

Abstract
Background
Inflammation is a major cellular strain causing increased risk of osteo-degenerative diseases. Omega-3 fatty acids have been great source in suppressing inflammation. We investigated the effect of α-linolenic acid (ALA) on RANKL-stimulated osteoclast differentiation, LPS-induced and ovariectomized bone loss in mice models.
Material/Methods
The bone marrow macrophages (BMMs) were isolated from femurs of ICR mice, stimulated with RANKL, and treated with ALA (100, 200, 300 μM). Major analytical methods include histological analysis, osteoclasts viability assay, serum cytokines and chemokines ELISA, and gene expression by qPCR.
Results
ALA intervention inhibited RANKL-induced osteoclasts proliferation and differentiation. ALA inhibited bone resorption activity as measured by materialization of F-actin ring structures as well. ALA suppressed the RANKL-induced osteoclast markers c-Fos, c-Jun and NFATc1 together with transcription factor proteins TRAP, OSCAR, cathepsin K and β3-integrin. ALA also suppressed the RANKL-stimulated phosphorylation of JNK, ERK, and AKT as well as NF-κB and BCL-2 proteins. ALA intervention (100 and 300 mg/kg) to LPS-challenged mice showed annulled morphometric changes induced by LPS by suppressing the levels of proinflammatory cytokines and chemokines. ALA (100 and 300 mg/kg) intervention to estrogen-deficiency induced bone loss mice (ovariectomized) showed reductions in TRAP+ osteoclasts count, CTX-I expression, levels of IL-1β, IL-2, IL-6, IL10, TNF-α and MCP-1 and iNOS and COX-2.
Conclusions
ALA suppresses RANKL-induced osteoclast differentiation and prevents inflammatory bone loss via downregulation of NF-κB-iNOS-COX-2 signaling. ALA is suggested to be a preventive herbal medicine against inflammatory bone disorders.
MeSH Keywords: alpha-Linolenic Acid, Anti-Inflammatory Agents, Apoptosis, Bone Diseases, NF-kappa B, Nitric Oxide Synthase Type II
Background
The skeletal disorder osteoporosis is characterized by abnormalities of bone structural strength, ultimately leading to the increased risk of fracture. Osteoporosis is the most prevalent metabolic disorder of bones especially in older age. Since treating osteoporotic fractures is a challenging task, restoring skeletal integrity is more important which can be achieved by increased bone formation (osteoblastogenesis) and by preventing or reducing bone loss by decreased bone resorption (osteoclastogenesis) [1]. Commonly used therapeutic interventions against osteoporosis mainly aim to block osteoclastic bone resorption. Osteoblastogenesis targeting therapeutic interventions may also be effective in reversing or preventing osteoporotic bone loss. Structural changes in bone like formation and resorption are equilibrated simultaneous processes under systemic regulation of factors like vitamin D and parathyroid hormone, cytokines and chemokines, prostaglandins (PGs), and nitric oxide (NO) [1–3]. Among the PGs, osteoblastic PGE2 is a potent local factor stimulating bone resorption both in vivo and in vitro [4]. In vitro studies demonstrated the biphasic and concentration-dependent effects of PGE2 on bone formation [5,6]. The effect of PGE2 supplementation is concentration dependent; low concentration PGE2 stimulates bone formation while high concentrations inhibits bone formation [5,7]. Apart from PGE2, NO is another signaling molecule playing various vital roles in biological processes including bone structure-function and resorption [8]. NO is produced from amino acid L-arginine and O2 by the action of NO synthase (NOS) in mostly mammalian cells. Among the 3 NOS isoforms, iNOS is induced by cytokines, which indeed is elevated in osteoblasts by cytokines in several animals. Thus osteoblast-derived NO may essentially contribute to localized inflammatory bone destruction diseases like rheumatoid arthritis [9]. The NO and PGE2 cross-talk in osteoblastic cells MC3T3-E1 demonstrated that iNOS-mediated NO production in response to the cytokine-activated cyclooxygenase-2 (COX-2) [10]. In response to proinflammatory cytokines, iNOS-COX-2 cross-talk causes overproduction of PGE2 and NO, which inhibits the activity of an osteoblast formation marker alkaline phosphatase.
Alternative and herbal medicines containing omega-3 (n−3) polyunsaturated fatty acids (PUFA) like eicosapentaenoic acid (EPA, 20: 5n−3) and docosahexaenoic acid (DHA, 22: 6n−3) have been used for their protective roles in inflammatory health conditions including bone degeneration [11,12]. Omega-3 PUFA have also been associated with improving protein synthesis in muscles [13,14], and alleviation of muscular atrophy in cancer and sepsis [15,16]. Omega-3 PUFA in general appears to be a novel drug agent in therapy of patients with severe injury and inflammatory weight loss problems. Several related studies have focused on the preventive use of n−3PUFA rich dietary fish oil supplementation [17]. Similarly, the n−6PUFA rich dietary safflower oil and menhaden oil used to manage aging-induced bone loss in middle-aged male rats showed reduced production of PGE2 and NO [18]. The preventive roles of PUFA against inflammatory disease is largely mediated via inhibition of overproduction of proinflammatory cytokines and chemokines [11–13,17,18], yet the specific signaling pathways involved remain unclear. The role of nuclear factor-κB (NF-κB), iNOS-COX-2 and other proinflammatory cytokines and chemokines are important considerations in understanding the molecular mechanisms of inflammatory bone disorders. Reports suggest that suppressing NF-κB signaling reduces inflammation of bones and prevents degenerative bone diseases such N-methyl pyrrolidone, a small bioactive molecule, stimulated bone formation and inhibited osteoclast differentiation and bone resorption by suppressing NF-κB signaling [19]. Recently an extract of Allium hookeri was found to have potent anti-inflammatory effects against lipopolysaccharide(LPS)-induced RAW264.7 cells by suppressing NF-κB [20].
Thus, the present study aimed to investigate the role of α-linolenic acid (ALA) in inflammatory bone diseased animal models and in receptor activator of nuclear factor kappa-B ligand (RANKL)-induced osteoclast cell cultures. ALA, a n3PUFA mainly found in natural resources, is classified as one of 2 essential fatty acids considered necessary for health. ALA might have anti-inflammatory effects like other O3FAs by decreasing the levels of proinflammatory cytokines and chemokines. The study intended to examine the interaction of NF-κB-iNOS-COX2 cross links and to explore the involvement of other osteo-inflammatory signaling molecules like RANKL. Primarily results of the study demonstrate that ALA prevents bone loss and degeneration caused by inflammation.
Material and Methods
Materials
The α-linolenic acid and lipopolysaccharide was purchased from Sigma-Aldrich (MO, USA). Most of the other chemicals and reagents of high purity-analytical grades were obtained from manufacturers like Sigma-Aldrich, Invitrogen-Thermo Fisher, Merck-Millipore etc., otherwise mentioned in methodology.
Experimental animals and models
The experimental procedures were conducted in accordance with the Institutional Animal Ethical Committee with grant of Animal Ethical Clearance for both animal model experiments in the study. Male ‘Institute for Cancer Research (ICR)’ mice (5 week) and C57BL/6J female mice (6 week) were housed under controlled conditions at 22–24°C temperature and 50–60% humidity with 12 h light and dark cycle. Mice were allowed to free to access of food and water unless specified in groups for specific dietary and living conditions.
Preparation of mouse BMMs, osteoclast differentiation and cytotoxicity assay
Femurs and tibiae were extracted from male 5-week-old ICR mice and flushed to isolate bone marrow cells (BMCs). Isolated BMCs were resuspended in α-MEM media supplemented with 10% FBS and 1% penicillin and streptomycin. Isolated nonadherent cells were cultured in presence of macrophage colony-stimulating factor (M-CSF, 30 ng/ml) for 3 days. Isolated floating cells content was discarded while adherent cells (adhered to the bottom of the culture dish) were designated as bone marrow macrophages (BMMs). The adherent BMMs were seeded at 2×105 density per well and cocultured with M-CSF (30 ng/mL) and RANKL (100 ng/mL) followed by treatment with or without ALA for 4 days. Cells were then fixed in formalin (3.7%) for 30 min then permeabilized with Triton X-100 (0.1%). Cells were then stained for tartrate-resistant acid phosphatase (TRAP) and TRAP positive multinucleated cells (MNCs) that contained more than 5 nuclei were counted and classified as osteoclasts. The cell viability of BMMs was assayed by seeding 1×104 cells/well density in 96-well plates followed by M-CSF (30 ng/mL) and ALA treatment at increasing concentrations for 3 days. Cytotoxicity was assayed by adding 50 μl of XTT (sodium 3′-[1-(phenyl-aminocarbonyl)-3,4-tetrazolium]-bis(4-methoxy-6-nitro) reagent following standard procedure. The optical density was recorded at 450 nm in an automated microplate reader (Molecular Devices, USA).
Bone resorption pit assay
Primary osteoblasts and BMCs were cocultured on a collagen gel-coated petri dish (90 mm) in the media containing Vitamin D3 and PGE2 for 6 days. Cocultured cells were detached by 1% trypsin-EDTA treatment with 0.1% collagenase at 37°C for 10 min, and then replated on hydroxyapatite-coated plates followed by treatment with or without ALA for 24 h. The cells were then removed and resorption pits were imaged and analyzed using Imaging Software.
F-Actin ring formation staining
BMMs were cocultured with M-CSF (30 ng/mL) and RANKL (100 ng/mL) with indicated concentration of ALA for 3 days. Cells were then fixed with phosphate-buffered saline (PBS) added with formalin (3.7%) for 30 min and permeabilized with PBST (0.1% Triton X-100) for 20 min. Then cells were blocked with 2.5% bovine serum albumin (BSA) for 60 min followed by staining with phalloidin (Thermofisher, CA, USA) and DAPI (4,6-diamidino-2-phenylindole) (Sigma, USA) for 30 min at 37°C. Cells were subjected to imaging sing a fluorescence microscope (Olympus, USA).
RNA isolation and quantitative real-time PCR
BMMs were seeded at 2×105/well density in 6-well plate and cocultured with M-CSF (30 ng/ml) and RANKL (100 ng/ml) with or without ALA for 4 days. Cells harvested from plates, washed with PBS, and pelleted by centrifugation. Total cellular RNA from BMMs was isolated using TRIzol reagent (Thermo Fisher Scientific Inc., OK, USA) as per manufacturer’s instructions. Total 1 μg cellular RNA was reverse transcribed using Superscript III Reverse Transcriptase Kit (Thermo Fisher, USA) according to manufacturer’s instruction. The RNA mixture was incubated at 65°C for 10 min and cDNA was synthesized by incubating with first strand buffer (50 mM Tris-HCl, pH 8.3; 75 mM KCl; 3 mM MgCl2), 100 mM dithiothreitol, RNase inhibitor with Superscript III reverse transcriptase at 45°C for 60 min. Quantitative Real-Time PCR was performed on ABI 7500 Fast Real-Time thermal cycler system (Applied Biosystems, USA) in a reaction mixture (20 μL) containing 1 μg cDNA, 10 pmol primers (forward and reverse each) and 10 μL of SYBR Green dye (Thermo Fischer, USA). The Q-RT-PCR amplification conditions were set at: 95°C for 5 min, 95°C for 1 min, 60°C for 30 s, and 72°C for 1 min, for 40 cycles. SYBR Green binding to double-stranded DNA resulted in the fluorescence that was measured for quantified using the threshold cycle (Ct) value. Relative levels of interleukins (IL-1β, IL-2, IL-6 and IL-10), iNOS and COX-2 were normalized to that of β-actin using primers as described earlier [21,22].
Protein isolation and western blotting
Total cellular protein was isolated from BMMs cultures by harvesting-pelleting the by centrifugation followed by lysis in cell lysis buffer containing 20 mM Tris pH 7.5, 1 mM EDTA, 150 mM NaCl, 2.5 mM sodium pyrophosphate, 1% Triton X-100, 1% sodium vanadate, 1 mM phenylmethane sulfonyl fluoride (PMSF) and freshly added protease inhibitor cocktail. Cells were lysed for 30 min at 4°C with intermittent vortex mixing followed by centrifugation at 10,000×g for 10 min at 4°C. The supernatant was collected, designated as total cellular protein, and protein estimation performed using BCA protein assay kit (Sigma, MO, USA). A total of 20 μg protein each sample was electrophoresed by SDS-PAGE and protein bands were transferred onto PVDF transfer membranes (Merck-Millipore, Darmstadt, Germany). Protein transferred membranes were blocked in fat-free skimmed milk followed by binding with protein specific primary antibodies following standard procedure. Membranes were then treated with respective secondary antibodies and protein bands were detected using Enhanced Chemi-Luminescence (ECL) solution (Thermo Fisher, USA). Primary antibodies for anti-caspase-3 (CST#9662)), anti-procaspase-9 (CST#9508), anti-PARP (CST#9542), anti-Bcl2 (CST#2876), anti-Bcl-xL (CST#2762), anti-ERK (sc-93), anti-pERK (CST#9106), anti-AKT (CST#4691), anti-pAKT (CST#9271), anti-JNK (sc-572), anti-pJNK (sc-6254), anti-NF-κB (p65) (NB100-2176), anti-β-actin (sc-1615), anti-cFos (ab184666), anti-cJun (CST#9165), anti-NFATc1 (NB100-56732), anti-TRAP (ab133238), anti-OSCAR (ab93817), anti-β3-integrin (ab119992), anti-cathepsin K (ab66237) were obtained from Cell Signaling Technology (CST); Santacruz Biotech. (sc); Abcam (ab); Novus Biologicals (NB).
Lipopolysaccharides (LPS)-induced bone loss mice model
The effect of ALA was examined also on LPS-induced inflammatory bone destruction in 5-week-old male ICR mice. Mice were randomly divided into 4 groups containing 5 mice each (n=5, each group). Mice were treated: control (PBS); LPS (10 mg/kg); LPS+ALA100 (100 mg/kg); LPS+ALA300 (300 mg/kg). LPS was injected intraperitoneally at 10 mg/kg dose on day one only while ALA and PBS were administered orally daily for 9 days. Mice were allowed to free access for food and water and sacrificed on day 10.
Ovariectomized bone loss mice model
The effect of ALA was examined on estrogen deficiency-induced bone loss in 8-week-old female C57BL/6 mice. Mice were randomly divided into 4 groups (n=3, each group): control; ovariectomized (OVX); OVX-ALA (100 and 300 mg/kg). Mice in all groups except control mice were subjected to removal of ovary. Bilateral ovariectomy was performed by CO2 anesthesia to mice followed by surgical incision and removal of ovaries and then suturing. Ovariectomized mice were divided in to 3 groups (OVX) for treatment with or without ALA. After 4 weeks of surgical recovery, mice were orally administered with ALA at 100 and 300 mg/kg or PBS in control mice daily for 4 weeks followed by sacrifice and further analyses.
High-resolution microcomputed tomography and histological analysis
The High-Resolution microcomputed tomography (micro-CT) Analysis (NFR-Polaris-S160) was performed to analyze the metaphysical changes and degenerative signs in bone. Female 8-week-old C57BL/6 mice were used to extract out intact left femur for h-micro-CT analysis. Scanning of femurs was performed over a 2-mm distance from the growth plate and a total of 200 sections per scan were obtained. Then 3D reconstruction was performed for bone morphological analysis for bone volume per tissue volume (BV/TV), trabecular separation (Tb.Sp), trabecular thickness (Tb.Th), and trabecular number (Tb.N) using Imaging tools. Extracted femurs were fixed in 4% neutral-buffered paraformaldehyde for 24 h for decalcification. Femurs were decalcified in 15% EDTA for 2 weeks followed by paraffin embedding. Bone sections of 5 μm thickness were cut using microtome and stained with hematoxylin and eosin (H&E) for histological examination, while other sections were stained for TRAP positivity. Bone resorption parameters including osteoclasts count per field of bone tissue were quantified using Imaging software (Pro-Plus).
Systemic inflammation assessment and serum analysis
The magnitude of inflammation was determined by measurement of the levels of proinflammatory cytokines and chemokines (IL-1β, IL-2, IL-6, IL10, TNF-α, and IFN-γ), monocyte chemoattractant protein 1 (MCP-1) from plasma (20 μL) using a multiplex Mouse Cytokine Immunoassay kit (Bio-Rad, USA) following the manufacturer’s protocols. Serum RANKL and OPG levels were detected using commercial ELISA Assay Kits according to the manufacturer’s protocol (R&D Systems, MN, USA). Serum cross linked C-telopeptide of type I collagen (CTX-I) levels, a specific marker of bone resorption, were determined using a mouse-specific ELISA Assay Kit as per the manufacturer’s protocol (Biomatik, DE, USA).
Statistical analyses
Experiments of the study were performed at least in triplicates. Data were quantitatively expressed mean±standard deviation (SD). Statistical analysis of results was performed using SPSS v.17 (SPSS Inc., USA). Analysis of variance followed by a Tukey’s post hoc test was used to compare parameters among 3 groups. P<0.05 was considered statistically significant.
Results
ALA modulates differentiation and function of RANKL-induced osteoclast
The effect of ALA was evaluated on osteoclast differentiation by stimulating mouse BMMs cocultured with M-CSF and RANKL followed by ALA treatment. The cell viability and toxicity assayed using XTT assay kit for osteoclast differentiation. Control group cell cultures showed high number of TRAP-positive osteoclasts while ALA treatment showed reduction in the number of TRAP-positive cells in a dose-dependent manner (Figure 1A). As compared to control (100%), ALA treatment showed 67, 42 and 28% TRAP-positive cells at 100, 200 and 300 μM concentrations, respectively. The XTT assay results showed that ALA had no significant effect on viability of cells thus no resultant toxicity at the indicated concentrations (data not presented). Resorption pits were analyzed on hydroxyapatite-coated plates seeded with mature osteoclasts. Control sets showed resorption pits in a high frequency which was subsequently inhibited by ALA in a dose-dependent manner (Figure 1B). As compared to control (100%), ALA treatment showed reduction in pit area to 66, 44 and 18% at 100, 200 and 300 μM concentrations, respectively. The materialization of F-actin (filamentous actin) ring structures in osteoclasts is a precise marker of the osteoclasts’ bone resorption activity. Thus, the effects of ALA on osteoclast function and bone resorption was examined. F-actin ring formation was inhibited in an ALA dose-dependent manner (Figure 1C). F-actin formation was inhibited in a similar pattern to TRAP-positive cells with 82, 66 and 34% F-actin ring formation at 100, 200 and 300 μM concentrations, respectively. These results collectively demonstrate that ALA reduced both the formation and bone resorption properties of osteoclasts.
Figure 1.

Effect of ALA on osteoclast differentiation and maturation. (A) BMMs were cocultured with M-CSF (30 ng/mL) and RANKL (100 ng/mL) with indicated concentrations of ALA for 3 days. TRAP-positive multinucleated cells were counted as osteoclasts (bar graph). * P<0.008 vs. control group. (B) Mature osteoclasts seeded on hydroxyapatite-coated plates were treated with indicated concentrations of ALA for 24 h. Cells were imaged and pit areas were visualized and quantified. * P<0.006, ** P=0.002 vs. control. (C) BMMs were cultured and treated with ALA and stained with DAPI with phalloidin. Fluorescence imaging was performed and for visualization and quantification of F-actin rings. * P<0.004, *** P=0.001 vs. control.
ALA inhibits RANKL-induced osteoclastogenesis
The effect of ALA on RANKL-mediated osteoclastogenesis was analyzed by assessing the levels of osteoclast marker, c-Fos, c-Jun and nuclear factor of activated T cells cytoplasmic (NFATc1). c-Fos, c-Jun and NFATc1 are reportedly involved in osteoclast differentiation [23,24]. BMMs were treated with RANKL for 24 h with or without ALA (100 and 200 μM) and total cellular protein were isolated and proteins expression level was analyzed by western blotting (Figure 2). Western blotting results show that RANKL-induced the levels of all 3 marker proteins in 24 h of treatment time, as compared to RANKL negative BMMs. Furthermore, BMMs treated with ALA showed suppressed levels of all 3 proteins in a dose-dependent manner. To analyze any contraindications, RANKL negative BMMs were treated with 200 μM ALA and the levels of c-Fos, c-Jun and NFATc1 were analyzed by western blotting, which shows that ALA has no effect on RANKL negative BMMs (data not shown). In addition to c-Fos, c-Jun and NFATc1, we also analyzed the levels of the transcription factor proteins TRAP, osteoclast-associated immunoglobulin-like receptor (OSCAR), β3-integrin and cathepsin K by western blotting. These proteins are reportedly involved in RANKL-induced osteoclastogenesis [23,24]. Results show that RANK induced the levels TRAP, OSCAR and, β3-integrin and cathepsin K in 24 h of treatment time which were subsequently suppressed by treatment with ALA in a dose-dependent manner (Figure 2). These results indicate that ALA could attenuate the protein levels of c-Fos, c-Jun and NFATc1 along with other transcriptional regulators in osteoclastogenesis. These results indicate too that ALA may be utilized in bone disorders featuring degeneration and inflammation.
Figure 2.
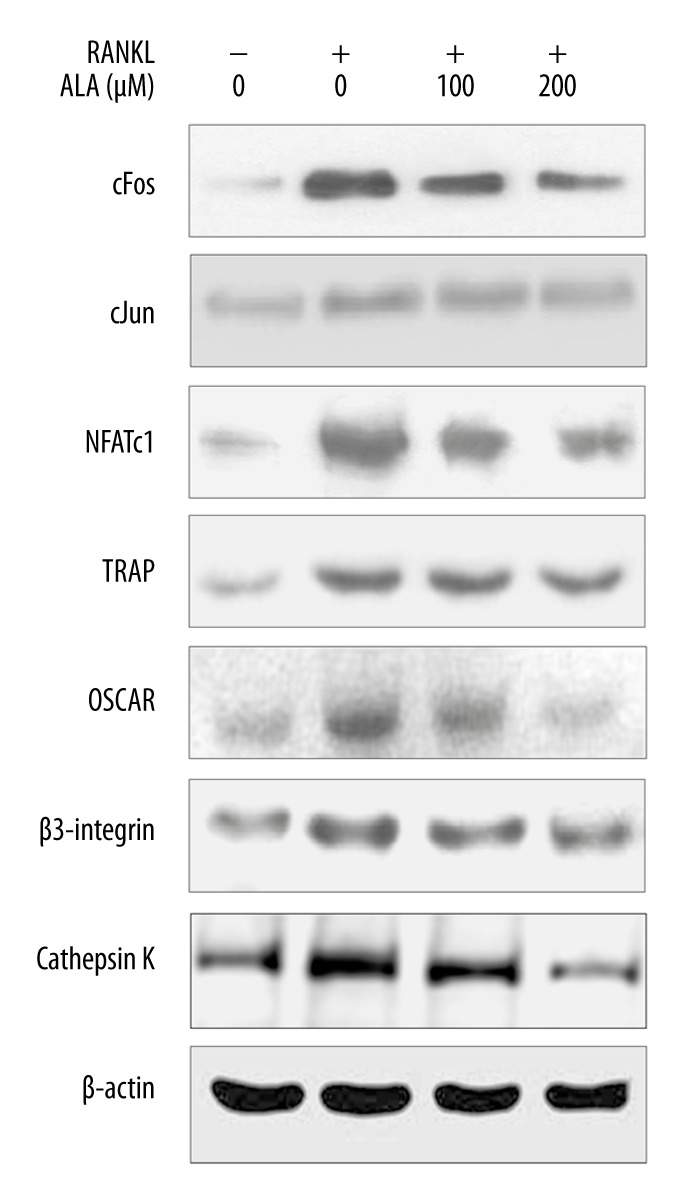
Effect of ALA on RANKL-induced osteoclastogenesis marker proteins. M-CSF (30 ng/mL) and RANKL (100 ng/mL) treated BMMs were cocultured with indicated concentrations of ALA for 3 days. Cells were then harvested, protein isolated, and western blotting was performed.
ALA regulates osteoclastogenesis by modulating cell growth and death signaling
To further investigate the mechanism of ALA-mediated inhibition of osteoclastogenesis, we examined the effects of ALA on RANKL-induced proteins in cell growth and death signaling pathways. BMMs were treated with RANKL for 24 h without or with ALA (100 and 200 μM) and total cellular protein were isolated and level of proteins was analyzed by western blotting (Figure 3). Western blotting results show that RANKL induced the levels of all 3 marker proteins in 24 h of treatment time, as compared to RANKL negative BMMs. BMMs treated with ALA showed suppressed levels of all 3 proteins in a dose-dependent manner. RANKL stimuli are supposed to enhance the levels of cell growth signaling molecules like c-Jun NH2-terminal kinases (JNK), extracellular signal-regulated kinases (ERK) and AKT (protein kinase B) by activating their phosphorylation. The phosphorylation of JNK and ERK is associated with enhanced cell growth; osteoclastogenesis in case of bones is apparently stimulated by RANKL. JNK-ERK-AKT signaling pathways activate cell growth signaling in general and as well as they are characterized by reduced cell death. Thus, we assessed the effect of ALA on cell growth and death signaling genes by analyzing their protein levels by western blotting (Figure 3). Results show that RANKL moderately induced the levels of total forms of JNK, ERK and AKT at 24 h of treatment of BMMs. Likewise, RANKL stimuli enhanced the phosphorylation of JNK, ERK and AKT, as demonstrated by high levels of pJNK, pERK and pAKT. Treatment of RANKL-stimulated BMMs by ALA showed dose-dependent slight suppression in total forms of JNK, ERK and AKT while phosphorylated form of JNK, ERK and AKT were fairly more suppressed. Along with suppression of cell growth signaling proteins, we also examined the level of cell death signaling components mainly apoptosis related proteins by western blotting (Figure 3). Results show that RANKL stimulation moderately suppressed the levels of caspase-3 and poly (ADP-ribose) polymerase (PARP) proteins. Furthermore, we observed that ALA treatment to RANKL-stimulated BMMs caused noticeable suppression in the levels of caspase-3 and PARP in a dose-dependent manner. Caspase-3 is an executioner caspase causing cell death by cleaving a nuclear substrate PARP. Suppression of caspase-3 and PARP, apoptosis inactivation, may also be among the causes of more cell growth, which was subsequently suppressed by treating with ALA. In addition to above proteins we also observed that ALA significantly suppressed the levels of NF-κB in a dose-dependent manner (Figure 3), which was elevated by RANKL stimulation to BMMs. RANKL-mediated activation of NF-κB may correlate with osteoclastogensis by the fact that NF-κB is a transcriptional upregulator of BCL-2 family anti-apoptotic proteins like BCL-2 and BCL-xL. Although western blotting demonstrated only fair decrease in BCL-2 and BCL-xL proteins by RANKL stimulation, ALA treatment clearly increased the inhibited levels of BCL-2 and BCL-xL proteins (Figure 3). The anti-apoptotic effect of RANKL in osteoclast is reported to be mediated by JNK/c-Jun signaling [25] and ERK and NF-κB pathways are reciprocally involved in survival and activation of osteoclasts [26]. These reports strongly correlate with our results as well as BCL-2 and BCL-xL results are complementary to the results of caspase-3 and PARP. These results collectively demonstrate that RANKL induced osteoclastogenesis by enhancing cell growth signaling and suppressing cell death signaling.
Figure 3.

Effect of ALA on cell growth signaling events and apoptosis. M-CSF (30 ng/mL) and RANKL (100 ng/mL) treated BMMs were cocultured with indicated concentrations of ALA for 3 days. Cells were then harvested, protein isolated, and western blotting was performed.
ALA prevents LPS-induced bone loss in mice
LPS is a proven inflammatory polysaccharide, a major cell wall component of Gram negative bacteria. LPS elicits the release of various proinflammatory molecules like cytokines and chemokines, metalloproteinases and other inflammatory molecules [27–29]. LPS enhances the rate of bone resorption by modulating the production of tumor necrosis actor-α (TNF-α), IL-1, prostaglandin E2 (PGE2) causing severe inflammatory bone degeneration [27–29]. LPS may stimulate osteoclast differentiation and activation by modulating RANKL-induced mitogen-activated protein kinases (MAPKs) signaling components and that suppression of these signaling molecules may suppress the inflammation and degeneration of bones [30]. In this order, we examined the effects of ALA on LPS (10 mg/kg) induced bone loss in mice by removing femur for subsequent analyses. Firstly, control and treated mice groups were analyzed for morphometric features which revealed that treatment of LPS-induced mice with ALA restored morphometric parameters in a dose-dependent manner. We assessed BV/TV percent ratio, Tb.Sp in mm, Tb.N per mm, and Tb.Th in mm and compared among various mice groups (Figure 4A). Control mice showed 40±4% BV/TV ratio that was inhibited by LPS stimulation to 23±5%. LPS-induced reduction in BV/TV ratio was further revoked by treatment with ALA in dose-dependent manner, 28±5 and 34±4% at 100 and 300 mg/kg respectively, with statistical significance (Figure 4A). Control mice showed 0.24±0.04 mm Tb.Sp ratio that was elevated to double (0.48±0.05 mm) by LPS stimulation. LPS-induced elevation of Tb.Sp was further suppressed by treatment with ALA in dose-dependent manner, 0.36±0.04 and 0.28±0.03 mm at 100 and 300 mg/kg respectively, with statistical significance (Figure 4A). The Tb.N parameter per mm was normal in control mice (3.4±0.4) that were reduced to 2.2±0.3 by LPS stimulation. LPS-induced reduction in Tb.N (per mm) was further revoked by treatment with ALA in dose-dependent manner, 2.6±0.2 and 3.3±0.3 at 100 and 300 mg/kg respectively, with statistical significance (Figure 4A). However, there was no significant change in Tb.Th (mm) by LPS stimulation and by treating with ALA. We further determined the effect of ALA to RANKL and osteoprotegerin (OPG) proteins expression by ELISA from serum. The serum levels of RANKL were normal in control mice (0.18±0.03 ng/ml) that were significantly elevated to 0.42±0.04 by LPS stimulation. LPS-induced elevated levels of RANKL were further suppressed by treatment with ALA in dose-dependent manner, 0.31±0.04 and 0.22±0.03 at 100 and 300 mg/kg respectively, with statistical significance (Figure 4B). The OPG expression level in control mice serum was 4.8±0.6 ng/ml that was significantly reduced by LPS stimulation to 0.31±0.06 ng/ml. LPS-induced reduction in OPG levels was further suppressed by treatment with ALA in dose-dependent manner, 0.31±0.04 and 0.22±0.03 at 100 and 300 mg/kg respectively, with statistical significance (Figure 4B). As observed the levels of RANKL and OPG, their ratio (RANKL/OPG) was significantly high in LPS-stimulated mice that were subsequently suppressed by ALA treatment in dose-dependent manner (Figure 4B). We further performed histological analysis of LPS-induced mice for osteoclast formation and bone loss and osteoclasts count (Figure 4C). The H&E stained plates show that LPS stimulation caused severe degenerative changes in bone sections that were revoked by ALA treatment. Similarly, the LPS stimulation caused remarkable increase in the TRAP+ osteoclasts count that were subsequently suppressed by ALA in a dose-dependent manner. The quantitative estimation of TRAP+ OC relative to control (100±14) show that LPS was 1.57-fold high in TRAP+ OC that was suppressed to 1.42- and 1.14-fold at 100 and 300 mg/kg of ALA, respectively (Figure 4C). We also estimated the levels of C-terminal telopeptide of type I collagen (CTX-I) concentration in serum of mice. CTX-I is a bone resorption marker that was noticeably elevated in LPS-stimulated mice (56±8 ng/ml) as compared to control (34±7 ng/ml). Furthermore, treatment of LPS-induced mice with ALA at 100 and 300 mg/kg showed 46±6 and 38±5 ng/ml CTX-I in serum, respectively. Collectively these results indicate that ALA treatment resulted in the downregulation of RANKL and up-regulation of OPG and suppression of the levels of CTX-I in LPS-treated mice.
Figure 4.

Effect of ALA on LPS-induced bone loss in mice. (A) 5-week-old ICR mice were stimulated with LPS injection (10 mg/kg) followed by treatment with indicated concentrations of ALA for 9 days. Femurs were radiographed by micro-CT and BV/TV, Tb.Sp, Tb.N and Tb.Th of each femur were recorded. ^ P<0.018 vs. control, * P<0.008 vs. LPS, ** P<0.032 vs. control, *** P<0.041 vs. control. (B) The levels of RANKL, OPG and CTX-I were measured from serum in ng/ml and RANKL/OPG ratio was presented. ^ P<0.012 vs. control, * P<0.028 vs. LPS, ** P<0.006 vs. LPS, *** P<0.046 vs. control. (C) Femurs were dissected, fixed and sectioned, stained with H&E (upper) and treated with TRAP reagent (lower) followed by counting of TRAP+ osteoclasts (OC) per field of tissue. ^ P<0.008 vs. control, * P<0.032 vs. LPS.
ALA prevents OVX-mediated bone loss in mice
The ovariectomized (OVX) mice model is a preclinical animal model for the study of osteoporosis, widely used for pharmacological interventions against estrogen-deficiency induced bone loss. OVX mice were treated with and without ALA at 100 and 300 mg/kg dose for 4 weeks followed by removal of femur and subsequent analyses as described in LPS-induced mice model. The morphometric analysis shows that BV/TV ratio in control mice was 6.4±1.2% that was inhibited by ovariectomy (OVX group) to 2.8±0.8%. OVX mice with reduced BV/TV ratio were further revoked by treatment with ALA in dose-dependent manner, 3.8±1.1 and 5.4±0.8% at 100 and 300 mg/kg respectively, with statistical significance (Figure 5A). Control mice showed 0.52±0.06 mm Tb.Sp ratio that was elevated to 0.66±0.07 mm in OVX mice, which was further suppressed by treatment with ALA with statistical significance at 300 mg/kg (P=0.036). The Tb.N parameter per mm from control mice (2.3±0.2) was reduced in OVX mice (1.8±0.3), which were further revoked by treatment with ALA with statistical significance at 300 mg/kg (P=0.042). The Tb.Th (mm) was marginally reduced in OVX mice (P=0.048) as compared to control that was further revoked by ALA treatment. The levels of CTX-I were estimated from serum that was moderately high in OVX mice (24 ng/ml) as compared to control (16 ng/ml). CTX-I levels were noticeably reduced in OVX mice treated with ALA especially at 300 mg/kg (17.5 ng/ml, P=0.022) (Figure 5B). We further performed histological analysis of OVX mice for osteoclast formation and bone loss (Figure 5C) and osteoclasts count (Figure 5D). The H&E stained plates from OVX mice showed remarkable degenerative signs that were revoked by ALA treatment. Similarly, the OVX mice showed a remarkable increase in TRAP+ osteoclasts count that were subsequently suppressed by ALA treatment. The quantitative estimation of TRAP+ OC was much high in OVX mice (88±14) as compared to control mice (48±11). The ALA intervention to OVX mice showed suppression in TRAP+ OC in a dose-dependent manner with statistical significance (Figure 5D). Collectively these results indicate that ALA intervention restored the OVX-mediated bone erosion.
Figure 5.

Effect of ALA on OVX-induced bone loss in mice. (A) 8-week-old C57BL/6 mice were ovariectomized followed by treatment with indicated concentrations of ALA for 4 weeks. Femurs were radiographed by micro-CT and BV/TV, Tb.Sp, Tb.N and Tb.Th of each femur were recorded. ^ P<0.038 vs. control, * P<0.026 vs. OVX. (B) The levels of CTX-I were measured from serum in ng/ml. ^ P=0.024 vs. control, * P=0.026 vs. OVX. (C, D) Femurs were dissected, fixed and sectioned, stained with H&E (upper) and treated with TRAP reagent (lower) followed by counting of TRAP+ osteoclasts (OC) per field of tissue. ^ P=0.007 vs. control, * P=0.026 vs. OVX, * P=0.014 vs. OVX.
ALA intervention suppressed gene expression of inflammation mediators
In order to delineate the mechanism, we assessed the levels of inflammation-related cytokines and chemokines from serum of LPS-induced mice. The gene expression analysis of RANKL-induced BMMs treated with ALA showed interesting result. Stimulating BMMs with RANKL intensely increased the expression levels of IL-1β, IL-2, IL-6, IL10, iNOS and COX-2 genes (Figure 6A). ALA intervention remarkably suppressed the levels of most of these immune components in plasma in dose-dependent manner. As compared to control (normalized to 1.0), RANKL stimulation to BMMSs caused increase in the expression of IL-1β, IL-2, IL-6, IL10, iNOS and COX-2 genes by 3.4, 2.9, 3.2, 2.8, 2.8 and 3.2 folds, respectively. These overexpressed gene levels were suppressed by ALA treatment in a dose-dependent manner, nearly to control (Figure 6A). LPS is known to readily interact with the abundant TLR4 receptors and eliciting inflammatory response that was measured from blood plasma by multiplex immunoassay. In in vivo experiments, challenging mice with LPS dramatically increased the levels of IL-1β, IL-2, IL-6, IL10, TNF-α and MCP-1 in plasma (Figure 6B). ALA intervention remarkably suppressed the levels of most of these immune components in plasma in dose-dependent manner. As compared to control (normalized to 1.0), LPS induced the levels of IL-2, IL-1β, IL-6, IL10, TNF-α and MCP-1 by 2.96, 2.24, 2.54, 1.79, 2.02 and 1.55 folds, respectively. These elevated levels were suppressed by ALA treatment to LPS-stimulated mice in a dose-dependent manner to a level close to control (Figure 6B). These results suggest that ALA protect against LPS-induced inflammation and damage in bones by suppressing the levels of proinflammatory molecules.
Figure 6.

Effect of ALA on proinflammatory cytokines and chemokines. (A) M-CSF (30 ng/mL) and RANKL (100 ng/mL) treated BMMs were cocultured with indicated concentrations of ALA for 3 days. Cells were then harvested, RNA isolated, cDNA synthesized, and qPCR performed. Quantification of gene was presented relative to normalized control (1.0 fold). ^ P<0.012 vs. control, * P<0.032 vs. RANKL. (B) Systemic inflammation assessment from LPS-challenged mice. A multiplex Mouse Cytokine Immunoassay was performed and quantity of proteins was presented in pg/ml serum. ^ P<0.012 vs. control, * P<0.032 vs. RANKL.
Discussion
Several bone degenerative disorders feature overproduction of inflammatory mediators causing weight loss, bone disorders, organ failure and death. LPS potentially activates the immune system releasing a variety of cellular inflammatory mediators like IL-1β, IL-2, IL-6, IL-10 and TNF-α resulting in severe bone degeneration. Ovariectomy causes estrogen imbalance and thus inflammatory bone disorder. Various anti-inflammatory therapeutic and preventive modalities are attempted yet the side-effect of chemical interventions remains a major concern. This could possibly be limited by using natural product with potent anti-inflammatory effects. In this study, we investigated the effect of α-linolenic acid on RANKL-mediated osteoclast differentiation, LPS-induced bone loss in mice, and on estrogen-deficiency induced bone loss in ovariectomized mice. Results of the study mainly advocate the use of ALA containing omega-3 fatty acids supplements with preventive properties.
Aging is a natural process but may occur with issues of metabolic bone diseases like rheumatoid arthritis, osteoporosis, and Paget’s disease [1]. The supreme difficulties associated with bone diseases are mainly diagnosis followed by therapy especially for osteoporosis. Osteoclasts are the major bone-resorbing multinucleated giant cell types produced by hematopoietic stem cells upon stimulation by the cytokines receptor activator of NF-κB ligand RANKL and M-CSF or CSF-1. RANKL is a member of the TNF family, an osteoclast marker causing exaggerated cell proliferation and differentiation. M-CSF is thus the major regulator of osteoclast progenitor and macrophage proliferation and survival [31,32]. The binding of RANKL to the RANK receptor activates multiple downstream signaling pathways including NF-κB, AKT and MAPKs (mainly ERK, p38 and JNK). It causes further activation of the transcription factors like c-Fos, cJun and NFATc1 inducing osteoclast differentiation [31]. In addition, Ca2+ signaling also plays important role in metabolic bone disorders that was modulated by oleanolic acid acetate in RANKL-induced osteoclast differentiation [33]. Natural omega-3 PUFAs like eicosapentaenoic acid, docosahexaenoic acid and ALA have shown anti-inflammatory effects by decreasing the levels of proinflammatory cytokines and chemokines [11,12]. ALA is an n−3 fatty acid classified as one of 2 essential fatty acids considered necessary for health. Since they are not produced within the human body, they must be acquired through diet such as ALA is obtained from seeds (like chia and flaxseed), nuts (especially walnuts), and many common vegetable oils. We have meticulously investigated the effect of ALA on inflammatory bone degeneration in mice models and on osteoclast differentiation.
The in vitro analyses performed in this study demonstrate that ALA intervention inhibits RANKL-induced osteoclastogenesis and differentiation function (Figure 1). The cytotoxicity assay showed RANKL-induced high number of TRAP-positive osteoclasts that were inhibited by ALA. The bone resorption activity as measured by materialization of F-actin ring structures was also inhibited by ALA. In continuation ALA inhibited the levels of formation of resorption pits formed by RANKL stimulation. The growth inhibitory effect of ALA on osteoclasts was possibly mediated by suppression of osteoclast markers c-Fos, c-Jun and NFATc1. RANKL stimulated BMMs when treated with ALA showed suppressed levels of all 3 proteins. ALA also inhibited the RANKL stimulated levels of the transcription factor proteins TRAP, OSCAR, β3-integrin and cathepsin K (Figure 2). Altogether these results indicated that ALA could suppress the activities of c-Fos, c-Jun and NFATc1 along with other transcriptional regulators in osteoclastogenesis. Modulating cFos has been interesting measure in osteoclastogenic problems. cFos and NFATc1 deficient mice have shown symptoms of osteopetrosis because of defects in osteoclastogenesis [34,35]. Furthermore, NFATc1 regulation by c-Fos during osteoclastogenesis is facilitated by the activity of osteoclast-specific genes such as TRAP, OSCAR, β3-integrin, DC-STAMP, CTR, and cathepsin K [35–38]. Thus, our findings infer that ALA could downregulate the expression of various genes and proteins in osteoclasts differentiation and function. RANKL is known to induce phosphorylation of MAPK proteins such as JNK, ERK and AKT as critical upstream events in bone formation. Thus, the p38 and JNK inhibition and apoptosis reactivation in osteoclasts have gained clinical interest [25,26,39]. In this study, ALA was found to regulate osteoclastogenesis by modulating the levels of cell growth and death signaling genes and proteins. RANKL-stimulated BMMs enhanced levels of cell growth signaling molecules like JNK, ERK and AKT especially their phosphorylated forms were elevated. It indicates that RANKL possibly induces phosphorylation of JNK, ERK and AKT which then causes enhanced osteoclastogenesis. ALA intervention to RANKL-stimulated BMMs altered the effects of RANKL and JNK, ERK and AKT phosphorylations were diminished (Figure 2). RANKL-stimulated BMMs were slow in apoptosis as observed by reduced levels pro-caspase-3 and PARP as well as high levels of anti-apoptosis proteins Bcl-2 and Bcl-xL. ALA intervention to these cells revoked the effects of RANKL and showed apoptosis reactivation activity (Figure 3). These results confirmed that ALA has preventive role in RANKL-induced osteoclastogenesis by suppressing cell growth signaling and by reactivating apoptosis. The anti-apoptotic effect of RANKL in osteoclast is reported to be mediated by JNK/c-Jun signaling [25] and ERK and NF-κB pathways are reciprocally involved in survival and activation of osteoclasts [26]. These reports strongly correlate with our results as well as BCL-2 and BCL-xL results are complementary to the results of caspase-3 and PARP.
The rationale behind using both the LPS challenged and ovariectomized mice model in this study was to ascertain the effect of ALA on diverse inflammatory conditions. As LPS directly alters bone environment, estrogen regulates osteoclast proliferation and differentiation activity by promoting TGF-β-induced osteoclast apoptosis. Thus, estrogen agonists could increase OPG production by activating estrogen receptor-α which subdues RANKL activity [40,41]. Ovariectomy causes estrogen deficiency and upregulates osteoclastogenesis by enhancing T cell TNF-α production. OVX also induces the Th17 cell differentiation secreting IL-7 that stimulates bone loss by enhancing osteoclast production and inhibiting osteoblast differentiation [41,42]. LPS challenged mice showed inflammatory bone loss by stimulating osteoclast differentiation, while ALA intervention to LPS-challenged mice prevented these effects (Figure 4). The morphometric analysis showed that LPS-stimulated mice had reduced BV/TV ratio, elevated Tb.Sp (mm), and reduced Tb.N (per mm) and that ALA intervention reverted these effects to significant levels. Also, ALA intervention suppressed the LPS-induced levels of RANKL, OPG and CTX-I in serum of mice. The histological features were also improved by ALA treatment to LPS challenged mice and LPS-stimulated TRAP positive osteoclast count was also suppressed by ALA. The mechanism behind anti-inflammatory effects of ALA was possibly mediated by suppression of proinflammatory cytokines and chemokines. In ovariectomized mice also ALA was preventive to the inflammatory response due to estrogen-dependent bone loss (Figure 5). ALA treated OVX mice showed improved morphometric features as compared to diseased mice. OVX mice showed reduced BV/TV ratio, elevated Tb.Sp ratio and reduced Tb.N, and these altered morphometric parameters were reverted to close to control in ALA treated OVX mice. The histological analysis of ALA treated OVX mice femur was comparatively improved showing reduced osteoclast formation and bone loss by ALA. ALA treatment showed suppressed TRAP+ osteoclasts count increased due to ovariectomy. ALA treated caused suppression of expression of CTX-I that were elevated by ovariectomy. Collectively these results indicate that ALA intervention restored the OVX-mediated bone erosion which may involve suppression of the expression of proinflammatory chemokines and cytokines. RANKL-stimulated BMMs and LPS-challenged mice showed an aberrant expression of IL-1β, IL-2, IL-6, IL10, TNF-α and MCP-1 and iNOS and COX-2 were remarkably suppressed to the level close to control by ALA treatment (Figure 6). These results demonstrate the anti-inflammatory role of ALA in LPS challenged mice which might be mediated by suppression of the expression of proinflammatory molecules. Reduction in IL-1β, IL-2 and IL-6 levels by ALA indicates the mechanistic approach involved in the suppression of inflammation as well as reduction in osteoclast differentiation. Notably, NF-κB is known to interact with iNOS-COX-2 as well as BCL-2 family, thus it plays pivotal role in bone inflammation and degeneration by transcriptional up-regulation of proinflammatory cytokines and chemokines. Recent reports suggest that downregulation of NF-κB, iNOS, COX-2, IL-1β, MMPs by plant extracts and single molecules prevents inflammatory bone loss and other degenerations of bone [19,20,43–46]. These reports strongly support our findings in concluding that downregulation of NF-κB, iNOS-COX2, and other inflammation mediators exerts anti-inflammatory effects in degenerative inflamed bones. Modulation of NF-κB-iNOS-COX-2 and associated molecules has been associated with prevention of inflammation in not only bones [28,30,47] but also in cancers especially intestinal inflammation and colon cancer [21,22,48] and their downregulation by natural resources has been potentially preventive.
Conclusions
Results of the study demonstrate that ALA suppresses RANKL-induced osteoclast differentiation by modulating expression of ERK, JNK, AKT and NF-κB signaling components and downstream activation of NFATc1 and target genes expression. ALA also causes apoptosis reactivation. ALA furthermore restored bone density in LPS challenged mice as well as OVX-induced bone loss mice model. The mechanism behind the effects of ALA is apparently by modulating NF-κB-iNOS-COX-2 and associated proinflammatory cytokines and chemokines that ultimately suppress inflammation and osteoclast differentiation, reactivates apoptosis. These findings are first to our knowledge for using ALA against inflammatory bone disorder. Findings of the study advocate the use of ALA as a promising therapeutic agent for preventing inflammation-driven bone diseases like osteoporosis and rheumatoid arthritis.
Footnotes
Source of support: Departmental sources
Conflict of interest
The authors declare no conflicts of interest.
References
- 1.Raisz LG. Pathogenesis of osteoporosis: Concepts, conflicts, and prospects. J Clin Invest. 2005;115:3318–25. doi: 10.1172/JCI27071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen S. Role of RANK ligand in normal and pathologic bone remodeling and the therapeutic potential of novel inhibitory molecules in musculoskeletal diseases. Arthritis Rheum. 2006;55:15–18. doi: 10.1002/art.21706. [DOI] [PubMed] [Google Scholar]
- 3.Ralston SH, Ho LP, Helfrich MH, et al. Nitric oxide: A cytokine-induced regulator of bone resorption. J Bone Miner Res. 1995;10:1040–49. doi: 10.1002/jbmr.5650100708. [DOI] [PubMed] [Google Scholar]
- 4.Coon D, Gulati A, Cowan C, He J. The role of cyclooxygenase-2 (COX-2) in inflammatory bone resorption. J Endod. 2007;33:432–36. doi: 10.1016/j.joen.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Watkins BA, Li Y, Allen KG, et al. Dietary ratio of (n−6)/(n−3) polyunsaturated fatty acids alters the fatty acid composition of bone compartments and biomarkers of bone formation in rats. J Nutr. 2000;130:2274–84. doi: 10.1093/jn/130.9.2274. [DOI] [PubMed] [Google Scholar]
- 6.Ono K, Kaneko H, Choudhary S, et al. Biphasic effect of prostaglandin E2 on osteoclast formation in spleen cell cultures: Role of the EP2 receptor. J Bone Miner Res. 2005;20:23–29. doi: 10.1080/14041040510033842. [DOI] [PubMed] [Google Scholar]
- 7.Shen CL, Yeh JK, Rasty J, et al. Protective effect of dietary long-chain n−3 polyunsaturated fatty acids on bone loss in gonad-intact middle-aged male rats. Br J Nutr. 2006;95:462–68. doi: 10.1079/bjn20051664. [DOI] [PubMed] [Google Scholar]
- 8.Zheng H, Yu X, Collin-Osdoby P, Osdoby P. RANKL stimulates inducible nitric-oxide synthase expression and nitric oxide production in developing osteoclasts. An autocrine negative feedback mechanism triggered by RANKL-induced interferon-beta via NF-kappaB that restrains osteoclastogenesis and bone resorption. J Biol Chem. 2006;281:15809–20. doi: 10.1074/jbc.M513225200. [DOI] [PubMed] [Google Scholar]
- 9.Hukkanen M, Hughes FJ, Buttery LD, et al. Cytokine-stimulated expression of inducible nitric oxide synthase by mouse, rat, and human osteoblast-like cells and its functional role in osteoblast metabolic activity. Endocrinology. 1995;136:5445–53. doi: 10.1210/endo.136.12.7588294. [DOI] [PubMed] [Google Scholar]
- 10.Kanematsu M, Ikeda K, Yamada Y. Interaction between nitric oxide synthase and cyclooxygenase pathways in osteoblastic MC3T3-E1 cells. J Bone Miner Res. 1997;12:1789–96. doi: 10.1359/jbmr.1997.12.11.1789. [DOI] [PubMed] [Google Scholar]
- 11.Calder PC. Omega-3 polyunsaturated fatty acids and inflammatory processes: Nutrition or pharmacology? Br J Clin Pharmacol. 2013;75:645–62. doi: 10.1111/j.1365-2125.2012.04374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simopoulos AP. Omega-3 fatty acids in inflammation and autoimmune diseases. J Am Coll Nutr. 2002;21:495–505. doi: 10.1080/07315724.2002.10719248. [DOI] [PubMed] [Google Scholar]
- 13.Smith GI, Atherton P, Reeds DN, et al. Dietary omega-3 fatty acid supplementation increases the rate of muscle protein synthesis in older adults: A randomized controlled trial. Am J Clin Nutr. 2011;93:402–12. doi: 10.3945/ajcn.110.005611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith GI, Atherton P, Reeds DN, et al. Omega-3 polyunsaturated fatty acids augment the muscle protein anabolic response to hyperinsulinaemia-hyperaminoacidaemia in healthy young and middle-aged men and women. Clin Sci (Lond) 2011;121:267–78. doi: 10.1042/CS20100597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whitehouse AS, Smith HJ, Drake JL, Tisdale MJ. Mechanism of attenuation of skeletal muscle protein catabolism in cancer cachexia by eicosapentaenoic acid. Cancer Res. 2001;61:3604–9. [PubMed] [Google Scholar]
- 16.Khal J, Tisdale MJ. Downregulation of muscle protein degradation in sepsis by eicosapentaenoic acid (EPA) Biochem Biophys Res Commun. 2008;375:238–40. doi: 10.1016/j.bbrc.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 17.Calder PC. Fatty acids and inflammation: The cutting edge between food and pharma. Eur J Pharmacol. 2011;668(Suppl 1):S50–58. doi: 10.1016/j.ejphar.2011.05.085. [DOI] [PubMed] [Google Scholar]
- 18.Hakeda Y, Nakatani Y, Kurihara N, et al. Prostaglandin E2 stimulates collagen and non-collagen protein synthesis and prolyl hydroxylase activity in osteoblastic clone MC3T3-E1 cells. Biochem Biophys Res Commun. 1985;126:340–45. doi: 10.1016/0006-291x(85)90611-4. [DOI] [PubMed] [Google Scholar]
- 19.Ghayor C, Gjoksi B, Siegenthaler B, Weber FE. N-methyl pyrrolidone (NMP) inhibits lipopolysaccharide-induced inflammation by suppressing NF-kappaB signaling. Inflamm Res. 2015;64:527–36. doi: 10.1007/s00011-015-0833-x. [DOI] [PubMed] [Google Scholar]
- 20.Jang JY, Lee MJ, You BR, et al. Allium hookeri root extract exerts anti-inflammatory effects by nuclear factor-kappaB down-regulation in lipopolysaccharide-induced RAW264.7 cells. BMC Complement Altern Med. 2017;17:126. doi: 10.1186/s12906-017-1633-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mishra SK, Kang JH, Kim DK, et al. Orally administered aqueous extract of Inonotus obliquus ameliorates acute inflammation in dextran sulfate sodium (DSS)-induced colitis in mice. J Ethnopharmacol. 2012;143:524–32. doi: 10.1016/j.jep.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 22.Mishra SK, Kang J-H, Song K-H, et al. Inonotus obliquus suppresses proliferation of colorectal cancer cells and tumor growth in mice models by downregulation of β-catenin/NF-κB-signaling pathways. European Journal of Inflammation. 2013;11:615–29. [Google Scholar]
- 23.Matsuo K, Owens JM, Tonko M, et al. Fosl1 is a transcriptional target of c-Fos during osteoclast differentiation. Nat Genet. 2000;24:184–87. doi: 10.1038/72855. [DOI] [PubMed] [Google Scholar]
- 24.Milde-Langosch K, Roder H, Andritzky B, et al. The role of the AP-1 transcription factors c-Fos, FosB, Fra-1 and Fra-2 in the invasion process of mammary carcinomas. Breast Cancer Res Treat. 2004;86:139–52. doi: 10.1023/B:BREA.0000032982.49024.71. [DOI] [PubMed] [Google Scholar]
- 25.Ikeda F, Matsubara T, Tsurukai T, et al. JNK/c-Jun signaling mediates an anti-apoptotic effect of RANKL in osteoclasts. J Bone Miner Res. 2008;23:907–14. doi: 10.1359/jbmr.080211. [DOI] [PubMed] [Google Scholar]
- 26.Miyazaki T, Katagiri H, Kanegae Y, et al. Reciprocal role of ERK and NF-kappaB pathways in survival and activation of osteoclasts. J Cell Biol. 2000;148:333–42. doi: 10.1083/jcb.148.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abu-Amer Y, Ross FP, Edwards J, Teitelbaum SL. Lipopolysaccharide-stimulated osteoclastogenesis is mediated by tumor necrosis factor via its P55 receptor. J Clin Invest. 1997;100:1557–65. doi: 10.1172/JCI119679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agarwal S, Piesco NP, Johns LP, Riccelli AE. Differential expression of IL-1 beta, TNF-alpha, IL-6, and IL-8 in human monocytes in response to lipopolysaccharides from different microbes. J Dent Res. 1995;74:1057–65. doi: 10.1177/00220345950740040501. [DOI] [PubMed] [Google Scholar]
- 29.Aznar C, Fitting C, Cavaillon JM. Lipopolysaccharide-induced production of cytokines by bone marrow-derived macrophages: Dissociation between intracellular interleukin 1 production and interleukin 1 release. Cytokine. 1990;2:259–65. doi: 10.1016/1043-4666(90)90026-p. [DOI] [PubMed] [Google Scholar]
- 30.Hou GQ, Guo C, Song GH, et al. Lipopolysaccharide (LPS) promotes osteoclast differentiation and activation by enhancing the MAPK pathway and COX-2 expression in RAW264.7 cells. Int J Mol Med. 2013;32:503–10. doi: 10.3892/ijmm.2013.1406. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka S, Nakamura K, Takahasi N, Suda T. Role of RANKL in physiological and pathological bone resorption and therapeutics targeting the RANKL-RANK signaling system. Immunol Rev. 2005;208:30–49. doi: 10.1111/j.0105-2896.2005.00327.x. [DOI] [PubMed] [Google Scholar]
- 32.Arai F, Miyamoto T, Ohneda O, et al. Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor kappaB (RANK) receptors. J Exp Med. 1999;190:1741–54. doi: 10.1084/jem.190.12.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim JY, Cheon YH, Oh HM, et al. Oleanolic acid acetate inhibits osteoclast differentiation by downregulating PLCgamma2-Ca(2+)-NFATc1 signaling, and suppresses bone loss in mice. Bone. 2014;60:104–11. doi: 10.1016/j.bone.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 34.Grigoriadis AE, Wang ZQ, Cecchini MG, et al. c-Fos: A key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science. 1994;266:443–48. doi: 10.1126/science.7939685. [DOI] [PubMed] [Google Scholar]
- 35.Winslow MM, Pan M, Starbuck M, et al. Calcineurin/NFAT signaling in osteoblasts regulates bone mass. Dev Cell. 2006;10:771–82. doi: 10.1016/j.devcel.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 36.Kanazawa K, Azuma Y, Nakano H, Kudo A. TRAF5 Functions in Both RANKL- and TNFα-Induced Osteoclastogenesis. J Bone Miner Res. 2003;18:443–50. doi: 10.1359/jbmr.2003.18.3.443. [DOI] [PubMed] [Google Scholar]
- 37.Lee ZH, Kim HH. Signal transduction by receptor activator of nuclear factor kappa B in osteoclasts. Biochem Biophys Res Commun. 2003;305:211–14. doi: 10.1016/s0006-291x(03)00695-8. [DOI] [PubMed] [Google Scholar]
- 38.Oursler MJ. Recent advances in understanding the mechanisms of osteoclast precursor fusion. J Cell Biochem. 2010;110:1058–62. doi: 10.1002/jcb.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X, Udagawa N, Itoh K, et al. p38 MAPK-mediated signals are required for inducing osteoclast differentiation but not for osteoclast function. Endocrinology. 2002;143:3105–13. doi: 10.1210/endo.143.8.8954. [DOI] [PubMed] [Google Scholar]
- 40.Hughes DE, Dai A, Tiffee JC, et al. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-beta. Nat Med. 1996;2:1132–36. doi: 10.1038/nm1096-1132. [DOI] [PubMed] [Google Scholar]
- 41.Zallone A. Direct and indirect estrogen actions on osteoblasts and osteoclasts. Ann NY Acad Sci. 2006;1068:173–79. doi: 10.1196/annals.1346.019. [DOI] [PubMed] [Google Scholar]
- 42.Tyagi AM, Srivastava K, Mansoori MN, et al. Estrogen deficiency induces the differentiation of IL-17 secreting Th17 cells: A new candidate in the pathogenesis of osteoporosis. PLoS One. 2012;7:e44552. doi: 10.1371/journal.pone.0044552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng W, Zhang H, Jin Y, et al. Butein inhibits IL-1beta-induced inflammatory response in human osteoarthritis chondrocytes and slows the progression of osteoarthritis in mice. Int Immunopharmacol. 2017;42:1–10. doi: 10.1016/j.intimp.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 44.Li L, Sapkota M, Kim SW, Soh Y. Herbacetin inhibits inducible nitric oxide synthase via JNK and nuclear factor-kappaB in LPS-stimulated RAW264.7 cells. Eur J Pharmacol. 2015;765:115–23. doi: 10.1016/j.ejphar.2015.08.032. [DOI] [PubMed] [Google Scholar]
- 45.Taraballi F, Corradetti B, Minardi S, et al. Biomimetic collagenous scaffold to tune inflammation by targeting macrophages. J Tissue Eng. 2016;7 doi: 10.1177/2041731415624667. 2041731415624667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ganesan R, Doss HM, Rasool M. Majoon ushba, a polyherbal compound ameliorates rheumatoid arthritis via regulating inflammatory and bone remodeling markers in rats. Cytokine. 2016;77:115–26. doi: 10.1016/j.cyto.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 47.Kim JB, Han AR, Park EY, et al. Inhibition of LPS-induced iNOS, COX-2 and cytokines expression by poncirin through the NF-kappaB inactivation in RAW 264.7 macrophage cells. Biol Pharm Bull. 2007;30:2345–51. doi: 10.1248/bpb.30.2345. [DOI] [PubMed] [Google Scholar]
- 48.Wang S, Liu Z, Wang L, Zhang X. NF-kappaB signaling pathway, inflammation and colorectal cancer. Cell Mol Immunol. 2009;6:327–34. doi: 10.1038/cmi.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]