

Abstract
Postnatal bone growth involves a dramatic increase in length and girth. Intriguingly, this period of growth is independent of growth hormone and the underlying mechanism is poorly understood. Recently, an IGF2 mutation was identified in humans with early postnatal growth restriction. Here, we show that IGF2 is essential for longitudinal and appositional murine postnatal bone development, which involves proper timing of chondrocyte maturation and perichondrial cell differentiation and survival. Importantly, the Igf2 null mouse model does not represent a simple delay of growth but instead uncoordinated growth plate development. Furthermore, biochemical and two-photon imaging analyses identified elevated and imbalanced glucose metabolism in the Igf2 null mouse. Attenuation of glycolysis rescued the mutant phenotype of premature cartilage maturation, thereby indicating that IGF2 controls bone growth by regulating glucose metabolism in chondrocytes. This work links glucose metabolism with cartilage development and provides insight into the fundamental understanding of human growth abnormalities.
KEY WORDS: IGF2, Postnatal, Cartilage, Growth plate, Endochondral ossification, Glucose metabolism
Summary: Attenuation of glucose consumption in Igf2 null mice rescued the mutant phenotype of premature cartilage maturation, revealing that IGF2 controls cartilage development in part by regulating glucose metabolism in chondrocytes.
INTRODUCTION
Endochondral ossification is an essential process for long bone growth, for which a cartilage template is used. Here, chondrocytes undergo proliferation and hypertrophic differentiation (i.e. maturation), forming a distinctive growth plate that drives longitudinal growth (Kronenberg, 2003). Newly formed chondrocytes are proliferative and morphologically round, but eventually become flat chondrocytes to form the ‘columnar zone’. Proliferating chondrocytes eventually exit the cell cycle to enter the prehypertrophic phase, which is followed by the hypertrophic phase. Such longitudinal growth is governed by the parathyroid hormone-related protein (PTHrP)/Ihh signaling loop, and mediated by the perichondrium (Kronenberg, 2003). The perichondrium is also crucial for the appositional growth that increases the girth of the developing bone and involves perichondrial cell proliferation and differentiation. A key feature of the postnatal period of bone growth is the rapid increase in girth, as well as the appearance of the secondary ossification center (SOC), a process that involves the hypertrophic differentiation of epiphyseal chondrocytes and subsequent ossification (Blumer et al., 2008).
Although embryonic long bone growth has been extensively studied, the mechanisms that govern postnatal bone development are still largely elusive. Growth hormone (GH) is a major regulator of postnatal bone growth, but GH receptor knockout mice grow normally until postnatal day (P) 10, suggesting that early postnatal growth is GH independent (Lupu et al., 2001). Insulin-like growth factors (IGF1 and IGF2) are important growth factors that govern both prenatal and postnatal body growth and development (Fisher et al., 2005; Schlegel et al., 2010). IGF2, but not IGF1, is strongly expressed in the proliferating zone of the growth plate (Reinecke et al., 2000; Shinar et al., 1993; Tsang et al., 2007). Although mutations in IGF1 and its receptor, IGF1R, have been reported in humans with stunted growth, mutations in IGF2 had not been identified until recently, when Begemann et al. showed that a nonsense IGF2 mutation is associated with human postnatal growth restriction (Begemann et al., 2015; Klammt et al., 2011; Netchine et al., 2011). This specifically highlights the importance of IGF2 in controlling skeletal development after birth, raising the need to understand how IGF2 exerts this activity.
In this article, we investigated the role of IGF2 in longitudinal and appositional bone growth using Igf2 null mice, and discovered that an underlying mechanism by which IGF2 controls cartilage development is through regulating glucose metabolism in chondrocytes.
RESULTS
The Igf2 null bone has an abnormal growth plate with a disproportionally larger hypertrophic zone and delayed secondary ossification center formation
To investigate the role of IGF2 on postnatal long bone growth, we analyzed Igf2 knockout mice. Igf2 is an imprinted gene that is only expressed from the paternally inherited allele (Baker et al., 1993). Thus, the Igf2 knockout (KO) phenotype is observed from a homozygote (Igf2−/−) as well as from a heterozygote with a KO allele from the father (Igf2+m/−p), which we refer to as an ‘Igf2 null’ or ‘Igf2 mutant’. We examined metatarsal bones of wild-type (WT) and Igf2 null mice, as the metatarsal bone is widely used as a model for studying long bone development (Reno et al., 2006; Stickens et al., 2004). Although Igf2 null embryos appear smaller than the WT, no significant differences in length or width were observed between the embryonic WT and Igf2 null metatarsal bones (Fig. 1A-C). However, starting from P7, Igf2 null bones showed a substantial reduction in the total length and width of the bone as well as the cartilage (Fig. 1D-G). We have also observed similar phenotypes in the femur and tibia (Fig. S1); however, these bones have a curvature associated with the growth plate in later stages, making quantification difficult. In addition, because metatarsal bones are more immature than the femur and tibia, they exhibit more significant postnatal growth, which allows us to capture bone development more easily. Therefore, we decided to continue analyzing metatarsal bones in subsequent studies. Alcian Blue staining confirmed that the Igf2 null growth plate cartilage was shorter and disproportionally thinner than the WT (Fig. 2A-F). The mutant growth plate was generally well formed, but its hypertrophic zone was disproportionally larger and the epiphyseal zone shorter (Fig. 2G,H). In addition, there was a clear delay in SOC formation in the mutant, which contributed to the shortened cartilage template (Fig. 2H).
Fig. 1.

Igf2 null mutants exhibit abnormalities in postnatal metatarsal bone growth. (A) Parameters used in metatarsal bone measurement. The darker area in the diaphysis indicates mineralized bone and calcified cartilage. Note that the same image is used in Fig. 5A. (B) Comparison of bone length between WT and Igf2 null bones at E15.5 and E17.5. (C) Comparison of bone width between WT and Igf2 null bones at E15.5 and E17.5. (D-G) Comparison of total bone length (D), bone width at diaphysis (E), length of the cartilage portion at metaphysis (F) and width of the cartilage portion at metaphysis (G) in WT and Igf2 null bones at postnatal stages P0-P21. At least eight embryonic bones/group and six postnatal bones/group were analyzed. Mean±s.d. *P<0.05 (unpaired t-tests between WT and Igf2 null).
Fig. 2.
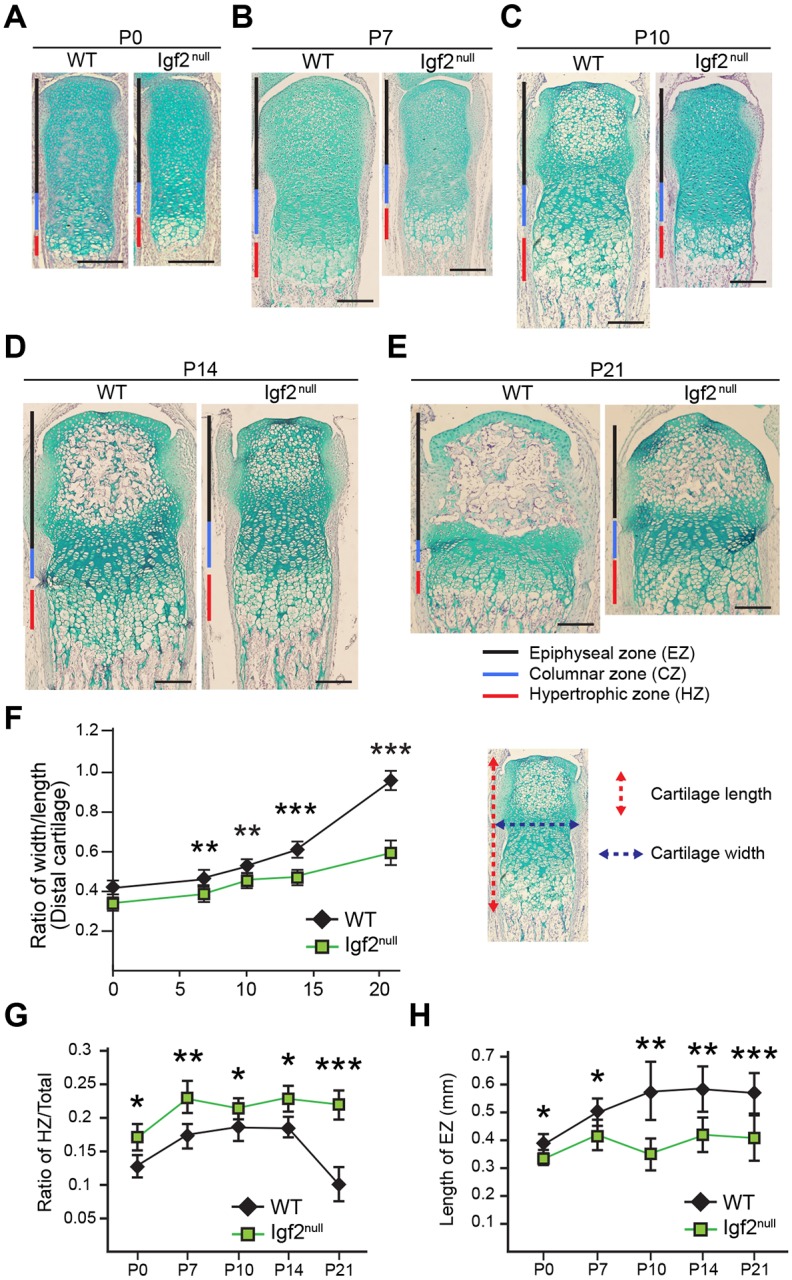
Igf2 null mutant metatarsal bones have abnormal growth plate architecture and delayed secondary ossification center formation in postnatal development. (A-E) Alcian Blue and Hematoxylin staining of the cartilage portion from the distal end of the middle metatarsal bone at P0, 7, 10, 14 and 21. Red, blue and black bars indicate the hypertrophic zone (HZ), the columnar zone (CZ) and the epiphyseal zone (EZ), respectively. (F) Comparison of width/length ratio of the cartilage template between WT and Igf2 null bones. The length is the sum of the lengths of HZ, CZ and EZ. (G) Comparison of the ratio of the HZ in the cartilage template (HZ/total) between WT and Igf2 null bones. (H) Comparison of EZ length between WT and Igf2 null bones. Scale bars: 200 µm. At least six bones/group were analyzed. Mean±s.d. *P<0.05, **P<0.01, ***P<0.001 (unpaired t-tests between WT and Igf2 null).
Immunohistochemical (IHC) analysis indicated that collagen type II (Col-II) and type X (Col-X) protein expression was comparable between the WT and the mutant, but there is a clear delay in Col-X expression in the SOC of the Igf2 null (Fig. 3A,B; Fig. S2). The master chondrogenic regulator Sox9 is normally expressed widely in columnar chondrocytes, but is reduced in hypertrophic chondrocytes (Akiyama et al., 2002; Amarilio et al., 2007; Huang et al., 2001; Saito et al., 2010) (Fig. 3C). In the Igf2 mutant, however, significantly higher levels of Sox9 persisted in hypertrophic chondrocytes (Fig. 3C, arrows) as well as in chondrocytes of the SOC, indicating a delay in repression of Sox9 expression upon chondrocyte hypertrophy (Fig. 3C). Ihh is another key regulator of cartilage development; Ihh mRNA is specifically expressed in the prehypertrophic zone, but its protein can diffuse into neighboring zones (Bastepe et al., 2004; Bitgood and McMahon, 1995; Kake et al., 2009). We found a shorter Ihh-positive prehypertrophic zone in the Igf2 mutant (Fig. 3D), indicating that IGF2 deficiency caused a shortening of the prehypertrophic zone (Fig. 3D). The key hypertrophic zone regulators Runx2, HIF1α and HIF2α were strongly expressed in the hypertrophic zone and the developing SOC of the WT bone, but their expression was clearly reduced in the hypertrophic zone of the Igf2 mutant, which might be related to increased Sox9 expression in this zone (Fig. 3D-G). Additionally, they were absent from the developing SOC of the Igf2 mutant, indicating a delay in SOC development (Fig. 3D-G). Interestingly, markers that are associated with terminal differentiation, such as alkaline phosphatase (ALP) activity and MMP13 expression, were not significantly affected in Igf2 mutants, suggesting that it is unlikely that the lack of IGF2 had affected mineralization or terminal differentiation of chondrocytes (Fig. S3).
Fig. 3.

Histological analysis of the growth plate of WT and Igf2 null metatarsal bones at P7. (A-G) Collagen II (Col-II) (A), collagen X (Col-X) (B), Sox9 (C), IHH (D), Runx2 (E), HIF1α (F) and HIF2α (G) IHC. Arrows in C indicate areas with abundant Sox9-positive cells in the Igf2 null compared with the WT [hypertrophic zone and the secondary ossification center (SOC)]. (H) EdU incorporation. The arrow indicates EdU-positive cells in the SOC of the Igf2 null bone. (I) TUNEL assay. Arrows indicate TUNEL-positive cells in the proliferating zone as well as the hypertrophic zone in the Igf2 null bone. Boxed areas are magnified as indicated. At least six bones/group were analyzed. Scale bars: 200 µm.
To assess chondrocyte proliferation and survival, we performed 5-ethynyl-2′-deoxyuridine (EdU) incorporation, Ki67 (Mki67) staining and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining. In the columnar zone, similar percentages of EdU-positive cells were observed between the WT and the mutant, despite an overall reduction in the total number of EdU-positive cells (Fig. 3H, Fig. S4), suggesting that the loss of IGF2 did not affect the rate of columnar chondrocyte proliferation. On the other hand, in the SOC, widespread EdU-positive cells were found in the Igf2 null, which is consistent with a delay in hypertrophic differentiation (Fig. 3H). Similar results were obtained with Ki67 staining (Fig. S4). By contrast, TUNEL staining revealed a significant increase in apoptosis (Fig. 3I), suggesting that IGF2 is required for chondrocyte survival.
The Igf2 mutant does not represent a simple delay in bone development
To address whether the observed phenotype represents a simple delay in development in the Igf2 null, we compared WT and mutant metatarsal bones of the same length. Based on the growth chart, WT at P12 and Igf2 null at P21 are of the same bone length (Fig. 4A); however, the Igf2 null growth plate was much smaller and resembled the growth plate of a more mature bone (Fig. 4B,C). As a result of a shortened growth plate, the mineralized diaphysis of the mutant was disproportionally longer (Fig. 4B,C). Consistent with a more mature phenotype in the Igf2 null, a stronger ALP activity was observed (Fig. 4C). Interestingly, the Igf2 null bone was significantly thinner than the length-matched WT bone (Fig. 4D), suggesting uncoordinated longitudinal and lateral growth. In addition, the Igf2 null bone had a disproportionally larger hypertrophic zone (Fig. 4E). Similar results were obtained when WT and mutant bones from two more pairs that were analyzed: P5 WT versus P12 Igf2 null and P9 WT versus P14 Igf2 null (Fig. S5). Thus, the Igf2 null does not represent a simple delay in bone development.
Fig. 4.
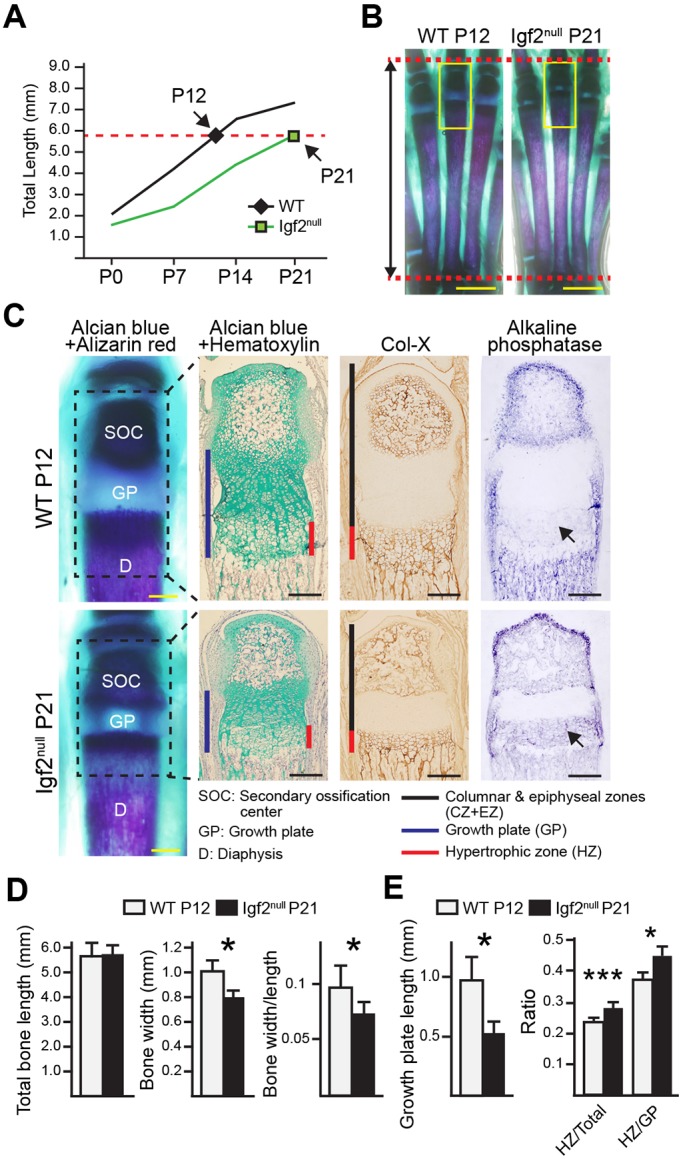
Histological analysis of length-matched Igf2 null and WT metatarsal bones. (A) P21 Igf2 null bones and P12 WT bones were length-matched pairs. (B) Whole-mount Alizarin Red/Alcian Blue staining images. Scale bars: 2 mm. (C) Magnified images of the boxed areas in B. Black dashed lined squares indicate the area for which Alcian Blue/Hematoxylin, collagen X and alkaline phosphatase (AP) staining is shown to the right. Arrows indicate areas with positive AP activity. Scale bars: 200 μm. (D) Analysis of total bone length, bone width, and the ratio of bone width/length in Igf2 null and WT bones. (E) Analysis of growth plate (GP) length and ratios of hypertrophic zone (HZ)/total (length of the cartilage portion) and HZ/GP in the cartilage template. At least six bones/group were analyzed. Mean±s.d. *P<0.05, ***P<0.001 (unpaired t-tests between WT and Igf2 null).
IGF2 is required for perichondrium growth and development
To investigate why the Igf2 null bone was thinner, we examined the perichondrium (Kobayashi et al., 2011; Kronenberg, 2006). For consistency, the thickness of the perichondrium juxtaposing the prehypertrophic zone where Ihh is expressed was quantified (Fig. 5A-C). Overall, the perichondrium grew thicker over time, but the mutant was consistently thinner (Fig. 5A-C). Although distinct perichondrium markers are not well established, Thy1 (also known as CD90) has been reported to be present in the outer layer of the perichondrium (Fig. 5D) (Nakamura et al., 2010). Significantly, Thy1 expression was diminished in the perichondrium of the Igf2 null bones (Fig. 5D). Interestingly, we did not observe a difference in CD44, which was expressed in the inner layer of the perichondrium (Fig. 5D) (Kobayashi et al., 2011). In addition to the change in Thy1 expression, there was also a substantial decrease in the number of Ki67- and EdU-positive cells and an increase in the number of TUNEL-positive cells in the perichondrium in the Igf2 null bone (Fig. 5E,F). As perichondrial cells can differentiate into osteoblasts and chondrocytes (Colnot et al., 2004; Kobayashi et al., 2011; Srour et al., 2015), the expression of Sox9 and Runx2 was assessed, and a reduction in these two proteins was observed (Fig. 5G). To determine whether the reduced thickness in perichondrium represents a simple delay in the development in the Igf2 null bones, we compared the thickness of the perichondrium in metatarsal bones of the same lengths. Although at the beginning, the perichondrium of bones of the same lengths (WT P5 versus Igf2 null P10, WT P9 versus Igf2 null P14) had similar thickness, later in development, the mutant perichondrium was thinner and had very little Ki67, Sox9 and Runx2 staining (WT P12 versus Igf2 null P21) (Fig. 5H,I). This result suggests that the Igf2 null bone does not represent a simple delay in perichondrium development. Together, these data suggest that IGF2 is required for perichondrial cell proliferation, survival and differentiation.
Fig. 5.

Histological analysis of the perichondrium in the Igf2 null and the WT metatarsal bone in postnatal stages. (A) Area of the perichondrium focused in histological analysis (red rectangle). Note that this is the same image as shown in Fig. 1A. (B) Comparison of perichondrium thickness between the Igf2 null and the WT using histological sections. Red brackets indicate the thickness of perichondrium. (C) Quantification of perichondrium thickness from P0 to P21. (D) IHC analysis of Thy1 and CD44 of the Igf2 and WT perichondrium at P7. Yellow brackets indicate the thickness of perichondrium. (E) IHC of Ki67, EdU incorporation, and TUNEL staining in the perichondrium. (F) Quantification on the percentage of Ki67-positive cells in the perichondrium. (G) IHC of Sox9 and Runx2 in the cartilage template. (H) Quantification of perichondrium thickness in metatarsal bones of the same length. (I) IHC of Ki67, Sox9 and Runx2 in metatarsal bones of P12 WT and P21 Igf2 null mice. Scale bars: 50 µm in all histographs. At least six bones/group were analyzed. At least three repeats were performed. Mean±s.d. *P<0.05 (unpaired t-tests between WT and Igf2 null).
IGF2 regulates chondrocyte gene expression in vitro
To determine whether IGF2 is required locally to regulate chondrocyte development, as the Igf2 null is a global knockout, we cultured isolated metatarsal bones. We found that the mutant bones grew more slowly in length and width, with a slightly larger hypertrophic zone and a shortened columnar zone (Fig. 6A-C), suggesting that IGF2 is required within the bone to regulate chondrocyte development. To confirm this finding, we isolated primary epiphyseal chondrocytes and cultured them in alginate beads to maintain a 3D environment (De Ceuninck et al., 2004). Igf2 null cells expressed significantly lower levels of the cartilage matrix genes aggrecan (Acan), Col2a1 (Col-II) and Col10a1 (Col-X), which is consistent with a delayed chondrocyte differentiation process (Fig. 6D). We also evaluated the expression of Igf1, but did not observe a significant difference. Treating cultured chondrocytes with IGF2 protein induced cartilage gene expression (Fig. 6E), suggesting that IGF2 is sufficient to promote chondrogenic gene expression. Interestingly, Sox9 mRNA expression was not altered in these manipulations (Fig. 6D,E), despite the protein expression being altered in the hypertrophic chondrocytes in our in vivo analysis. This suggests a potential difference in the regulation of SOX9 protein and mRNA by IGF2. Because a key regulator of chondrocyte hypertrophy, HIF2α, was the most upregulated by IGF2 compared with other genes in our in vitro study (Fig. 6E, Hif2a), we blocked HIF2α activity by using a chemical inhibitor (Pfander et al., 2003; Regan et al., 2014; Scheuermann et al., 2013). A significant reduction in Runx2 expression was observed, suggesting that IGF2 requires HIF2α to regulate Runx2 expression (Fig. 6F).
Fig. 6.

Ex vivo bone organ culture and in vitro chondrocyte analyses of the Igf2 null bone. (A) Representative images of metatarsal bones of WT and Igf2 null after 7 days of ex vivo culture. (B) Percentage of growth in length and width of metatarsal bones after 7 days of culture. (C) Alcian Blue and Hematoxylin staining of the metatarsal bones after 7 days of culture. The ratio of the lengths of hypertrophic zone (HZ)/total length of the cartilage template, as well as the ratio of the columnar zone (CZ)/HZ between the WT and Igf2 null bones were compared. For A-C, at least ten bones were analyzed for each group (WT and Igf2 null). Scale bars: 200 μm. (D) RT-qPCR analysis of Igf2 null and WT epiphyseal chondrocytes after 1 week of culturing in alginate beads. Data are presented as the ratio of Igf2 null/WT gene expression. (E) RT-qPCR analysis of epiphyseal chondrocytes treated with exogenous 10 ng/ml IGF2 and cultured in 3D alginate beads for 1 week. Data are presented as the ratio of gene expression from IGF2 treatment versus non-treated control in WT chondrocytes. (F) RT-qPCR analysis of selected genes after the treatment with an HIF2α inhibitor, HIF2 antagonist 2. For D-F, the experiments were conducted with biological triplicates and repeated three times. Mean±s.d. *P<0.05, **P<0.01, ***P<0.001 (D,E: unpaired t-test between WT and Igf2 null; F: ANOVA followed by Dunnett's test).
IGF2 regulates glucose metabolism in growth plate chondrocytes
To investigate the underlying mechanisms further, we tested the possible role of IGF2 in regulating glucose metabolism, based on prior reports showing IGF2 promoting glucose uptake in embryonic limb bud chondrocytes (Bhaumick and Bala, 1991), and altering glucose metabolism impaired extracellular matrix (ECM) production (Heywood et al., 2014; Martin et al., 2012; Nishida et al., 2013). We found that IGF2 treatment of WT postnatal epiphyseal chondrocytes in 3D alginate beads significantly increased glucose uptake by the end of the 7 day culture period, but did not significantly affect the total amount of glucose consumed in this period (Fig. 7A). Typically, as chondrocytes reside in a hypoxic environment, they are known to rely mostly on glycolysis to generate energy (Rajpurohit et al., 1999). On the other hand, chondrocytes consume a basal level of oxygen to sustain the mitochondrial respiratory chain to generate oxidants such as reactive oxygen species (ROS) as electron acceptors to maintain cellular redox balance. As such, glycolysis and electron transport are coupled (Lane et al., 2015; Martin et al., 2012). To investigate how these processes are changed in the Igf2 null chondrocytes, we assayed the lactate level as a readout of glycolysis, and oxygen consumption rate (OCR) and ROS production as indicators of cellular respiration. We did not observe any significant changes in lactate production, OCR or ROS levels after IGF2 treatment, which is consistent with the observation that the ATP production level was also not significantly changed (Fig. 7A).
Fig. 7.

Igf2 null chondrocytes have altered glucose metabolism. (A,B) Analysis of glucose uptake in chondrocytes at day 7 of culture, total glucose consumption after 7 days of culture, L-lactate levels, oxygen consumption rates (OCR), reactive oxygen species (ROS), and intracellular ATP levels at day 7 of culture in WT and Igf2 null in the absence or presence of IGF2. (C) PAS staining images of the metatarsal bones of P10 WT and Igf2 null sections. Arrows indicate punctate PAS-positive staining areas indicating glycogen deposits. The diffuse staining reflects other polysaccharides in the cartilage matrix (see Fig. S6). The magnified areas indicate intense PAS staining in the prehypertrophic zone. (D) RT-qPCR analysis of glucose transporters GLUT1 and GLUT4 in Igf2 null and WT epiphyseal chondrocytes after 1 week of culturing in alginate beads, or WT chondrocytes treated with 10 ng/ml of IGF2. Data are presented as the ratio of Igf2 null/WT gene expression, and the ratio of gene expression from IGF2 treatment versus non-treated control in WT chondrocytes, respectively. (E) Analysis of optic redox ratios FAD/(FAD+NADH) of epiphyseal chondrocytes. (F) Analysis of optic redox ratios FAD/(FAD+NADH) in the columnar zone (CZ), hypertrophic zone (HZ) and secondary ossification center (SOC) using sections of the WT and Igf2 null bones. Mineralized areas in SOC and HZ were not analyzed owing to their endogenous fluorescence. (G) Analysis of optical redox ratios FAD/(FAD+NADH) of chondrocytes using sections of WT and Igf2 null newborn bones cultured in the absence or presence of 10 ng/ml IGF2 for 7 days. Three mice/genotype and four sections/mouse were analyzed. Mean±s.d. *P<0.05 (A,B,D,E,G: ANOVA followed by Dunnett's test; F: unpaired t-test between WT and Igf2 null). Scale bars: 200 µm (C,F,G); 50 µm (E).
However, these metabolic indicators were significantly different between the Igf2 null and the WT. The Igf2 null chondrocytes had consumed twice the amount of glucose compared with WT cells after 7 days of culture, which was restored to basal levels if these cells were cultured with exogenous IGF2 (Fig. 7B). The level of lactate was also elevated in the Igf2 null (Fig. 7B). In addition, OCR and ROS levels were increased in the Igf2 null, and were restored to WT levels if these mutant cells were cultured with exogenous IGF2 (Fig. 7B). Consistent with these observations, Igf2 null chondrocytes exhibited an increased intracellular ATP level compared with WT (Fig. 7B). These results indicate that there is an overall increase in glucose metabolism in the Igf2 null cells, including both glycolysis and oxidative phosphorylation. Because in our culture conditions glycolysis took place even when there was abundant oxygen, we consider chondrocytes to have used aerobic glycolysis, as in the case of cancer cells (Lunt and Vander Heiden, 2011).
Another branch of glucose metabolism is glycogenesis, in which glucose is converted to glycogen rather than continuing with the rest of the glycolysis pathways to generate lactate or go through oxidative phosphorylation (Adeva-Andany et al., 2016). As glycogen has been found to be present in growth plate cartilage (Daimon, 1977; Long et al., 2006; Wang et al., 1999), we also evaluated glycogen levels in the WT and Igf2 metatarsal bones by performing Periodic Acid Schiff (PAS) staining. It is evident that in the WT bone, abundant glycogen was present in the proliferating chondrocytes, especially in the columnar zone (Fig. 7C, Fig. S6). Much less glycogen was detected in the Igf2 null growth plate (Fig. 7C). Thus, it is possible that the lack of IGF2 caused a shift between glycogen storage and glucose usage.
To further probe the underlying mechanisms that caused the difference in glucose uptake and consumption between the WT and the Igf2 null chondrocytes, we examined glucose transporters Glut1 (Slc2a1) and Glut4 (Slc2a4) mRNA expression. Glut4 mRNA levels were slightly decreased in the Igf2 null, and significantly increased in the WT upon IGF2 protein administration (Fig. 7D). IGF2 treatment slightly decreased the Glut1 mRNA level, but no significant difference was observed between WT and Igf2 null, suggesting that IGF2 regulates different glucose transporters in different ways (Fig. 7D). We also performed western blot analysis on growth plate chondrocytes to examine the activation of Akt and AMP-activated kinase (AMPK), which are known to regulate glucose metabolism and can act downstream of IGF signaling (Greene and Loeser, 2015; Xi et al., 2016) but did not observe any differences between the WT and the Igf2 null cells upon IGF2 treatment (Fig. S7). Our prior study on human adult articular chondrocytes also failed to detect Akt activation by IGF2, suggesting that IGF2 may act differently than IGF1 in chondrocytes (Uchimura et al., 2015).
To investigate glucose metabolism in live cells, we assessed the redox levels of growth plate chondrocytes from WT and Igf2 null bones by two-photon fluorescence imaging. This imaging method capitalizes on the endogenous fluorescence emitted by two key co-enzymes in glucose metabolism: the oxidized form of flavin adenine dinucleotide (FAD) and reduced form of nicotinamide dinucleotide (NADH) (Huang et al., 2002). NADH is generated early during glycolysis in the cytoplasm, whereas both NADH and FADH2 are generated in the mitochondria during the Krebs cycle. Then in the electron transport chain, NADH and FADH2 are oxidized back to NAD+ and FAD, respectively (Huang et al., 2002). Because the only significant endogenous fluorescence observed are those from NADH and FAD, which represent opposite metabolic states (reduction versus oxidation) (redox), the balance of glycolysis and oxidative phosphorylation can be reflected by the relative fluorescence of these two molecules. As such, the optical ratio is defined as FAD/(FAD+NADH) (Hassinen and Chance, 1968; Heikal, 2010; Huang et al., 2002; Koenig and Schneckenburger, 1994; Kunz and Kunz, 1985; Masters, 1984; Scholz et al., 1969). For example, under hypoxic conditions, a decrease in oxidative phosphorylation in reference to anaerobic glycolysis is expected, resulting in reduced conversion of NADH to NAD+, thus a surplus of NADH, and ultimately a decrease in the optical redox ratio (Quinn et al., 2013; Varone et al., 2014).
In our analysis, Igf2 null chondrocytes in 3D cultures showed a lower redox ratio than the WT cells, but adding IGF2 to the mutant cells restored the redox ratio to WT levels (Fig. 7E). Whereas our biochemical analysis indicates an increase in both oxidative phosphorylation and glycolysis in the Igf2 null, this optical redox analysis suggests a shift in balance towards glycolysis. To determine whether this is the case in vivo, we analyzed sections of the WT and mutant metatarsal bones. Igf2 null chondrocytes in all zones of the growing cartilage had a lower redox ratio, consistent with the analysis on in vitro cultured chondrocytes (Fig. 7F). As a gain-of-function experiment, we analyzed IGF2-treated metatarsal bones in ex vivo cultures and found that exogenous IGF2 elevated the optical redox ratios to WT levels in the Igf2 null (Fig. 7G). Thus, these analyses indicate that loss of IGF2 causes an overall increase in glucose metabolism, which can be restored by exogenous IGF2.
Attenuating glycolysis restores normal longitudinal growth rate and chondrocyte maturation in Igf2 null bones
To address whether increased glucose metabolism in the Igf2 null causes the chondrocyte maturation phenotype in longitudinal bone growth, we applied a widely used glycolysis inhibitor to Igf2 null bones in ex vivo cultures. This inhibitor, 3-bromopyruvate (3-BrPA), specifically inhibits hexokinase II, a key enzyme that converts glucose to glucose-6-phosphate (Geschwind et al., 2002; Jones et al., 1995; Ko et al., 2004; Sugita et al., 2011). Significantly, although the Igf2 null bones were consistently smaller than the WT bone, the rate of longitudinal growth of the Igf2 null bone after 3-BrPA treatment became similar to that of the WT bone (Fig. 8A,B). Histological analysis demonstrated that 3-BrPA caused a decrease in the length of the hypertrophic zone and an increase in the ratio of the columnar zone over the hypertrophic zone (Fig. 8C,D), thus rescuing the defect in chondrocyte maturation of the Igf2 null bone. Interestingly, we did not observe significant differences in WT chondrocytes caused by 3-BrPA administration. It is possible that the amount of 3-BrPA used in our experiments was not high enough to lower glycolysis to a level that affected WT development; but was high enough to reduce elevated glucose metabolism in the Igf2 null. There was no significant difference in the intensity of Col-X staining between the WT and the mutant (Fig. 8E). However, very little Ki67 staining was observed in the mutant, and treatment with 3-BrPA significantly increased the number of Ki67-positive cells, suggesting of an enhancement of chondrocyte proliferation (Fig. 8E). Therefore, this study indicates that the regulatory role of IGF2 on chondrocyte maturation and matrix production in endochondral ossification is mediated by its activity on glucose metabolism in chondrocytes.
Fig. 8.

Attenuating glycolysis rescues the phenotype of Igf2 null bones. (A) Representative images of ex vivo WT and Igf2 null metatarsal bones cultured in the absence or presence of 1 µM glycolysis inhibitor 3-BrPA for 7 days. (B) Percentage of growth of metatarsal bones after 7 days of culture. (C) Alcian Blue and Hematoxylin staining of the metatarsal bones after 7 days of culture. Magnified views of the proliferation and hypertrophic zones of the growth plate are presented on the right. (D) The ratio of the lengths of hypertrophic zone (HZ)/total length of the cartilage template, as well as the ratio of the columnar zone (CZ)/HZ between the WT and Igf2 null bones were compared. Six bones/group were analyzed. (E) Images of IHC for Col-X and Ki67 in Igf2 null metatarsal bones grown the absence or presence of 3-BrPA. In C and E, red, blue and black bars indicate the hypertrophic zone (HZ), the columnar zone (CZ) and the epiphyseal zone (EZ), respectively. Scale bars: 200 µm. Mean±s.d. At least three repeats were performed. *P<0.05 (ANOVA followed by Dunnett's test).
DISCUSSION
In this study, we show that IGF2 is an essential factor for coordinated longitudinal and appositional growth as well as glucose metabolism in early postnatal cartilage development (Fig. 9). Earlier studies have indicated that IGF2 is widely expressed in many tissues during embryonic and early postnatal stages prior to the weaning age (DeChiara et al., 1991; Liu, 1993), but its function on bone growth has not been defined. Our age-matched and length-matched analyses of WT and mutant metatarsal bones demonstrate that IGF2 regulates chondrocyte development in several ways.
Fig. 9.
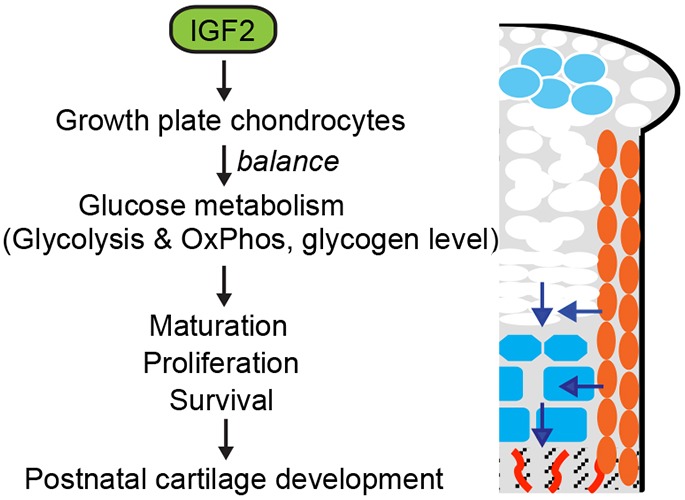
Model of the role of IGF2 in regulating postnatal bone growth. IGF2 maintains the balance of glycolysis and glucose metabolism, as wells as glycogen levels, controlling the pace of hypertrophic differentiation and chondrocyte cell survival and cell proliferation during postnatal long bone growth. Blue represents hypertrophic chondrocytes; orange represents periochondrial cells. Arrows indicate direction of movement as cells mature.
First, IGF2 regulates the progression of chondrocytes from the proliferating to the hypertrophic phase. As indicated by Ihh staining, the Igf2 mutant has a shorter prehypertrophic phase than the WT. Perhaps as a result, mutant hypertrophic chondrocytes retained higher levels of Sox9 expression, which may explain the abnormal expression of many of the hypertrophic markers in the mutant (Runx2, HIF2α). Although matrix protein expression (Col-II and Col-X) generally did not appear altered when detected by IHC, a reduction in their mRNA expression in isolated epiphyseal chondrocytes was indeed observed. The shortened prehypertrophic phase and retained Sox9 expression in the hypertrophic zone of the mutant are consistent with the notion of an accelerated pace toward bone maturation during longitudinal growth in the absence of IGF2. As a result, by the time the Igf2 null bone reaches the same length as the WT bone, it shows features of a more mature bone in terms of longitudinal bone growth compared with age-matched WT, including a shortened growth plate and higher levels of ALP expression (Fig. 4C). It has been reported that SOC formation is dependent on thyroid hormone-induced Ihh and osterix (Sp7) activation (Xing et al., 2014). As Igf2 null bones have smaller Ihh-positive prehypertrophic chondrocytes, it is also possible that a delay in SOC formation is caused by altered Ihh expression. Additionally, because the Igf2 null bones are smaller and narrower, the center of the epiphysis could have higher oxygen tension, which would delay HIF1α expression and vascular invasion (Araldi and Schipani, 2010; Maes et al., 2012).
Second, IGF2 regulates perichondrial cell proliferation and differentiation. Perichondrium is plays a crucial role in long bone elongation by facilitating the PTHrP/Ihh signaling loop as well as providing a source of osteoblasts (Colnot et al., 2004; Derks et al., 2013; Hill et al., 2005; Hojo et al., 2013; Kobayashi et al., 2011; Srour et al., 2015; Van Osch et al., 2000). However, mechanisms to ensure perichondrial cell proliferation and survival during rapid postnatal bone growth are not well understood. Our data suggest that IGF2 is required for perichondrial cell proliferation and survival as well as for Sox9 and Runx2 expression in the inner perichondrium, which may account for the reduced perichondrium and bone thickness in the Igf2 null bones. It will be interesting to investigate further the relationship between IGF2 and these other signaling pathways in perichondrial cell proliferation and chondrogenic differentiation. As perichondrial cells are heterogeneous (Hojo et al., 2013), it will also be interesting to determine whether IGF2 promotes chondrogenic and osteogenic differentiation from the same progenitor cells or simply maintains the progenitor population.
It has been reported that IGF2 is required for embryonic growth as well as overall postnatal body growth (Baker et al., 1993; Burns and Hassan, 2001; DeChiara et al., 1990, 1991; Louvi et al., 1997). However, we have analyzed at least eight metatarsal bones per stage, and did not observe significant differences in the length and width of the mutant and WT embryonic bones. It is possible that a study involving a much larger number of embryos might reveal statistical differences, or that the differences between the mutant and the WT might be more pronounced in larger bones. Nevertheless, our data suggests that IGF2 has a relatively subtle activity on embryonic metatarsal bone growth.
It is not clear how IGF2 exerts its activity on cartilage development. IGF2 is capable of binding to IGF1R, IGF2R and the insulin receptor (IR; INSR), although its affinity to IR is low (Hartmann et al., 1992). Additionally, their activities are modulated by various IGF-binding proteins (Fisher et al., 2005). The Igf1r null mouse has shorter bones, but the IR null mouse has a normal bone length (Wang et al., 1999; Zhang et al., 2014). However, both knockouts had a reduced hypertrophic zone (Wang et al., 1999, 2015; Zhang et al., 2014), which is consistent with the phenotype of the Igf1 knockout (Wang et al., 1999). Igf2 null bones, on the other hand, exhibit a disproportionally larger hypertrophic zone. These data indicate different roles of IGF1 and IGF2 on cartilage development. Consistent with this notion, although IGF2 overexpression promoted postnatal growth, it failed to compensate the phenotype caused by the loss of IGF1 (Moerth et al., 2007). IGF2 is unique in its ability to bind to IGF2R (Takigawa et al., 1997), and it has been shown that the direct binding of IGF2 to IGF2R stimulated proteoglycan synthesis and induced calcium influx in chondrocytes, as it occurs even in the presence of an anti-IGF-IR antibody (Poiraudeau et al., 1997; Sessions et al., 1987; Takigawa et al., 1997). On the other hand, knockout of Igf2r exhibits increased skeletal growth (Eggenschwiler et al., 1997). Thus, none of the single knockout of the potential receptors exhibits the same phenotype as that of the Igf2 null mouse. However, it is possible that the phenotype of the Igf2 null mouse is a result of its binding to multiple receptors, together with interaction of multiple IGF-binding proteins and subsequent complex downstream signaling. Thus, it will be important to evaluate these IGF signaling components in the context of IGF2 in future experiments.
Although it has been recently discovered that glucose metabolism regulates osteogenesis (Esen et al., 2013; Wei et al., 2015), its role in cartilage development is largely unknown. Significantly, our study indicates that IGF2 is required to regulate glucose metabolism, possibly by keeping both anaerobic glycolysis and oxidative phosphorylation under control. It is not clear why both Igf2 null chondrocytes and IGF2-treated chondrocytes had increased glucose uptake, and why such increased glucose uptake is coupled to increased glucose metabolism in the Igf2 null bone. It is plausible that IGF2 alters the presence of glucose transporters on the cell membrane and the balance of glucose breakdown and glycogen storage. Perhaps an optimal amount of glucose metabolism is required for proper cartilage development. Our optical imaging data on live cells as well as tissue sections indicates that substrate utilization can be very dynamic. It suggests a shift toward anaerobic glycolysis over oxidative phosphorylation in the Igf2 null chondrocytes, even though both were apparently increased in Igf2 null chondrocytes based on results from biochemical testing. Shapiro et al. showed in the growing tibial cartilage of 4-week-old chicks a higher NADH fluorescence in the hypertrophic zone and an increased NAD+/NADH fluorescence ratio when cartilage became calcified, although no signals that regulate this process have been elucidated (Shapiro et al., 1982). We did not observe such a dramatic difference in the redox ratio, perhaps because we also have taken the endogenous fluorescence of FAD into account, or because of a difference in species and age of the animals as well as imaging methods. Shapiro et al. employed scanning microfluorimetry to record fluorescence signals with a resolution of 100 µm, whereas we used two-photon excited fluorescence microscopy with a resolution in the order of a micron. Nevertheless, both data support the notion that regulation of glucose metabolism plays a role in cartilage development. Although current technology does not allow precise measurement of each aspect of glucose metabolism (such as glucose uptake and consumption) in the whole animal as in in vitro situations, we have used the best available in vivo assessment method to indicate an altered glucose metabolism in vivo in the Igf2 null mouse, thus complementing the biochemical analysis.
Altered levels of glucose metabolism in chondrocytes cause multiple effects. Pathways regulating glucose metabolism are known to influence body growth (Baker et al., 1993; Zhu et al., 2011). It was reported that culturing chondrocytes in high-glucose medium led to reduced chondrogenic phenotypes (Heywood et al., 2014), and that inhibiting glycolysis impaired ECM production (Nishida et al., 2013). It has also been reported that higher ATP production in chondrocytes led to increased chondrocyte cell death in a knockout mouse with reduced phosphate level (Hyp mouse) (Sugita et al., 2011). Interestingly, in these mutant bones, an elongated hypertrophic zone was found, which we also observed in the Igf2 null mouse. Strikingly, attenuating glycolysis with the hexokinase II inhibitor 3-BrPA in growing Igf2 null bones rescued this phenotype, suggesting that enhanced chondrocyte hypertrophy in the Igf2 null cells results from higher glucose levels and that elevated glucose metabolism promotes chondrocyte maturation. It is not clear how IGF2 elicits its activity on glucose metabolism. It has been reported that ROS promote chondrocyte hypertrophy and cell death during endochondral ossification (Del Carlo and Loeser, 2002; Morita et al., 2007; Zuscik et al., 2008). However, it is unclear whether it is the case for the Igf2 null bone. Future studies will focus on determining the underlying mechanisms, including the receptors and downstream pathways.
In mice, it has been demonstrated that neonatal or early postnatal growth takes place in a growth hormone-independent manner (Stratikopoulos et al., 2008). In humans, it was also reported that some infants with growth hormone deficiency grow normally in the first 6 to 9 months (Ogilvy-Stuart, 2003; Wit and van Unen, 1992). Altered IGF2 activity is associated with Beckwith–Wiedemann syndrome and Silver–Russell syndrome (Bartholdi et al., 2009; Poole et al., 2012). Most recently, a nonsense mutation (p.Ser64Ter) was identified in a multi-generational family of postnatal growth restriction, resembling Silver–Russell syndrome (Begemann et al., 2015). Very similar growth curves were observed in these patients compared with the growth curve of Igf2 null mice. This mutation is expected to result in a truncated IGF2 protein that is devoid of binding sites to the receptor and IGF-binding proteins, rendering a loss-of-function phenotype. Although the underlying mechanisms for IGF2-associated human conditions are not completely understood, these studies indicate a clinical relevance of our investigation and support the notion that Igf2 null mice can be used to model growth deficiency conditions in humans.
MATERIALS AND METHODS
Experimental animals
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committees of Tufts University. Mice were housed with standard chow diet under 14 h light/10 h dark cycle.
Murine epiphyseal chondrocytes in vitro culture
Murine epiphyseal chondrocytes were isolated based on a previously published protocol (Gosset et al., 2008). P1 chondrocytes were encapsulated in 1.2% alginate beads at a density of 8×105 cells/ml and cultured in DMEM/F-12, 10% fetal bovine serum (FBS), 25.5 μg/ml ascorbic acid for 7 days before being subjected to RT-qPCR and glucose metabolism assays. Beads were treated with 10 ng/ml IGF2 (Peprotech) and/or 10 µM HIF-2 antagonist 2 (Sigma). For collecting encapsulated cells, alginate beads were dissolved in 55 mM sodium citrate buffer supplemented with 10 mM HEPES.
Metatarsal bones ex vivo culture
The middle three developing metatarsal bones were isolated from newborn mice and cultured based on a protocol adapted from other published reports (Mårtensson et al., 2004; Minina et al., 2001). Briefly, isolated bones were cultured in DMEM supplemented with 0.25% FBS, 50 μg/ml ascorbic acid and 1.25 mM sodium pyruvate (Sigma) for 7 days in the absence or presence of 10 mg/ml IGF2 or 1 µM 3-bromopyruvate (3-BrPA).
Tissue preparation for cryo/paraffin sectioning
Samples were fixed in 1% paraformaldehyde and then decalcified in 0.33 M EDTA. Samples were embedded in OCT (Tissue-Tek Sakura) for cryosections and paraffin (Paraplast Plus, McCormick Scientific) for paraffin sections. All were sectioned at 5 µm thickness.
Histological analysis
For Hematoxylin and Eosin staining, sections were stained with Hematoxylin for 10 min and 1% Eosin for 30 s. For Alcian Blue staining, sections were stained with 1% (w/v) Alcian Blue and counterstained with Hematoxylin. Whole-mount Alcian Blue/Alizarin Red staining was performed based on a previously published protocol (Ovchinnikov, 2009). For collagen II (Col-II), collagen X (Col-X) and MMP13 IHC, enzymatic antigen retrieval was used (0.3% hyaluronidase and 0.15% trypsin at 37°C, 12 min). For CD44, Ki67, Runx2, Sox9, HIF1α, HIF2α and Ihh staining, heat-induced antigen retrieval was used (10 mM citric acid buffer, pH 6.0 for 10 min in steam). Information regarding primary antibody sources and dilutions is listed in Table S1. All antibodies were validated by western blots and IHC analyses with positive and negative controls, in reference to literature. For fluorescence staining, 15 µg/ml Dylight594 Streptavidin (Vector Laboratories) was sometimes used for Ki67 and Sox9 staining. For chromogenic staining, the VECTASTAIN ABC Elite kit (Vector Laboratories) was used. For staining involving mouse primary antibodies, the M.O.M. kit (Vector Laboratories) was used. Alkaline phosphatase activity was assayed by washing cryosections with NTMT (100 mM NaCl, 100 mM Tris-HCl pH 9.5, 50 mM MgCl2, 0.1% Tween-20) and incubating with NBT/BCIP (Roche). TUNEL assay was performed using the TUNEL in situ cell death detection kit (Roche). EdU assay involves the intraperitoneal injection of EdU (50 µg/g body weight) and analyzing the tissues using the Click-iT EdU imaging kit (Invitrogen) 2 h later. For PAS staining, cryosections were rehydrated in water for 1 h, and incubated with 0.5% periodic acid for 5 min, then incubated in Schiff reagent for 15 min. As controls for specificity for glycogen staining, sections were either treated with 100% saliva or 10% α-amylase (Sigma) for 1 h before periodic acid treatment.
RT-qPCR analysis
Total RNA was isolated using the RNeasy mini kit (Qiagen) and reverse transcribed using the M-MLV reverse transcriptase (Invitrogen). qPCR was carried out using the iQ5 Real Time Detection System (Bio-Rad). TATA-binding protein (Tbp) served as a reference gene.
Western blot analysis
Isolated murine epiphyseal chondrocytes at passage 2 were grown to confluency in DMEM/F12, 10% FBS before culturing in serum-free DMEM/F12 medium for 16 h. The cells were subsequently treated with or without 10 mg/ml IGF2 in serum-free medium for 1 h, and then lysed in the ‘Phosphatase inhibition’ protein isolation buffer (300 mM NaCl, 5 mM EDTA, 50 mM NaF, 40 mM sodium pyrophosphate, 50 nM K2PO4, 10 mM sodium molybdate) (Fagotto et al., 1999) supplemented with 2 mM orthovanadate, 0.5% Triton X-100, 0.6 mM DTT, 0.2 M Tris-HCl pH 7.5, and 0.1% Protease inhibitor cocktail set III (Millipore). Protein concentrations were determined using the DC Protein Assay (Bio-Rad). Primary and secondary antibody information can be found in Table S1. Bands were visualized using the Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific).
Glucose metabolism assays
For glucose uptake, glucose-free DMEM with 100 μg/ml 2-NBDG was applied to the chondrocytes, and the uptake of 2-NBDG was assayed 30 min later (Glucose Uptake Cell-based Assay Kit, Cayman Chemical) using a plate reader (FlexStation II, Molecular Devices). For glucose consumption, 3 μl of the media was incubated with 250 μl assay reagent [Glucose (HK) assay kit (Sigma)], and read in a plate reader (Bio-Rad Benchmark Plus). For the L-lactate assay, the medium was de-proteinized with a 10 kDa MWCO spin filter to remove lactate dehydrogenase, and then incubated with the assay buffer (L-lactate assay kit I, Eton Biosciences), and read in a plate reader (Bio-Rad Benchmark Plus). For intracellular ATP, chondrocytes cultured in alginate beads for 7 days were lysed in 200 mM Tris pH 7.5, 2 M NaCl, 20 mM EDTA and 0.2% Triton X-100 for 5 min, and assayed using the ATP Determination Kit (Invitrogen), and read in microplate luminometer (Perkin Elmer, 1450 Microbeta TriLux). For OCR and ROS assays, chondrocytes cultured in alginate beads were extracted and plated in 96-well plates at 5×104 cells/well. OCR was assayed using the OCR kit (Cayman Chemical), and ROS was assayed using the Image-iT Live Green Reactive Oxygen Species Detection Kit (Invitrogen). Both were read in a plate reader (FlexStation II, Molecular devices). In all assays, the total cell numbers based on Hoechst reading were used for normalization.
Microscopy
Metatarsal bones were viewed under the Leica MZ16F stereomicroscope. Sections were viewed under an Olympus IX71 inverted microscope. Images were acquired using Olympus DP70 digital camera and associated software. For redox imaging, two-photon excited fluorescence microscopy was performed on a Leica TCS SP2 confocal microscope equipped with a tunable (710-920 nm) titanium-sapphire laser (Mai Tai; Spectra Physics). NADH fluorescence images were acquired with an excitation 755 nm and emission 460 (±20) nm filter. FAD images were acquired with an excitation 860 nm and emission 525 (±25) nm filter (Huang et al., 2002; Quinn et al., 2013).
Statistical analysis
Calculations of power and sample size were performed using Intercooled Stata version 14 (College Station, TX, USA). For all experiments, at least three independent experiments were conducted, with triplicate biological repeats/experiment. Outlier analysis was performed using the GraphPad outlier calculator program (Burns et al., 2005). Data were reported as mean±s.d., and analyzed using an unpaired t-test or one-way analysis of variance followed by post hoc Dunnett's test using GraphPad Prism. P<0.05 is considered significant.
Acknowledgements
We thank Dr Margaret Goodell (Baylor College of Medicine) for providing the Igf2 null mice; Dr Thomas Linsenmayer (Tufts University) for the collagen antibodies; and Dr Brent Cochran (Tufts University) for the HIF1α and HIF2α antibodies. We thank Dr Heber Nielsen (Tufts Medical Center) for advice on statistical analysis. We thank Dr Henry Kronenberg (Harvard Medical School) for fruitful discussions and review of this work, and Averi Gibson, and Carrie Hui Mingalone, Nicholas Spetko, and Ramesh Govindan (Tufts University) for critically reading the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: T.U.; Methodology: T.U.; Software: T.U.; Validation: T.U., J.M.H.; Formal analysis: T.U., J.M.H., D.S.N., Z.L., I.G.; Investigation: T.U., J.M.H., D.S.N., I.G., L.Z.; Resources: T.U., Z.L., I.G.; Data curation: T.U., J.M.H., D.S.N., Z.L., I.G., L.Z.; Writing - original draft: T.U., D.S.N., C.J.R., I.G., L.Z.; Writing - review & editing: T.U., J.M.H., D.S.N., C.J.R., L.Z.; Visualization: T.U., L.Z.; Supervision: C.J.R., L.Z.; Project administration: L.Z.; Funding acquisition: L.Z.
Funding
This work has been supported by a Collaborates Grant from Tufts University School of Medicine (M250001 to L.Z.), grants from the National Institutes of Health (AR054611 and AR069278 to L.Z.), the National Science Foundation (CBET-0966920) and a grant from the Tufts Clinical Translational Science Institute (UL1TR001064). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.155598.supplemental
References
- Adeva-Andany M. M., González-Lucán M., Donapetry-García C., Fernández-Fernández C. and Ameneiros-Rodriguez E. (2016). Glycogen metabolism in humans. BBA Clin 5, 85-100. 10.1016/j.bbacli.2016.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H., Chaboissier M. C., Martin J. F., Schedl A. and de Crombrugghe B. (2002). The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 16, 2813-2828. 10.1101/gad.1017802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarilio R., Viukov S. V., Sharir A., Eshkar-Oren I., Johnson R. S. and Zelzer E. (2007). HIF1alpha regulation of Sox9 is necessary to maintain differentiation of hypoxic prechondrogenic cells during early skeletogenesis. Development 134, 3917-3928. 10.1242/dev.008441 [DOI] [PubMed] [Google Scholar]
- Araldi E. and Schipani E. (2010). Hypoxia, HIFs and bone development. Bone 47, 190-196. 10.1016/j.bone.2010.04.606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker J., Liu J.-P., Robertson E. J. and Efstratiadis A. (1993). Role of insulin-like growth factors in embryonic and postnatal growth. Cell 75, 73-82. 10.1016/S0092-8674(05)80085-6 [DOI] [PubMed] [Google Scholar]
- Bartholdi D., Krajewska-Walasek M., Ounap K., Gaspar H., Chrzanowska K. H., Ilyana H., Kayserili H., Lurie I. W., Schinzel A. and Baumer A. (2009). Epigenetic mutations of the imprinted IGF2-H19 domain in Silver-Russell syndrome (SRS): results from a large cohort of patients with SRS and SRS-like phenotypes. J. Med. Genet. 46, 192-197. 10.1136/jmg.2008.061820 [DOI] [PubMed] [Google Scholar]
- Bastepe M., Weinstein L. S., Ogata N., Kawaguchi H., Juppner H., Kronenberg H. M. and Chung U.-I. (2004). Stimulatory G protein directly regulates hypertrophic differentiation of growth plate cartilage in vivo. Proc. Natl. Acad. Sci. USA 101, 14794-14799. 10.1073/pnas.0405091101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begemann M., Zirn B., Santen G., Wirthgen E., Soellner L., Buttel H. M., Schweizer R., van Workum W., Binder G. and Eggermann T. (2015). Paternally inherited IGF2 mutation and growth restriction. N. Engl. J. Med. 373, 349-356. 10.1056/NEJMoa1415227 [DOI] [PubMed] [Google Scholar]
- Bhaumick B. and Bala R. M. (1991). Differential effects of insulin-like growth factors I and II on growth, differentiation and glucoregulation in differentiating chondrocyte cells in culture. Acta Endocrinol. 125, 201-211. 10.1530/acta.0.1250201 [DOI] [PubMed] [Google Scholar]
- Bitgood M. J. and McMahon A. P. (1995). Hedgehog and Bmp genes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Dev. Biol. 172, 126-138. 10.1006/dbio.1995.0010 [DOI] [PubMed] [Google Scholar]
- Blumer M. J. F., Longato S. and Fritsch H. (2008). Structure, formation and role of cartilage canals in the developing bone. Ann. Anat. 190, 305-315. 10.1016/j.aanat.2008.02.004 [DOI] [PubMed] [Google Scholar]
- Burns J. L. and Hassan A. B. (2001). Cell survival and proliferation are modified by insulin-like growth factor 2 between days 9 and 10 of mouse gestation. Development 128, 3819-3830. [DOI] [PubMed] [Google Scholar]
- Burns M. J., Nixon G. J., Foy C. A. and Harris N. (2005). Standardisation of data from real-time quantitative PCR methods - evaluation of outliers and comparison of calibration curves. BMC Biotechnol. 5, 31 10.1186/1472-6750-5-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colnot C., Lu C., Hu D. and Helms J. A. (2004). Distinguishing the contributions of the perichondrium, cartilage, and vascular endothelium to skeletal development. Dev. Biol. 269, 55-69. 10.1016/j.ydbio.2004.01.011 [DOI] [PubMed] [Google Scholar]
- Daimon T. (1977). The presence and distribution of glycogen particles in chondrogenic cells of the tibiotarsal anlage of developing chick embryos. Calcif. Tissue Res. 23, 45-51. 10.1007/BF02012765 [DOI] [PubMed] [Google Scholar]
- De Ceuninck F., Lesur C., Pastoureau P., Caliez A. and Sabatini M. (2004). Culture of chondrocytes in alginate beads. Methods Mol. Med. 100, 15-22. 10.1385/1-59259-810-2:015 [DOI] [PubMed] [Google Scholar]
- DeChiara T. M., Efstratiadis A. and Robertsen E. J. (1990). A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 345, 78-80. 10.1038/345078a0 [DOI] [PubMed] [Google Scholar]
- DeChiara T. M., Robertson E. J. and Efstratiadis A. (1991). Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64, 849-859. 10.1016/0092-8674(91)90513-X [DOI] [PubMed] [Google Scholar]
- Del Carlo M. Jr and Loeser R. F. (2002). Nitric oxide-mediated chondrocyte cell death requires the generation of additional reactive oxygen species. Arthritis. Rheum. 46, 394-403. 10.1002/art.10056 [DOI] [PubMed] [Google Scholar]
- Derks M., Sturm T., Haverich A. and Hilfiker A. (2013). Isolation and chondrogenic differentiation of porcine perichondrial progenitor cells for the purpose of cartilage tissue engineering. Cells Tissues Organs 198, 179-189. 10.1159/000354897 [DOI] [PubMed] [Google Scholar]
- Eggenschwiler J., Ludwig T., Fisher P., Leighton P. A., Tilghman S. M. and Efstratiadis A. (1997). Mouse mutant embryos overexpressing IGF-II exhibit phenotypic features of the Beckwith-Wiedemann and Simpson-Golabi-Behmel syndromes. Genes Dev. 11, 3128-3142. 10.1101/gad.11.23.3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esen E., Chen J., Karner C. M., Okunade A. L., Patterson B. W. and Long F. (2013). WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab. 17, 745-755. 10.1016/j.cmet.2013.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagotto F., Jho E., Zeng L., Kurth T., Joos T., Kaufmann C. and Costantini F. (1999). Domains of axin involved in protein-protein interactions, Wnt pathway inhibition, and intracellular localization. J. Cell Biol. 145, 741-756. 10.1083/jcb.145.4.741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher M. C., Meyer C., Garber G. and Dealy C. N. (2005). Role of IGFBP2, IGF-I and IGF-II in regulating long bone growth. Bone 37, 741-750. 10.1016/j.bone.2005.07.024 [DOI] [PubMed] [Google Scholar]
- Geschwind J. F., Ko Y. H., Torbenson M. S., Magee C. and Pedersen P. L. (2002). Novel therapy for liver cancer: direct intraarterial injection of a potent inhibitor of ATP production. Cancer Res. 62, 3909-3913. [PubMed] [Google Scholar]
- Gosset M., Berenbaum F., Thirion S. and Jacques C. (2008). Primary culture and phenotyping of murine chondrocytes. Nat. Protoc. 3, 1253-1260. 10.1038/nprot.2008.95 [DOI] [PubMed] [Google Scholar]
- Greene M. A. and Loeser R. F. (2015). Function of the chondrocyte PI-3 kinase-Akt signaling pathway is stimulus dependent. Osteoarthritis Cartilage 23, 949-956. 10.1016/j.joca.2015.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann H., Meyer-Alber A. and Braulke T. (1992). Metabolic actions of insulin-like growth factor II in cultured adult rat hepatocytes are not mediated through the insulin-like growth factor II receptor. Diabetologia 35, 216-223. 10.1007/BF00400920 [DOI] [PubMed] [Google Scholar]
- Hassinen I. and Chance B. (1968). Oxidation-reduction properties of the mitochondrial flavoprotein chain. Biochem. Biophys. Res. Commun. 31, 895-900. 10.1016/0006-291X(68)90536-6 [DOI] [PubMed] [Google Scholar]
- Heikal A. A. (2010). Intracellular coenzymes as natural biomarkers for metabolic activities and mitochondrial anomalies. Biomark Med. 4, 241-263. 10.2217/bmm.10.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heywood H. K., Nalesso G., Lee D. A. and Dell'accio F. (2014). Culture expansion in low-glucose conditions preserves chondrocyte differentiation and enhances their subsequent capacity to form cartilage tissue in three-dimensional culture. Biores. Open Access 3, 9-18. 10.1089/biores.2013.0051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill T. P., Später D., Taketo M. M., Birchmeier W. and Hartmann C. (2005). Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 8, 727-738. 10.1016/j.devcel.2005.02.013 [DOI] [PubMed] [Google Scholar]
- Hojo H., Ohba S., Taniguchi K., Shirai M., Yano F., Saito T., Ikeda T., Nakajima K., Komiyama Y., Nakagata N. et al. (2013). Hedgehog-Gli activators direct osteo-chondrogenic function of bone morphogenetic protein toward osteogenesis in the perichondrium. J. Biol. Chem. 288, 9924-9932. 10.1074/jbc.M112.409342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Chung U.-I., Kronenberg H. M. and de Crombrugghe B. (2001). The chondrogenic transcription factor Sox9 is a target of signaling by the parathyroid hormone-related peptide in the growth plate of endochondral bones. Proc. Natl. Acad. Sci. USA 98, 160-165. 10.1073/pnas.98.1.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S., Heikal A. A. and Webb W. W. (2002). Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys. J. 82, 2811-2825. 10.1016/S0006-3495(02)75621-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A. R., Gillan L. and Milmlow D. (1995). The anti-glycolytic activity of 3-bromopyruvate on mature boar spermatozoa in vitro. Contraception 52, 317-320. 10.1016/0010-7824(95)00217-X [DOI] [PubMed] [Google Scholar]
- Kake T., Kitamura H., Adachi Y., Yoshioka T., Watanabe T., Matsushita H., Fujii T., Kondo E., Tachibe T., Kawase Y. et al. (2009). Chronically elevated plasma C-type natriuretic peptide level stimulates skeletal growth in transgenic mice. Am. J. Physiol. Endocrinol. Metab. 297, E1339-E1348. 10.1152/ajpendo.00272.2009 [DOI] [PubMed] [Google Scholar]
- Klammt J., Kiess W. and Pfäffle R. (2011). IGF1R mutations as cause of SGA. Best Pract. Res. Clin. Endocrinol. Metab. 25, 191-206. 10.1016/j.beem.2010.09.012 [DOI] [PubMed] [Google Scholar]
- Ko Y. H., Smith B. L., Wang Y., Pomper M. G., Rini D. A., Torbenson M. S., Hullihen J. and Pedersen P. L. (2004). Advanced cancers: eradication in all cases using 3-bromopyruvate therapy to deplete ATP. Biochem. Biophys. Res. Commun. 324, 269-275. 10.1016/j.bbrc.2004.09.047 [DOI] [PubMed] [Google Scholar]
- Kobayashi S., Takebe T., Inui M., Iwai S., Kan H., Zheng Y.-W., Maegawa J. and Taniguchi H. (2011). Reconstruction of human elastic cartilage by a CD44+ CD90+ stem cell in the ear perichondrium. Proc. Natl. Acad. Sci. USA 108, 14479-14484. 10.1073/pnas.1109767108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig K. and Schneckenburger H. (1994). Laser-induced autofluorescence for medical diagnosis. J. Fluoresc. 4, 17-40. 10.1007/BF01876650 [DOI] [PubMed] [Google Scholar]
- Kronenberg H. M. (2003). Developmental regulation of the growth plate. Nature 423, 332-336. 10.1038/nature01657 [DOI] [PubMed] [Google Scholar]
- Kronenberg H. M. (2006). PTHrP and skeletal development. Ann. N. Y. Acad. Sci. 1068, 1-13. 10.1196/annals.1346.002 [DOI] [PubMed] [Google Scholar]
- Kunz W. S. and Kunz W. (1985). Contribution of different enzymes to flavoprotein fluorescence of isolated rat liver mitochondria. Biochim. Biophys. Acta 841, 237-246. 10.1016/0304-4165(85)90064-9 [DOI] [PubMed] [Google Scholar]
- Lane R. S., Fu Y., Matsuzaki S., Kinter M., Humphries K. M. and Griffin T. M. (2015). Mitochondrial respiration and redox coupling in articular chondrocytes. Arthritis Res. Ther. 17, 54 10.1186/s13075-015-0566-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. (1993). Mice carrying null mutations of the genes encoding insulinlike growth factor I (lgt-1) and type 1 IGF receptor (lgf1r). Cell 75, 59-72. 10.1016/0092-8674(93)90679-K [DOI] [PubMed] [Google Scholar]
- Long F., Joeng K.-S., Xuan S., Efstratiadis A. and McMahon A. P. (2006). Independent regulation of skeletal growth by Ihh and IGF signaling. Dev. Biol. 298, 327-333. 10.1016/j.ydbio.2006.06.042 [DOI] [PubMed] [Google Scholar]
- Louvi A., Accili D. and Efstratiadis A. (1997). Growth-promoting interaction of IGF-II with the insulin receptor during mouse embryonic development. Dev. Biol. 189, 33-48. 10.1006/dbio.1997.8666 [DOI] [PubMed] [Google Scholar]
- Lunt S. Y. and Vander Heiden M. G. (2011). Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27, 441-464. 10.1146/annurev-cellbio-092910-154237 [DOI] [PubMed] [Google Scholar]
- Lupu F., Terwilliger J. D., Lee K., Segre G. V. and Efstratiadis A. (2001). Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev. Biol. 229, 141-162. 10.1006/dbio.2000.9975 [DOI] [PubMed] [Google Scholar]
- Maes C., Araldi E., Haigh K., Khatri R., Van Looveren R., Giaccia A. J., Haigh J. J., Carmeliet G. and Schipani E. (2012). VEGF-independent cell-autonomous functions of HIF-1alpha regulating oxygen consumption in fetal cartilage are critical for chondrocyte survival. J. Bone Miner. Res. 27, 596-609. 10.1002/jbmr.1487 [DOI] [PubMed] [Google Scholar]
- Mårtensson K., Chrysis D. and Sävendahl L. (2004). Interleukin-1beta and TNF-alpha act in synergy to inhibit longitudinal growth in fetal rat metatarsal bones. J. Bone Miner. Res. 19, 1805-1812. 10.1359/JBMR.040805 [DOI] [PubMed] [Google Scholar]
- Martin J. A., Martini A., Molinari A., Morgan W., Ramalingam W., Buckwalter J. A. and McKinley T. O. (2012). Mitochondrial electron transport and glycolysis are coupled in articular cartilage. Osteoarthritis Cartilage 20, 323-329. 10.1016/j.joca.2012.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters B. R. (1984). Noninvasive corneal redox fluorometry. Curr. Top Eye Res. 4, 139-200. [PubMed] [Google Scholar]
- Minina E., Wenzel H. M., Kreschel C., Karp S., Gaffield W., McMahon A. P. and Vortkamp A. (2001). BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development 128, 4523-4534. [DOI] [PubMed] [Google Scholar]
- Moerth C., Schneider M. R., Renner-Mueller I., Blutke A., Elmlinger M. W., Erben R. G., Camacho-Hübner C., Hoeflich A. and Wolf E. (2007). Postnatally elevated levels of insulin-like growth factor (IGF)-II fail to rescue the dwarfism of IGF-I-deficient mice except kidney weight. Endocrinology 148, 441-451. 10.1210/en.2006-0385 [DOI] [PubMed] [Google Scholar]
- Morita K., Miyamoto T., Fujita N., Kubota Y., Ito K., Takubo K., Miyamoto K., Ninomiya K., Suzuki T., Iwasaki R. et al. (2007). Reactive oxygen species induce chondrocyte hypertrophy in endochondral ossification. J. Exp. Med. 204, 1613-1623. 10.1084/jem.20062525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H., Yukita A., Ninomiya T., Hosoya A., Hiraga T. and Ozawa H. (2010). Localization of Thy-1-positive cells in the perichondrium during endochondral ossification. J. Histochem. Cytochem. 58, 455-462. 10.1369/jhc.2010.955393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netchine I., Azzi S., Le Bouc Y. and Savage M. O. (2011). IGF1 molecular anomalies demonstrate its critical role in fetal, postnatal growth and brain development. Best Pract. Res. Clin. Endocrinol. Metab. 25, 181-190. 10.1016/j.beem.2010.08.005 [DOI] [PubMed] [Google Scholar]
- Nishida T., Kubota S., Aoyama E. and Takigawa M. (2013). Impaired glycolytic metabolism causes chondrocyte hypertrophy-like changes via promotion of phospho-Smad1/5/8 translocation into nucleus. Osteoarthritis Cartilage 21, 700-709. 10.1016/j.joca.2013.01.013 [DOI] [PubMed] [Google Scholar]
- Ogilvy-Stuart A. L. (2003). Growth hormone deficiency (GHD) from birth to 2 years of age: diagnostic specifics of GHD during the early phase of life. Horm. Res. 60, 2-9. 10.1159/000071219 [DOI] [PubMed] [Google Scholar]
- Ovchinnikov D. (2009). Alcian blue/alizarin red staining of cartilage and bone in mouse. Cold Spring Harb. Protoc. 2009, pdb prot5170 10.1101/pdb.prot5170 [DOI] [PubMed] [Google Scholar]
- Pfander D., Cramer T., Schipani E. and Johnson R. S. (2003). HIF-1alpha controls extracellular matrix synthesis by epiphyseal chondrocytes. J. Cell Sci. 116, 1819-1826. 10.1242/jcs.00385 [DOI] [PubMed] [Google Scholar]
- Poiraudeau S., Lieberherr M., Kergosie N. and Corvol M.-T. (1997). Different mechanisms are involved in intracellular calcium increase by insulin-like growth factors 1 and 2 in articular chondrocytes: voltage-gated calcium channels, and/or phospholipase C coupled to a pertussis-sensitive G-protein. J. Cell. Biochem. 64, 414-422. 10.1002/(SICI)1097-4644(19970301)64:3<414::AID-JCB8>3.0.CO;2-Y [DOI] [PubMed] [Google Scholar]
- Poole R. L., Leith D. J., Docherty L. E., Shmela M. E., Gicquel C., Splitt M., Temple I. K. and Mackay D. J. G. (2012). Beckwith-Wiedemann syndrome caused by maternally inherited mutation of an OCT-binding motif in the IGF2/H19-imprinting control region, ICR1. Eur. J. Hum. Genet. 20, 240-243. 10.1038/ejhg.2011.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn K. P., Sridharan G. V., Hayden R. S., Kaplan D. L., Lee K. and Georgakoudi I. (2013). Quantitative metabolic imaging using endogenous fluorescence to detect stem cell differentiation. Sci. Rep. 3, 3432 10.1038/srep03432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajpurohit R., Mansfield K., Ohyama K., Ewert D. and Shapiro I. M. (1999). Chondrocyte death is linked to development of a mitochondrial membrane permeability transition in the growth plate. J. Cell. Physiol. 179, 287-296. 10.1002/(SICI)1097-4652(199906)179:3<287::AID-JCP6>3.0.CO;2-T [DOI] [PubMed] [Google Scholar]
- Regan J. N., Lim J., Shi Y., Joeng K. S., Arbeit J. M., Shohet R. V. and Long F. (2014). Up-regulation of glycolytic metabolism is required for HIF1alpha-driven bone formation. Proc. Natl. Acad. Sci. USA 111, 8673-8678. 10.1073/pnas.1324290111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinecke M., Schmid A. C., Heyberger-Meyer B., Hunziker E. B. and Zapf J. (2000). Effect of growth hormone and insulin-like growth factor I (IGF-I) on the expression of IGF-I messenger ribonucleic acid and peptide in rat tibial growth plate and articular chondrocytes in vivo. Endocrinology 141, 2847-2853. 10.1210/endo.141.8.7624 [DOI] [PubMed] [Google Scholar]
- Reno P. L., McBurney D. L., Lovejoy C. O. and Horton W. E. Jr (2006). Ossification of the mouse metatarsal: differentiation and proliferation in the presence/absence of a defined growth plate. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 288A, 104-118. 10.1002/ar.a.20268 [DOI] [PubMed] [Google Scholar]
- Saito T., Fukai A., Mabuchi A., Ikeda T., Yano F., Ohba S., Nishida N., Akune T., Yoshimura N., Nakagawa T. et al. (2010). Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat. Med. 16, 678-686. 10.1038/nm.2146 [DOI] [PubMed] [Google Scholar]
- Scheuermann T. H., Li Q., Ma H.-W., Key J., Zhang L., Chen R., Garcia J. A., Naidoo J., Longgood J., Frantz D. E. et al. (2013). Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat. Chem. Biol. 9, 271-276. 10.1038/nchembio.1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel W., Halbauer D., Raimann A., Albrecht C., Scharmer D., Sagmeister S., Helmreich M., Häusler G. and Egerbacher M. (2010). IGF expression patterns and regulation in growth plate chondrocytes. Mol. Cell. Endocrinol. 327, 65-71. 10.1016/j.mce.2010.06.005 [DOI] [PubMed] [Google Scholar]
- Scholz R., Thurman R. G., Williamson J. R., Chance B. and Bucher T. (1969). Flavin and pyridine nucleotide oxidation-reduction changes in perfused rat liver. I. Anoxia and subcellular localization of fluorescent flavoproteins. J. Biol. Chem. 244, 2317-2324. [PubMed] [Google Scholar]
- Sessions C. M., Emler C. A. and Schalch D. S. (1987). Interaction of insulin-like growth factor II with rat chondrocytes: receptor binding, internalization, and degradation. Endocrinology 120, 2108-2116. 10.1210/endo-120-5-2108 [DOI] [PubMed] [Google Scholar]
- Shapiro I. M., Golub E. E., Kakuta S., Hazelgrove J., Havery J., Chance B. and Frasca P. (1982). Initiation of endochondral calcification is related to changes in the redox state of hypertrophic chondrocytes. Science 217, 950-952. 10.1126/science.7112108 [DOI] [PubMed] [Google Scholar]
- Shinar D. M., Endo N., Halperin D., Rodan G. A. and Weinreb M. (1993). Differential expression of insulin-like growth factor-I (IGF-I) and IGF-II messenger ribonucleic acid in growing rat bone. Endocrinology 132, 1158-1167. 10.1210/endo.132.3.8440176 [DOI] [PubMed] [Google Scholar]
- Srour M. K., Fogel J. L., Yamaguchi K. T., Montgomery A. P., Izuhara A. K., Misakian A. L., Lam S., Lakeland D. L., Urata M. M., Lee J. S. et al. (2015). Natural large-scale regeneration of rib cartilage in a mouse model. J. Bone Miner. Res. 30, 297-308. 10.1002/jbmr.2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickens D., Behonick D. J., Ortega N., Heyer B., Hartenstein B., Yu Y., Fosang A. J., Schorpp-Kistner M., Angel P. and Werb Z. (2004). Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development 131, 5883-5895. 10.1242/dev.01461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratikopoulos E., Szabolcs M., Dragatsis I., Klinakis A. and Efstratiadis A. (2008). The hormonal action of IGF1 in postnatal mouse growth. Proc. Natl. Acad. Sci. USA 105, 19378-19383. 10.1073/pnas.0809223105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita A., Kawai S., Hayashibara T., Amano A., Ooshima T., Michigami T., Yoshikawa H. and Yoneda T. (2011). Cellular ATP synthesis mediated by type III sodium-dependent phosphate transporter Pit-1 is critical to chondrogenesis. J. Biol. Chem. 286, 3094-3103. 10.1074/jbc.M110.148403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takigawa M., Okawa T., Pan H.-O., Aoki C., Takahashi K., Zue J.-D., Suzuki F. and Kinoshita A. (1997). Insulin-like growth factors I and II are autocrine factors in stimulating proteoglycan synthesis, a marker of differentiated chondrocytes, acting through their respective receptors on a clonal human chondrosarcoma-derived chondrocyte cell line, HCS-2/8. Endocrinology 138, 4390-4400. 10.1210/endo.138.10.5265 [DOI] [PubMed] [Google Scholar]
- Tsang K. Y., Chan D., Cheslett D., Chan W. C. W., So C. L., Melhado I. G., Chan T. W. Y., Kwan K. M., Hunziker E. B., Yamada Y. et al. (2007). Surviving endoplasmic reticulum stress is coupled to altered chondrocyte differentiation and function. PLoS Biol. 5, e44 10.1371/journal.pbio.0050044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchimura T., Foote A. T., Smith E. L., Matzkin E. G. and Zeng L. (2015). Insulin-like growth factor II (IGF-II) inhibits IL-1beta-induced cartilage matrix loss and promotes cartilage integrity in experimental osteoarthritis. J. Cell. Biochem. 116, 2858-2869. 10.1002/jcb.25232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Osch G. J. V. M., Van Der Veen S. W., Burger E. H. and Verwoerd-Verhoef H. L. (2000). Chondrogenic potential of in vitro multiplied rabbit perichondrium cells cultured in alginate beads in defined medium. Tissue Eng. 6, 321-330. 10.1089/107632700418047 [DOI] [PubMed] [Google Scholar]
- Varone A., Xylas J., Quinn K. P., Pouli D., Sridharan G., McLaughlin-Drubin M. E., Alonzo C., Lee K., Munger K. and Georgakoudi I. (2014). Endogenous two-photon fluorescence imaging elucidates metabolic changes related to enhanced glycolysis and glutamine consumption in precancerous epithelial tissues. Cancer Res. 74, 3067-3075. 10.1158/0008-5472.CAN-13-2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Zhou J. and Bondy C. A. (1999). Igf1 promotes longitudinal bone growth by insulin-like actions augmenting chondrocyte hypertrophy. FASEB J 13, 1985-1990. [DOI] [PubMed] [Google Scholar]
- Wang Y., Menendez A., Fong C., ElAlieh H. Z., Kubota T., Long R. and Bikle D. D. (2015). IGF-I signaling in osterix-expressing cells regulates secondary ossification center formation, growth plate maturation, and metaphyseal formation during postnatal bone development. J. Bone Miner. Res. 30, 2239-2248. 10.1002/jbmr.2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J., Shimazu J., Makinistoglu M. P., Maurizi A., Kajimura D., Zong H., Takarada T., Lezaki T., Pessin J. E., Hinoi E. et al. (2015). Glucose uptake and Runx2 synergize to orchestrate osteoblast differentiation and bone formation. Cell 161, 1576-1591. 10.1016/j.cell.2015.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wit J. M. and van Unen H. (1992). Growth of infants with neonatal growth hormone deficiency. Arch. Dis. Child. 67, 920-924. 10.1136/adc.67.7.920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi G., Rosen C. J. and Clemmons D. R. (2016). IGF-I and IGFBP-2 stimulate AMPK activation and autophagy, which are required for osteoblast differentiation. Endocrinology 157, 268-281. 10.1210/en.2015-1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing W., Cheng S., Wergedal J. and Mohan S. (2014). Epiphyseal chondrocyte secondary ossification centers require thyroid hormone activation of Indian hedgehog and osterix signaling. J. Bone Miner. Res. 29, 2262-2275. 10.1002/jbmr.2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., He Q., Tsang W. P., Garvey W. T., Chan W. Y. and Wan C. (2014). Insulin exerts direct, IGF-1 independent actions in growth plate chondrocytes. Bone Res. 2, 14012 10.1038/boneres.2014.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H., Shyh-Chang N., Segrè A. V., Shinoda G., Shah S. P., Einhorn W. S., Takeuchi A., Engreitz J. M., Hagan J. P., Kharas M. G. et al. (2011). The Lin28/let-7 axis regulates glucose metabolism. Cell 147, 81-94. 10.1016/j.cell.2011.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuscik M. J., Hilton M. J., Zhang X., Chen D. and O'Keefe R. J. (2008). Regulation of chondrogenesis and chondrocyte differentiation by stress. J. Clin. Invest. 118, 429-438. 10.1172/JCI34174 [DOI] [PMC free article] [PubMed] [Google Scholar]