

ABSTRACT
E3 ubiquitin ligase recognizes its protein substrates via specific molecular signatures for ubiquitin proteasomal degradation. However, the role of acetylation/deacetylation in the process of E3 ubiquitin ligase recognizing its protein substrates is not fully studied. Here, we report that a tandem IK motif in protein arginine methyltransferase 1 (PRMT1) forms an acetyldegron to recruit the F-box/LRR-repeat protein 17 (FBXL17), a component of the SKP1–CUL1–F-box protein (SCF)-type E3 ubiquitin ligase complex. PRMT1 is polyubiquitylated for proteasome degradation with a half-life of approximately 4 h in lung epithelial cells. SCFFbxl17 mediates PRMT1 polyubiquitylation at K117. SCFFbxl17 specifically binds PRMT1 via a unique motif IKxxxIK. Strikingly, the acetylation/deacetylation status of the lysine residues within the motif determines Fbxl17 binding. Deacetylation on both K200 and K205 by Sirtuin 1 (Sirt1) and acetylation of p300 (EP300) on K205 collaboratively prepare the motif for SCFFbxl17 binding thereby triggering PRMT1 protein degradation. Pathogen-derived lipopolysaccharide (LPS) downregulates Sirt1 and p300 to protect PRMT1 from degradation. This study demonstrates that LPS promotes PRMT1 stability by blockade of PRMT1 and SCFFbxl17 binding via an acetylation/deacetylation-modified acetyldegron; and LPS-elevated levels of PRMT1 lead to bronchial epithelial cell overgrowth in pulmonary inflammatory diseases.
KEY WORDS: PRMT1, SCFFbxl17 E3 ligase, p300, EP300, Sirt1, Ubiquitylation, Proteasome, Protein degradation, Acetylation/deacetylation, Acetyldegron, Lipopolysaccharide
Summary: Bacteria-derived endotoxin LPS downregulates both the acetyltransferase p300 and the deacetylase Sirt1 to promote PRMT1 protein stability via an SCFFbxl17 E3 ligase-recognized acetydegron that augments bronchial epithelial cell overgrowth.
INTRODUCTION
Cellular proteins are dynamic and stringently monitored in responding to endogenous and exogenous pathophysiological inputs. The ubiquitin proteasome system is a machinery that degrades most of the cellular proteins. In orchestration with gene transcriptional machinery, the ubiquitin proteasome system-mediated proteolysis governs protein availability to regulate or re-programme a defined cellular function. Therefore, ubiquitin proteasome-mediated proteolysis is involved in a vast range of life processes in living cells. In general, proteins destined for proteasomal degradation are polyubiquitylated (Komander and Rape, 2012). Protein ubiquitylation is an enzymatic cascade involving an E1 ubiquitin-activating enzyme, an E2 ubiquitin-conjugating enzyme and an E3 ubiquitin ligase (Hochstrasser, 2009; Varshavsky, 2006). Among them, the E3 ubiquitin ligase specifically recognizes and binds its protein substrate for ubiquitylation. Each E3 ubiquitin ligase may recognize and bind a group of protein substrates for ubiquitylation. The molecular mechanism(s) of how an E3 ubiquitin ligase recognizes the protein substrates for ubiquitylation is an interesting area of study.
Mounting evidence proves that protein ubiquitylation is not a random event but a stringently controlled process (Komander and Rape, 2012). The molecular signatures, usually referred to as ‘degron’, are uncovered for substrate recognition by E3 ubiquitin ligases. Post-translational modifications including phosphorylation, glycosylation, palmitoylation as well as other less characterized modifications are the important components of such molecular signatures (Pejaver et al., 2014). Generally, protein substrates contain defined degrons that are specific for E3 ubiquitin ligase docking. The amino acid residue(s) in the degron often undergoes appropriate post-translational modifications to facilitate or impact E3 ubiquitin ligase docking. Phosphorylation is a well-known post-translational modification that regulates E3 ubiquitin ligase docking to a protein substrate. One such modification forms a phosphodegron, in which the serine/threonine phosphorylation status within the degron determines its binding capacity of an E3 ubiquitin ligase (Liu et al., 2010; Ye et al., 2004). Acetylation has long been recognized as an important post-translational modification in protein stability. Acetylation of the N-terminal residues protects the protein from degradation (Hwang et al., 2010). However, the function of acetylation/deacetylation in E3 ubiquitin ligase recognition of protein substrates is yet to be studied.
There are approximately 600–1000 genes encoding E3 ubiquitin ligases in the human genome. Based on their primary sequence homology and functional domains, E3 ubiquitin ligases are categorized into three major families, including the RING (really interesting new gene) finger family of E3 ubiquitin ligases, HECT (the homologous to E6-AP C-terminus) E3 ubiquitin ligases and complex E3 ubiquitin ligases (Bhattacharyya et al., 2014). The SCF–F-box complex is one subfamily of the complex E3 ubiquitin ligase, which contains a key substrate receptor component, the F-box protein (Skaar et al., 2014). The F-box protein has an N-terminal F-box domain, which functions to bind Skp1 and a C-terminal domain for substrate recruiting. According to the substrate-binding domains, F-box proteins are categorized into Fbxw, Fbxl and Fbxo subfamilies (Skaar et al., 2013). Among the Fbxl subfamily, Fbxl17 has been considered as a potentially useful biomarker for breast cancer therapy (Guan et al., 2014; Xiao et al., 2008). Genome-wide association analysis discovered that the FBXL17 gene may play a role in coronary heart disease (Domarkiene et al., 2013). Fbxl17-mediated ubiquitylation of Sufu (suppressor of fused homolog) regulates hedgehog signaling that is involved in medulloblastoma tumor growth (Raducu et al., 2016). In addition, Fbxl17 acts as a regulator of the NFR2 (nuclear factor erythroid-derived 2-related factor 2) oxidative stress pathway where Fbxl17 turnover of the transcriptional repressor BACH1 controls the transcription of NRF2 (Tan et al., 2013). The molecular mechanism(s) of how Fbxl17 recognizes the protein substrates is unknown.
Protein arginine methyltransferases (PRMTs) are a family of enzymes that catalyze histone and non-histone protein asymmetric methylation. Methylated arginine residues have been characterized into three types in mammalian cells: ω–NG–monomethylarginine (MMA); ω–NG, NG–asymmetric dimethylarginine (ADMA); and ω–NG, N′G–symmetric dimethylarginine (SDMA). Each type of arginine methylation is catalyzed by one of 11 PRMTs. PRMTs can be further classified as type I–IV according to the methylarginine products (Wei et al., 2014). PRMT1 is the most common and major form of type I PRMTs, which leads to the formation of ADMA (Tang et al., 2000). Altered ADMA has been detected in patients with lung cancer (Yoshimatsu et al., 2011), pulmonary arterial hypertension (Gorenflo et al., 2001; Kielstein et al., 2005; Pullamsetti et al., 2005), asthma (Scott et al., 2011) and in various end-stage organ failure patients. The ADMA metabolite might be a consequence of enhanced type I PRMT expression. Dysregulation of PRMT1 has been reported to be involved in the pathogenesis of many human diseases. For instance, PRMT1 has been found to be upregulated in various types of lung cancer (Parry and Ward, 2010). In the antigen-induced pulmonary inflammation rat asthma model, the expression of PRMT1 was significantly elevated, which might be induced by Th2 cytokine IL-4 (Sun et al., 2012). But the mechanism underlying the enhanced PRMT1 expression or stability still needs further research.
In this study, we identified that SCFFbxl17 specifically interacts with PRMT1 via a previously uncharacterized acetyldegron to ubiquitylate PRMT1 for proteasomal degradation. Both acetylation and deacetylation of the lysine residues within the degron are crucial in Fbxl17 recruitment. Deacetylase Sirtuin 1 (Sirt1) activity contributes to K200 and K205 deacetylation. The subsequent acetylation of K205 mediated by p300 (officially known as EP300) prepares the acetyldegron for Fbxl17 binding that leads to PRMT1 ubiquitin–proteasomal degradation. Aberrant expression of PRMT1 has been reported in various diseases that may be the result of dysregulated Sirt1 and/or p300.
RESULTS
PRMT1 is a labile protein degraded via the ubiquitin–proteasome machinery
Protein stability of PRMT1 is yet to be studied. We studied protein stability of PRMT1 using protein biosynthesis inhibitor cycloheximide (CHX) in murine lung epithelial MLE12 cells. PRMT1 immunoblotting results showed that the half-life of PRMT1 in MLE12 cells was ∼4 h (Fig. 1A,D). To investigate the degradation pathway involved in PRMT1 degradation, cells were treated with a proteasome inhibitor MG132 or a lysosome inhibitor E64D. Treatment with MG132 but not E64D resulted in PRMT1 accumulation, suggesting that the ubiquitin–proteasomal pathway mediates PRMT1 degradation (Fig. 1B–D). Consistent with this observation, overexpression of ubiquitin decreased PRMT1 protein in a ubiquitin-dependent manner (Fig. 1E). These results indicate that PRMT1 is a labile protein degraded via ubiquitin proteasomal machinery.
Fig. 1.

PRMT1 undergoes proteasomal degradation. (A–D) Cells were treated with (A) CHX (40 µg ml–1), (B) MG132 (20 µM) or (C) E64D (20 µM) for the indicated times, and cell lysates were subjected to immunoblotting analysis with antibodies against PRMT1 and α-tubulin. The relative PRMT1 protein levels from densitometry analysis of the immunoblots were plotted and the half-life of PRMT1 was calculated as previously described (Li et al., 2017) (D). (E) Cells were transfected with indicated amounts of HA-tagged ubiquitin plasmid, and the cell lysates were immunoblotted with anti-PRMT1, HA and β-actin antibodies. The results are representative of n=3 experiments.
E3 ubiquitin ligase SCFFbxl17 specifically ubiquitylates PRMT1
Ubiquitin proteasomal degradation is an enzymatic culmination that involves an E3 ubiquitin ligase to specifically recognize its substrate. We screened potential SCF complex protein(s) that target PRMT1 for degradation by introducing V5-tagged F-box proteins into MLE12 cells. PRMT1 immunoblotting results showed that Fbxl17 selectively reduced PRMT1 protein as compared with other Fbxl subfamily members (Fig. 2A). Fbxl17 overexpression decreased PRMT1 protein level in a concentration-dependent manner (Fig. 2B, left panels). As a control, Fbxl19 showed less effect on PRMT1 protein stability (Fig. 2B, right panels). Analysis of PRMT1 immunoprecipitates showed that PRMT1 associates with components of the SCF-E3 ligase complex Fbxl17, Cullin1 and Skp1 (Fig. 2C). Knockdown of Fbxl17 by a short-hairpin RNA (shRNA) increased PRMT1 stability (Fig. 2D,E). In addition, knockdown of Fbxl17 by shRNA decreased PRMT1 polyubiquitylation (Fig. 2F). These observations indicate that SCFFbxl17 targets PRMT1 for ubiquitylation and degradation.
Fig. 2.

SCFFbxl17 targets PRMT1 for polyubiquitylation and degradation. (A) Equivalent amounts of plasmids (2 µg) encoding V5-tagged F-box proteins were introduced into cells by transfection as indicated. Cell lysates were analyzed with PRMT1, V5 and β-actin antibodies. (B) Cells were transfected with increasing amounts of V5-tagged Fbxl17 or Fbxl19 plasmids, and cell lysates were immunoblotted with PRMT1, V5 and β-actin antibodies. (C) Cell lysates were subjected to PRMT1 immunoprecipitation. The PRMT1 immunoprecipitates were analyzed by immunoblotting with Fbxl17, Cullin1, Skp1 and PRMT1 antibodies. (D) MLE12 cells were transfected with different Fbxl17 shRNA constructs for 72 h as indicated. Cell lysates were analyzed by immunoblotting with Fbxl17 and β-actin antibodies. (E) Cells were transfected with scrambled shRNA (control) or effective shRNA construct (shFbxl17-2 in D). After 72 h, the cells were treated with CHX (40 µg ml–1) at the indicated times, and the cell lysates were processed for PRMT1, Fbxl17 and β-actin immunoblotting. (F) The lysates from scrambled shRNA or shFbxl17-transfected cells were subjected to PRMT1 immunoprecipitation. The precipitates were probed with ubiquitin (Ub) and PRMT1 antibodies. The input lysates were immunoblotted with Fbxl17 and α-tubulin antibodies. The results are representative of n=3 experiments.
SCFFbxl17 ubiquitylates PRMT1 at K117
Mass spectrometry studies have revealed that PRMT1 is ubiquitylated at multiple lysine residues (Mertins et al., 2013). To identify which lysine residues(s) is the acceptor site within PRMT1 for SCFFbxl17-mediated ubiquitylation, we constructed a series of V5-tagged PRMT1 lysine point mutants. First, we assessed the protein stability of mutants by CHX exposure. Results showed a tendency that several mutants including K117R, K184R and K328R were more stable than other mutant and wild-type (WT) PRMT1 (Fig. S1). Second, we assessed the accumulation of these mutants after MG132 treatment. V5 immunoblotting results show that WT and all other lysine point mutants except for K117R accumulated after MG132 treatment, suggesting that K117 is the ubiquitin acceptor site (Fig. 3A; Fig. S2). Third, we then co-expressed WT and K117R mutant PRMT1 with Fbxl17. V5 immunoblotting analysis showed that K117R was resistant to Fbxl17-induced PRMT1 degradation (Fig. 3B). Ectopic expression of increasing amounts of Fbxl17 did not reduce the protein level of K117R mutant as compared with WT and K298R mutant PRMT1 (Fig. 3C,D). We further compared the polyubiquitylation levels between WT and K117R PRMT1 by V5 immunoprecipitation followed by ubiquitin immunoblotting analyses. As shown (Fig. 3E), K117R mutant showed greatly reduced polyubiquitylation compared with that of the WT. Taken together, these data indicate that K117 within PRMT1 is the SCFFbxl17-dependent ubiquitin acceptor site.
Fig. 3.

K117 is the SCFFbxl17-dependent ubiquitin acceptor site within PRMT1. (A) MLE12 cells were transfected with V5-tagged WT or single lysine mutant PRMT1. After 24 h of transfection, the cells were treated with or without MG132 (20 µM) for 4 h. Cell lysates were blotted with V5 and α-tubulin antibodies. (B) Empty vector or Fbxl17 plasmids (2 µg well–1) were co-transfected with WT or mutant PRMT1 plasmids as indicated for 24 h. The cell lysates were proceeded with V5, Fbxl17 and α-tubulin immunoblotting. (C,D) MLE12 cells were co-transfected with V5-tagged WT or single lysine mutant PRMT1 and a variety amount of Fbxl17 as indicated for 24 h. Cell lysates were analyzed by immunoblotting with V5, Fbxl17 and α-tubulin antibodies. The results from densitometry analysis of the V5 immunoblots of C were plotted in D. (E) K117R mutant decreases the polyubiquitylation of PRMT1. MLE12 cells were transfected with V5-tagged PRMT1 (WT) or K117R mutant, then the cell lysates were immunoprecipitated with V5 antibody. The precipitates and input lysates (from WT) were processed for ubiquitin and V5 immunoblotting. The results are representative of n=3 experiments.
Fbxl17 docks on an IKxxxIK motif within PRMT1 and the lysine acetylation status in the motif determines Fbxl17 binding capacity
Because SCFFbxl17 specifically ubiquitylates and degrades PRMT1, we were intrigued to study the molecular signature(s) within PRMT1 that directs Fbxl17 binding. We constructed a series of V5-tagged truncated PRMT1 expression plasmids (Fig. 4A). The truncated mutant proteins were synthesized in vitro via a TnT-coupled reticulocyte system. Fbxl17 was obtained from MLE12 cells by Fbxl17 immunoprecipitation. Results from a pull-down assay showed that an N-terminal truncate (aa1–180) did not bind to Fbxl17, suggesting that amino acid residues between aa181 and 210 were crucial for Fbxl17 binding (Fig. 4B). The primary sequence of aa181–210 (Fig. 4C) contains a tandem IK motif that is an important molecular signature for F-box protein binding (Chen et al., 2015). To check if these residues contributed to Fbxl17 binding, we conducted site-directed mutagenesis and pull-down assays. Results showed that mutation of hydrophobic isoleucine residue to negatively charged hydrophilic aspartic acid (I204D) impaired Fbxl17 binding (Fig. 4D). Lysine residue is prone to post-translational modification in living cells; one such post-translational modification is acetylation. To study if the acetylation status of K200 and K205 affects Fbxl17 binding, K200 and K205 were replaced by either a glutamine (acetylation mimic) or an arginine (non-acetylation mimic). Pull-down assay results showed that acetylation of K200 (K200Q) or non-acetylation of K205 (K205R) mutants hampered their in vitro binding of Fbxl17. K200R or K205Q mutants showed a higher binding capacity to Fbxl17 (Fig. 4E). These findings revealed that two adjoining IK residues within PRMT1 play key roles in Fbxl17 recruitment. To confirm above observations, we tested the half-life of acetylation and non-acetylation PRMT1 mimics (Fig. 4F,G). Immunoblotting results showed that I199D and I204D mutants slightly reduced their degradation. Consistent with above observation, K200Q slowed down and K200R accelerated the protein degradation. K205Q enhanced the protein degradation and K205R stabilized the protein from degradation. In addition, double mutation of K200A/K205A stabilized the protein from degradation. These data indicate that Fbxl17 selectively docked on an acetylation-modified IKxxxIK motif within PRMT1 that led to PRMT1 ubiquitylation and proteasomal degradation. K200 deacetylation and K205 acetylation showed higher capacity of Fbxl17 binding, indicating that PRMT1 degradation requires a coordinated function of both acetyltransferase(s) and deacetylase(s).
Fig. 4.

Fbxl17 docks on an IKxxxIK motif within PRMT1 and the acetylation status of the lysine residues in the motif is crucial for Fbxl17 binding. (A) Schematic presentation of truncated PRMT1 mutants. (B) V5-tagged PRMT1 truncated mutants were synthesized in TnT reticulocyte lysate systems. HeLa cell lysates were immunoprecipitated with Fbxl17 antibody. In vitro binding assays were performed by mixing PRMT1 mutants and Fbxl17 beads for 2 h, and Fbxl17 pulling down precipitates were analyzed with V5 and Fbxl17 immunoblotting. (C) Amino acid sequence 181–210 of PRMT1. The IK residue sequences are indicated in red font. (D) Cell lysates were immunoprecipitated with Fbxl17 antibody and then mixed with in vitro-synthesized V5-tagged WT or mutant (I199D and I204D) PRMT1 proteins for 2 h. The eluted proteins were subjected to V5 and Fbxl17 immunoblotting. The in vitro-synthesized recombinant proteins were analyzed with V5 antibody. (E) Endogenous Fbxl17 were obtained with Fbxl17 antibody immunoprecipitation. The protein-A/G agarose beads associated Fbxl17 were incubated with in vitro-synthesized V5-tagged WT, K200Q, K200R, K205Q and K205R mutant PRMT1 for 2 h. The pull-down proteins were immunoblotted with V5 and Fbxl17 antibodies. The in vitro-synthesized recombinants were analyzed with V5 antibody. (F,G) V5-tagged WT and mutant PRMT1 were introduced in to MLE12 cells for 24 h. The cells were treated with CHX (40 µg ml–1) for the indicated times and cell lysates were subjected to immunoblotting analysis with antibodies against V5 and α-tubulin. The immunoblots were analyzed by densitometry and plotted, respectively, in G. The results are representative of n=3 experiments.
Acetyltransferase p300 preferably acetylates K205 within the IKxxxIK motif
To identify the enzymes that determine lysine acetylation status within the IKxxxIK motif, we conducted immunoprecipitation studies. Results from PRMT1 immunoprecipitation studies showed that PRMT1 associated with acetyltransferase p300 (Fig. 5A). To identify the potential p300-catalyzed acetylation site(s) within the IKxxxIK motif, we checked p300-mediated acetylation of PRMT1 in endogenous cells by co-expression of WT and mutant PRMTs with or without p300. Ectopic expression of p300 increased acetylation level of PRMT1 in MLE12 cells (Fig. 5B). Expression of p300 increased K200A acetylation level but not that of K205A, indicating that p300 preferably acetylated K205. To confirm this observation, we conducted in vitro p300-mediated acetylation assay of WT, K200A, K205A and K200A/K205A mutant PRMT1s. Similar to the results from endogenous cells, acetyl-K immunoblotting results showed that WT PRMT1 was highly acetylated by p300 (Fig. 5C). The acetylation level of dual alanine mutant K200A/K205A was greatly reduced to the input level. Both K200A and K205A showed a decreased acetylation level, with a marked reduction in K205A, suggesting that K205 is the major p300-catalyzed acetylation site. We then observed if p300 affected PRMT1 protein stability. Interestingly, both ectopic expression of p300 or knockdown of p300 by shRNA did stabilize PRMT1 protein stability, indicating that p300 is not the sole component in governing PRMT1 degradation (Fig. 5D). These data indicate that p300 preferably acetylated K205, which constitutes one of the components of the degron.
Fig. 5.

Acetyltransferase p300 preferably acetylates K205 within the motif. (A) Cell lysates were immunoprecipitated with PRMT1 and then immunoblotted with p300, GCN5 and PRMT1 antibodies. (B) WT, K200A or K205A mutant PRMT1 were co-transfected with empty vector or p300 plasmids into MLE12 cells as indicated. After 48 h of transfection, cell lysates were immunoprecipitated with V5 antibody and the precipitates were immunoblotted with acetyl-K and V5 antibodies. Cell lysates were analyzed by immunoblotting with p300, V5 and α-tubulin antibodies. (C) V5-tagged PRMT1 recombinants (WT, K200A, K205A and K200A/K205A mutant PRMT1) were synthesized with TnT reticulocyte lysate systems. The recombinants were subjected to p300-mediated acetylation for 1 h at 25°C. The mixtures were proceeded with acetyl-K and V5 immunoblotting. (D,E) PRMT1 half-life was determined in cells with empty vector and p300 overexpression (D), or scramble and p300 knockdown by shRNA (E). The results are representative of n=3 experiments.
Deacetylation enzyme Sirt1 acts on both K200 and K205 within the IKxxxIK motif
Considering that p300 acetylated K205 but altering p300 protein did not degrade PRMT1, and Fbxl17 selectively bound non-acetylated K200, we studied the potential deacetylase(s) involved in K200 deacetylation. It is reported that HDAC5 interacts with PRMT1 (Greco et al., 2011), so we tested whether HDAC5 affects PRMT1 degradation. PRMT1 immunoblotting results showed that neither overexpression nor knockdown of HDAC5 affected PRMT1 degradation (Fig. S3), indicating that a deacetylase(s) but not HDAC5 is involved in PRMT1 degradation. Therefore, we conducted PRMT1 immunoprecipitation studies and identified that PRMT1 associated with a deacetylase Sirt1 (Fig. 6A). We performed an in vitro deacetylation assay to determine if Sirt1 contributes to K200 or K205 deacetylation. Results showed that Sirt1 reduced the acetylation level of WT but not K200A/K205A mutant PRMT1 (Fig. 6B). Sirt1 reduced the acetylation level of both K200A and K205A mutant PRMT1, indicating that K200 and K205 were Sirt1-dependent deacetylation acceptor sites (Fig. 6B). We then investigated the role of Sirt1 in PRMT1 protein stability. Ectopic expression of Sirt1 slowed down PRMT1 degradation (Fig. 6C,D) and knockdown of Sirt1 greatly increased PRMT1 stability (Fig. 6E,F). These data indicate that Sirt1 contributed to the deacetylation of both K200 and K205, which constitutes another component of the degron.
Fig. 6.

Deacetylase Sirt1 deacetylates K200 and K205 in the motif. (A) Cell lysates were immunoprecipitated with PRMT1 and the immunoprecipitates were immunoblotted with Sirt1, HDAC2 and PRMT1 antibodies. (B) TnT-synthesized proteins (WT, K200A, K205A and K200A/K205A mutant PRMT1) were subjects of Sirt1-mediated deacetylation for 1 h at 25°C. The proteins were immunoblotted with acetyl-K and V5 antibodies. (C–F) Cell transfected with empty vector and Sirt1 overexpression plasmid (C), or scramble and Sirt1 shRNA constructs (E) were treated with CHX to determine PRMT1 half-life. Cell lysates were analyzed by immunoblotting with PRMT1, Sirt1 and α-tubulin antibodies. The densitometry results of C and E were plotted in D and F, respectively. The results are representative of n=3 experiments.
Coordinately ordered Sirt1-catalyzed deacetylation and p300-catalyzed acetylation in the IKxxxIK motif prepare PRMT1 for degradation
As both deacetylase Sirt1 and acetyltransferase p300 are involved in the formation of an IKxxxIK acetyldegron, we attempted to dissect the possible sequential order of the enzymes in the acetyldegron-forming process. First, we studied if the acetylation level of K200 or K205 affects Sirt1-mediated deacetylation of PRMT1. We conducted in vitro p300-mediated acetylation to obtain hyper-acetylated WT and mutant PRMT1 and applied the hyper-acetylated PRMT1 for Sirt1-mediated deacetylation assay. Results showed that Sirt1 did not reduce the acetylation levels of hyper-acetylated WT or hypo-acetylated K200A/K205A double-mutant PRMT1, but reduced the acetylation levels of K200A and slightly reduced those of K205A singly mutated PRMT1 (Fig. 7A). These data suggest a possibility that Sirt1-mediated deacetylation is prior to the event of p300-mediated acetylation. Second, we tested the protein stability of K200 acetylation/deacetylation mimics in the presence of p300. Ectopic expression of p300 accelerated K200R degradation but did not affect K200Q stability (Fig. 7B). Furthermore, ectopic expression of Sirt1 slowed down K205Q but did not affect K205R degradation (Fig. 7C), suggesting that the action of Sirt1 is before K205 acetylation as well. In all, these data suggest that PRMT1 degradation is under the control of a highly ordered coordination of p300 and Sirt1. PRMT1 IKxxxIK acetyldegron formation is probably the result of a surge of Sirt1-mediated deacetylation on K200 and K205 followed by a p300-catalyzed K205 acetylation.
Fig. 7.
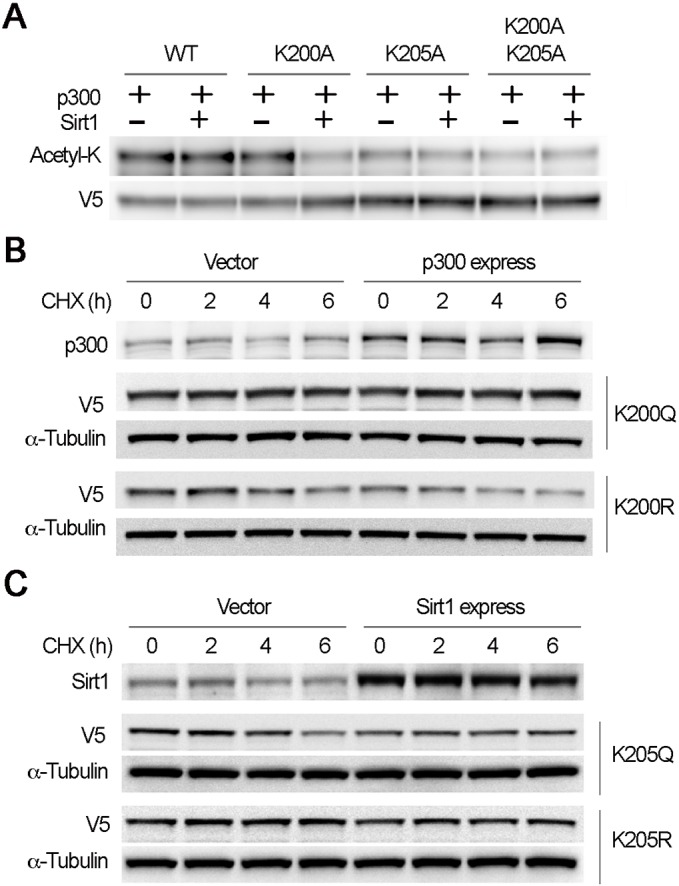
Sequential Sirt1-mediated deacetylation on K200 and p300-mediated acetylation on K205 prepare the IKxxxIK motif for Fbxl17 binding. (A) TnT-synthesized proteins (WT, K200A, K205A and K200A/K205A mutant PRMT1) were preacetylated with recombinant p300 for 1 h at 25°C. The WT and mutant PRMT1 proteins were pulled down by V5 antibody. The pulled-down products were subjected to Sirt1-mediated deacetylation for 1 h at 25°C. The proteins were immunoblotted with acetyl-K and V5 antibodies. (B) Acetylation mimic (K200Q) or deacetylation mimic (K200R) of mutant PRMT1 were co-transfected with empty vector (right panels) or p300 expression constructs (left panels) as indicated. After 24 h the cells were treated with CHX for different time phases. Cell lysates were analyzed by immunoblotting with V5, p300 and α-tubulin antibodies. (C) The half-life of acetylation mimic (K205Q) and deacetylation mimic (K205R) of mutant PRMT1 were co-transfected with empty vector (right panels) or Sirt1 expression constructs (left panels) as indicated. After 24 h, the cells were treated with CHX for various time points. Cell lysates were analyzed by immunoblotting with V5, Sirt1 and α-tubulin antibodies. The results are representative of n=3 experiments.
LPS-induced PRMT1 accumulation by downregulation of both p300 and Sirt1
It has been reported that PRMT1 expression is upregulated in the pulmonary inflammation model. To determine whether bacterial infection upregulates PRMT1 stability, we treated bronchial epithelial BEAS 2B (BS2B) cells with Gram-negative bacteria-derived endotoxin LPS overnight. Results showed that LPS elevated PRMT1 protein in a concentration-dependent pattern (Fig. 8A,B). We then assessed the acetylation level of PRMT1 under LPS treatment. LPS overnight treatment induced PRMT1 accumulation, and both p300 and Sirt1 were downregulated (Fig. 8C, input). The interaction of PRMT1 with both p300 and Sirt1 was interrupted with LPS, which resulted in a large reduction of PRMT1 acetylation (Fig. 8C). To confirm the role PRMT1 has in bronchial epithelial cell proliferation, we overexpressed or knocked down PRMT1 and observed proliferation of the BS2B cells. Results showed that PRMT1 promotes BS2B cell proliferation, whereas silencing of PRMT1 by shRNA slowed down cell proliferation (Fig. 8D). We further determined BS2B proliferation in the presence of LPS. LPS treatment accelerated BS2B cell proliferation (Fig. 8E). Overexpression of PRMT1 enhanced LPS-stimulated BS2B growth, but the growth was suppressed by overexpression of Fbxl17 because it degrades PRMT1. As predicted, overexpression of Fbxl17 did not inhibit K200A/K205A double-mutant PRMT1-mediated cell proliferation. Fbxl17 itself is believed to promote proliferation (Raducu et al., 2016). Co-expression of Fbxl17 and K200A/K205A mutant PRMT1 augmented LPS-induced cell proliferation. These data indicate that LPS deregulates p300 and Sirt1 to change the acetylation status within the IKxxxIK acetyldegron. Impaired SCFFbxl17-mediated PRMT1 degradation thus contributes to bronchial epithelial overgrowth.
Fig. 8.

LPS downregulates both p300 and Sirt1 to accumulate PRMT1 in MLE12 cells. (A,B) Cells were treated with the indicated amounts of LPS in HITES medium without FCS for 16 h and the levels of endogenous PRMT1 and α-tubulin proteins were measured by immunoblotting. Bar charts show densitometric analysis of immunoblots in A. *P<0.05 (LPS treated versus untreated). (C) WT PRMT1 was introduced into MLE12 cells for 24 h. The cells were treated with or without LPS (2 µg ml–1) for another 16 h and the cell lysates were immunoprecipitated with V5 antibody. The precipitates were immunoblotted with p300, Sirt1, acetyl-K and V5 antibodies. Cell lysates (Input) were immunoblotted with p300, Sirt1, V5 and α-tubulin antibodies. (D) PRMT1 was expressed for 24 h or silenced by shRNA for 48 h in human bronchial BS2B cells. Then, equivalent amounts of cells were cultured in FBS-free medium for another 24 h. The cell growth data were plotted. (E) WT or K200A/K205A double-mutant PRMT1 were co-expressed with or without Fbxl17 in BS2B cells for 24 h. The cells were treated with LPS (2 μg ml–1) for another 24 h. Live cells were counted and normalized with untreated cells. *P<0.05, compared with empty vector (EV) without LPS. **P<0.01, compared with EV without LPS. #P<0.05, compared with LPS treated EV without Fbxl17. ##P<0.01, compared with LPS treated EV with Fbxl17. The results are representative of n=3 experiments. (F) Schematic presentation of the mechanism of E3 ubiquitin ligase SCFFbxl17-mediated PRMT1 degradation via an acetyldegron. PRMT1 contains an IKxxxIK motif in which the lysine residues are acetylated in MLE12 cells. The acetylation status of the lysine residues within the IKxxxIK motif is governed by deacetylase Sirt1 and acetyltransferase p300. Deacetylase Sirt1 acts on both K200 and K205. Acetyltransferase p300 preferably catalyzes K205 acetylation. Highly coordinated sequential deacetylation and acetylation are required for optimal Fbxl17 binding. A surge of Sirt1 deacetylates both K200 and K205 followed by p300-catalyzed K205 acetylation leads to SCFFbxl17-mediated PRMT1 ubiquitylation and thereafter proteasomal degradation. Pathological stimulus LPS downregulates both Sirt1 and p300 at protein level that results in PRMT1 accumulation in MLE12 cells.
DISCUSSION
The primary findings of this study are: (i) PRMT1 is a labile protein degraded via the ubiquitin proteasomal pathway; (ii) SCFFbxl17 ubiquitylates PRMT1 at K117; (iii) Fbxl17 interacts with PRMT1 via an uncharacterized IKxxxIK motif; (iv) acetylation status of the lysine residues in the acetyldegron determines Fbxl17 docking on PRMT1, and highly ordered action of Sirt1 followed by p300 results in K200 deacetylation and K205 acetylation in the acetyldegron that determines PRMT1 stability; and (v) pathological stimulus LPS deregulates both p300 and Sirt1 to accumulate PRMT1 to enhance small airway epithelial cell growth (Fig. 8F). Mass spectrometry studies have revealed that more than 90% of the nuclear proteins are post-translationally modified by acetylation (Choudhary et al., 2014). It is well known that acetylation and deacetylation are rigorously involved in the regulation of gene transcriptional activities (Glozak et al., 2005; Struhl, 1998). In general, histone acetylation results in gene transcriptional activation and histone deacetylation is related to gene transcriptional suppression (Grunstein, 1997). In this study, we report that acetylation/deacetylation regulates a histone modification enzyme PRMT1 at the level of protein stability. Recent studies have established that ubiquitin proteasomal degradation of histone modification enzymes is an important mechanism to regulate transcriptional activity (Kristeleit et al., 2004; Zou and Mallampalli, 2014). Acetylation/deacetylation may control a histone modification enzyme not only at transcriptional level but also at protein stability level. The role of acetylation/deacetylation in the modulation of histone modification enzymes at protein stability increases the complexity of gene transcription regulation.
Acetylation/deacetylation is a crucial post-translational modification that acts as a signal for protein degradation as well (Asher et al., 2008; Deribe et al., 2010; Ito et al., 2002; Kong et al., 2006). Acetylation/deacetylation may regulate protein stability by distinct mechanisms. It is well known that N-terminal acetylation of amino acids affects protein stability (Polevoda and Sherman, 2002), yet acetylation may shift protein cellular compartmentalization to regulate protein stability as well (Inuzuka et al., 2012). p300-mediated acetylation of lysine residues in nuclear localization signal may relocate Skp2 in the cellular compartments to escape from Cdh1-mediated degradation (Inuzuka et al., 2012). More importantly, lysine residue is the acceptor for both acetylation and ubiquitylation. The interplay between acetylation and ubiquitylation at the same amino acid affects protein stability (Caron et al., 2005; Grönroos et al., 2002). Acetyltransferase GCN5-mediated acetylation competes with SCFFbxl18-catalyzed ubiquitylation at K143 of Morf4l1 and elevates Morf4l1 protein stability to increase cell death in acute lung injury (Zou et al., 2015). However, deacetylation enzymes such as HDAC2 take part in the regulation of this process too. Deacetylase HDAC2 facilitates Alcat1 ubiquitylation and lysosomal degradation to impair mitochondrial function (Zou et al., 2016). Here, we demonstrated that acetylation/deacetylation affects PRMT1 protein degradation via E3 ubiquitin ligase docking. Like a phosphodegron, acetylation modifies the lysine residues in the docking site of an E3 ubiquitin ligase to form an acetyldegron IKxxxIK motif. A recent independent study reports that p300/CREB-binding protein (CBP)-mediated tandem acetylation of K11 and K14 facilitates glutamine synthetase binding to CRL4CRBN E3 ubiquitin ligase. It demonstrates a third pattern of acetyldegron that p300/CBP-mediated acetylation of a tandem KxxK motif leads to glutamine synthetase ubiquitylation and proteasomal degradation (Van Nguyen et al., 2016). GCN5L1 acetylates K505 within KIF1-binding protein (KBP) to promote SCFFbxo15 binding, which is important in mitochondrial biogenesis (Donato et al., 2017). Our previous observation reported that a tandem IK motif in nucleoside diphosphate kinase A (NDPK-A) – especially the second IK in the motif – is crucial for Fbxo24 binding (Chen et al., 2015). Deacetylation of the second K facilitates Fbxo24 docking on NAPK-A and GCN5-mediated acetylation abrogates this action. In this study, both IK pairs in the IKxxxIK motif are crucial for Fbxl17 binding; deacetylation of the first K and acetylation of second K prepares the motif for Fbxl17 binding. These studies suggest distinct substrate-recognizing patterns for Fbxo and Fbxl subfamily members.
In this acetyldegron, two important elements, K200 and K205, are differentially modified by acetylation and deacetylation. In MLE12 cells, both K200 and K205 are acetylated. A mutation of either site reduces the total acetylation level of PRMT1. Acetylation or deacetylation of both sites may attribute to PRMT1 homeostasis in the cells. For an optimal Fbxl17 binding, K200 must be deacetylated and K205 be acetylated. Results from in vitro acetylation/deacetylation studies indicate that Sirt1 deacetylates both K200 and K205, but p300 preferably acetylates K205. Sirt1 deacetylates baseline but not hyper-acetylated K200 and K205. Manipulation of protein levels of either Sirt1 or p300 results in an abrogation of PRMT1 degradation. These observations suggest a model that Sirt1 and p300 sequentially act on the motif to achieve an optimal SCFFbxl17 recruitment. It is interesting that both acetylation and deacetylation determine the binding capacity of an E3 ubiquitin ligase. The relationship of p300 and Sirt1 is complicated and distinct in different model systems. Here, LPS treatment increased PRMT1 stability in MLE12 cells by downregulating both p300 and Sirt1 and interrupting the interaction of PRMT1 with p300 and/or Sirt1.
PRMT1 is a multifunctional enzyme that modifies histone and non-histone protein substrates by asymmetric methylation (Tang et al., 2000). Aberrant expression of PRMT1 at the transcriptional level has been reported in many diseases including chronic pulmonary diseases and lung cancers (Scott et al., 2011; Yoshimatsu et al., 2011). High protein level in living cells is the result of an orchestrated action of both gene transcription and protein stability. This study demonstrated that protein stability is also a crucial factor in aberrant PRMT1 expression. A highly coordinated action of both an acetylation enzyme p300 and a deacetylation enzyme Sirt1 stringently govern the protein stability of PRMT1. Deregulation of either p300 or Sirt1 increases the stability of PRMT1. This may explain the fact that the aberrant expression of PRMT1 is observed in many diseased conditions. In this study, we identified that LPS downregulates both p300 and Sirt1 to protect PRMT1 from Fbxl17-mediated degradation, thus attributing to bronchial epithelial overgrowth, such as in asthma. The mechanism of how p300 and Sirt1 are regulated in terms of protein stability in this scenario is yet to be studied.
MATERIALS AND METHODS
Cell cultures and reagents
Murine lung epithelial (MLE12) cells and human bronchial BS2B cells were cultured in HITES medium (500 ml DMEM/F12, 2.5 mg insulin, 2.5 mg transferrin, 2.5 mg sodium selenite, 2.5 mg transferrin, 10 μM hydrocortisone, 10 μM β-estradiol, 10 mM HEPES and 2 mM L-glutamine) containing 10% fetal bovine serum (FBS) as previously described (Zou et al., 2013). HeLa cells were maintained with Eagle's minimal essential medium (EMEM) containing 10% FBS. Cells were maintained in a 37°C incubator in the presence of 5% CO2. V5 antibody, pcDNA3.1D-His-V5-TOPO cloning kit (Cat#: K490001) and Escherichia coli Top10 competent cells (C404006) were from Invitrogen (St Louis, MO, USA). Hemagglutinin (HA) tag (3724S, lot: 5), acetylated-lysine (9441S, lot: 12), HDAC2 (5113S, lot: 1), HDAC5 (20458S, lot: 1) and α-tubulin (2144S, lot: 5) antibodies were from Cell Signaling (Danvers, MA, USA). PRMT1 (sc-166963, lot: H2013), p300 (sc-585, lot: K0711), Sirt1 (sc-15404, lot: H1914), GCN5 (sc-20698, lot: B1011) and ubiquitin (sc-166553, lot: A2710) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Fbxl17 (ab111683, lot: GR65075-7) antibody was from Abcam (Cambridge, MA, USA). The PRMT1 cDNA and shRNA, Fbxl17 cDNA and shRNA, p300 shRNA, Sirt1 shRNA were from Origene (Rockville, MD, USA). p300 cDNA plasmid was deposited by Warner Greene (AddGene plasmid #23252; Chen et al., 2002) and Sirt1 by Toren Finkel (AddGene plasmid #10962; Nemoto et al., 2005). Recombinant enzymatic active p300 (31124) and Sirt1 (31340) proteins were from Active Motif (Carlsbad, CA, USA). Cycloheximide (ALX-380-269-G001, lot: 01061518), E64D (BML-PI107-0001, Lot# 08181537) and ubiquitin aldehyde (BML-UW8450-0050, lot: 07021447) were from Enzo Life Sciences (Farmingdale, NY, USA). β-actin (A3853) antibody and bacterial LPS from E. coli O111:B4 (L4391, lot: 115M4090V) were from Sigma (Carlsbad, CA, USA). MG132 (F1101, lot: F11052079) was from UBPBio (Aurora, CO, USA). QuikChange II XL site-directed mutagenesis kits (200522) were from Agilent Technologies (Santa Clara, CA, USA). TnT Quick Coupled Transcription/Translation Systems (L1170) were from Promega (Madison, WI, USA). Immobilized protein-A/G agarose beads (20421) were from Pierce (Rockford, IL, USA). All other reagents were of the highest grade available commercially.
Cloning and mutagenesis
V5-tagged PRMT1 truncations were cloned into pcDNA3.1D-His-V5-TOPO plasmid using PCR-based approaches as previously described (Zou et al., 2016). Mutagenesis was introduced by using a QuikChange II XL site-directed mutagenesis kit (Agilent Technologies, CA, USA) according to the manufacturer's instructions. The primers used in the construction of PRMT1 truncations and site-directed mutagenesis are listed in Table S1.
Plasmid transfection
All plasmids were introduced into MLE12 cells using electroporation executed with a nuclear transfection apparatus (Amaxa Biosystems, Gaithersburg, MD, USA) with a preset program T-013, following the manufacturer's instructions as previously described (Zou et al., 2016). Briefly, one million cells in 100 μl of transfection buffer (20 mM HEPES in PBS buffer) were mixed with 3 μg of plasmids (including expression and shRNA constructs). After electroporation, the cells were cultured with 2 ml conditional HITES medium in 6-well plates for 24 h for further analyses.
Immunoblotting and co-immunoprecipitation
Immunoblotting and co-immunoprecipitation were conducted as previously described (Zou et al., 2016). Briefly, for immunoblotting, whole-cell extracts (normalized to total protein concentration) were resolved by SDS-PAGE and transferred to membranes by electroblotting. The membranes were blocked with 5% (w/v) non-fat milk in Tris-buffered saline and probed with primary antibodies as indicated. Membranes were developed by an enhanced chemiluminescence (ECL) system. For immunoprecipitation, 1 mg of cell lysates (in PBS with 0.5% Tween 20 plus protease inhibitors) were incubated with specific primary antibodies for 2 h at room temperature. The mixture was added to 35 µl of protein A/G-agarose beads for an additional 2 h at room temperature. The precipitated complex was washed three times with 0.5% Tween 20 in PBS and analyzed by immunoblotting with ECL system.
In vitro pull-down assay
We conducted in vitro binding assays to identify the Fbxl17-binding domain within PRMT1. V5-tagged PRMT1 truncated or site-directed mutant proteins were in vitro expressed using a TnT-coupled reticulocyte system. Endogenous Fbxl17 protein was obtained by Fbxl17 immunoprecipitation from HeLa cell lysate. Fbxl17-precipitated protein A/G-agarose beads were incubated with a variety of PRMT1 truncations or mutants for 2 h. The beads were washed extensively with 0.5% Tween 20 in PBS and analyzed by V5 and Fbxl17 immunoblotting.
In vitro acetylation assay
V5-tagged WT, K200A, K205A and K200A/K205A mutant PRMT1 proteins were in vitro expressed by a TnT-coupled reticulocyte system and incubated with 100 ng recombinant p300 protein (or dilution buffer); 5 µg of acetyl-CoA in a 30 µl reaction mixture contained 50 mM Tris-HCl (pH 8.0), 10% glycerol, 0.1 mM EDTA, 1 mM DTT and 50 mM KCl. The reaction mixtures were incubated 1 h at 30°C. The reaction was stopped by adding protein loading buffer and the samples were analyzed with immunoblotting.
In vitro deacetylation assay
TnT-synthesized V5-tagged WT, K200A, K205A and K200A/K205A mutant PRMT1 proteins were mixed with 100 ng recombinant Sirt1 in a 70 µl reaction mixture, respectively. The mixture was incubated for 1 h at 30°C in the deacetylase buffer [final concentration: 25 mM Tris-HCl (pH 8.0), 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 0.2 mM DTT and 1 mM NAD+]. The reaction was terminated by the addition of SDS-PAGE loading dye and the proteins were analyzed by immunoblotting. For p300 pre-treatment, TnT-synthesized V5-tagged PRMT1 WT and K200A, K205A and K200A/K205A proteins were mixed with 100 ng recombinant p300 in a 70 µl reaction mixture. After 1 h incubation at 30°C, each reaction mixture was divided equally into two parts and diluted with 300 µl of PBS with 0.3% Tween 20 and incubated with 2 µg of anti-V5 antibody for 1 h at room temperature followed by the addition of protein A/G-agarose for another 1 h and subsequently preceded in deacetylation assay.
Proliferation assay
Human bronchial BS2B cells were transfected with an indicated plasmid. Equivalent number of cells (1×105) was grown to 90% confluence in 6-well plates in FBS-free medium for 24 h. Total cell numbers were measured with a TC10 automatic cell counter (Bio-Rad). The velocity of cell growth was normalized by that in PRMT1 expression cells. For LPS treatment, BS2B cells were transfected with an empty vector, or co-transfected in the presence or absence of Fbxl17 plasmid with a plasmid encoding WT PRMT1 protein or K200A/K205A double-mutant PRMT1 constructs. After 24 h, the cells were treated with 2 μg ml–1 of LPS for another 20 h. Cells were harvested and counted with a TC10 automatic cell counter (Bio-Rad). The data were normalized with untreated BS2B cells and plotted in a bar graph.
Statistical analysis
Densitometric quantification of the bands was achieved using ImageJ. Data are presented as means±standard deviation (s.d.). All results were analyzed by an analysis of variance test or a Student’s t-test.
Supplementary Material
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: C.Z.; Methodology: Y.L., J.L., X.L.; Validation: Y.L.; Formal analysis: X.L.; Investigation: Y.L., J.L., X.L.; Data curation: Y.L., J.L., C.Z.; Writing - original draft: Y.L.; Writing - review & editing: Y.L., J.L., X.L., C.Z.; Supervision: C.Z.; Project administration: C.Z.; Funding acquisition: C.Z.
Funding
This work was supported by a National Institutes of Health R01 grant HL125435 (to C.Z.). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.206904.supplemental
References
- Asher G., Gatfield D., Stratmann M., Reinke H., Dibner C., Kreppel F., Mostoslavsky R., Alt F. W. and Schibler U. (2008). SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 134, 317-328. 10.1016/j.cell.2008.06.050 [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S., Yu H., Mim C. and Matouschek A. (2014). Regulated protein turnover: snapshots of the proteasome in action. Nat. Rev. Mol. Cell Biol. 15, 122-133. 10.1038/nrm3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron C., Boyault C. and Khochbin S. (2005). Regulatory cross-talk between lysine acetylation and ubiquitination: role in the control of protein stability. BioEssays 27, 408-415. 10.1002/bies.20210 [DOI] [PubMed] [Google Scholar]
- Chen L.-F., Mu Y. and Greene W. C. (2002). Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-κB. EMBO J. 21, 6539-6548. 10.1093/emboj/cdf660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., Xiong S., Li J., Li X., Liu Y., Zou C. and Mallampalli R. K. (2015). The ubiquitin E3 ligase SCF-FBXO24 recognizes deacetylated nucleoside diphosphate kinase A to enhance its degradation. Mol. Cell. Biol. 35, 1001-1013. 10.1128/MCB.01185-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C., Weinert B. T., Nishida Y., Verdin E. and Mann M. (2014). The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536-550. 10.1038/nrm3841 [DOI] [PubMed] [Google Scholar]
- Deribe Y. L., Pawson T. and Dikic I. (2010). Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 17, 666-672. 10.1038/nsmb.1842 [DOI] [PubMed] [Google Scholar]
- Domarkiene I., Pranculis A., Germanas S., Jakaitiene A., Vitkus D., Dzenkeviciute V., Kucinskiene Z. and Kucinskas V. (2013). RTN4 and FBXL17 genes are associated with coronary heart disease in genome-wide association analysis of Lithuanian families. Balkan J. Med. Genet. 16, 17-22. 10.2478/bjmg-2013-0026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato V., Bonora M., Simoneschi D., Sartini D., Kudo Y., Saraf A., Florens L., Washburn M. P., Stadtfeld M., Pinton P. et al. (2017). The TDH-GCN5L1-Fbxo15-KBP axis limits mitochondrial biogenesis in mouse embryonic stem cells. Nat. Cell Biol. 19, 341-351. 10.1038/ncb3491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glozak M. A., Sengupta N., Zhang X. and Seto E. (2005). Acetylation and deacetylation of non-histone proteins. Gene 363, 15-23. 10.1016/j.gene.2005.09.010 [DOI] [PubMed] [Google Scholar]
- Gorenflo M., Zheng C., Werle E., Fiehn W. and Ulmer H. E. (2001). Plasma levels of asymmetrical dimethyl-L-arginine in patients with congenital heart disease and pulmonary hypertension. J. Cardiovasc. Pharmacol. 37, 489-492. 10.1097/00005344-200104000-00016 [DOI] [PubMed] [Google Scholar]
- Greco T. M., Yu F., Guise A. J. and Cristea I. M. (2011). Nuclear import of histone deacetylase 5 by requisite nuclear localization signal phosphorylation. Mol. Cell. Proteomics 10, M110.004317 10.1074/mcp.M110.004317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grönroos E., Hellman U., Heldin C.-H. and Ericsson J. (2002). Control of Smad7 stability by competition between acetylation and ubiquitination. Mol. Cell 10, 483-493. 10.1016/S1097-2765(02)00639-1 [DOI] [PubMed] [Google Scholar]
- Grunstein M. (1997). Histone acetylation in chromatin structure and transcription. Nature 389, 349-352. 10.1038/38664 [DOI] [PubMed] [Google Scholar]
- Guan Y.-P., Yang X.-X., Yao G.-Y., Qiu F., Chen J., Chen L.-J., Ye C.-S. and Li M. (2014). Breast cancer association studies in a Han Chinese population using 10 European-ancestry-associated breast cancer susceptibility SNPs. Asian Pac. J. Cancer Prev. 15, 85-91. 10.7314/APJCP.2014.15.1.85 [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. (2009). Origin and function of ubiquitin-like proteins. Nature 458, 422-429. 10.1038/nature07958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang C.-S., Shemorry A. and Varshavsky A. (2010). N-terminal acetylation of cellular proteins creates specific degradation signals. Science 327, 973-977. 10.1126/science.1183147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inuzuka H., Gao D., Finley L. W. S., Yang W., Wan L., Fukushima H., Chin Y. R., Zhai B., Shaik S., Lau A. W. et al. (2012). Acetylation-dependent regulation of Skp2 function. Cell 150, 179-193. 10.1016/j.cell.2012.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A., Kawaguchi Y., Lai C.-H., Kovacs J. J., Higashimoto Y., Appella E. and Yao T.-P. (2002). MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 21, 6236-6245. 10.1093/emboj/cdf616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielstein J. T., Bode-Boger S. M., Hesse G., Martens-Lobenhoffer J., Takacs A., Fliser D. and Hoeper M. M. (2005). Asymmetrical dimethylarginine in idiopathic pulmonary arterial hypertension. Arterioscler. Thromb. Vasc. Biol. 25, 1414-1418. 10.1161/01.ATV.0000168414.06853.f0 [DOI] [PubMed] [Google Scholar]
- Komander D. and Rape M. (2012). The ubiquitin code. Annu. Rev. Biochem. 81, 203-229. 10.1146/annurev-biochem-060310-170328 [DOI] [PubMed] [Google Scholar]
- Kong X., Lin Z., Liang D., Fath D., Sang N. and Caro J. (2006). Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1α. Mol. Cell. Biol. 26, 2019-2028. 10.1128/MCB.26.6.2019-2028.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristeleit R., Stimson L., Workman P. and Aherne W. (2004). Histone modification enzymes: novel targets for cancer drugs. Expert Opin. Emerg. Drugs 9, 135-154. 10.1517/14728214.9.1.135 [DOI] [PubMed] [Google Scholar]
- Li X., Lai Y., Li J., Zou M. and Zou C. (2017). Oxidative stress destabilizes protein arginine methyltransferase 4 via glycogen synthase kinase 3beta to impede lung epithelial cell migration. Am. J. Physiol. Cell Physiol. 313, 285-294. 10.1152/ajpcell.00073.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.-Y., Zha Z.-Y., Zhou X., Zhang H., Huang W., Zhao D., Li T., Chan S. W., Lim C. J., Hong W. et al. (2010). The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCFβ-TrCP E3 ligase. J. Biol. Chem. 285, 37159-37169. 10.1074/jbc.M110.152942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P., Qiao J. W., Patel J., Udeshi N. D., Clauser K. R., Mani D. R., Burgess M. W., Gillette M. A., Jaffe J. D. and Carr S. A. (2013). Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 10, 634-637. 10.1038/nmeth.2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto S., Fergusson M. M. and Finkel T. (2005). SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J. Biol. Chem. 280, 16456-16460. 10.1074/jbc.M501485200 [DOI] [PubMed] [Google Scholar]
- Parry R. V. and Ward S. G. (2010). Protein arginine methylation: a new handle on T lymphocytes? Trends Immunol. 31, 164-169. 10.1016/j.it.2010.01.006 [DOI] [PubMed] [Google Scholar]
- Pejaver V., Hsu W.-L., Xin F., Dunker A. K., Uversky V. N. and Radivojac P. (2014). The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci. 23, 1077-1093. 10.1002/pro.2494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polevoda B. and Sherman F. (2002). The diversity of acetylated proteins. Genome Biol. 3, 1 10.1186/gb-2002-3-5-reviews0006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullamsetti S., Kiss L., Ghofrani H. A., Voswinckel R., Haredza P., Klepetko W., Aigner C., Fink L., Muyal J. P., Weissmann N. et al. (2005). Increased levels and reduced catabolism of asymmetric and symmetric dimethylarginines in pulmonary hypertension. FASEB J. 19, 1175-1177. 10.1096/fj.04-3223fje [DOI] [PubMed] [Google Scholar]
- Raducu M., Fung E., Serres S., Infante P., Barberis A., Fischer R., Bristow C., Thézénas M. L., Finta C., Christianson J. C. et al. (2016). SCF (Fbxl17) ubiquitylation of Sufu regulates Hedgehog signaling and medulloblastoma development. EMBO J. 35, 1400-1416. 10.15252/embj.201593374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott J. A., North M. L., Rafii M., Huang H., Pencharz P., Subbarao P., Belik J. and Grasemann H. (2011). Asymmetric dimethylarginine is increased in asthma. Am. J. Respir. Crit. Care. Med. 184, 779-785. 10.1164/rccm.201011-1810OC [DOI] [PubMed] [Google Scholar]
- Skaar J. R., Pagan J. K. and Pagano M. (2013). Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. Mol. Cell Biol. 14, 369-381. 10.1038/nrm3582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar J. R., Pagan J. K. and Pagano M. (2014). SCF ubiquitin ligase-targeted therapies. Nat. Rev. Drug Discov. 13, 889-903. 10.1038/nrd4432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K. (1998). Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 12, 599-606. 10.1101/gad.12.5.599 [DOI] [PubMed] [Google Scholar]
- Sun Q., Yang X., Zhong B., Jiao F., Li C., Li D., Lan X., Sun J. and Lu S. (2012). Upregulated protein arginine methyltransferase 1 by IL-4 increases eotaxin-1 expression in airway epithelial cells and participates in antigen-induced pulmonary inflammation in rats. J. Immunol. 188, 3506-3512. 10.4049/jimmunol.1102635 [DOI] [PubMed] [Google Scholar]
- Tan M.-K. M., Lim H.-J., Bennett E. J., Shi Y. and Harper J. W. (2013). Parallel SCF adaptor capture proteomics reveals a role for SCFFBXL17 in NRF2 activation via BACH1 repressor turnover. Mol. Cell 52, 9-24. 10.1016/j.molcel.2013.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J., Frankel A., Cook R. J., Kim S., Paik W. K., Williams K. R., Clarke S. and Herschman H. R. (2000). PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J. Biol. Chem. 275, 7723-7730. 10.1074/jbc.275.11.7723 [DOI] [PubMed] [Google Scholar]
- Van Nguyen T., Lee J. E., Sweredoski M. J., Yang S.-J., Jeon S.-J., Harrison J. S., Yim J.-H., Lee S. G., Handa H., Kuhlman B. et al. (2016). Glutamine triggers acetylation-dependent degradation of glutamine synthetase via the thalidomide receptor cereblon. Mol. Cell 61, 809-820. 10.1016/j.molcel.2016.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. (2006). The early history of the ubiquitin field. Protein Sci. 15, 647-654. 10.1110/ps.052012306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H., Mundade R., Lange K. and Lu T. (2014). Protein arginine methylation of non-histone proteins and its role in diseases. Cell Cycle 13, 32-41. 10.4161/cc.27353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao G. G., Zhou B.-S., Somlo G., Portnow J., Juhasz A., Un F., Chew H., Gandara D. and Yen Y. (2008). Identification of F-box/LLR-repeated protein 17 as potential useful biomarker for breast cancer therapy. Cancer Genomics Proteomics 5, 151-160. [PubMed] [Google Scholar]
- Ye X., Nalepa G., Welcker M., Kessler B. M., Spooner E., Qin J., Elledge S. J., Clurman B. E. and Harper J. W. (2004). Recognition of phosphodegron motifs in human cyclin E by the SCFFbw7 ubiquitin ligase. J. Biol. Chem. 279, 50110-50119. 10.1074/jbc.M409226200 [DOI] [PubMed] [Google Scholar]
- Yoshimatsu M., Toyokawa G., Hayami S., Unoki M., Tsunoda T., Field H. I., Kelly J. D., Neal D. E., Maehara Y., Ponder B. A. J. et al. (2011). Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int. J. Cancer 128, 562-573. 10.1002/ijc.25366 [DOI] [PubMed] [Google Scholar]
- Zou C. and Mallampalli R. K. (2014). Regulation of histone modifying enzymes by the ubiquitin–proteasome system. Biochim. Biophys. Acta 1843, 694-702. 10.1016/j.bbamcr.2013.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou C., Chen Y., Smith R. M., Snavely C., Li J., Coon T. A., Chen B. B., Zhao Y. and Mallampalli R. K. (2013). SCF(Fbxw15) mediates histone acetyltransferase binding to origin recognition complex (HBO1) ubiquitin-proteasomal degradation to regulate cell proliferation. J. Biol. Chem. 288, 6306-6316. 10.1074/jbc.M112.426882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou C., Li J., Xiong S., Chen Y., Wu Q., Li X., Weathington N. M., Han S., Snavely C., Chen B. B. et al. (2015). Mortality factor 4 like 1 protein mediates epithelial cell death in a mouse model of pneumonia. Sci. Transl. Med. 7, 311-171 10.1126/scitranslmed.aac7793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou C., Synan M. J., Li J., Xiong S., Manni M. L., Liu Y., Chen B. B., Zhao Y., Shiva S., Tyurina Y. Y. et al. (2016). LPS impairs oxygen utilization in epithelia by triggering degradation of the mitochondrial enzyme Alcat1. J. Cell Sci. 129, 51-64. 10.1242/jcs.176701 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.