

ABSTRACT
The domain within the otic vesicle (OV) known as the neurosensory domain (NSD), contains cells that will give rise to the hair and support cells of the otic sensory organs, as well as the neurons that form the cochleovestibular ganglion (CVG). The molecular dynamics that occur at the NSD boundary relative to adjacent OV cells is not well defined. The Tbx1 transcription factor gene expression pattern is complementary to the NSD, and inactivation results in expansion of the NSD and expression of the Notch ligand, Jag1 mapping, in part of the NSD. To shed light on the role of Jag1 in NSD development, as well as to test whether Tbx1 and Jag1 might genetically interact to regulate this process, we inactivated Jag1 within the Tbx1 expression domain using a knock-in Tbx1Cre allele. We observed an enlarged neurogenic domain marked by a synergistic increase in expression of NeuroD and other proneural transcription factor genes in double Tbx1 and Jag1 conditional loss-of-function embryos. We noted that neuroblasts preferentially expanded across the medial-lateral axis and that an increase in cell proliferation could not account for this expansion, suggesting that there was a change in cell fate. We also found that inactivation of Jag1 with Tbx1Cre resulted in failed development of the cristae and semicircular canals, as well as notably fewer hair cells in the ventral epithelium of the inner ear rudiment when inactivated on a Tbx1 null background, compared to Tbx1Cre/− mutant embryos. We propose that loss of expression of Tbx1 and Jag1 within the Tbx1 expression domain tips the balance of cell fates in the NSD, resulting in an overproduction of neuroblasts at the expense of non-neural cells within the OV.
KEY WORDS: Otic vesicle, Inner ear, Notch, Tbx1, Jag1, Neurosensory
Summary: Normal dosages of Tbx1 and Jag1 are required to maintain a proper balance of cell types within the neurosensory domain of the otic vesicle to form the inner ear.
INTRODUCTION
The inner ear forms from the otic vesicle (OV), which is a closed, continuous epithelium consisting of cells that will differentiate to form specialized cell types (Bok et al., 2007). The T-box transcription factor gene, Tbx1, plays a major role in the OV for inner ear morphogenesis. Tbx1 is expressed in the posterior-lateral OV, mostly complementary to the region that forms the cochleovestibular ganglion (CVG) (Ma et al., 1998; Raft et al., 2004). When Tbx1 is inactivated in the mouse, the CVG is duplicated in size, marked by an expansion in expression of Atonal-related basic helix-loop-helix (bHLH) proneural transcription factors Neurogenin1 (Ngn1; also known as Neurog1) and NeuroD (Neurod1), in the neurogenic domain (Raft et al., 2004). In addition, the cochlea and vestibular system do not develop (Vitelli et al., 2003; Raft et al., 2004). However, failure of otic epithelial cells to proliferate normally in Tbx1 null mice may render it difficult to identify the range of its specific requirements (Xu et al., 2007). Overexpression (Funke et al., 2001) or constitutive expression of Tbx1 (Freyer et al., 2013), on the other hand, has the opposite effect for neurogenesis, meaning that the neurogenic domain is reduced in area (Freyer et al., 2013). In addition to this defect, the utricle and the saccule, containing hair cells derived from prosensory patches during early development, do not form. This suggests that Tbx1 may restrict neurogenesis and sensorigenesis. We are interested in understanding the mechanism for this, so we considered other genes that are required for these processes.
The Notch ligand, Jagged1 (Jag1), and group B Sox SRY-related HMG box transcription factor (Sox2) are broadly co-expressed in the ventral region of the OV. Within these expression domains, exists a subdomain known as the neurosensory domain (NSD), the cells of which have the potential to differentiate into neurons, hair cells or support cells (Adam et al., 1998; Cole et al., 2000; Daudet et al., 2007; Fekete and Wu, 2002; Kiernan et al., 2006, 2005; Morsli et al., 1998; Neves et al., 2011; Pan et al., 2010; Raft and Groves, 2014). Mouse genetic studies show that inactivation of Jag1 results in failed development of prosensory patches early in development, and a reduction in the size of the neurogenic zone of the CVG (Kiernan et al., 2006; Pan et al., 2010), suggesting that it may be required for neurosensory competence or maintenance. Sox2 expression is maintained by Jag1 and is also required for neurosensory specification (Kiernan et al., 2006; Neves et al., 2011; Pan et al., 2010). Tbx1 appears to function in a complementary manner to Jag1.
Little is known about the cellular and molecular dynamic requirements of cells in the NSD for proper inner ear development. Notch pathway genes have been implicated in tissue boundary formation during development in various contexts. Since loss of Tbx1 results in an expansion of the CVG and inactivation of Jag1 results in a somewhat smaller CVG (Kiernan et al., 2006; Pan et al., 2010), we wanted to test whether the two genes might act antagonistically in the NSD for inner ear development. In this study, we utilized Tbx1 (Huynh et al., 2007) and Jag1 (Kiernan et al., 2006) conditional loss-of-function mouse mutants to evaluate their functions in the NSD, and found a surprising novel function in neurogenesis.
RESULTS
Expression of Tbx1 and Jag1 around the NSD
Both Jag1 and Sox2 are markers of the NSD-containing cells required for neurogenesis and sensorigenesis (Raft and Groves, 2014). We performed immunofluorescence studies to determine where and when Tbx1, Jag1 and Sox2 are expressed with respect to each other and to the NSD (Fig. 1). At embryonic day (E) 9.5, Tbx1 was broadly expressed in the posterolateral wall of the OV (Fig. 1A, b-c′; Fig. S1). As expected, Jag1and Sox2 proteins were expressed in a similar pattern to one another. Expression of the two was complementary to Tbx1 in the anterior domain, while there was some overlap in expression with Tbx1 in the posterior lateral OV (Fig. 1A, a-c′). By E10.5, expression of all three genes became more restricted within their respective complementary domains, such that Jag1 and Sox2 had less overlap in expression with Tbx1 (Fig. 1A, d-f; Fig. S1). The complementary expression patterns between Tbx1 and Sox2/Jag1 and known loss-of-function phenotypes (Raft et al., 2004; Pan et al., 2010; Puligilla et al., 2010) suggest that there might be an antagonistic relationship between them. We decided to focus on evaluating the relationship between Tbx1 and Jag1 in more detail, since there appeared to be a sharper border between Jag1 and Tbx1, than Sox2 and Tbx1, expression (Fig. 1A), suggesting that the two may interact genetically to form an important boundary within the OV. Another reason to focus our study on Jag1 is that Jag1 has been shown to function upstream of Sox2 during inner ear development (Pan et al., 2010; Neves et al., 2011). Finally, Notch1 has been shown to function downstream of Tbx1 during neurogenesis in the OV (Xu et al., 2007), providing additional rationale to further study its ligand, Jag1, which had never been linked to Tbx1 up until this point.
Fig. 1.

NSD gene expression in wild-type and Tbx1 null embryos. (A) Immunofluorescence was performed using antibodies to Jag1, Tbx1 and Sox2 on tissue sections from mouse embryos at E9.5 (a-c′) and E10.5 (d-f′). Jag1 and Sox2 are expressed broadly in the otic epithelium at E9.5, overlapping with Tbx1 expression in the PVL OV, but are largely complementary. By E10.5, Jag1 and Sox2 expression is mainly restricted to the anterior ventral lateral and posterior medial OV, with less overlap with Tbx1 expression, which is restricted to the posterior dorsal lateral OV. (B) Immunofluorescence was performed using antibodies to Jag1, Tbx1, Isl1 and NeuroD, as indicated (g-i′). Jag1 is expressed in the NSD, where neuroblasts expressing both Isl1 (g) and NeuroD (h) are derived. Tbx1 is expressed complementary to Jag1 (i,i′), as well as NeuroD (h), at the same border. The diagram below depicts the planes of sections used. (C) Schematic summarizing the wild-type expression of the genes examined in B, with respect to domains. (D) Immunofluorescence for Jag1 (red) on transverse sections in embryos at E9.5 and E10.5 was performed. The sections progress from the anterior to posterior domains of the OV. At E9.5, Jag1 expression expands in both the medial and lateral domain (white brackets), while at E10.5, Jag1 expression expands in the lateral domain (white brackets). (E) Immunofluorescence for Jag1 in Tbx1 GOF and littermate control embryos showing a shift in the expression from lateral (white arrow) to medial (yellow arrow) in anterior sections (p′,q′).
At this point, in order to evaluate Tbx1 and Jag1 expression with respect to regions of neurogenesis, the expression of Jag1, Tbx1, Isl1, and NeuroD were compared (Fig. 1B). Jag1 was expressed in the region where neuroblasts expressing both Isl1 (Fig. 1B, g) and NeuroD (Fig. 1B, h) are derived, while Tbx1 was expressed in a largely complementary manner to these genes (Fig. 1B, i,i′). This indicates that Tbx1 is excluded from the neurogenic domain within the NSD, marked by overlapping expression of Jag1 and NeuroD. A schematic of expression patterns is shown in Fig. 1C, showing the expression patterns of these genes, with respect to one another and the NSD border.
We also wanted to determine what effect loss or gain of Tbx1 would have on Jag1 expression and, indirectly, the NSD. At E9.5, Jag1 expression expanded anteriorly and posteriorly on the medial and lateral walls of the OV when Tbx1 was globally inactivated (Fig. 1D, j′,k′,l′). By E10.5, Jag1 expression in wild-type embryos became restricted mostly to the posteromedial OV, with small regions of expression in the anterolateral wall of the OV (Fig. 1D, m,n,o). When Tbx1 was inactivated, Jag1 expression again expanded across the medial-lateral axis of the posterior OV (Fig. 1D, m′,n′,o′). The expansion in Jag1 expression into the posterior OV correlated with the region of ectopic neurogenesis in Tbx1−/− OVs that has been previously described (Fig. 1B) (Raft et al., 2004; Xu et al., 2007). We also examined the expression of Jag1 in Tbx1 GOF (Pax2-Cre/+; Tbx1-GFP/+) (Freyer et al., 2013) embryos (Fig. 1E), and found that the expression domain shifted from the anteroventral lateral to anteroventral medial OV (Fig. 1E, p,p′). In control embryos, Jag1 is expressed in the medial OV in more posterior sections (Fig. 1E, q,r), whereas in in anterior sections, Jag1 expression is exclusively lateral (Fig. 1E, p). In contrast, in Tbx1 GOF embryos, Jag1 expression is almost exclusively medial throughout the OV, with significantly more coverage of the medial wall of the OV compared to controls, particularly in anterior sections (Fig. 1E, p′,q′,r′). These findings indicate a change in the position of the medial-lateral border and shift of the NSD. Of note, cells within the anteroventral lateral region of the OV contribute to the CVG and utricle (Fekete and Wu, 2002), the cells of which are derived from a shared lineage within the NSD (Raft et al., 2007) We further asked whether there was mutual antagonism between Tbx1 and Jag1, by examining Tbx1 expression in Jag1 null mice in the OV. There was some decrease in the area of Tbx1 expression, although this loss is likely secondary to the reduction in size of the OV (Fig. S2). The position of the expression domain with respect to the OV appeared unaltered. These findings suggest that there is a unidirectional pathway by which Tbx1 restricts the Jag1 expression and the size and location of the NSD.
Next, we performed cell lineage tracing of Tbx1 using a Tbx1Cre mouse line (Huynh et al., 2007) crossed with a GFP reporter line (Batista-Brito et al., 2009) in order to map the overlap between the Tbx1 lineage and Jag1 expression to know where Jag1 might be getting inactivated. We found that Jag1 partially colocalized with the Tbx1 cell lineage, marked by GFP in mostly the posterior ventral lateral (PVL) OV at E10 (Fig. 2A), but broadened to include the posterior medial domain at E10.5 (Fig. 2B). There was less overlap in the anteroventral region, adjacent to the CVG (Fig. 3A-C). On a separate note, there is another distinct Tbx1 lineage within the mesoderm surrounding the OV as well (Fig. 2B). The OV from the E10.5 embryo shown in Fig. 2B was used for serial section 3D reconstruction (Fig. 2C). The purpose was to better visualize the areas of overlap between Jag1 and the Tbx1 cell lineage. This clarified that Jag1 expression occurred mostly at the border of the Tbx1 lineage, with overlap occurring primarily in the PVL and posterior ventral medial (PVM) domains, which contribute to the lateral SCC and, in part, the saccule and cochlea, respectively (Fig. 2C) (Fekete and Wu, 2002). A very clear border between the Tbx1 lineage and Jag1 expression can been seen at the dorsal-ventral (white arrow) and medial-lateral (black arrow) axis (Fig. 2C). We also performed Tbx1 lineage tracing when Tbx1 was homozygously inactivated, in conjunction with antibody staining for NeuroD. Interestingly, this revealed that while the ectopic CVG that forms in Tbx1 null embryos is composed largely of cells derived from the Tbx1 cell lineage, a number of neuroblasts positive for NeuroD are GFP-negative (GFP−), suggesting they are not derived from the Tbx1 lineage and could indicate nonautonomous roles (Fig. S3). Previous findings that Jag1-Notch1 signaling plays a role in CVG formation (Pan et al., 2010) and loss of Tbx1 affects aNotch expression (Xu et al., 2007), provide a reasonable basis for speculating that Tbx1 may regulate CVG formation nonautonomously by affecting this cell signaling pathway. Another point worth noting is that it has been previously shown (and confirmed in this report) that the CVG and distal ganglia fuse in Tbx1 null embryos (Xu et al., 2007). Thus, one might speculate that GFP− cells from this experiment are derived from epibranchial ganglia; however, these authors also show contribution of the Tbx1 cell lineage to epibranchial ganglia, particularly the IXth ganglion, when one allele of Tbx1 is inactivated (Xu et al., 2007). Thus, some GFP+ cells in this experiment may also derive from epibranchial ganglia.
Fig. 2.

Tbx1 lineage is largely complementary to the NSD. (A,B) Immunofluorescence for Jag1 (red) and GFP (green) on transverse sections of a Tbx1Cre/+;CMV-GFP flox/+ embryo at E10 and E10.5, showing largely complementary expression between Jag1 and the Tbx1 cell lineage in more anterior sections of the OV, but some colocalization (white arrows) in more posterior ventral regions. (C) 3D reconstruction of serial sections of the E10.5, Tbx1Cre/+;CMV-GFP flox/+ embryo shown in Fig. 3C, showing the position of expression of the Tbx1 lineage and Jag1 as well as co-expression of both genes. The Tbx1 cell lineage is shown in pink, Jag1 protein expression is shown in yellow, and the overlap between the two is shown in orange. A dorsal-ventral (white arrow) and medial-lateral (black arrow) border between the Tbx1 lineage and Jag1 expression is shown.
Fig. 3.

The CVG is significantly smaller in size in Jag1−/− embryos compared to wild-type controls at E10.5. (A) Immunofluorescence using a Tuj1 antibody was performed to mark the CVG (traced with a white dashed line). (B) Schematic depicting the phenotype in A. (C) The average volume of the CVG marked by Tuj1 expression was measured and averaged per ear per genotype. Average OV volume was also calculated. Both CVG and OV volume in Jag1−/− embryos were significantly lower than in Jag1+/+ embryos. Average neural tube length was also calculated to serve as a control for overall embryo size; there was no significant difference between genotypes. (Jag1+/+ embryos, n=3; Jag1−/− embryos, n=4). ***P<0.001; *P<0.05. Data are mean±s.e.m. Values were normalized to those of wild-type levels.
To further delineate Tbx1 and Jag1 domains with respect to the NSD border, we also performed lineage tracing using a Pax3Cre (Paired Box Gene 3) mouse line crossed with GFP reporter mice to mark the neural crest cell lineage. This lineage is present in the neurosensory regions of the inner ear, except for the cristae (Freyer et al., 2011). Immunofluorescence on alternating serial sections at E9.5 demonstrated that Jag1 was expressed throughout much of the Pax3 lineage (Fig. S4, top; white brackets), while Tbx1 was complementary to this (Fig. S4, bottom; white arrowheads). When taken together, these data show that Tbx1 expression borders and restricts Jag1 expression, primarily in regions within the NSD that give rise to the utricle and CVG.
Inactivation of Jag1 results in a significant reduction in the CVG size
We evaluated E10.5 Jag1−/− versus Jag1+/+ embryos using an anti-Tuj1 antibody, which marks differentiated neurons. We measured the area around the Tuj1 staining marking the CVG and found that there was a significant decrease in the average area of the CVG in Jag1 null embryos compared to wild-type controls, to ∼50% of wild-type levels (P<0.001) (Fig. 3). This is a more drastic reduction than previously reported in Jag1 conditional loss-of-function embryos (Pan et al., 2010). We also measured the area of the OV in these embryos and found that it was reduced in size to a similar degree in Jag1 null embryos (P<0.05) (Fig. 3). To ensure that differences were not due to changes in the overall embryo size, we also measured the length of the neural tube in these embryos and found that there was no significant difference on average (Fig. 3).
Dual inactivation of Tbx1 and Jag1 causes a synergistic expansion of neuroblasts in the absence of increased cell proliferation
The above data indicate that Tbx1 and Jag1 are expressed in a largely complementary manner in the OV, but there is some overlap in expression in NSD border cells as well as in the posterior OV. Further, loss of Jag1 results in a 60% reduction in the size of the CVG, opposite to the phenotype resulting from a loss of Tbx1. To shed light on the requirement of Jag1 at the NSD border, as well as to test whether Tbx1 and Jag1 might genetically interact, we inactivated Jag1 within the Tbx1 expression domain using a knock-in Tbx1Cre allele.
We first performed in situ hybridization using a Jag1 RNA probe showing that Jag1 expression was reduced in posterior and, to a lesser extent, anterior domains of the OV at E10.5 in Tbx1Cre/+;Jag1flox/− as well as Tbx1Cre/−;Jag1floxflox mutant embryos, when compared to Tbx1Cre/+;Jag1flox/+ and Tbx1Cre/− littermate control embryos, respectively (Fig. S5). These findings confirmed that Jag1 was inactivated within the Tbx1 lineage as expected. Note that Jag1 expression was also reduced in the pharyngeal apparatus, where the two genes also overlap in expression, but not in regions where they are not co-expressed, such as the forelimb (Fig. S4). It is also important to note that resulting Tbx1Cre/+;Jag1flox/+ conditional double heterozygous embryos also have reduced dosage of Tbx1 since the Cre is knocked into the endogenous Tbx1 locus, producing a functionally null allele (Huynh et al., 2007).
To test whether CVG development would be affected in resulting double mutants, RNA in situ hybridization was performed for NeuroD at E10 and E10.5. At E10, Tbx1Cre/+ embryos exhibited a small, but visible, increase in NeuroD expression along the anterior-posterior (A-P) axis of the ventral OV, compared to wild-type controls (Fig. 4A). Double heterozygous Tbx1Cre/+;Jag1flox/+ embryos at E10, compared to Tbx1Cre/+ embryos, exhibited a greater expansion in NeuroD expression. This expansion was along the dorsal-ventral (D-V) axis of the anterior OV, with the CVG rudiment appearing larger (Fig. 4A). We then inactivated both alleles of Jag1 by generating Tbx1Cre/+;Jag1flox/− embryos. These embryos exhibited an even greater expansion in NeuroD expression along the D-V axis and posterior ventral domain of the OV. A large increase of NeuroD expression occurred in Tbx1Cre/− embryos. In these embryos, an ectopic ganglion formed in the posterior domain of the OV, at the same stages (Fig. 4A,B). Further, the anterior and posterior ectopic CVG became fused, along with cranial ganglia VII and IX (Fig. 4A,B). This phenotype has previously been described (Raft et al., 2004; Xu et al., 2007) and is shown for the sake of comparison. When one or both copies of Jag1 were inactivated on a Tbx1Cre/− background, NeuroD expression expanded to an even greater degree than in Tbx1Cre/− embryos at both E10 and E10.5 (Fig. 4A,B). Histological analysis was performed of whole-mount in situ hybridization experiments on both E10 and E10.5 embryos, though only sections at E10.5 are shown (Fig. 4B). The histological sections further illustrated the expansion of the NeuroD domain, as well as expansion of distal IXth and Xth ganglia, and fusion of these three ganglia with the Vth ganglion, in the case of the double homozygous mutant embryos (Fig. 4B, yellow arrow). Further, the area of NeuroD expression was quantified in tissue sections for both E10 and E10.5 mutant embryos (Fig. S6A). Indeed, we found that the area of expression was significantly greater in double mutant embryos in than in Tbx1 single mutant embryos at both stages. This expansion appeared to be dose-dependent, as decreasing dosage of Tbx1 and Jag1 correlated with increasing expression levels of NeuroD.
Fig. 4.

Inactivation of Tbx1 and Jag1 with Tbx1Cre results in expanded proneural gene expression. (A,B) Whole-mount in situ hybridization was performed on genotypes shown, using an antisense probe for NeuroD at E10 (A) and E10.5 (B). There is a synergistic increase in expression in the CVG (cranial ganglion VIII) as Tbx1 and Jag1 dosage decreases. There is a similar increase in expression in the other cranial ganglia as well (VII, IX, X), shown in both whole mounts and sections. Fusion of all the ganglia with the Vth ganglion occurs in Tbx1Cre/−;Jag1flox/flox mutants, indicated by the yellow arrow. Adjacent is a schematic depicting the phenotype observed in Tbx1 single and Tbx1;Jag1 double mutants. (C) Immunofluorescence on transverse sections with a NeuroD antibody. Both the CVG and OV epithelium are outlined with a white dashed line. The mean total numbers of NeuroD+ cells in the OV epithelium alone (D) and the OV epithelium in combination with the CVG (E) were significantly greater in double mutant OVs as compared to Tbx1Cre/− OVs. Note: three embryos and six OVs per genotype were used for each group. Embryos were stage-matched at 27ss. Data are mean±s.e.m. **P<0.005; ***P<0.001.
Because whole-mount in situ hybridization is somewhat limited in the resolution of expression it provides, we also performed immunofluorescence studies on tissue sections using an antibody to NeuroD on double conditional null mutant embryos (Tbx1Cre/−;Jag1flox/+ and Tbx1Cre/−;Jag1flox/flox) and Tbx1Cre/− embryos (Fig. 4C). We used this to quantify the number of NeuroD positive (NeuroD+) cells within the OV epithelium alone, as well as the total number of NeuroD+ cells within the epithelium, plus cells that have delaminated to form the CVG (Fig. 4D,E) at E9.5. Because the epibranchial ganglia appear to fuse with the CVG in these mutants, it is important that we quantify cells within the OV epithelium alone, in order to isolate the otic epithelial-specific function of Jag1 and Tbx1 from potential function in other cranial placodes or ganglia. More distal ganglia can be easily distinguished as discrete patches of cells in many of the transverse sections; therefore, we are confident that a majority of the cells present in the VIIIth ganglion/CVG region are composed of otic epithelial cells. However, a fraction of those ventral-most cells may likely originate from the epibranchial ganglia, and so, two distinct quantifications are needed. The mean total number of NeuroD+ cells from both analyses were significantly greater in double mutant OVs as compared to Tbx1Cre/− OVs (Fig. 4D). We additionally quantified neuroblast numbers in the CVG in double conditional null mutant embryos (Tbx1Cre/−;Jag1flox/flox) and Tbx1Cre/− embryos using an antibody to Isl1 on serial sections at E10.5 (Fig. S6B). Similarly, we found that the mean total number of Isl1+ cells from both analyses were significantly greater in double mutants as compared to Tbx1Cre/− mutants (Fig. S6B). Interestingly, we noticed from both NeuroD and Isl1 immunofluorescence experiments that neuroblasts preferentially expanded into the lateral domain of the CVG in double mutants (Fig. 4C; Fig. S6B).
One explanation for the expansion of the NSD region in Tbx1;Jag1 double conditional null mutant embryos, is that there is an increase in cell proliferation of neural progenitor cells. To address this, we performed immunofluorescence on tissue sections at E9.5 using antibodies to phospho-Histone H3 (pHH3) to mark proliferating cells, and NeuroD to mark the neuroblasts (Fig. 5D). We calculated the mitotic index of proliferating neuroblasts from the OV by dividing the total number of proliferating cells (pHH3+) that colocalized with NeuroD by the total number of NeuroD+ cells, and these numbers were averaged per OV. We found that the mitotic indices were not higher, but in fact they were lower in both Tbx1Cre/−;Jag1flox/+ and Tbx1Cre/−;Jag1floxflox embryos compared to Tbx1Cre/− embryos (Fig. 5A,B). These differences were statistically significant: P<0.05 for both. We also compared the average number of pHH3+ cells that colocalized with NeuroD per ear and found consistent results (Fig. 5B). Thus, proliferation alone cannot account for the changes in neural precursor expression observed. We also noted that the OV was noticeably smaller in Tbx1;Jag1 double conditional null mutant embryos compared to Tbx1Cre/− embryos, and wondered if this could be due to a reduction in the number of non-neural proliferating cells. Indeed, we found that there was a significant reduction (P<0.05 for both double mutants) in the average number of proliferating cells that were negative for NeuroD expression within the OV at E9.5 (Fig. 5C). One possibility is that the smaller OV arises due to a change in cell fate from non-neural to neural and, subsequently, cells delaminated from the OV to form the CVG. These findings were somewhat surprising since inactivation of Jag1 is associated with a decrease in CVG size (Fig. 3) (Pan et al., 2010). Nonetheless, our findings show that the expansion of the neurogenic region observed in Tbx1;Jag1 double mutant embryos is not merely additive, nor synthetic, but rather it is due to a synergistic expansion of neuroblasts when both genes are lost at the NSD border. Taken together, these findings suggest that Tbx1 and Jag1 genetically interact at the NSD border to regulate neurosensory patterning across the medial-lateral axis of the OV, and that loss of both genes brings about an overproduction of neuroblasts contributing to the CVG that is not caused by increased cell proliferation.
Fig. 5.

An increased number of NeuroD+ cells is unaccompanied by changes in neuroblast proliferation in Tbx1Cre/−;Jag1flox/+ and Tbx1Cre/−;Jag1flox/flox mutants at E9.5. (A) Bar graphs plotting the mean mitotic index per ear within the neuroblast population as defined by NeuroD expression. Mitotic index was calculated by dividing the total number of proliferating cells (pHH3+) that colocalized with NeuroD by the total number of NeuroD+ cells. Values were markedly lower in Tbx1Cre/−;Jag1 flox/+ and Tbx1Cre/−;Jag1 flox/flox OVs and the differences reached statistical significance. (B) Bar graphs plotting the mean total number of pHH3+ cells in the OV within NeuroD+ domain per OV. Values were statistically significantly lower in Tbx1Cre/−;Jag1 flox/+ and Tbx1Cre/−;Jag1 flox/flox OVs compared to Tbx1Cre/− OVs. (C) Bar graphs plotting the mean total number of pHH3+ cells in the OV outside the NeuroD+ domain per OV. Values were statistically significantly lower in Tbx1Cre/−;Jag1 flox/+ and Tbx1Cre/−;Jag1 flox/flox OVs compared to Tbx1Cre/− OVs. (D) Examples of the dual immunofluorescence experiment using antibodies to NeuroD and pHH3, used to calculate the above data. There are visibly fewer proliferating cells throughout the OV of Tbx1Cre/−;Jag1 flox/+ embryos compared to Tbx1Cre/ embryos. The VIIIth ganglion/CVG is markedly larger in Tbx1Cre/−;Jag1 flox/+ mutants compared to Tbx1Cre/− mutants. A distinct portion of the fused distal epibranchial ganglia (IX/X) is also outlined below. Note: three embryos and six OVs per genotype were used for each group. Embryos were stage-matched at 27ss. Data are mean±s.e.m. *P<0.05; **P<0.005.
Later embryonic defects in the inner ear in Tbx1 and/or Jag1 mutant embryos
Paintfilling and histological analysis of embryos was performed to examine the morphology of the inner ear during later embryonic stages (Fig. 6; Fig. S7). We first compared Tbx1Cre;Jag1flox/flox and Tbx1Cre;Jag1flox/− embryos at E15.5 to littermate controls. We found that Tbx1Cre;Jag1flox/flox mutant embryos had similar inner malformations as observed when Jag1 was conditionally inactivated using the Foxg1Cre allele (Kiernan et al., 2006). The Foxg1Cre allele is frequently used to inactivate genes throughout the otic epithelium (Hébert and McConnell, 2000), presumably resulting in a broader inactivation of Jag1 than would a Tbx1Cre-mediated inactivation. The major phenotype observed was incomplete formation of the SCCs and ampullae, as well as a slightly smaller saccule (Fig. 6). Closer observation with histological analysis on E15.5 and adult inner ears (Fig. 6B, a-d′; Fig. S7), revealed that in Tbx1Cre;Jag1flox/flox or Tbx1Cre;Jag1flox/− mutants, all three cristae were missing and the saccule was shortened. The rest of the sensory structures developed normally. Hair cells in the sensory structures that formed appeared histologically normal (Fig. 6B, a-d′; Fig. S7). Previously published Foxg1Cre/+;Jag1flox/flox mutant embryos (Kiernan et al., 2006) shared the cristae and SCC phenotype; however, they also had abnormal cochleae, utricles and associated hair and support cells. The saccules were slightly smaller as in the Tbx1Cre-mediated mutant embryos, and hair cells were present. Tbx1Cre/+;Jag1flox/− mutants did not exhibit obvious abnormalities in the cochlea or utricles; however, we did not quantify hair or support cell numbers, which may be required in order to reveal subtle differences. We also examined the spiral/cochlear ganglia and vestibular ganglia, and noticed that while there was no significant difference in size in the spiral ganglia between mutants, the vestibular ganglia were much smaller in Tbx1Cre/+;Jag1flox/− mutants compared to Tbx1Cre/+;Jag1flox/+ mutants. Since all three cristae are not present in Tbx1Cre;Jag1flox/− mutants, it is likely that lack of hair cell innervation caused the vestibular ganglion to be either underdeveloped or partially degenerate (Fig. 6B, c,c′).
Fig. 6.

Gross morphological inner ear defects in Tbx1;Jag1 compound mutant embryos at E15.5. (A) Paintfilling of E15.5, Tbx1Cre/+;Jag1flox/+, Tbx1Cre/+;Jag1flox/flox, Tbx1Cre/−, Tbx1Cre/−;Jag1flox/+ and wild-type control inner ears. Tbx1Cre/+;Jagflox/+ ears are relatively normal but present with narrowing of the canals, particularly the anterior canal (white asterisk). Tbx1Cre/+;Jagflox/flox ears display incomplete development of the anterior semicircular canal (ac), posterior semicircular canal (pc) and lateral semicircular canal (lc), as well as their associated ampullae, respectively (aa, pa, la) (white asterisks). Tbx1Cre/− mutant ears have an enlarged endolymphatic duct (ed) (white tracing), while the rest of the inner ear does not fully develop and remains a vesicle with a posterior protrusion. Tbx1Cre/−;Jagflox/+ mutant ears are similar to Tbx1Cre/− ears but the vesicle is smaller and the protrusion is extended ventrally. (B) Transverse histological sections stained by H&E. All three cristae develop normally in Tbx1Cre/+;Jag1flox/+ embryos (a,b). The anterior crista (ac) and lateral crista (lc) attached to the utricle (u) are shown. An image of b at a higher magnification in the inset shows rows of hair cells (hc) and support cells (sc) that develop normally. All three cristae are missing from Tbx1Cre/+;Jag1flox/flox ears (a′,b′). Vestibular (vg) and spiral ganglia (sg) appear to form normally in Tbx1Cre/+;Jag1flox/+ embryos (c,d). The vg is noticeably smaller in Tbx1Cre/+;Jag1flox/flox embryos while the sg appears to develop normally (c′,d′). The inner ear rudiment (the vesicle ventral of the endolymphatic duct) in Tbx1Cre/− mutants overall are mostly lacking in hair cells, particularly in lateral regions (red arrowhead); however, the medial ventral wall of the vesicle is fairly densely populated with hair cells (black arrowhead) (e). An image of e at a higher magnification is shown in f. Tbx1Cre/−;Jag1flox/+ ears are similar to Tbx1Cre/− ears in that they are mostly lacking hair cells, particularly in lateral regions (red arrowhead) (e′). Medial regions are populated by some hair cells, but much more sparsely. An image of e′ at a higher magnification is shown in f′.
Next, we examined Tbx1Cre/−;Jag1flox/+ embryos compared with Tbx1Cre/− embryos at E15.5 in the same manner. Paintfilling of Tbx1Cre/− mutant embryos revealed the endolymphatic duct is greatly enlarged while the rest of the inner ear has been reduced to a vesicle with a small posterior protrusion on the medial side of the ear (Fig. 6A). This is a very similar, though slightly less severe phenotype, to that which has been published (Freyer et al., 2013). Paintfilling of Tbx1Cre/−;Jag1flox/+ inner ears revealed a similar phenotype to Tbx1Cre/− mutants, with an enlarged endolymphatic duct while none of the other structures form (Fig. 6A). The remaining cystic OV in compound mutants was smaller and different in shape compared to Tbx1Cre/− inner ears. There is also a protrusion from the OV, but it extended ventrally as opposed to posteriorly (n=6). Histological analysis of these ears was more telling. Interestingly, although Tbx1Cre/− ears were severely malformed, the ventral medial portion of the vesicle contained a row of hair cells (Fig. 6B, e,f, black arrowheads). Most of the OV was unpopulated with hair cells outside of this domain (Fig. 6B, e,f, red arrowheads). The Tbx1Cre/−;Jag1flox/+ OV contained much fewer hair cells in the same domain (Fig. 6B, e′,f′). Serial sections of mutant embryos confirmed this throughout most of the abnormal OV. In addition, the cochlear and vestibular ganglia were absent in Tbx1Cre/− and Tbx1Cre/−;Jag1flox/+ mutant embryos. Because there are so few hair cells in both mutant embryos, this is again, likely to be due to lack of innervation and, consequently, underdevelopment and/or degeneration of the ganglia and neurons within.
DISCUSSION
Tbx1 modulates neurosensory cell fate in a domain-specific manner
In this study, we found that loss of one allele of Jag1 in the Tbx1 expression domain exacerbated the expanded neurogenesis phenotype of Tbx1 loss-of-function embryos. This co-occurred with increased proneural gene expression in the OV, unaccompanied by changes in cell proliferation. Based upon all this, we created a model that summarizes our findings, shown in Fig. 7. Despite the fact that loss of Jag1 alone resulted in a smaller CVG than in wild-type embryos, when one or both alleles of Jag1 was inactivated with Tbx1, more neuroblasts were present than in global Tbx1 null mutant embryos (Fig. 7). This revealed that Jag1 plays a complex role in neural development that is unmasked when inactivated within the Tbx1 expression domain together with Tbx1, and that Tbx1 and Jag1 may genetically interact in this process.
Fig. 7.
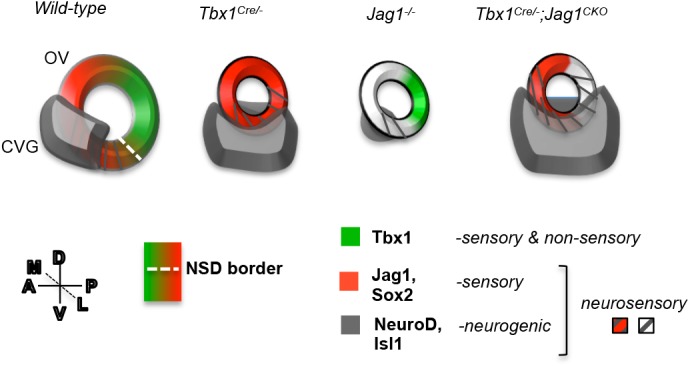
Model summary. A schematic of the OV summarizing the expression of the key genes of this study in the OV that is oriented as shown (dorsal-ventral, D-V; medial-lateral, M-L; anterior-posterior, A-P). In wild-type embryos at E10-10.5, Tbx1 is expressed at the NSD border marked by overlapping expression of neurogenic genes (grey stripes) and Jag1/Sox2 (red) in the anteroventral OV. This NSD is positioned adjacent to the developing CVG, marked by NeuroD and Isl1 expression (grey), and contributes to it some of its progenitor cells fated to become neurons. When Tbx1 is homozygously inactivated (Tbx1Cre/−), Jag1 expression is expanded throughout the OV, disrupting the NSD border. The CVG also expands into the posterior and lateral domain in Tbx1Cre/− (or Tbx1−/−) OVs. When Jag1 is inactivated (Jag1−/−), the Tbx1 expression domain is unchanged, and the CVG is significantly smaller in size. When Jag1 is inactivated together with Tbx1 in the Tbx1 expression domain (Tbx1Cre/−;Jag1f/+ or Tbx1Cre/−;Jag1f/f – termed Tbx1Cre/−;Jag1CKO), the NSD border is disrupted and the CVG is further enlarged.
In this work, we found that inactivation of Tbx1 and Jag1 within the Tbx1 expression domain particularly affected the M-L boundary, in addition to the already disrupted A-P boundary seen in Tbx1−/− OVs (Fig. 4) (Raft et al., 2004). Relevant to these findings, Sapede et al. (2012) performed cell lineage analysis in zebrafish that identified a population of common progenitors that can give rise to neurons and hair cells that reside in the posteromedial part of the OV. In this study, we found Jag1 and Sox2, required for sensorigenesis and neurogenesis, to be expressed in the posteromedial region of the OV, forming a border with lateral-expressed Tbx1 at the NSD border. One interpretation of these data is that regulation of the M-L boundary is important for the maintenance of this domain and Jag1, along with Sox2, serves as marker of this common pool of cells, the fate of which is restricted by Tbx1.
Tbx1 fate mapping in Tbx1 null mutant embryos also provided evidence suggesting that Tbx1 may regulate CVG formation in a cell-nonautonomous (as well as cell-autonomous) manner. Based upon these findings, we further speculate that there might be an intermediate gene or genes between Tbx1 and master proneural transcription factors NeuroD and Ngn1, one of which may be Jag1.
The role of Jag1 in NSD development
Our data suggest that Tbx1 restricts neurosensory cell formation by acting at the NSD border of the OV either directly or indirectly, restricting Jag1 and Sox2 expression. Previous findings in chick show that overexpression of human JAG1 in the OV induces expression of Sox2 in a Notch-dependent manner (Neves et al., 2011), but that this only occurs for specification of the sensory patches. This study concluded that Sox2 establishes early neurosensory competence, which is independent of Jag1 (Neves et al., 2011). While this is the situation in chick, we speculate that in mouse, Jag1 plays a role in the specification or maintenance of the fate of cell subtypes within the NSD and not just the sensory patches. Mouse studies showed that Jag1 is required for the specification of sensory precursors and a subset of neural precursors (Pan et al., 2010). This supports the hypothesis that Jag1 plays a role in NSD (Pan et al., 2010). Our findings with respect to Tbx1 also support a new possible role of Jag1 in the NSD. Furthermore, our studies show that Jag1 expression may be broader in the mouse OV than in chick and more closely resemble Sox2 expression (Neves et al., 2011). If the expression of Jag1 in mouse more closely follows that of Sox2, this could indicate that, like Sox2, it plays a role in the development of the NSD. Finally, studies in mouse also suggest that the clonal population of progenitors that can give rise to neurons and hair cells may consist of a larger pool of cells than in chick (Fritzsch et al., 2006). If this is the case, regulation of this cell population may necessitate a larger network of genes, such as Tbx1. While these are all plausible hypotheses, it is also possible that the differences we see in mouse and chick are due to differences in experimental strategy. Since Jag1 was overexpressed in chick studies and inactivated in mouse studies, it is possible that Jag1 is required, but not sufficient, for the development of the NSD.
Tbx1 and Jag1 may be required to maintain a balance of cell types within the NSD
Loss of Tbx1 may affect neurosensory precursor cell fate via Jag1, as evidenced by the further expansion of proneural gene expression when Jag1 is concomitantly inactivated. Since sensory structures do not develop in Tbx1−/− embryos, but the CVG is duplicated (Arnold et al., 2006; Raft et al., 2004; Vitelli et al., 2003), we surmise that the expansion of the area of expression of NSD genes indicates a switch in cell fate from sensory to neural. Additional inactivation of Jag1 may enhance this switch, based on our findings of a synergistic expansion in proneural gene expression in the OV, as well as failure of cristae and some hair cells to form properly at later stages, in Tbx1;Jag1 compound mutants. We cannot exclude the possibility that nonsensory cells may also change their fate to form neuroblasts, as the semicircular canals and ampullae also do not fully form in Tbx1Cre/+;Jag1flox/flox mutant inner ears.
Expansion in the neuroblast population observed in the compound mutant embryos appears contradictory to the perceived role of Jag1 in neurosensory development; however, Notch1 is known to have contrasting roles during neurogenesis and sensorigenesis (Adam et al., 1998; Daudet et al., 2007; Pan et al., 2010; Petrovic et al., 2014), making the interpretation of such experiments complex. One potential explanation for our findings is that Jag1 may define which cells within the NSD become sensory or neural by repressing neural fate within the boundary cells where it is co-expressed with Tbx1. Outside this domain and perhaps earlier in development, Jag1 may play a role in neurosensory competence together with Sox2. This may explain why both the CVG and sensory organs are hypoplastic when Jag1 is globally inactivated (Pan et al., 2010). Thus, we suggest that Tbx1 and Jag1 function in opposing pathways during NSD development, but we speculate that they can also repress neural differentiation within the otic epithelium by acting similarly on common downstream factors.
One of the main strengths of this study is that we had optimal Cre alleles to inactivate Tbx1 and Jag1 in the same expression domains. This allowed us to discover an interaction between the two genes in neurosensory patterning in which loss of Jag1 enhances the effect of loss of Tbx1 in restricting neurogenesis. We performed in situ hybridization of many molecular markers but could not observe a difference in the pattern of expression between the Tbx1 loss-of-function mutant and the double Tbx1;Jag1 loss-of-function mutant embryos. This is one of the limitations in the study, and we believe this is due to the choice of genes evaluated or lack of resolution of in situ hybridization methods we used. To determine the mechanism for the Tbx1;Jag1 interaction and to gain further insights into cell fate changes in the NSD, we suggest that unbiased genome wide expression profiling approaches are required. This could be coupled with single cell RNA-sequencing of NSD cells in wild-type and mutant OVs in order to fully elucidate the molecular mechanism.
Conclusions
In conclusion, Tbx1 and Jag1 act in discrete regions of the OV to limit the size of the CVG in mouse embryos. Inactivating Jag1 with Tbx1Cre reveals a novel role for Jag1, whereby in this context Jag1 acts to repress neural fate, in contrast to its known function. We suggest that normal Tbx1 and Jag1 dosage may be required to maintain a proper balance of cell types within the NSD of the OV to form the inner ear. This knowledge could have important implications for future stem cell therapies designed to treat sensorineural hearing loss and vestibular disorders.
MATERIALS AND METHODS
Mice
Mice used in this study comply with the regulatory standards of the Institutional Animal Care and Use Committee (IACUC, #20160507) of Albert Einstein College of Medicine. All mouse models have been previously described. The CMV-GFP reporter mice were obtained from Dr. Gordon Fishell at New York University Langone Medical Center (Batista-Brito et al., 2009). Pax2-Cre mice were provided by Dr. Andrew K. Groves (Ohyama and Groves, 2004). Tbx1Cre/+ mice were provided by Dr. Antonio Baldini (Huynh et al., 2007). Tbx1-GFP mice, termed ‘Tbx1 GOF’, were engineered in our laboratory, in which a Tbx1-GFP fusion protein was added downstream of a loxP-STOP-loxP site in the Rosa26 locus and used as previously described (Freyer et al., 2013). Tbx1+/− and Tbx1flox mice (Arnold et al., 2006) were engineered in our laboratory and used as previously described. Jag1 null (stock number 010616), Jag1flox (stock number 010618) and Pax3Cre/+ (stock number 005549) mice were obtained from Jackson Laboratories. Embryos were dissected according to date of vaginal plug (E0.5). Embryonic stages <E11.5 were confirmed by counting pairs of somites. Animals were maintained in a 12 h dark/12 h light cycle in compliance with the Albert Einstein College of Medicine Institutional Animal Care and IACUC. Mice were genotyped for Cre alleles using the following primers: (5′-CAATGCTGTTTCACTGGTTATG-3′) and (5′-CATTGCCCCTGTTTCACTATC-3′). The Tbx1flox allele was detected using primers that have been previously described (Braunstein et al., 2009). CMV-GFP reporter and Tbx1-GFP mice were genotyped for the GFP allele using the following primers: GFP-Fwd (5′-TAAACGGCCACAAGTTCAGC-3′) and GFP-Rev (5′-GAACTCCAGCAGGACCATG-3′), while the wild-type allele was detected using the following primers: RO1F (5′-GCAATACCTTTCTGGGAGTT-3′) and GFP-wt-R (5′-CAATGCTCTGTCTAGGGGTT-3′). The interval within the Jag1flox allele was amplified as described by Jackson Laboratories using the following primers: 10092 Jag1 Forward (5′-TCAGGCATGATAAACCCTAGC-3′) and 10093 Jag1 Reverse (5′-CTACATACAGCATCTACATGC-3′). Jag1 null mice were also genotyped as described by Jackson Laboratories using the following primers: 10089 Forward (5′-TCTCACTCAGGCATGATAAACC-3′), 10090 Wild-type Reverse (5′ TAACGGGGACTCCGGACAGGG-3′), and oIMR8162 Mutant Reverse (5′- TGGATGTGGAATGTGTGCGAG-3′).
Immunofluorescence on tissue sections
Embryos were fixed in 4% paraformaldehyde at 4°C. Fixation times varied according to embedding method: 2 h for frozen embryos and overnight for paraffin-embedded embryos. Frozen embryos were washed in 0.1 M phosphate buffered saline (PBS) and embedded in 30% sucrose in PBS at 4°C overnight. Embryos were embedded in O.C.T. compound (Tissue-Tek) on dry ice and stored at −80°C. Transverse cryosections were generated at 10 μm in thickness. Tissue sections were washed in PBS and permeabilized in PBS/0.5% Triton X-100 for 5 min. They were then washed in PBS followed by PBS/0.1% Triton X-100 and blocked in 5% goat or donkey serum (G9023 and D9663, Sigma-Aldrich) in PBS/0.1% Triton X-100 for 1 h at room temperature (RT) followed by incubation with primary antibodies diluted in block for 1 h at RT. Primary antibodies used were as follows: rabbit polyclonal α-Tbx1 (1:500; Zymed, San Francisco, CA, USA), goat polyclonal α-Jag1 (1:100; C-20, Santa Cruz Biotechnology), goat polyclonal α-Sox2 (1:500; Santa Cruz Biotechnology), goat polyclonal α-NeuroD (1:500; sc-1084, Santa Cruz Biotechnology), goat polyclonal α-GFP (1:500; Abcam), rabbit polyclonal α-GFP (1:500; ab290, Abcam), rabbit polyclonal α-phospho-histone H3 (1:500; Ser 10, 06-570, Emd Millipore, Billerica, MA, USA), and mouse monoclonal α-Tuj1 (1:1000; MMS-435P, Covance, Princeton, NJ, USA). Tissue sections were then washed three times in PBS/0.1% Triton X-100 and incubated with secondary antibodies diluted in block together with DAPI (4′,6-diamidine-2-phenylidole-dihydrochloride; 1:500) for 1 h at RT. Secondary antibodies were as follows: Alexa Fluor 568 goat α-rabbit IgG (A-11011, Invitrogen), Alexa Fluor 568 goat α-mouse IgG (A-11004, Invitrogen), Alexa Fluor 488 goat α-rabbit IgG (A-11008, Invitrogen), Alexa Fluor 488 donkey α-goat (A-11055, Invitrogen) and Alexa Fluor 568 donkey α-goat IgG (A-11057, Invitrogen). All secondary antibodies were used at a dilution of 1:500. Sections were washed three times in PBS/0.1% TritonX-100 followed by brief washes in PBS and then water. For dual color immunofluorescence, primary antibodies were incubated on tissue sections at the same time, and secondary antibodies were subsequently incubated on sections at the same time. Slides were mounted in Vectashield hard-set mounting medium (H-1400, Vector Laboratories) and stored at 4°C. Images were captured using an Axio Observer (Zeiss, Oberkochen, Germany).
Whole-mount RNA in situ hybridization
Embryos were fixed in 4% paraformaldehyde at 4°C overnight. They were then dehydrated in a series of methanol/PBS/0.1% Tween-20 dilutions to 100% methanol and stored at −20°C. Upon rehydration to 0.1%PBS/0.1% Tween-20, in situ hybridization was carried out as previously described (Franco et al., 2001). Antisense digoxigenin-labeled RNA probes to Tbx1 (Funke et al., 2001), NeuroD (Lee et al., 1995), and Jag1 (Jayasena et al., 2008) were used as described. The Hes6 RNA probe template was generated from amplified E9.5 mouse cDNA using the following primers: 5′-GGGGAATTAACCCTCACTAAAGGGAACGAGAGT CTTCAGGAGCT-3′ and 5′-GGGGTAATACGACTCACTATAGGGTACAAACGA GGAGCAGCTTC-3′.
Quantitative analyses
Cell counting and area measurements were performed using ImageJ software. Mitotic index was calculated by dividing the total number of pHH3+ cells that colocalized with NeuroD, divided by the total number of NeuroD+ cells (within the OV and CVG). OV area was calculated by subtracting the area of the inner OV by the area of the outer OV. Investigators were blinded to group allocation during all analyses. For all analyses, n≥6 ears (from at least three different animals) per group. Statistical analysis was performed using Microsoft Excel. Groups were compared using a two-tailed, Independent Samples (unequal variance) t-test. Data met the assumption of the test (i.e. data within groups fell under a normal distribution).
3D reconstruction
Serial sections of the otic vesicle were aligned using AutoAligner software (Bitplane AG) and regions of interest were traced and reconstructed using BioVis3D software.
Histology
Embryos were fixed in 4% paraformaldehyde overnight at 4°C. They were then dehydrated to 70% ethanol and embedded in paraffin. Tissue sectioning was performed at 10-12 μm thicknesses. Tissues were cleared in xylene, stained with hematoxylin and eosin (H&E) and then mounted in Permount.
Paintfilling
Embryos were cut below the forelimbs and fixed in 5% glacial acetic acid, 2% formaldehyde and 75% ethanol overnight. This was followed by dehydration to ethanol and clearing in methyl salicylate. Embryos were bisected dorsally and the brain was removed. A micropipette was used to microinject 0.2% correction fluid diluted in methyl salicylate into the utricle. Paintfilled inner ears were imaged and stored in methyl salicylate.
Acknowledgements
We thank Kevin Fisher for help with the 3D reconstruction of the otic vesicle; Drs Laina Freyer and Silvia Racedo for helpful scientific discussion and critical reading of the manuscript; and Drs Raquel Castellanos, Dennis Monks and Jonathan Chung for helpful scientific discussion.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
Conceptualization: S.M., B.E.M.; Methodology: S.M., B.E.M.; Formal analysis: S.M., B.E.M.; Investigation: S.M.; Resources: B.E.M.; Writing - original draft: S.M.; Writing - review & editing: S.M., B.E.M.; Visualization: S.M., B.E.M.; Supervision: B.E.M.; Project administration: B.E.M.; Funding acquisition: B.E.M.
Funding
This work was supported by the National Institute on Deafness and other Communication Disorders [DC05186 to B.E.M.].
Supplementary information
Supplementary information available online at http://bio.biologists.org/lookup/doi/10.1242/bio.027359.supplemental
References
- Adam J., Myat A., Le Roux I., Eddison M., Henrique D., Ish-Horowicz D. and Lewis J. (1998). Cell fate choices and the expression of Notch, Delta and Serrate homologues in the chick inner ear: parallels with Drosophila sense-organ development. Development 125, 4645-4654. [DOI] [PubMed] [Google Scholar]
- Arnold J. S., Braunstein E. M., Ohyama T., Groves A. K., Adams J. C., Brown M. C. and Morrow B. E. (2006). Tissue-specific roles of Tbx1 in the development of the outer, middle and inner ear, defective in 22q11DS patients. Hum. Mol. Genet. 15, 1629-1639. 10.1093/hmg/ddl084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bok J., Chang W. and Wu D. K. (2007). Patterning and morphogenesis of the vertebrate inner ear. Int. J. Dev. Biol. 51, 521-533. 10.1387/ijdb.072381jb [DOI] [PubMed] [Google Scholar]
- Braunstein E. M., Monks D. C., Aggarwal V. S., Arnold J. S. and Morrow B. E. (2009). Tbx1 and Brn4 regulate retinoic acid metabolic genes during cochlear morphogenesis. BMC Dev. Biol. 9, 31 10.1186/1471-213X-9-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole L. K., Le Roux I., Nunes F., Laufer E., Lewis J. and Wu D. K. (2000). Sensory organ generation in the chicken inner ear: contributions of bone morphogenetic protein 4, serrate1, and lunatic fringe. J. Comp. Neurol. 424, 509-520. [DOI] [PubMed] [Google Scholar]
- Daudet N., Ariza-McNaughton L. and Lewis J. (2007). Notch signalling is needed to maintain, but not to initiate, the formation of prosensory patches in the chick inner ear. Development 134, 2369-2378. 10.1242/dev.001842 [DOI] [PubMed] [Google Scholar]
- Fekete D. M. and Wu D. K. (2002). Revisiting cell fate specification in the inner ear. Curr. Opin. Neurobiol. 12, 35-42. 10.1016/S0959-4388(02)00287-8 [DOI] [PubMed] [Google Scholar]
- Franco D., de Boer P. A., de Gier-de Vries C., Lamers W. H. and Moorman A. F. (2001). Methods on in situ hybridization, immunohistochemistry and betagalactosidase reporter gene detection. Eur. J. Morphol. 39, 3-25. [DOI] [PubMed] [Google Scholar]
- Freyer L., Aggarwal V. and Morrow B. E. (2011). Dual embryonic origin of the mammalian otic vesicle forming the inner ear. Development 138, 5403-5414. 10.1242/dev.069849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyer L., Nowotschin S., Pirity M. K., Baldini A. and Morrow B. E. (2013). Conditional and constitutive expression of a Tbx1-GFP fusion protein in mice. BMC Dev. Biol. 13, 33 10.1186/1471-213X-13-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch B., Beisel K. W. and Hansen L. A. (2006). The molecular basis of neurosensory cell formation in ear development: a blueprint for hair cell and sensory neuron regeneration? BioEssays 28, 1181-1193. 10.1002/bies.20502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funke B., Epstein J. A., Kochilas L. K., Lu M. M., Pandita R. K., Liao J., Bauerndistel R., Schüler T., Schorle H., Brown M. C. et al. (2001). Mice overexpressing genes from the 22q11 region deleted in velo-cardio-facial syndrome/DiGeorge syndrome have middle and inner ear defects. Hum. Mol. Genet. 10, 2549-2556. 10.1093/hmg/10.22.2549 [DOI] [PubMed] [Google Scholar]
- Hébert J. M. and McConnell S. K. (2000). Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev. Biol. 222, 296-306. 10.1006/dbio.2000.9732 [DOI] [PubMed] [Google Scholar]
- Huynh T., Chen L., Terrell P. and Baldini A. (2007). A fate map of Tbx1 expressing cells reveals heterogeneity in the second cardiac field. Genesis 45, 470-475. 10.1002/dvg.20317 [DOI] [PubMed] [Google Scholar]
- Jayasena C. S., Ohyama T., Segil N. and Groves A. K. (2008). Notch signaling augments the canonical Wnt pathway to specify the size of the otic placode. Development 135, 2251-2261. 10.1242/dev.017905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan A. E., Xu J. and Gridley T. (2006). The notch ligand JAG1 is required for sensory progenitor development in the mammalian inner ear. PLoS Genet. 2, e4 10.1371/journal.pgen.0020004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. E., Hollenberg S. M., Snider L., Turner D. L., Lipnick N. and Weintraub H. (1995). Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helixloop-helix protein. Science 268, 836-844. [DOI] [PubMed] [Google Scholar]
- Ma Q., Chen Z., Barrantes I. D. B., Luis de la Pompa J. and Anderson D. J. (1998). neurogenin1 is essential for the determination of neuronal precursors for proximal cranial sensory ganglia. Neuron 20, 469-482. 10.1016/S0896-6273(00)80988-5 [DOI] [PubMed] [Google Scholar]
- Morsli H., Choo D., Ryan A., Johnson R. and Wu D. K. (1998). Development of the mouse inner ear and origin of its sensory organs. J. Neurosci. 18, 3327-3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves J., Parada C., Chamizo M. and Giráldez F. (2011). Jagged 1 regulates the restriction of Sox2 expression in the developing chicken inner ear: a mechanism for sensory organ specification. Development 138, 735-744. 10.1242/dev.060657 [DOI] [PubMed] [Google Scholar]
- Ohyama T. and Groves A. K. (2004). Generation of Pax2-Cre mice by modification of a Pax2 bacterial artificial chromosome. Genesis 38, 195-199. 10.1002/gene.20017 [DOI] [PubMed] [Google Scholar]
- Pan W., Jin Y., Stanger B. and Kiernan A. E. (2010). Notch signaling is required for the generation of hair cells and supporting cells in the mammalian inner ear. Proc. Natl. Acad. Sci. USA 107, 15798-15803. 10.1073/pnas.1003089107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovic J., Formosa-Jordan P., Luna-Escalante J. C., Abelló G., Ibañes M., Neves J. and Giraldez F. (2014). Ligand-dependent Notch signaling strength orchestrates lateral induction and lateral inhibition in the developing inner ear. Development 141, 2313-2324. 10.1242/dev.108100 [DOI] [PubMed] [Google Scholar]
- Puligilla C., Dabdoub A., Brenowitz S. D. and Kelley M. W. (2010). Sox2 induces neuronal formation in the developing mammalian cochlea. J. Neurosci. 30, 714-722. 10.1523/JNEUROSCI.3852-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raft S. and Groves A. K. (2014). Segregating neural and mechanosensory fates in the developing ear: patterning, signaling, and transcriptional control. Cell Tissue Res. 359, 315-332. 10.1007/s00441-014-1917-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raft S., Nowotschin S., Liao J. and Morrow B. E. (2004). Suppression of neural fate and control of inner ear morphogenesis by Tbx1. Development 131, 1801-1812. 10.1242/dev.01067 [DOI] [PubMed] [Google Scholar]
- Raft S., Koundakjian E. J., Quinones H., Jayasena C. S., Goodrich L. V., Johnson J. E., Segil N. and Groves A. K. (2007). Cross-Regulation of Ngn1 and Math1 coordinates the production of neurons and sensory hair cells during inner ear development. Development 134, 4405-4415. 10.1242/dev.009118 [DOI] [PubMed] [Google Scholar]
- Sapede D., Dyballa S. and Pujades C. (2012). Cell lineage analysis reveals three different progenitor pools for neurosensory elements in the otic vesicle. J. Neurosci. 32, 16424-16434. 10.1523/JNEUROSCI.3686-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitelli F., Viola A., Morishima M., Pramparo T., Baldini A. and Lindsay E. (2003). TBX1 is required for inner ear morphogenesis. Hum. Mol. Genet. 12, 2041-2048. 10.1093/hmg/ddg216 [DOI] [PubMed] [Google Scholar]
- Xu H., Viola A., Zhang Z., Gerken C. P., Lindsay-Illingworth E. A. and Baldini A. (2007). Tbx1 regulates population, proliferation and cell fate determination of otic epithelial cells. Dev. Biol. 302, 670-682. 10.1016/j.ydbio.2006.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]