

ABSTRACT
Tau normally associates with and stabilizes microtubules (MTs), but is hyperphosphorylated and aggregated into neurofibrillary tangles in Alzheimer's disease and related neurodegenerative diseases, which are collectively known as tauopathies. MTs are regulated by different forms of post-translational modification, including acetylation; acetylated MTs represent a more stable microtubule population. In our previous study, we showed that inhibition of histone deacetylase 6 (HDAC6), which deacetylates tubulin at lysine 40, rescues defects in MTs and in neuromuscular junction growth caused by tau overexpression. However, HDAC6 also acts on other proteins that are involved in distinct biological processes unrelated to tubulins. In order to examine directly the role of increased tubulin acetylation against tau toxicity, we generated a site-directed α-tubulinK40Q mutation by CRISPR/Cas9 technology to mimic the acetylated MTs and found that acetylation-mimicking α-tubulin rescued tau-induced MT defects and neuromuscular junction developmental abnormalities. We also showed that late administration of ACY-1215 and tubastatin A, two potent and selective inhibitors of HDAC6, rescued the tau-induced MT defects after the abnormalities had already become apparent. Overall, our results indicate that increasing MT acetylation by either genetic manipulations or drugs might be used as potential strategies for intervention in tauopathies.
KEY WORDS: Acetylation, Drosophila, Microtubule, Tauopathy
Highlighted Article: Increased acetylation of microtubules by genetic and pharmacological approaches rescues human tau overexpression-induced toxicity in Drosophila muscles and neurons.
INTRODUCTION
The microtubule-associated protein tau stabilizes microtubules (MTs). However, in tauopathies, including Alzheimer's disease (AD) and frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), tau is hyperphosphorylated, leading to the aggregation of tau and MT destabilization, and finally resulting in neuronal death and a reduction in brain weight and volume (Ballatore et al., 2007; Gomez-Isla et al., 1996; Huang and Mucke, 2012).
MTs are regulated by different forms of post-translational modification, including phosphorylation, glutamylation, tyrosination and acetylation. For example, acetylation occurs on lysine 40 (K40) of α-tubulin inside the MT lumen (Janke and Bulinski, 2011; Nogales et al., 1998), which is controlled by a balance of acetyltransferases and deacetylases. Two histone deacetylase-related enzymes, histone deacetylase 6 (HDAC6) and sirtuin type 2, have been found to deacetylate α-tubulin in vivo and in vitro (Hubbert et al., 2002; North et al., 2003). In the brains of AD patients, HDAC6 is significantly increased compared with the normal brain, and the tubulin acetylation is reduced in neurons carrying the tau neurofibrillary tangles (Hempen and Brion, 1996). We previously showed that ectopically expressed human tau in Drosophila results in decreased MT density, increased MT fragments, and more satellite boutons at neuromuscular junctions (NMJs) (Xiong et al., 2013). In Drosophila, HDAC6 regulates the deacetylation of α-tubulin, and we showed that null mutants of HDAC6 attenuate the tau toxicity in Drosophila (Xiong et al., 2013).
Specifically, loss of deacetylase activity conferred by the tubulin-specific deacetylase domain of HDAC6 is crucial for the rescue of tau-mediated MT defects (Xiong et al., 2013). HDAC6 has two deacetylase domains, DD1 and DD2. DD2 has been shown to deacetylate tubulin specifically (Haggarty et al., 2003; Kaluza et al., 2011). Introducing a mutation of H664A in HDAC6 DD2 is shown to rescue tau-mediated MT abnormalities, suggesting that amelioration of MT defects is dependent on increased MT acetylation (Xiong et al., 2013). However, HDAC6 can deacetylate multiple substrates involved in distinct biological processes unrelated to tubulins, such as tau, cortactin and the crucial chaperone, heat shock protein 90 (Cook et al., 2014; Kaluza et al., 2011; Valenzuela-Fernández et al., 2008). In addition to its deacetylase activity, HDAC6 also interacts directly with multiple proteins, including tau, the molecular chaperone p97, mDia2, ubiquitin, p150Glued and protein phosphatase 1 (Boyault et al., 2006; Brush et al., 2004; Chen et al., 2005; Destaing et al., 2005; Ding et al., 2008; Seigneurin-Berny et al., 2001). Taken together, it is of importance to examine directly the role of tubulin acetylation against the toxicity of tau by mutating the K40 site of tubulin.
In this study, we aimed to determine the effect of acetylated tubulin on tauopathy. We generated site-directed α-tubulinK40Q and α-tubulinK40R mutations to mimic acetylated and non-acetylated MTs, respectively, in Drosophila and showed that increasing α-tubulin acetylation by either mutation or drugs can rescue tau-mediated MT defects in muscles and NMJ abnormalities in the neuronal system. Our findings suggest new targets for the development of therapeutic drugs for tauopathies.
RESULTS
Site-directed mutation of α-tubulin to mimic acetylated and non-acetylated MTs
There are four α-tubulin genes in Drosophila, referred to as α1 (CG1913), α2 (CG9476), α3 (CG2512) and α4 (CG8308), at chromosomal locations 84B, 85E, 84D and 67C, respectively. The genes α1 and α3 are constitutively expressed and differ from each other by only two amino acid substitutions (Theurkauf et al., 1986). However, α1 is highly expressed at all developmental stages, whereas α3 is expressed at low levels at most developmental stages (Matthews et al., 1989). By contrast, α2 is testes specific, whereas α4 transcripts accumulate only in ovarian nurse cells, eggs and early embryos (Theurkauf et al., 1986). Thus, αl-tubulin (hereafter referred to as α-tubulin) may be the major source for microtubules in most cells. In mammals, there are 15-20 distinct α-tubulin-encoding genes (Hall and Cowan, 1985). Thus, owing to functional redundancy, it is difficult to determine the effects of acetylated MT on physiological development and pathology in vivo.
To define the role of acetylated tubulin on MT dynamics better, we set out to make point mutations of the widely and abundantly expressed α-tubulin. Glutamine is hydrophilic and uncharged, and similar to acetylated lysine, whereas arginine has a positively charged side chain like lysine but cannot be acetylated (Kim et al., 2006; Li et al., 2012). Using CRISPR/Cas9-mediated targeted mutagenesis, we generated two different α-tubulin mutations, in which α-tubulin K40 was substituted with glutamine (K40Q) or arginine (K40R) to mimic acetylated and non-acetylated tubulin, respectively (Fig. 1A,B). Both mutants showed normal development and fertility in both sexes. Staining with antibodies against total α-tubulin in α-tubK40Q mutants revealed that there were more MT bundles in the perinuclear area of muscle cells compared with wild type (Fig. 1C,D). However, we did not find an apparent difference in MT network in muscle cells between α-tubK40R and wild type (Fig. 1C,E). In both α-tubK40Q and α-tubK40R mutants, very weak or no staining for acetylated α-tubulin was detected in muscles (Fig. 1D,E) or within the neuronal system (data not shown). The results of western blotting did not reveal any signals for expected acetylated α-tubulin in α-tubK40Q or α-tubK40R mutant muscles (Fig. 1F). Notably, the α-tubulin band of α-tubK40Q was slightly higher than the wild type. These results demonstrate that the antibody against acetylated tubulin did not recognize either α-tubK40Q or α-tubK40R mutants, and that α-tubulin encoded by CG1913 remained the major, if not the only tubulin within the muscles.
Fig. 1.
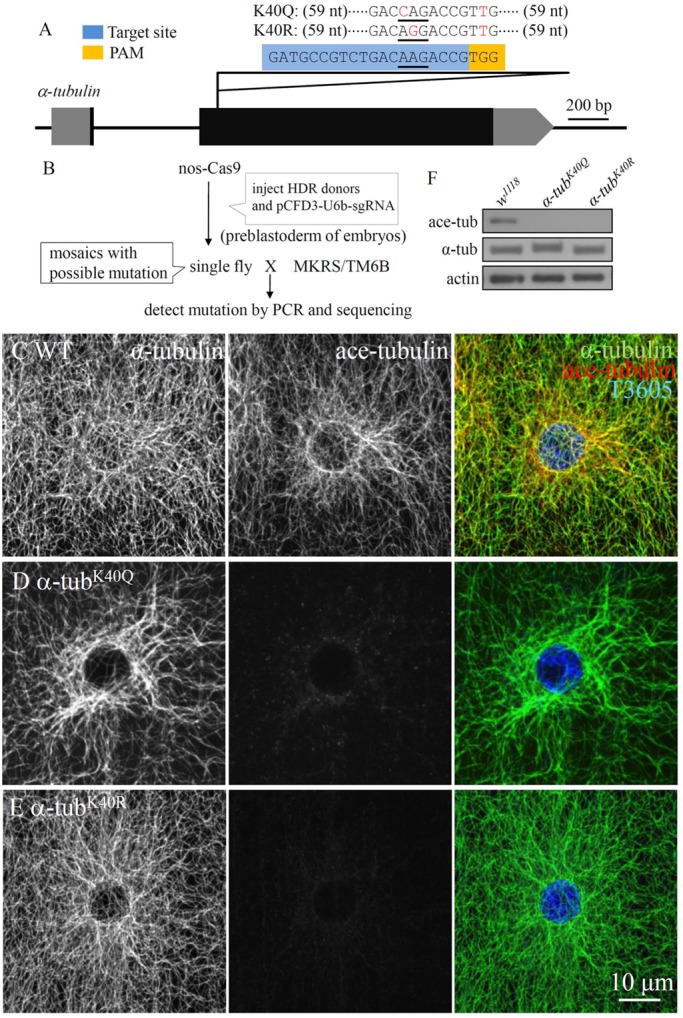
CRISPR/Cas9-mediated mutagenesis of α-tubulin. (A) The intron-exon organization of α-tubulin (CG1913). Exons and untranslated region (UTR) are shown as black and grey blocks, respectively. The sgRNA target site is in blue. PAM, protospacer-adjacent motif highlighted in yellow. For targeted point mutation, the lysine (K) 40 codon, AAG, was replaced with the glutamine (Q) codon, CAG, or arginine (R) codon, AGG. (B) Cross scheme and screening procedures. HDR, homology-directed repair. (C-E) The larval muscles co-stained with antibodies against total tubulin, acetylated tubulin and T3605, which labels the nucleus in wild type (C), α-tubK40Q (D) and α-tubK40R (E). Scale bar: 10 μm. (F) Western blot analysis of acetylated and total α-tubulin in larval muscles of wild type, α-tubK40Q and α-tubK40R mutants. Actin was used as a loading control.
MTs carrying α-tubK40Q mutation are more resistant to cold treatment
Increased acetylation has been associated with MT stabilization (Janke and Bulinski, 2011). However, it was found by kinetic analysis of polymerization and depolymerization that isolated acetylated and non-acetylated MTs have similar stabilities in vitro (Maruta et al., 1986). Thus, the effects of acetylation on MT dynamics remain uncertain.
Cold treatment is widely used to induce MT depolymerization in live cells (Chu et al., 2011; Tala et al., 2014). To determine whether the α-tubK40Q mutation increased the stability of MTs, we treated live larvae on ice for 30 min before dissection and immunostaining. At room temperature, the MT network appeared largely normal in different genotypes except that there were thick MT bundles in HDAC6 null (HDAC6KO) mutant cells, in which the perinuclear MT density was mildly but significantly increased compared with wild type (Fig. 2A–F). Upon cold treatment, the MT network in muscle cells of wild type was greatly reduced, and only a residual MT network could be observed in muscles (Fig. 2A,A′). Compared with wild type, HDAC6KO and α-tubK40Q mutants exhibited significantly denser perinuclear MTs after cold treatment (∼23% increase, P<0.01 for HDAC6KO and 11% increase, P<0.05 for α-tubK40Q; Fig. 2A′,B′,D′,G). On the contrary, in HDAC6-overexpressing larvae and α-tubK40R mutants, the density of the MT network was significantly reduced compared with the wild type after cold treatment (36 and 48% of the wild-type density, respectively; P<0.001 for both), and the MT fibres were much shorter and sparser (Fig. 2A′,C′,E′,G). In summary, the MTs in HDAC6KO and α-tubK40Q mutants, which have a higher level of acetylation and acetylation-mimicking MTs, respectively, are more resistant to cold-induced MT depolymerization, whereas the MTs in HDAC6OE and α-tubK40R mutants, with un-acetylated α-tubulin, are more labile on cold treatment.
Fig. 2.

Acetylated MTs are more resistant to cold treatment. Third instar larval muscles were co-stained with antibodies against total tubulin (green) and a nuclear dye T3605 (blue). (A,B,C,D,E) Muscle cells of wild type (A), HDAC6KO null mutants (B), HDAC6-overexpressing animals (C), α-tubK40Q (D) and α-tubK40R (E) mutant larvae maintained at 25°C. (A′,B′,C′,D′,E′) Muscle cells from larvae of the different genotypes treated on ice for 30 min to depolymerize MTs and stained with anti-α-tubulin (green) and T3605 (blue). Scale bar: 10 μm. (F,G) Quantification of perinuclear MT densities in muscle cells of different genotypes treated at room temperature (F) or on ice for 30 min (G) before dissection. n=12 muscle cells for each genotype. *P<0.05, **P<0.01, ***P<0.001 (one-way ANOVA). Error bars indicate s.e.m.
α-tubK40Q mutation rescues the MT defects and NMJ growth anomalies caused by tauV337M overexpression
We previously reported that HDAC6 mutations and inhibitors increase α-tubulin acetylation and rescue the tau toxicity induced by overexpression of tauV337M (Xiong et al., 2013), demonstrating the importance of α-tubulin acetylation in inhibiting tau toxicity. We previously showed that HDAC6 mutations do not affect tau phosphorylation in Drosophila (Xiong et al., 2013), although HDAC6 inhibition reduces tau hyperphosphorylation in mice (Zhang et al., 2014). Given that HDAC6 has multiple substrates and interacting proteins (Cook et al., 2014; Ding et al., 2008; Kaluza et al., 2011; Valenzuela-Fernández et al., 2008), it remains possible that mechanisms other than tubulin acetylation might contribute to the rescue effect.
To define the effects of MT acetylation on MT defects caused by tau overexpression, we expressed human FTDP-17-associated V337M mutant tau in Drosophila and analysed genetic interactions between α-tubK40Q and the ectopically expressed tau by quantifying the density of perinuclear MT in larval muscle cells. Muscle-specific overexpression of tauV337M (C57-Gal4/+, tauV337M) led to significantly reduced perinuclear MT density, with sparser and shorter fibres compared with the genetic control (C57-Gal4/+; Fig. 3A,A′,B,B′). Specifically, the percentage of the tubulin-positive area in the defined perinuclear area was 29% in muscle cells overexpressing tauV337M (Fig. 3B,B′,D), compared with 46% in the genetic control (P<0.001; Fig. 3A,A′,D). The decrease of perinuclear MT density caused by tauV337M overexpression was fully rescued by the α-tubK40Q mutation (47%; P<0.001; Fig. 3C,C′,D), demonstrating that the tauV337M-induced MT defects could be fully rescued by acetylation-mimicking α-tubulin.
Fig. 3.

α-tubK40Q antagonizes tau overexpression in regulating MT network formation in muscle cells and NMJ growth. (A-C′) Third instar larval muscles double-labelled with anti-tubulin (green) to reveal MT network and nuclear T3605 (blue). A′-C′ are magnified images of the top-right of the corresponding A-C. (A,A′) Genetic control C57-Gal4/+. (B,B′) Muscular overexpression of tauV337M (C57-Gal4/tauV337M) resulted in decreased MT density. (C,C′) Overexpression of tauV337M in α-tubK40Q background (C57-Gal4/tauV337M; α-tubK40Q). α-tubK40Q mutation rescued MT defects caused by tauV337M overexpression. (D) Quantification of MT densities in different genotypes. n=15 cells for each genotype. (E-H) Representative muscle four NMJs from wandering third instar larvae double-labelled with anti-CSP (red) and anti-HRP (green) to reveal synaptic vesicles and the neuronal membrane, respectively. (E) Genetic control elav-Gal4/+. (F) Neuronal overexpression of tauV337M (elav-Gal4/tauV337M). (G) α-tubK40Q. (H) Overexpression of tauV337M in α-tubK40Q background (elav-Gal4/tauV337M; α-tubK40Q). The α-tubK40Q mutation rescued the increased number of satellite boutons caused by neuronal overexpression of tauV337M. Arrows in F indicate satellite boutons. Scale bars: 20 μm in A; 10 μm in C and H; 5 μm in C′. (I) Quantification of the number of satellite boutons in different genotypes. n=12 NMJs for each genotype. ***P<0.001 (one-way ANOVA). Error bars indicate s.e.m.
In order to investigate the effect of acetylated microtubules on tau toxicity in other cell types, we ectopically expressed tauV337M in neurons driven by the pan-neuronal elav-Gal4. Our results showed that neuronal overexpression of tauV337M led to more satellite boutons at the NMJ (6.55±1.32) compared with the genetic control (1.33±0.31; P<0.001; Fig. 3E,F,I), consistent with previous reports (Chee et al., 2005; Xiong et al., 2013). The excess satellite boutons caused by neuronal overexpression of tauV337M were rescued by the homozygous α-tubK40Q mutation (2.64±0.43; P<0.001; Fig. 3F,H,I), which alone showed normal NMJs (1.14±0.29; Fig. 3G,I). Together, these results show that acetylation-mimicking α-tubulin mutation rescues tau-induced toxicity in both neuronal and non-neuronal cells.
We also performed a genetic interaction assay between tauV337M and α-tubK40R. To our surprise, α-tubK40R partly rescued MT defects, and completely reversed more satellite boutons induced by tau overexpression (Fig. S3). Increased MT acetylation and α-tubK40Q mutation might rescue tau-induced defects, probably by promoting motor-based transport (Reed et al., 2006; Song and Brady, 2015), whereas α-tubK40R rescued tau-induced defects by a presently unknown mechanism. We note that α-tubK40R has a positively charged guanidino group, which might affect the in vivo function of microtubules, leading to the rescue of tau-induced MT defects. In summary, the mimic of non-acetylated tubulin, α-tubK40R, rendered MT more labile on cold treatment (Fig. 2E′), but it did not perform as non-acetylated tubulins in enhancing tau toxicity in muscles (Fig. S3G).
Pharmacological inhibition of the tubulin-specific deacetylase activity of HDAC6 rescues MT defects caused by tauV337M overexpression
Mounting evidence supports the notion that administration of HDAC6 inhibitors and MT-stabilizing drugs improve transport defects and cognition in transgenic mouse models of tauopathy (Zhang et al., 2005, 2014). In Drosophila, the tau-induced MT defects are significantly rescued by tubacin, an inhibitor of the tubulin-specific deacetylase activity of HDAC6 (Xiong et al., 2013). Although tubacin was previously added to medium where eggs were laid and larvae developed (Xiong et al., 2013), it was not known whether the drug protects MT integrity, reverses MT defects, or a combination of both. To examine the possibility of a potential use in medicine, we set out to determine whether the tau toxicity could be rescued by HDAC6 inhibitors after the MT defects had already formed.
The use of tubacin in disease models has helped to validate HDAC6 as a drug target, but its non-drug-like structure, high lipophilicity and tedious synthesis together make it more useful as a research tool than a drug (Butler et al., 2010; Haggarty et al., 2003). ACY-1215 and tubastatin A are newly developed potent and highly selective inhibitors of the tubulin-specific deacetylase activity of HDAC6 and are more readily accessible than tubacin (Butler et al., 2010; Santo et al., 2012). We examined whether ACY-1215 and tubastatin A could rescue the already formed MT defects caused by tauV337M overexpression. Overexpression of tauV337M by C57-Gal4 led to apparent MT defects in the second instar larvae (Fig. 4A,B,G). As expected, treatment with 100 μM ACY-1215 or tubastatin A significantly increased the levels of acetylated α-tubulin in wild-type and tauV337M overexpression animals (Fig. 4I). Feeding larvae with 100 μM ACY-1215 and tubastatin A from the second larval stage mildly but significantly rescued the MT defects (P<0.05 and P<0.01, respectively, compared with the vehicle-fed larvae; Fig. 4C–F,H). As a control, the drug treatment of wild-type larvae did not affect the MT network or density (Fig. S1). In addition, a lower dose of 50 μM tubastatin A led to no obvious rescue, suggesting that the rescue effect of MT defects caused by tauV337M overexpression was dose dependent (Fig. S2). Together, our results suggest that ACY-1215 and tubastatin A might be therapeutically beneficial in tauopathies.
Fig. 4.

Inhibition of HDAC6 by ACY-1215 or tubastatin A increases acetylation of tubulin and rescues MT defects caused by tau overexpression. (A-F) Drosophila second or third instar larval muscles co-stained with anti-α-tubulin (green) to show MT network and T3605 (blue) to show the nucleus. (A) The MT network at second instar larval stage. (B) The MT network was disrupted at second instar larval stage in tauV337M-overexpressing larvae. Scale bar: 5 μm. (C-F) The third instar larval of different genotypes were fed cornmeal medium containing vehicle DMSO (C,D), ACY-1215 (E) or tubastatin A (F) from second instar larval stage. Scale bar: 10 μm. (G) Quantification of MT densities in the perinuclear area of second instar larvae in wild-type and tauV337M-overexpressing larvae. n=8 cells for each genotype. ***P<0.001 (t-test). Error bars indicate s.e.m. (H) Quantification of MT densities in the perinuclear area of third instar larval muscles from different genotypes treated with or without drugs. Comparisons were made between each genotype and the wild-type controls, as well as between specific genotypes (bracketed). n=16 cells for each genotype. *P<0.05, **P<0.01, ***P<0.001 (one-way ANOVA). Error bars indicate s.e.m. (I) ACY-1215 or tubastatin A treatment did not alter the level of total α-tubulin but increased the level of acetylated-tubulin in both wild-type and tau-overexpressing muscles.
ACY-1215 and tubastatin A rescue NMJ abnormality caused by tauV337M overexpression
As described above, ACY-1215 and tubastatin A rescued the already formed MT defects in muscles caused by muscle-specific tauV337M overexpression (Fig. 4). We next examined the rescue effects of the two drugs in the neuronal system. We fed larvae overexpressing tauV337M in neurons with ACY-1215 and tubastatin A from the second instar larval stage, and checked the phenotype of the NMJ at the third instar larval stage. Although no obvious defects were observed in second instar larvae expressing tauV337M by elav-Gal4 (data not shown), there were significantly more satellite boutons at the NMJs of third instar larvae overexpressing tauV337M pan-neuronally compared with the genetic controls (Fig. 3E,F,I). The anomaly of excess satellite boutons attributable to neuronal overexpression of tauV337M was fully reversed by ACY-1215 (1.91±0.45; P<0.001) and tubastatin A (1.91±0.39; P<0.001) compared with the vehicle-treated mutants (Fig. 5A–D). These results show that inhibition of HDAC6 and increased acetylation of MTs by ACY-1215 or tubastatin A rescue NMJ growth defects by inhibiting the toxicity caused by neuronal overexpression of tauV337M.
Fig. 5.
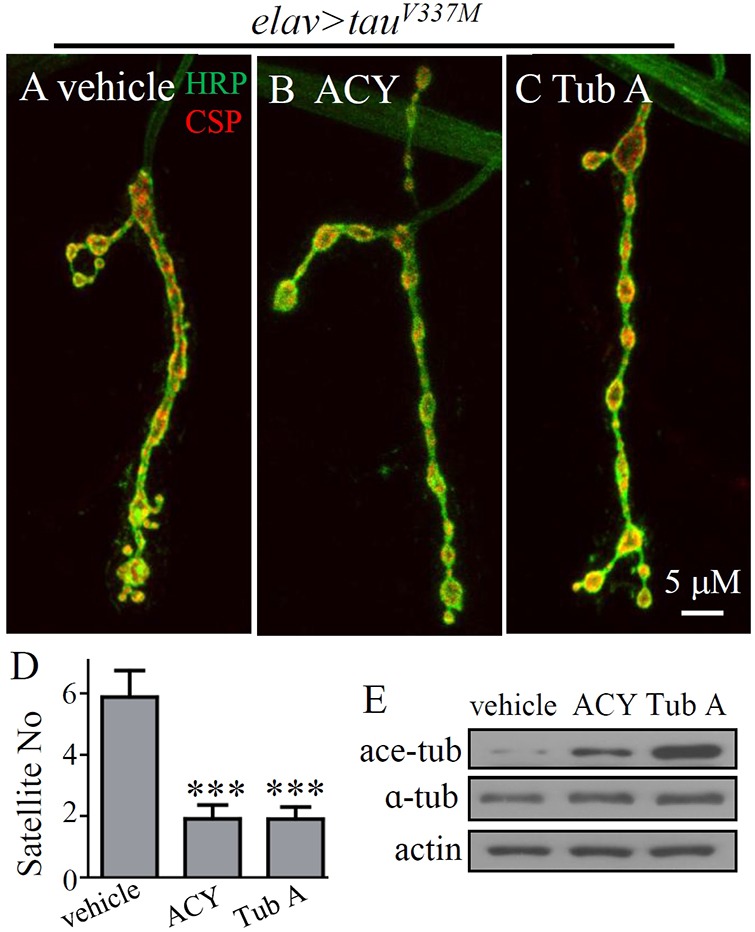
Inhibition of HDAC6 by ACY-1215 and tubastatin A rescues the increased number of satellite boutons induced by neuronal overexpression of tau. (A-C) Larvae overexpressing tauV337M were fed cornmeal medium containing vehicle (DMSO), ACY-1215 or tubastatin A (100 μM) from second instar larval stage and examined at late third instar larval stage. The increased satellite boutons caused by tauV337M overexpression were partly reversed by both ACY-1215 (B) and tubastatin A (C). Scale bar: 5 μm. (D) Quantification of the number of satellite boutons in tauV337M-overexpressing larvae with or without drug treatment. n=16 NMJs for each genotype. ***P<0.001 (one-way ANOVA). Error bars indicate s.e.m. (E) In larval brains, treatment with ACY-1215 or tubastatin A did not alter the level of total α-tubulin but increased the level of acetylated tubulin.
DISCUSSION
Increased acetylation of α-tubulin K40 promotes MT stability
Acetylation of α-tubulin K40 on the luminal face of MTs is an important post-translational modification of tubulin and is common on stable MTs in most cell types, yet the physiological role of α-tubulin K40 acetylation remains elusive. Besides α-tubulin K40, there are several additional lysine residues (K394, K60 and K370 of α-tubulin and K58, K103, K154 and K252 of β-tubulin) reported to be putative sites of acetylation (Liu et al., 2015). However, the functions of these seven sites have not been identified. Acetylation of α-tubulin K40 is not required for survival in protists, such as Tetrahymena and Chlamydomonas, but it is important in various biological processes (Akella et al., 2010; Gaertig et al., 1995; Kozminski et al., 1993). For example, MTs with reduced acetylation or carrying a K40R substitution in α-tubulin of Tetrahymena are more dynamic (Akella et al., 2010). In cultured fibroblasts, acetylation of α-tubulin K40 is required for contact inhibition of proliferation and cell-substrate adhesion (Aguilar et al., 2014). Neurons overexpressing a K40A mutant, which mimic non-acetylated α-tubulin, show altered motor-based trafficking and cell differentiation in mice (Creppe et al., 2009).
Our mutational analysis showed that acetylation of α-tubulin is not essential for survival in Drosophila. There were no obvious differences in lifespan, locomotion or fertility between wild type and α-tubK40Q or α-tubK40R mutants. However, there were increased MT bundles in the perinuclear area in α-tubK40Q muscle cells (Fig. 1D), and α-tubK40Q MTs were more resistant to cold-induced disassembly (Fig. 2D′,G). On the contrary, MT fibres were much shorter and sparser in α-tubK40R mutants upon cold treatment (Fig. 2E′,G), consistent with a previous report (Akella et al., 2010), although there was no significant difference in MT network between α-tubK40R mutants and wild type at room temperature (Fig. 1E; Fig. 2E,F). Thus, our findings support the hypothesis that acetylation of α-tubulin K40 promotes MT bundle formation and enhances MT stability upon cold treatment.
α-tubK40Q mutation rescues MT defects caused by tauV337M overexpression
MT defects, a hallmark of tauopathies, contribute directly to neurodegeneration (Ballatore et al., 2007). Previous studies in both cell cultures and primary culture neurons reveal that overexpressed tau shows reduced MT binding to motor proteins and inhibits transport of cellular components, which lead to MT disruption and synaptic decay (Mandelkow et al., 2004; Thies and Mandelkow, 2007). However, how the overexpressed tau leads to MT defects in vivo remains poorly understood. Our previous work showed that increased MT acetylation in HDAC6 null mutants rescued tau-induced MT defects in both muscles and neurons (Xiong et al., 2013). In mammals, HDAC6 binds with tau directly, maintains site-specific tau phosphorylation and promotes tau accumulation (Cook et al., 2012; Ding et al., 2008). Thus, it is possible that HDAC6 mutations rescue tau-mediated MT defects by promoting degradation of phosphorylated tau. However, HDAC6 null mutation does not reduce the level of phosphorylated tau in Drosophila (Xiong et al., 2013).
Our previous study supports the hypothesis that increased MT acetylation by inhibition of HDAC6 activity rescues tau-induced defects (Xiong et al., 2013), but other functions of HDAC6 might also have contributed to this rescue ability. Here we show, for the first time, that the acetylation-mimicking α-tubK40Q mutation rescues tau-induced pathologies (Fig. 3). How do the acetylation-mimicking α-tubK40Q mutation and increased MT acetylation rescue tau-induced MT defects? Neurons with long axons are likely to be highly sensitive to defects in motor proteins and their microtubule tracks. Trafficking defects have been implicated in various neurodegenerative diseases, including AD, amyotrophic lateral sclerosis and lissencephaly (Chevalier-Larsen and Holzbaur, 2006; Duncan and Goldstein, 2006; Keays et al., 2007). In cultured cells, tau overexpression results in blockage of transport, most probably as a result of excessive binding of tau to MTs, which not only prevents the transport of vesicles, but also hinders the binding of motor proteins to MTs (Ballatore et al., 2007; Dixit et al., 2008; Thies and Mandelkow, 2007). MT acetylation was reported to enhance kinesin-based transport, as inhibition of tubulin deacetylase modestly enhances kinesin binding to MTs and redistributes a putative kinesin cargo (Reed et al., 2006; Song and Brady, 2015). However, acetylation and kinesin motility and accumulation are not shown to correlate in vivo (Hammond et al., 2010), and tubulin acetylation alone does not affect kinesin-1 velocity and run length in vitro (Walter et al., 2012). Thus, whether increased MT acetylation rescues tau-induced MT defects by regulating motor-based transport remains to be investigated further.
Alternatively, MT acetylation might affect protein interactions with MTs, as acetylated MTs are favoured for severing by katanin (Mao et al., 2014; Sudo and Baas, 2010). By contrast, axonal MTs are less sensitive to katanin, as a result of the fact that axonal tau protects acetylated MTs from katanin (Yu et al., 2008). Thus, the acetylation level and the extent to which MTs are bound by MT-associated proteins determine the stability and dynamics of MTs in physiological and pathological conditions. It will be interesting to test whether katanin plays a role in the MT acetylation-mediated rescue of tau toxicity in both neuronal and non-neuronal systems.
MATERIALS AND METHODS
Drosophila stocks and husbandry
Flies were cultured on standard cornmeal medium at 25°C unless otherwise specified. w1118 was used as the wild-type control. Other stocks used included the muscle-specific C57-Gal4 (from V. Budnik, Univeristy of Massachusetts, Worcester, MA, USA), pan-neuronal elav-Gal4 and nos-Cas9 (both from Bloomington Stock Center). UAS–tauV337M was from M. Feany (Harvard Medical School, Boston, MA, USA). HDAC6 null mutant (HDAC6KO) and UAS-HDAC6 were from R. Jiao (Institute of Biophysics, CAS, Beijing, China; Du et al., 2010).
Site-directed mutation of the α-tubulin locus
We generated two site-directed mutations into α-tubulin (CG1393) from wild-type K40 to glutamine (Q) and arginine (R) to mimic the acetylated and non-acetylated protein, respectively (Kim et al., 2006; Li et al., 2012). The CRISPR/Cas9-mediated targeted mutagenesis of α-tubulin was performed largely according to previously published homology-directed repair procedures (Port et al., 2014). Efficient target recognition by the CRISPR/Cas9 system requires 20 nucleotides (nt) of homology between the single-guide RNA (sgRNA) and its genomic target (Gratz et al., 2013). Cleavage also requires that the 3′ end of the genomic target sequence contains a 3 bp proto-spacer adjacent motif (PAM) sequence, TGG, which differentiates self from invading DNA (Fig. 1A) (Jinek et al., 2012). Generation of α-tubulin sgRNA was performed by cloning sgRNA (a single synthetic guide RNA, gatgccgtctgacaagaccg) into the pCFD3-dU6 vector (49410; Addgene). This designed sgRNA recognized the α-tubulin DNA sequence at the target site nearby to the codon of α-tubulin K40 (Fig. 1A). The 131 bp single-stranded DNA oligonucleotide donor (ssODN) containing the site-directed mutation was synthesized. The ssODN sequence, cctgctgggagctctactgcttggagcacggcatccagcccgatggccagatgccgtctgaccag (or agg) accgttggcggaggtgatgactcgttcaacaccttcttcagcgagactggagctggcaagcacgtg (131 bp, single-stranded DNA), harbours a site-directed mutation within the sgRNA-α-tubulin target site, and a same-sense mutation in the PAM sequence (Fig. 1A). The donor DNA with the site-directed mutation (1 µg/µl) and the pCFD3-U6b-sgRNA-α-tubulin (400 ng/µl) were injected into the nos-Cas9 transgenic line (Fig. 1B), and the eclosed adults were crossed with MKRS/TM6B. The α-tubulin in the offspring was sequenced to identify whether the flies carried a mutation or not (Fig. 1B).
Immunochemical analyses and confocal microscopy
Western analysis was performed as described previously (Jin et al., 2009; Mao et al., 2014). Third instar larval carcasses, larval brains and ventral nerve cords were dissected in PBS, followed by homogenization in a lysis buffer [50 mM Tris-HCl, (pH 7.4), 150 mM NaCl, 1% NP-40 and 0.1% SDS]. Blots were first probed with primary antibodies anti-α-tubulin (1:10,000; mAb B-5-1-2; Sigma-Aldrich), anti-acetylated tubulin (1:10,000; mAb 6-11B-1; Sigma-Aldrich) and anti-actin (1:50,000; mAb 1501; Chemicon), followed by incubation with horseradish peroxidase (HRP)-coupled secondary antibodies. Protein bands were visualized by a chemiluminescence method (ECL Kit; Amersham).
For immunostaining of muscles and NMJs, we used the following primary antibodies: anti-α-tubulin (1:800; mAb B-5-1-2; Sigma-Aldrich), anti-acetylated tubulin (1:800; mAb 6-11B-1; Sigma-Aldrich), anti-cysteine string protein [CSP; 1:200; 6D6; Developmental Studies Hybridoma Bank (DSHB)], and FITC-conjugated goat anti-HRP (1:100; Jackson ImmunoResearch). To examine the MT network in muscles, muscle 2 in abdominal segment A4 was analysed, as it has fewer tracheal branches to obscure the viewing of MTs (Mao et al., 2014). Nuclei were visualized by TO-PRO(R) 3 iodide (T3605; 1:1000; Invitrogen) staining for 1 h at room temperature. Images were collected with an Olympus FV10-ASW confocal microscope and analysed using projections from complete z-stacks through the entire muscle four NMJ of the abdominal segment A3. Synaptic boutons were defined according to co-staining signals by anti-HRP, which labels neuronal membranes, and anti-CSP, which detects presynaptic vesicles. Satellite boutons were defined as boutons of smaller size than the adjacent mature boutons along NMJ branches.
Quantification of perinuclear MT density
Quantification of MT density in muscles was performed as previously described (Jin et al., 2009; Mao et al., 2014; Xiong et al., 2013). All images were projections of serial stacks through the muscle cell. The perinuclear areas were defined as the coverage that spans 5 or 20 μm around T3605-labelled nuclei. Tubulin staining signals within the perinuclear area from muscle 2 of abdominal segment A4 were calculated using ImageJ 3.0. The software reports the ratio of the tubulin-positive area divided by the total perinuclear area.
MT cold treatment assay
Live third instar larvae were put on ice for 30 min to depolymerize MTs. After dissection in cold Ca2+-free HL3.1 saline (70 mM NaCl, 5 mM KCl, 20 mM MgCl2, 10 mM NaHCO3, 115 mM sucrose, 5 mM trehalose and 5 mM HEPES) and fixation by paraformaldehyde, immunofluorescence staining was performed as previously described (Jin et al., 2009; Mao et al., 2014).
Pharmacological treatment of larvae with ACY-1215 and tubastatin A
Viable second instar larvae (48-72 h after egg laying) of elav-Gal4>UAS-tauV337M or control were removed from normal medium to the pre-prepared medium containing drug or DMSO vehicle control. ACY-1215 and tubastatin A (Selleck Company) were prepared as 10 mM stocks in DMSO and then added at a final concentration of 100 μM. Wandering late third instar larvae were dissected for immunostaining or western analysis.
Statistical analyses
All statistical comparisons were performed using GraphPad InStat 5 software. P-values were calculated by one-way ANOVA. Comparisons were made between a specific genotype and the wild-type control (asterisks are located above a column) or between two specific genotypes (asterisks are located above a bracket).
Supplementary Material
Acknowledgements
We thank M. Feany, R. J. Jiao, the Bloomington Stock Center and the Developmental Studies Hybridoma Bank, for fly stocks and antibodies. We are grateful to Drs Y. Xiong, L. Bao and Y.H. Chen for discussions.
Footnotes
This article is part of a special subject collection ‘Neurodegeneration: from Models to Mechanisms to Therapies’, which was launched in a dedicated issue guest edited by Aaron Gitler and James Shorter. See related articles in this collection at http://dmm.biologists.org/collection/neurodegenerative-disorders.
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: S.J., Y.Q.Z.; Methodology: C.-X.M., X.W.; Formal analysis: C.-X.M., X.W.; Investigation: C.-X.M., X.W., S.J.; Writing - original draft: C.-X.M., S.J., Y.Q.Z.; Writing - review & editing: C.-X.M., S.J., Y.Q.Z.; Supervision: S.J., Y.Q.Z.; Funding acquisition: S.J., Y.Q.Z.
Funding
This work was supported by the National Natural Science Foundation of China [81070909 and 31230045 to S.J.; 31110103907 and 31490590 to Y.Q.Z.; 31500839 to C.M.], the Ministry of Science and Technology of the People's Republic of China [2014CB942803 and 2016YFA0501000], and the Chinese Academy of Sciences [XDB02020400].
Supplementary information
Supplementary information available online at http://dmm.biologists.org/lookup/doi/10.1242/dmm.028316.supplemental
References
- Aguilar A., Becker L., Tedeschi T., Heller S., Iomini C. and Nachury M. V. (2014). Alpha-tubulin K40 acetylation is required for contact inhibition of proliferation and cell-substrate adhesion. Mol. Biol. Cell 25, 1854-1866. 10.1091/mbc.E13-10-0609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akella J. S., Wloga D., Kim J., Starostina N. G., Lyons-Abbott S., Morrissette N. S., Dougan S. T., Kipreos E. T. and Gaertig J. (2010). MEC-17 is an alpha-tubulin acetyltransferase. Nature 467, 218-222. 10.1038/nature09324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C., Lee V. M.-Y. and Trojanowski J. Q. (2007). Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 8, 663-672. 10.1038/nrn2194 [DOI] [PubMed] [Google Scholar]
- Boyault C., Gilquin B., Zhang Y., Rybin V., Garman E., Meyer-Klaucke W., Matthias P., Müller C. W. and Khochbin S. (2006). HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J. 25, 3357-3366. 10.1038/sj.emboj.7601210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brush M. H., Guardiola A., Connor J. H., Yao T.-P. and Shenolikar S. (2004). Deactylase inhibitors disrupt cellular complexes containing protein phosphatases and deacetylases. J. Biol. Chem. 279, 7685-7691. 10.1074/jbc.M310997200 [DOI] [PubMed] [Google Scholar]
- Butler K. V., Kalin J., Brochier C., Vistoli G., Langley B. and Kozikowski A. P. (2010). Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 132, 10842-10846. 10.1021/ja102758v [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee F. C., Mudher A., Cuttle M. F., Newman T. A., MacKay D., Lovestone S. and Shepherd D. (2005). Over-expression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Neurobiol. Dis. 20, 918-928. 10.1016/j.nbd.2005.05.029 [DOI] [PubMed] [Google Scholar]
- Chen C.-S., Weng S.-C., Tseng P.-H., Lin H.-P. and Chen C.-S. (2005). Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J. Biol. Chem. 280, 38879-38887. 10.1074/jbc.M505733200 [DOI] [PubMed] [Google Scholar]
- Chevalier-Larsen E. and Holzbaur E. L. F. (2006). Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta 1762, 1094-1108. 10.1016/j.bbadis.2006.04.002 [DOI] [PubMed] [Google Scholar]
- Chu C.-W., Hou F., Zhang J., Phu L., Loktev A. V., Kirkpatrick D. S., Jackson P. K., Zhao Y. and Zou H. (2011). A novel acetylation of beta-tubulin by San modulates microtubule polymerization via down-regulating tubulin incorporation. Mol. Biol. Cell 22, 448-456. 10.1091/mbc.E10-03-0203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C., Gendron T. F., Scheffel K., Carlomagno Y., Dunmore J., DeTure M. and Petrucelli L. (2012). Loss of HDAC6, a novel CHIP substrate, alleviates abnormal tau accumulation. Hum. Mol. Genet. 21, 2936-2945. 10.1093/hmg/dds125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C., Carlomagno Y., Gendron T. F., Dunmore J., Scheffel K., Stetler C., Davis M., Dickson D., Jarpe M., DeTure M. et al. (2014). Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum. Mol. Genet. 23, 104-116. 10.1093/hmg/ddt402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creppe C., Malinouskaya L., Volvert M.-L., Gillard M., Close P., Malaise O., Laguesse S., Cornez I., Rahmouni S., Ormenese S. et al. (2009). Elongator controls the migration and differentiation of cortical neurons through acetylation of alpha-tubulin. Cell 136, 551-564. 10.1016/j.cell.2008.11.043 [DOI] [PubMed] [Google Scholar]
- Destaing O., Saltel F., Gilquin B., Chabadel A., Khochbin S., Ory S. and Jurdic P. (2005). A novel Rho-mDia2-HDAC6 pathway controls podosome patterning through microtubule acetylation in osteoclasts. J. Cell Sci. 118, 2901-2911. 10.1242/jcs.02425 [DOI] [PubMed] [Google Scholar]
- Ding H., Dolan P. J. and Johnson G. V. W. (2008). Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 106, 2119-2130. 10.1111/j.1471-4159.2008.05564.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit R., Ross J. L., Goldman Y. E. and Holzbaur E. L. F. (2008). Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086-1089. 10.1126/science.1152993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du G., Liu X., Chen X., Song M., Yan Y., Jiao R. and Wang C.-C. (2010). Drosophila Histone Deacetylase 6 Protects Dopaminergic Neurons against alpha-Synuclein Toxicity by Promoting Inclusion Formation. Mol. Biol. Cell 21, 2128-2137. 10.1091/mbc.E10-03-0200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan J. E. and Goldstein L. S. B. (2006). The genetics of axonal transport and axonal transport disorders. PLoS Genet. 2, e124 10.1371/journal.pgen.0020124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaertig J., Cruz M. A., Bowen J., Gu L., Pennock D. G. and Gorovsky M. A. (1995). Acetylation of lysine 40 in alpha-tubulin is not essential in tetrahymena thermophila. J. Cell Biol. 129, 1301-1310. 10.1083/jcb.129.5.1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Isla T., Price J. L., McKeel D. W. Jr, Morris J. C., Growdon J. H. and Hyman B. T. (1996). Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J. Neurosci. 16, 4491-4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratz S. J., Cummings A. M., Nguyen J. N., Hamm D. C., Donohue L. K., Harrison M. M., Wildonger J. and O'Connor-Giles K. M. (2013). Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics 194, 1029-1035. 10.1534/genetics.113.152710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggarty S. J., Koeller K. M., Wong J. C., Grozinger C. M. and Schreiber S. L. (2003). Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 100, 4389-4394. 10.1073/pnas.0430973100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J. L. and Cowan N. J. (1985). Structural features and restricted expression of a human alpha-tubulin gene. Nucleic Acids Res. 13, 207-223. 10.1093/nar/13.1.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond J. W., Huang C.-F., Kaech S., Jacobson C., Banker G. and Verhey K. J. (2010). Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol. Biol. Cell 21, 572-583. 10.1091/mbc.E09-01-0044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempen B. and Brion J.-P. (1996). Reduction of acetylated alpha-tubulin immunoreactivity in neurofibrillary tangle-bearing neurons in Alzheimer's disease. J. Neuropathol. Exp. Neurol. 55, 964-972. 10.1097/00005072-199609000-00003 [DOI] [PubMed] [Google Scholar]
- Huang Y. and Mucke L. (2012). Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204-1222. 10.1016/j.cell.2012.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbert C., Guardiola A., Shao R., Kawaguchi Y., Ito A., Nixon A., Yoshida M., Wang X.-F. and Yao T.-P. (2002). HDAC6 is a microtubule-associated deacetylase. Nature 417, 455-458. 10.1038/417455a [DOI] [PubMed] [Google Scholar]
- Janke C. and Bulinski J. C. (2011). Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat. Rev. Mol. Cell Biol. 12, 773-786. 10.1038/nrm3227 [DOI] [PubMed] [Google Scholar]
- Jin S., Pan L., Liu Z., Wang Q., Xu Z. and Zhang Y. Q. (2009). Drosophila Tubulin-specific chaperone E functions at neuromuscular synapses and is required for microtubule network formation. Development 136, 1571-1581. 10.1242/dev.029983 [DOI] [PubMed] [Google Scholar]
- Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A. and Charpentier E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816-821. 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaluza D., Kroll J., Gesierich S., Yao T.-P., Boon R. A., Hergenreider E., Tjwa M., Rössig L., Seto E., Augustin H. G. et al. (2011). Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. EMBO J. 30, 4142-4156. 10.1038/emboj.2011.298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keays D. A., Tian G., Poirier K., Huang G.-J., Siebold C., Cleak J., Oliver P. L., Fray M., Harvey R. J., Molnár Z. et al. (2007). Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell 128, 45-57. 10.1016/j.cell.2006.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. Y., Woo E. M., Chong Y. T. E., Homenko D. R. and Kraus W. L. (2006). Acetylation of estrogen receptor alpha by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol. Endocrinol. 20, 1479-1493. 10.1210/me.2005-0531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozminski K. G., Diener D. R. and Rosenbaum J. L. (1993). High level expression of nonacetylatable alpha-tubulin in Chlamydomonas reinhardtii. Cell Motil. Cytoskeleton 25, 158-170. 10.1002/cm.970250205 [DOI] [PubMed] [Google Scholar]
- Li L., Wei D., Wang Q., Pan J., Liu R., Zhang X. and Bao L. (2012). MEC-17 deficiency leads to reduced alpha-tubulin acetylation and impaired migration of cortical neurons. J. Neurosci. 32, 12673-12683. 10.1523/JNEUROSCI.0016-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N., Xiong Y., Li S., Ren Y., He Q., Gao S., Zhou J. and Shui W. (2015). New HDAC6-mediated deacetylation sites of tubulin in the mouse brain identified by quantitative mass spectrometry. Sci. Rep. 5, 16869 10.1038/srep16869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow E.-M., Thies E., Trinczek B., Biernat J. and Mandelkow E. (2004). MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. J. Cell Biol. 167, 99-110. 10.1083/jcb.200401085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C.-X., Xiong Y., Xiong Z., Wang Q., Zhang Y. Q. and Jin S. (2014). Microtubule-severing protein Katanin regulates neuromuscular junction development and dendritic elaboration in Drosophila. Development 141, 1064-1074. 10.1242/dev.097774 [DOI] [PubMed] [Google Scholar]
- Maruta H., Greer K. and Rosenbaum J. L. (1986). The acetylation of alpha-tubulin and its relationship to the assembly and disassembly of microtubules. J. Cell. Biol. 103, 571-579. 10.1083/jcb.103.2.571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews K. A., Miller D. F. B. and Kaufman T. C. (1989). Developmental distribution of RNA and protein products of the Drosophila alpha-tubulin gene family. Dev. Biol. 132, 45-61. 10.1016/0012-1606(89)90203-0 [DOI] [PubMed] [Google Scholar]
- Nogales E., Wolf S. G. and Downing K. H. (1998). Structure of the αβ tubulin dimer by electron crystallography. Nature 391, 199-203. 10.1038/34465 [DOI] [PubMed] [Google Scholar]
- North B. J., Marshall B. L., Borra M. T., Denu J. M. and Verdin E. (2003). The human Sir2 ortholog, SIRT2, is an NAD(+)-dependent tubulin deacetylase. Mol. Cell 11, 437-444. 10.1016/S1097-2765(03)00038-8 [DOI] [PubMed] [Google Scholar]
- Port F., Chen H.-M., Lee T. and Bullock S. L. (2014). Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila. Proc. Natl. Acad. Sci. USA 111, E2967-E2976. 10.1073/pnas.1405500111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed N. A., Cai D., Blasius T. L., Jih G. T., Meyhofer E., Gaertig J. and Verhey K. J. (2006). Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 16, 2166-2172. 10.1016/j.cub.2006.09.014 [DOI] [PubMed] [Google Scholar]
- Santo L., Hideshima T., Kung A. L., Tseng J.-C., Tamang D., Yang M., Jarpe M., van Duzer J. H., Mazitschek R., Ogier W. C. et al. (2012). Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 119, 2579-2589. 10.1182/blood-2011-10-387365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneurin-Berny D., Verdel A., Curtet S., Lemercier C., Garin J., Rousseaux S. and Khochbin S. (2001). Identification of components of the murine histone deacetylase 6 complex: Link between acetylation and ubiquitination signaling pathways. Mol. Cell Biol. 21, 8035-8044. 10.1128/MCB.21.23.8035-8044.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y. Y. and Brady S. T. (2015). Post-translational modifications of tubulin: pathways to functional diversity of microtubules. Trends Cell Biol. 25, 125-136. 10.1016/j.tcb.2014.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudo H. and Baas P. W. (2010). Acetylation of microtubules influences their sensitivity to severing by katanin in neurons and fibroblasts. J. Neurosci. 30, 7215-7226. 10.1523/JNEUROSCI.0048-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tala, Sun X., Chen J., Zhang L., Liu N., Zhou J., Li D. and Liu M. (2014). Microtubule stabilization by mdp3 is partially attributed to its modulation of HDAC6 in addition to its association with tubulin and microtubules. PLoS ONE 9, e90932 10.1371/journal.pone.0090932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theurkauf W. E., Baum H., Bo J. Y. and Wensink P. C. (1986). Tissue-specific and constitutive alpha-tubulin genes of Drosophila melanogaster code for structurally distinct proteins. Proc. Natl. Acad. Sci. USA 83, 8477-8481. 10.1073/pnas.83.22.8477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thies E. and Mandelkow E.-M. (2007). Missorting of Tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J. Neurosci. 27, 2896-2907. 10.1523/JNEUROSCI.4674-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela-Fernández A., Cabrero J. R., Serrador J. M. and Sánchez-Madrid F. (2008). HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 18, 291-297. 10.1016/j.tcb.2008.04.003 [DOI] [PubMed] [Google Scholar]
- Walter W. J., Beránek V., Fischermeier E. and Diez S. (2012). Tubulin acetylation alone does not affect kinesin-1 velocity and run length in vitro. PLoS ONE 7, e42218 10.1371/journal.pone.0042218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y., Zhao K., Wu J., Xu Z., Jin S. and Zhang Y. Q. (2013). HDAC6 mutations rescue human tau-induced microtubule defects in Drosophila. Proc. Natl. Acad. Sci. USA 110, 4604-4609. 10.1073/pnas.1207586110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W., Qiang L., Solowska J. M., Karabay A., Korulu S. and Baas P. W. (2008). The microtubule-severing proteins spastin and katanin participate differently in the formation of axonal branches. Mol. Biol. Cell 19, 1485-1498. 10.1091/mbc.E07-09-0878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Maiti A., Shively S., Lakhani F., McDonald-Jones G., Bruce J., Lee E. B., Xie S. X., Joyce S., Li C. et al. (2005). Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc. Natl. Acad. Sci. USA 102, 227-231. 10.1073/pnas.0406361102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Liu C., Wu J., Tao J. J., Sui X. L., Yao Z. G., Xu Y. F., Huang L., Zhu H., Sheng S. L. et al. (2014). Tubastatin A/ACY-1215 improves cognition in Alzheimer's disease transgenic mice. J. Alzheimers Dis. 41, 1193-1205. 10.3233/JAD-140066 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.