in triplicate.
Statistics
Statistical comparisons of data were performed using paired or unpaired, two tailed, Student's t-test, ANOVA with Tukey's post hoc test, or Chi-Square tests for binary data, as appropriate. Significant differences were taken as P<0.05. Error bars represent mean±s.e.m.
Supplementary Material
Acknowledgements
The authors wish to thank Jose Ek Vitorin, MD, PhD, and John Kanady, PhD for their assistance and many helpful suggestions throughout the course of this work.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: N.L.J., T.K.P., J.B.; Methodology: N.L.J., T.K.P., H.L., J.L.S., P.D.L., P.L.S., J.M.B.; Validation: N.L.J., T.K.P., J.M.B.; Formal analysis: N.L.J., T.K.P., P.L.S., J.M.B.; Investigation: N.L.J., T.K.P., H.L., J.L.S., P.D.L., P.L.S., J.M.B.; Resources: N.L.J., T.K.P., P.L.S., J.M.B.; Data curation: J.M.B.; Writing - original draft: N.L.J.; Writing - review & editing: T.K.P., P.D.L., J.M.B.; Supervision: T.K.P., P.D.L., J.M.B.; Project administration: J.M.B.; Funding acquisition: P.D.L., J.M.B.
Funding
These studies were supported by the National Institutes of Health [HL056732 to J.M.B., HL007249 to J.M.B. and N.L.J., HL131712 to J.M.B. and P.L.S., GM072631 to P.L.S., GM55632 to P.D.L. and CCSG – CA023074] and American Heart Association [16PRE27500011 to N.L.J.]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.202572.supplemental
References
- Allagnat F., Dubuis C., Lambelet M., Le Gal L., Alonso F., Corpataux J.-M., Déglise S. and Haefliger J.-A. (2017). Connexin37 reduces smooth muscle cell proliferation and intimal hyperplasia in a mouse model of carotid artery ligation. Cardiovasc. Res. 113, 805-816. 10.1093/cvr/cvx079 [DOI] [PubMed] [Google Scholar]
- Bao X., Altenberg G. A. and Reuss L. (2004). Mechanism of regulation of the gap junction protein connexin 43 by protein kinase C-mediated phosphorylation. Am. J. Physiol. Cell Physiol. 286, C647-C654. 10.1152/ajpcell.00295.2003 [DOI] [PubMed] [Google Scholar]
- Breitkopf S. B. and Asara J. M. (2012). Determining in vivo phosphorylation sites using mass spectrometry. Curr. Protoc. Mol. Biol. Chapter 18, Unit18.19.1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt J. M., Nelson T. K., Simon A. M. and Fang J. S. (2008). Connexin 37 profoundly slows cell cycle progression in rat insulinoma cells. Am. J. Physiol. Cell Physiol. 295, C1103-C1112. 10.1152/ajpcell.299.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W.-J., Koltai S., Kocsis E., Scholz D., Schaper W. and Schaper J. (2001). Connexin37, not Cx40 and Cx43, is induced in vascular smooth muscle cells during coronary arteriogenesis. J. Mol. Cell Cardiol. 33, 957-967. 10.1006/jmcc.2001.1360 [DOI] [PubMed] [Google Scholar]
- Cooper C. D. and Lampe P. D. (2002). Casein kinase 1 regulates connexin-43 gap junction assembly. J. Biol. Chem. 277, 44962-44968. 10.1074/jbc.M209427200 [DOI] [PubMed] [Google Scholar]
- Dang X., Jeyaraman M. and Kardami E. (2006). Regulation of connexin-43-mediated growth inhibition by a phosphorylatable amino-acid is independent of gap junction-forming ability. Mol. Cell Biochem. 289, 201-207. 10.1007/s11010-006-9162-2 [DOI] [PubMed] [Google Scholar]
- Dyce P. W., Norris R. P., Lampe P. D. and Kidder G. M. (2012). Phosphorylation of serine residues in the C-terminal cytoplasmic tail of connexin43 regulates proliferation of ovarian granulosa cells. J. Membr. Biol. 245, 291-301. 10.1007/s00232-012-9450-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eghbali B., Kessler J. A., Reid L. M., Roy C. and Spray D. C. (1991). Involvement of gap junctions in tumorigenesis: transfection of tumor cells with connexin 32 cDNA retards growth in vivo. Proc. Natl. Acad. Sci. USA 88, 10701-10705. 10.1073/pnas.88.23.10701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ek-Vitorin J. F., King T. J., Heyman N. S., Lampe P. D. and Burt J. M. (2006). Selectivity of connexin 43 channels is regulated through protein kinase C-dependent phosphorylation. Circ. Res. 98, 1498-1505. 10.1161/01.RES.0000227572.45891.2c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ek Vitorín J. F., Pontifex T. K. and Burt J. M. (2016). Determinants of Cx43 channel gating and permeation: the amino terminus. Biophys. J. 110, 127-140. 10.1016/j.bpj.2015.10.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J. S., Angelov S. N., Simon A. M. and Burt J. M. (2011). Cx37 deletion enhances vascular growth and facilitates ischemic limb recovery. Am. J. Physiol. Heart Circ. Physiol. 301, H1872-H1881. 10.1152/ajpheart.00683.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriels J. E. and Paul D. L. (1998). Connexin43 is highly localized to sites of disturbed flow in rat aortic endothelium but connexin37 and connexin40 are more uniformly distributed [see comments]. Circ. Res. 83, 636-643. 10.1161/01.RES.83.6.636 [DOI] [PubMed] [Google Scholar]
- Good M. E., Nelson T. K., Simon A. M. and Burt J. M. (2011). A functional channel is necessary for growth suppression by Cx37. J. Cell Sci. 124, 2448-2456. 10.1242/jcs.081695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good M. E., Ek-Vitorín J. F. and Burt J. M. (2012). Extracellular loop cysteine mutant of cx37 fails to suppress proliferation of rat insulinoma cells. J. Membr. Biol. 245, 369-380. 10.1007/s00232-012-9459-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good M. E., Ek Vitorín J. F. and Burt J. M. (2014). Structural determinants and proliferative consequences of connexin 37 hemichannel function in insulinoma cells. J. Biol. Chem. 289, 30379-30386. 10.1074/jbc.M114.583054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough D. A. and Paul D. L. (2003). Beyond the gap: functions of unpaired connexon channels. Nat. Rev. Mol. Cell Biol. 4, 285-295. 10.1038/nrm1072 [DOI] [PubMed] [Google Scholar]
- Groban E. S., Narayanan A. and Jacobson M. P. (2006). Conformational changes in protein loops and helices induced by post-translational phosphorylation. PLoS Comput. Biol. 2, e32 10.1371/journal.pcbi.0020032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosely R., Kopanic J. L., Nabors S., Kieken F., Spagnol G., Al-Mugotir M., Zach S. and Sorgen P. L. (2013). Effects of phosphorylation on the structure and backbone dynamics of the intrinsically disordered connexin43 C-terminal domain. J. Biol. Chem. 288, 24857-24870. 10.1074/jbc.M113.454389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haefliger J. A., Nicod P. and Meda P. (2004). Contribution of connexins to the function of the vascular wall. Cardiovasc. Res. 62, 345-356. 10.1016/j.cardiores.2003.11.015 [DOI] [PubMed] [Google Scholar]
- Hirschi K. K., Xu C. E., Tsukamoto T. and Sager R. (1996). Gap junction genes Cx26 and Cx43 individually suppress the cancer phenotype of human mammary carcinoma cells and restore differentiation potential. Cell Growth Differ. 7, 861-870. [PubMed] [Google Scholar]
- Johnstone S. R., Kroncke B. M., Straub A. C., Best A. K., Dunn C. A., Mitchell L. A., Peskova Y., Nakamoto R. K., Koval M., Lo C. W. et al. (2012). MAPK phosphorylation of connexin 43 promotes binding of cyclin E and smooth muscle cell proliferation. Circ. Res. 111, 201-211. 10.1161/CIRCRESAHA.112.272302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanady J. D., Dellinger M. T., Munger S. J., Witte M. H. and Simon A. M. (2011). Connexin37 and Connexin43 deficiencies in mice disrupt lymphatic valve development and result in lymphatic disorders including lymphedema and chylothorax. Dev. Biol. 354, 253-266. 10.1016/j.ydbio.2011.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemitsu M. Y. and Lau A. F. (1993). Epidermal growth factor stimulates the disruption of gap junctional communication and connexin43 phosphorylation independent of 12-0- tetradecanoylphorbol 13-acetate-sensitive protein kinase C: the possible involvement of mitogen-activated protein kinase. Mol. Biol. Cell 4, 837-848. 10.1091/mbc.4.8.837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardami E., Dang X., Iacobas D. A., Nickel B. E., Jeyaraman M., Srisakuldee W., Makazan J., Tanguy S. and Spray D. C. (2007). The role of connexins in controlling cell growth and gene expression. Prog. Biophys. Mol. Biol. 94, 245-264. 10.1016/j.pbiomolbio.2007.03.009 [DOI] [PubMed] [Google Scholar]
- Kurjiaka D. T., Steele T. D., Olsen M. V. and Burt J. M. (1998). Gap junction permeability is diminished in proliferating vascular smooth muscle cells. Am. J. Physiol. 275, C1674-C1682. [DOI] [PubMed] [Google Scholar]
- Kwak B. R., Mulhaupt F., Veillard N., Gros D. B. and Mach F. (2002). Altered pattern of vascular connexin expression in atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 22, 225-230. 10.1161/hq0102.104125 [DOI] [PubMed] [Google Scholar]
- Lampe P. D., TenBroek E. M., Burt J. M., Kurata W. E., Johnson R. G. and Lau A. F. (2000). Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J. Cell Biol. 149, 1503-1512. 10.1083/jcb.149.7.1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe P. D., Cooper C. D., King T. J. and Burt J. M. (2006). Analysis of Connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J. Cell Sci. 119, 3435-3442. 10.1242/jcs.03089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille B. L. and O'Donnell F. (1986). Reductions in arterial diameter produced by chronic decreases in blood flow are endothelium-dependent. Science 231, 405-407. 10.1126/science.3941904 [DOI] [PubMed] [Google Scholar]
- Larson D. M., Wrobleski M. J., Sagar G. D. V., Westphale E. M. and Beyer E. C. (1997). Differential regulation of connexin43 and connexin37 in endothelial cells by cell density, growth, and TGF-Beta-1. Am. J. Physiol. 272, C405-C415. [DOI] [PubMed] [Google Scholar]
- Larson D. M., Seul K. H., Berthoud V. M., Lau A. F., Sagar G. D. V. and Beyer E. C. (2000). Functional expression and biochemical characterization of an epitope-tagged connexin37. Mol. Cell Biol. Res. Commun. 3, 115-121. 10.1006/mcbr.2000.0200 [DOI] [PubMed] [Google Scholar]
- Li T. Y., Colley D., Barr K. J., Yee S.-P. and Kidder G. M. (2007). Rescue of oogenesis in Cx37-null mutant mice by oocyte-specific replacement with Cx43. J. Cell Sci. 120, 4117-4125. 10.1242/jcs.03488 [DOI] [PubMed] [Google Scholar]
- Loewenstein W. R. (1973). Membrane junctions in growth and differentiation. Fed. Proc. 32, 60-64. [PubMed] [Google Scholar]
- Loewenstein W. R. and Azarnia R. (1990). Regulation of intercellular communication and growth by the cellular src gene. Ann. N. Y. Acad. Sci. 337-346. [DOI] [PubMed] [Google Scholar]
- McKinnon R. L., Bolon M. L., Wang H.-X., Swarbreck S., Kidder G. M., Simon A. M. and Tyml K. (2009). Reduction of electrical coupling between microvascular endothelial cells by NO depends on connexin37. Am. J. Physiol. Heart Circ. Physiol. 297, H93-H101. 10.1152/ajpheart.01148.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel S., Burnier L., Roatti A., Chassot A., Roth I., Sutter E., Galan K., Pfenniger A., Chanson M. and Kwak B. R. (2010). Unexpected role for the human Cx37 C1019T polymorphism in tumour cell proliferation. Carcinogenesis 31, 1922-1931. 10.1093/carcin/bgq170 [DOI] [PubMed] [Google Scholar]
- Moreno A. P., Chanson M., Elenes S., Anumonwo J., Scerri I., Gu H., Taffet S. M. and Delmar M. (2002). Role of the carboxyl terminal of connexin43 in transjunctional fast voltage gating. Circ. Res. 90, 450-457. 10.1161/hh0402.105667 [DOI] [PubMed] [Google Scholar]
- Nakamura K. and Shibata Y. (1999). Connexin43 expression in network-forming cells at the submucosal- muscular border of guinea pig and dog colon. Cells. Tissues. Organs 165, 16-21. 10.1159/000016669 [DOI] [PubMed] [Google Scholar]
- Nelson T. K., Sorgen P. L. and Burt J. M. (2013). Carboxy terminus and pore-forming domain properties specific to Cx37 are necessary for Cx37-mediated suppression of insulinoma cell proliferation. Am. J. Physiol. Cell Physiol. 305, C1246-C1256. 10.1152/ajpcell.00159.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori Y., Zaidan Dagli M. L., Yamakage K. and Yamasaki H. (2001). Involvement of gap junctions in tumor suppression: analysis of genetically-manipulated mice. Mutat. Res. 477, 191-196. 10.1016/S0027-5107(01)00120-8 [DOI] [PubMed] [Google Scholar]
- Remo B. F., Qu J., Volpicelli F. M., Giovannone S., Shin D., Lader J., Liu F. Y., Zhang J., Lent D. S., Morley G. E. et al. (2011). Phosphatase-resistant gap junctions inhibit pathological remodeling and prevent arrhythmias. Circ. Res. 108, 1459-1466. 10.1161/CIRCRESAHA.111.244046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts N. A., Marber M. S. and Avkiran M. (2004). Specificity of action of bisindolylmaleimide protein kinase C inhibitors: do they inhibit the 70kDa ribosomal S6 kinase in cardiac myocytes? Biochem. Pharmacol. 68, 1923-1928. 10.1016/j.bcp.2004.07.040 [DOI] [PubMed] [Google Scholar]
- Saez J. C., Retamal M. A., Basilio D., Bukauskas F. F. and Bennett M. V. (2005). Connexin-based gap junction hemichannels: gating mechanisms. Biochim. Biophys. Acta 1711, 215-224. 10.1016/j.bbamem.2005.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seul K. H., Kang K. Y., Lee K. S., Kim S. H. and Beyer E. C. (2004). Adenoviral delivery of human connexin37 induces endothelial cell death through apoptosis. Biochem. Biophys. Res. Commun. 319, 1144-1151. 10.1016/j.bbrc.2004.05.097 [DOI] [PubMed] [Google Scholar]
- Shin J.-L., Solan J. L. and Lampe P. D. (2001). The regulatory role of the C-terminal domain of connexin43. Cell Commun. Adhes 8, 271-275. 10.3109/15419060109080736 [DOI] [PubMed] [Google Scholar]
- Simon A. M., Goodenough D. A., Li E. and Paul D. L. (1997). Female infertility in mice lacking connexin 37. Nature 385, 525-529. 10.1038/385525a0 [DOI] [PubMed] [Google Scholar]
- Simon A. M. and McWhorter A. R. (2002). Vascular abnormalities in mice lacking the endothelial gap junction proteins connexin37 and connexin40. Dev. Biol. 251, 206-220. 10.1006/dbio.2002.0826 [DOI] [PubMed] [Google Scholar]
- Simon A. M. and McWhorter A. R. (2003). Role of connexin37 and connexin40 in vascular development. Cell Commun. Adhes. 10, 379-385. 10.1080/cac.10.4-6.379.385 [DOI] [PubMed] [Google Scholar]
- Simon A. M., Chen H. and Jackson C. L. (2006). Cx37 and Cx43 localize to zona pellucida in mouse ovarian follicles. Cell Commun. Adhes. 13, 61-77. 10.1080/15419060600631748 [DOI] [PubMed] [Google Scholar]
- Smith C. L., Debouck C., Rosenberg M. and Culp J. S. (1989). Phosphorylation of serine residue 89 of human adenovirus E1A proteins is responsible for their characteristic electrophoretic mobility shifts, and its mutation affects biological function. J. Virol. 63, 1569-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohl G. and Willecke K. (2004). Gap junctions and the connexin protein family. Cardiovasc. Res. 62, 228-232. 10.1016/j.cardiores.2003.11.013 [DOI] [PubMed] [Google Scholar]
- Solan J. L. and Lampe P. D. (2008). Connexin 43 in LA-25 cells with active v-src is phosphorylated on Y247, Y265, S262, S279/282, and S368 via multiple signaling pathways. Cell Commun. Adhes. 15, 75-84. 10.1080/15419060802014016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan J. L. and Lampe P. D. (2009). Connexin43 phosphorylation: structural changes and biological effects. Biochem. J. 419, 261-272. 10.1042/BJ20082319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan J. L. and Lampe P. D. (2017). Spatio-temporal regulation of connexin43 phosphorylation and gap junction dynamics. Biochim. Biophys. Acta. doi: 10.1016/j.bbamem.2017.04.008 [Epub ahead of print] 10.1016/j.bbamem.2017.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan J. L., Fry M. D., Tenbroek E. M. and Lampe P. D. (2003). Connexin43 phosphorylation at S368 is acute during S and G2/M and in response to protein kinase C activation. J. Cell Sci. 116, 2203-2211. 10.1242/jcs.00428 [DOI] [PubMed] [Google Scholar]
- Traub O., Hertlein B., Kasper M., Eckert R., Krisciukaitis A., Hülser D. and Willecke K. (1998). Characterization of the gap junction protein connexin37 in murine endothelium, respiratory epithelium, and after transfection in human HeLa cells. Eur. J. Cell Biol. 77, 313-322. 10.1016/S0171-9335(98)80090-3 [DOI] [PubMed] [Google Scholar]
- Warn-Cramer B. J., Lampe P. D., Kurata W. E., Kanemitsu M. Y., Loo L. W. M., Eckhart W. and Lau A. F. (1996). Characterization of the mitogen-activated protein kinase phosphorylation sites on the connexin43 gap junction protein. J. Biol. Chem. 271, 3779-3786. 10.1074/jbc.271.7.3779 [DOI] [PubMed] [Google Scholar]
- Yamaoka K. and Kameyama M. (2003). Regulation of L-type Ca2+ channels in the heart: overview of recent advances. Mol. Cell. Biochem. 253, 3-13. 10.1023/A:1026036931170 [DOI] [PubMed] [Google Scholar]
- Zhu D., Caveney S., Kidder G. M. and Naus C. C. G. (1991). Transfection of C6 glioma cells with connexin 43 cDNA: analysis of expression,intercellular coupling, and cell proliferation. Proc. Natl. Acad. Sci. USA 88, 1883-1887. 10.1073/pnas.88.5.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.