

Abstract
Purpose of the Review
This review has two purposes: 1) To provide an updated review of the genetic causes of congenital heart disease (CHD) and the clinical implications of these genetic mutations; 2) To provide a clinical algorithm for clinicians considering a genetics evaluation of a CHD patient.
Recent Findings
A large portion of congenital heart disease is thought to have a significant genetic contribution, and at this time a genetic cause can be identified in approximately 35% of patients. Through the advances made possible by next generation sequencing, many of the comorbidities that are frequently seen in patients with genetic congenital heart disease patients can be attributed to the genetic mutation that caused the congenital heart disease. These comorbidities are both cardiac and non-cardiac and include: neurodevelopmental disability, pulmonary disease, heart failure, renal dysfunction, arrhythmia and an increased risk of malignancy. Identification of the genetic cause of congenital heart disease helps reduce patient morbidity and mortality by improving preventive and early intervention therapies to address these comorbidities.
Summary
Through an understanding of the clinical implications of the genetic underpinning of congenital heart disease, clinicians can provide care tailored to an individual patient and continue to improve the outcomes of congenital heart disease patients.
Keywords: Genetics, Congenital Heart Disease, Clinical Outcomes
Introduction
The prognosis for children born with congenital heart disease (CHD) has improved dramatically over the last century. Prior to the availability of surgical techniques to palliate complex CHD, the estimated 1 year survival for these patients was between 0–36%, and very few patients survived to adulthood (1). Today, the expected long term survival of CHD patients is greater than 90% (2). Pediatric cardiologists must now not only provide early, post-natal care for CHD patients, but must also address issues that affect long term morbidity and mortality, such as: neurodevelopmental disability, respiratory disease, renal dysfunction, arrhythmia, heart failure, pulmonary hypertension and an increased risk of cancer(3–7). The increased incidence of these comorbidities in CHD patients compared to the non-CHD population has prompted research into genetic causes of CHD that may also contribute to these comorbid conditions.
Studies of CHD in monozygotic vs. dizygotic twins, the recurrence of CHD in siblings of patients affected by CHD, and pedigree analyses of families with recurrent CHD across generations all suggest a genetic contribution to CHD (8–10). Approximately 35% of CHD cases can now be attributed to genetic factors (11). Advances in large-scale human genomics and next generation sequencing have identified mutations in critical genes associated with CHD that may also influence CHD comorbidities. These discoveries are expanding the role of genetics evaluation in the care of CHD patients. The following review summarizes the current understanding of the genetic causes of CHD and provides a clinical algorithm to assist clinicians in utilizing available genetic testing modalities and genetics consultation services to improve outcomes in CHD patients.
I. The Genetics of CHD
Genetic changes associated with CHD include aneuploidy, copy number variations, and point mutations. These mutations can be inherited within a family following the laws of Mendel, known as Mendelian inheritance, or occur for the first time in the affected patient (sporadic or de novo mutations). Inherited mutations have differing degrees of penetrance, a term that refers to the proportion of individuals carrying a mutation that also demonstrate the associated disease, and varying expressivity, referring to the range of phenotypes associated with a given mutation. Each of these types of genetic change, as well as the effect of inheritance and penetrance are depicted in Figure 1 and are discussed more fully below.
Figure 1. The Genetic Causes and Inheritance Patterns of CHD.
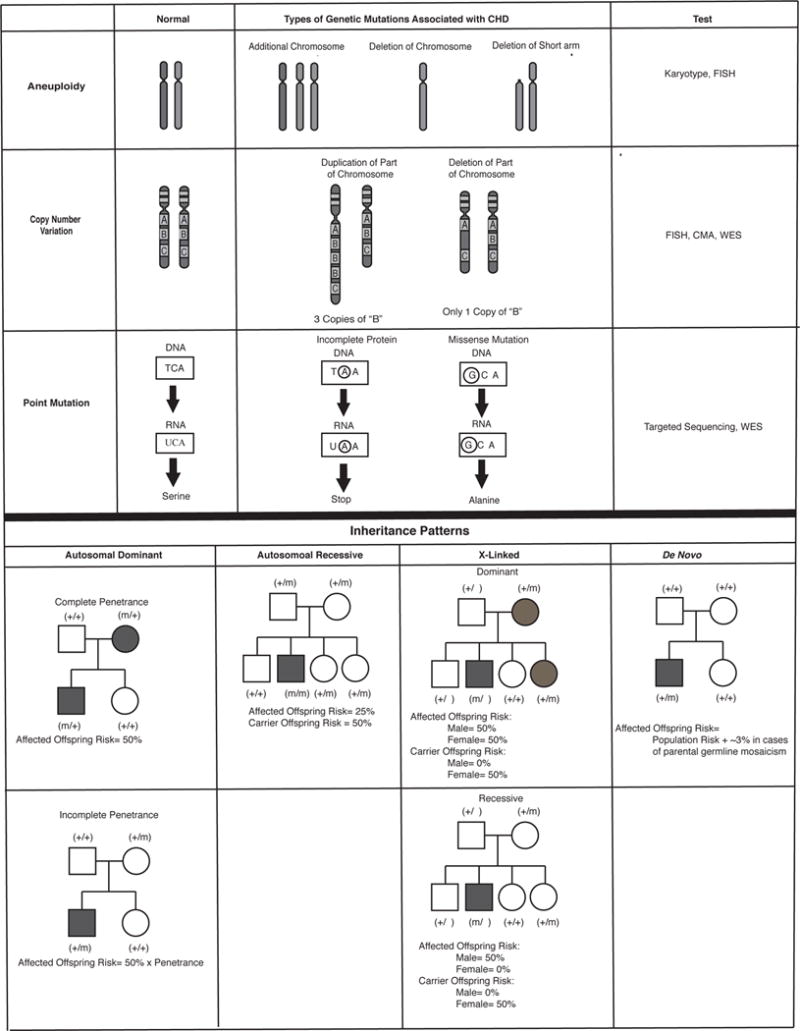
A) The genetic causes of CHD; B) Inheritance patterns of CHD
*** Both A and B are original figures
A. Aneuploidy and Copy Number Variant Mutations
In cases of aneuploidy, the genetic defect is an abnormal number of chromosomes or segments of chromosomes. This can be either due to the presence of an additional chromosome, such as Down Syndrome (Trisomy 21), the absence of a chromosome, such as Turner Syndrome (monosomy X), or deletion or duplication of one arm of a chromosome such as Cri du chat syndrome (5p-). The incidence in liveborn infants with CHD is 9–18% (12, 13). Although some aneuploidies have strong associations with particular malformations (such as atrioventricular septal defects with Trisomy 21 (14)), the range of CHD associated with aneuploidy is quite broad. Common aneuploidies associated with CHD are detailed in Table 1.
Table 1.
CHD Due to Common Aneuploidy and CNV Mutations
| Mutation | Incidence of CHD | Associated CHD* | Non-cardiac Malformations ** | Comorbidities** | |
|---|---|---|---|---|---|
| Aneuploidy | Trisomy 8 (15) | Insufficient Data | Insufficient Data | FD, CNS, MSK | IUGR, NDD, DLE |
| Trisomy 9 (16) | 61–80% | VSD | FD, MSK, GU, Renal, CNS | IUGR, NDD, Renal, GR, DLE | |
| Trisomy 13 (Patau Syndrome) (17, 18) |
60–80% | ASD, VSD, PDA, TOF | Dysmorphism, MSK, GI, GU, Renal, CNS | IUGR, NDD, CNS, DLE | |
| Trisomy 18 (Edward Syndrome) (17–19) |
60–80% | ASD, VSD, PDA | Dysmorphism, MSK, Renal, GI | IUGR, Feeding issues, apnea, NDD, DLE | |
| Trisomy 21 (Down Syndrome) (20) |
35–50% | Endocardial Defect (Primum ASD, Cleft MV, Inlet VSD, AVSD), VSD, ASD, PDA | Dysmorphism, MSK, GI, GU | IUGR, GR, NDD, HM, EN, Pulm, ID, DLE | |
| Monosomy X (Turner Syndrome) (21) |
33% | Left-sided heart defects (CoA, BAV, AS) | MSK, Renal, GU | IUGR, GR, En, CV, Onc, RH | |
| 4p- (Wolf-Hirschhorn Syndrome) (22) |
30–79% | ASD | FD, CNS, MSK, Renal | GR, NDD, ID | |
| 5p- (Cri du Chat) (23) |
5–29% | VSD, TOF, PA, DORV | Dysmorphism, CNS | NDD, MSK | |
| CNV Mutations | Del22q11 (DiGeorge Syndrome) (24) |
74% | Conotruncal anomalies (TOF, IAA, VSD, Truncus) | CNS, FD, renal | EN, ID, NDD |
| 1q21.1(del and dup) (25–27) |
74% | Left heart obstruction, conotruncal anomalies, ASD, VSD, PS | CNS, FD | CNS, NDD | |
| Del1p36 (28) |
71% | ASD, VSD, Valular anomalies, PDA, TOF, CoA, Cardiomyopathy | CNS, FD, GU, MSK, Renal | NDD | |
| Del3p25.1 (29,30) |
25% | ASD, AVSD, PDA, VSD | FD, GI, MSK, Renal | NDD | |
| Del7q11 (William Syndrome) (31) |
80% | Supravalvar AS, Branch PS, VSD | Arterial stenoses, FD | EN, GD, NDD | |
| Del8p23 (32) |
Insufficient Data | ASD, AVSD, PS | CNS, FD, Diaphragmatic hernia, GU | CNS, NDD | |
| Del11q24-25 (Jacobsen Syndrome) (33) |
56% | VSD, Left sided lesions | FD, GI, MSK, Renal, GU | HM, NDD | |
| Del15q11.2 (34) |
15% | Broad range: VSD, TOF, CoA, APVR, Pulm Atr | CNS, ENT, FD, GI | CNS, NDD | |
| Dup16p13.1 (35) |
40% | ASD, TOF | CNS, FD, MSK, GU | EN, NDD |
Abbreviations: APVR= anomalous pulmonary venous return, ASD= atrial septal defect, AS= aortic stenosis, AVSD= atrioventricular septal defect, BAV= bicuspid aortic valve, CoA= coarctation of the aorta, DORV= Double Outlet Right Ventricle, IAA= Interrupted aortic arch, MV= mitral valve, PA= pulmonary atresia, PDA= patent ductus arteriosus, PS= pulmonary stenosis, TGA= Transposition of the Great Arteries, TOF= tetralogy of fallot, Truncus= Truncus Arteriosus, VSD= ventricular septal defect
Abbreviations: Defined in text with the following additions: CNS= central nervous system, CV= cardiovascular, DLE= decreased life expectancy, EN= endocrine, FD= facial dysmorphism, GI= gastrointestinal, GU= genitourinary, GR= growth restriction, HM= hematologic, IUGR= intrauterine growth restriction, MSK= musculoskeletal, Onc= oncologic, Pulm= pulmonary, RH= rheumatologic, ID= Immunodeficiency
Copy number variation (CNV) refers to deletions or duplications of segments of chromosomes. They are estimated to cause 10–15% of CHD and common CNV mutations associated with CHD are listed in table 1 (36). They are usually de novo mutations, but can also follow patterns of Mendelian inheritance. Del22q11 (also known as DiGeorge Syndrome or Velocardio-facial syndrome) was one of the first copy number variation mutations to be associated with CHD. The deletion alters the expression of a key transcription factor TBX1, which results in the abnormal development of the pharyngeal arches and pouches leading to the clinical manifestations of the syndrome (37,38). These manifestations include CHD, hypocalcemia due to absence of the parathyroid glands, immunodeficiency due to thymic aplasia, characteristic facial features and neurodevelopmental abnormalities. There is significant heterogeneity, however, of clinical phenotypes among patients with del22q11, including between monozygotic twins with identical deletions (39).
B. Point Mutations, Mendelian Inheritance and De Novo Mutations
Point mutations alter the nucleotide sequence of a particular gene. In some cases, the change is inconsequential, causing no significant change to the amino-acid coded for by a particular codon (series of 3 nucleotides within a gene that identify which amino acid is inserted during protein synthesis). Some point mutations, however, dramatically alter protein structure and function by inserting either: 1) a “stop” codon that prematurely terminates gene transcription, 2) a “loss of function” mutation that significantly decreases the function of the protein through the insertion of a different amino acid in a critical region of the protein or 3) a “gain of function” mutation that results in abnormal upregulation of protein function.
It is estimated that approximately 2% of CHD is due to inherited point mutations. Such mutations most commonly affect genes for cardiac transcription factors, signaling molecules or cellular structures without significantly reducing reproductive potential (40, 41). For example, mutations in the NKX2.5 gene, a critical transcription factor that specifies cardiac mesoderm during embryogenesis, were some of the first mutations associated with inherited CHD. Its role in CHD, was discovered through pedigree analysis of families with atrial septal defects (ASD) and conduction disorders. Genotype-positive individuals, that is those who carried this mutation, had one of three phenotypes: 1) isolated ASD, 2) isolated conduction defects, or 3) ASD with conduction defects (42).
De novo mutations account for approximately 10% of CHD and their incidence has been stable over time. These mutations confer a reproductive disadvantage to affected individuals, with new mutations appearing at the same rate as previous generations of carriers with sporadic mutations fail to reproduce. In general, de novo mutations are more deleterious and cause more significant comorbidities than the mutations seen in Mendelian CHD (43). De novo mutations primarily cause CHD by altering critical developmental pathways shared by multiple developing organ systems, such as: chromatin remodeling (a critical process for regulating gene expression) and Notch signaling (instrumental for local intercellular communication in a variety of organ systems), (11, 44–48). The next section will show that mutations affecting these pathways provide a genetic explanation not only of CHD but also for the of some of the comorbidities that affect long term outcomes in CHD patients.
II. Clinical Implications of Genetic Causes of CHD
Three representative examples of comorbidities seen in CHD that have a common genetic origin with CHD are: Neurodevelopmental disability (NDD), respiratory disease and myocardial dysfunction. This section explores each of these comorbidities in detail with a focus on the clinical implications of their genetic origin.
A. Neurodevelopmental Outcomes in CHD
Epidemiological studies have found that 10% of all CHD patients and 50% of patients with severe CHD have NDD (4). Although clinical factors such as duration of deep hypothermic circulatory arrest at the time of surgical repair, duration of ICU stay, and pre-operative compromise of cerebral perfusion and oxygenation have been causally implicated in CHD-NDD, there is increasing evidence of a genetic contribution to neurodevelopmental outcomes. For example, Limperopolous, et al, found that preoperative NDD and microcephaly, both of which may be caused by a genetic syndrome, independently predicted neurodevelopmental outcomes in CHD patients (49).
One potential explanation for the dual effect of a genetic mutation causing both CHD and NDD, is altered chromatin remodeling, a critical process in regulating gene expression. Mutations affecting this process have the potential to affect the formation of multiple organ systems. Homsy et al, showed this in a study of 1,213 performed CHD trios (patient + unaffected parents) inclusive of CHD patients with and without NDD. They performed whole exome sequencing (WES) on the trios and identified de novo mutations in the CHD patients. They then compared these mutations to de novo mutations identified in cohorts of patients with NDD but without CHD. This identified 69 overlapping mutated genes that were present in both the CHD and the isolated NDD cohorts, many of which were mutations in genes affecting chromatin remodeling. When they compared the CHD patients with NDD to those without NDD, CHD patients with mutations in one of the overlapping 69 genes had a higher incidence of comorbid NDD thus identifying a potential genetic link between CHD and NDD (43).
The potential for a common genetic mutation to cause both CHD and NDD has significant implications for neurodevelopmental screening and intervention in CHD patients. Current guidelines for neurodevelopmental screening in CHD patients recommended that clinicians risk stratify CHD patients into high and low risk groups (Table 2). CHD patients with suspected genetic abnormalities or syndromes associated with NDD are stratified to the high risk group and should be referred for both early intervention services and formal developmental assessment. Thus appropriate genetic testing has the potential to improve outcomes for patients whose CHD is due to a genetic cause that also increases their risk of NDD by allowing for early intervention services that have a clear documented benefit in improving patient outcomes and decreasing the morbidity associated with NDD (50).
Table 2.
Categories of Pediatric CHD Patients at High Risk for Developmental Disorders or Disabilities
| 1. Neonates or infants requiring open heart surgery (cyanotic and acyanotic types), for example, HLHS, IAA, PA/IVS, TA, TAPVC, TGA, TOF, tricuspid atresia. |
| 2. Children with other cyanotic heart lesions not requiring open heart surgery during the neonatal or infant period, for example, TOF with PA and MAPCA(s), TOF with shunt without use of CPB, Ebstein anomaly. |
| 3. Any combination of CHD and the following comorbidities: |
| 3.1. Prematurity (<37 wk) |
| 3.2. Developmental delay recognized in infancy |
| 3.3. Suspected genetic abnormality or syndrome associated with DD |
| 3.4. History of mechanical support (ECMO or VAD use) |
| 3.5. Heart transplantation |
| 3.6. Cardiopulmonary resuscitation at any point |
| 3.7. Prolonged hospitalization (postoperative LOS >2-wk in the hospital) |
| 3.8. Perioperative seizures related to CHD surgery |
| 3.9. Significant abnormalities on neuroimaging or microcephaly* |
| 4. Other conditions determined at the discretion of the medical home providers |
CHD indicates congenital heart disease; HLHS, hypoplastic left heart syndrome; IAA, interrupted aortic arch; PA/IVS, pulmonary atresia with intact ventricular septum; TA, truncus arteriosus; TAPVC, total anomalous pulmonary venous connection; TGA, transposition of the great arteries; TOF, tetralogy of Fallot; PA, pulmonary atresia; MAPCA, major aortopulmonary collateral arteries; CPB, cardiopulmonary bypass; DD, developmental disorder or disability; ECMO, extracorporeal membrane oxygenation; VAD, ventricular assist device; and LOS, length of stay.
B. Respiratory Disease and CHD
Respiratory comorbidities cause significant morbidity and mortality in the CHD population. CHD patients demonstrate both an increased risk of death due to pneumonia and an increased incidence of moderate to severe impairment in lung function (3, 51). Similar to NDD associated with CHD, recent research has demonstrated common genetic pathways affecting both cardiac formation and respiratory status, particularly the role of cilia in embryonic development and post-natal organ function.
Cilia are hair-like structures found on the surface of most cells. They have a variety of functions both in development and in post-natal life, including: left-right organization of the embryo, development of the cardiovascular, renal, and neurologic systems, and post-natal function of the neurologic, reproductive and respiratory systems. During embryogenesis, mutations that compromise cilia function result in heart defects within the heterotaxy spectrum of CHD, as well as a number of other congenital malformations (48). During post-natal life cilia mutations affect respiratory function through impaired clearance of respiratory secretions, a condition known as primary ciliary dyskinesia (PCD). This leads to recurrent pulmonary infections and a progressive decline in pulmonary function. 6.5% of patients clinically diagnosed with PCD have concomitant cardiac defects within the heterotaxy spectrum of CHD.
Recognition of PCD in the context of CHD is challenging and requires a high degree of clinical suspicion. Patient history can suggest the diagnosis, particularly if there is a history of unexplained neonatal respiratory distress, situs inversus with respiratory symptoms, unexplained bronchiectasis or recurrent otitis media occurring with lower respiratory infections (52). Testing for PCD includes: electron microscopy, video microscopy and genetic evaluation. Each of these modalities can provide a component of the diagnosis, but each has limitations. Electron and video microscopy evaluate a patient’s cilia for structural and functional abnormalities. Electron microscopy is useful if patients have abnormal cilia ultrastructure, however it fails to make a diagnosis in the 30% of PCD patients who have normal cilia structure. The combination of electron microscopy and high speed video microscopy can definitively rule out PCD if ciliary function is normal, but cannot differentiate between PCD and secondary ciliary abnormalities due to inflammation or infection of the nasal or bronchial epithelium. Genetic testing looks for mutations in one of 35 genes known to cause PCD and makes a definitive diagnosis of PCD in 65% of PCD patients(52, 53). Given the limitations of each testing modality, clinicians must make the diagnosis by considering not only testing results but also the clinical history of the patient.
Once PCD is identified, a number of interventions are available to improve long term outcomes in affected CHD patients. For example, PCD patients undergoing cardiac surgery are known to have an increased risk of post-operative pulmonary complications (pneumonia, atelectasis, failure to wean from a ventilator, etc). This risk can be minimized by ensuring optimization of pre-operative pulmonary function, aggressive post-operative pulmonary toilet and (when possible) early extubation following surgery (54). Similarly vaccination and the early use of appropriate antibiotics can lessen the severity of pulmonary infection in patients with PCD and potentially slow the decline in pulmonary function in these patients (3).
C. Heart Failure in CHD
Approximately 25% of CHD patients have significant heart failure by the age of 30 (55). The mechanisms and clinical circumstances that lead to heart failure in CHD patients differ from those leading to heart failure in patients without CHD. There are very few studies examining the etiology and optimal management of CHD related heart failure. The absence of such studies, has left clinicians to extrapolate treatment decisions for CHD patients from guidelines developed from studies of patients with non-CHD heart failure (56). Unfortunately, the benefits demonstrated in the non-CHD heart failure management trials have not been replicated in the few studies of these management strategies for CHD-heart failure (57, 58).
The need for treatment specific to CHD-related heart failure has prompted studies of genetic mutations that contribute both to CHD and cardiac dysfunction (6). These studies have focused on genes with critical roles in embryonic cardiac formation and post-natal cardiac function such as MYH6. The MYH6 gene codes for the sarcomeric protein, myosin heavy chain 6 and is expressed during both fetal and post-natal life. MYH6 mutations are increasingly recognized to contribute to heart failure in the context of CHD (59, 60). Theis, et al, studied a cohort patients with hypoplastic left heart syndrome (HLHS) who developed systemic right ventricular failure following Fontan palliation. Sequencing affected patients revealed compound heterozygosity for recessive MYH6 mutations, whereas unaffected family members carried only one copy of the mutated gene (61). As more mutations are identified that cause both CHD and cardiac dysfunction, clinicians will be able to better identify CHD patients at high risk for subsequent heart failure, who will benefit from increased surveillance for heart failure and early interventions to address complications of heart failure.
To summarize, this section examined three CHD- related comorbidities that share a common genetic origin with CHD: NDD, respiratory comorbidities and CHD-related heart failure. Many other comorbidities are also seen in CHD patients, and investigations into genetic causes of these conditions is ongoing. The next section provides a clinical algorithm for clinicians to use when considering a genetic evaluation of a CHD patient.
III. Genetic Testing in CHD Patients
In order to initiate appropriate and cost efficient genetic testing of CHD patients clinicians must be familiar with available genetic tests and their clinical indications.
A. Currently Available Genetic Testing Modalities
Genetic tests can be divided into two categories: 1) general tests that look broadly to see if any genetic mutation is present and, 2) targeted tests that look for specific mutations. General testing modalities include: karyotype analysis, chromosomal microarray (CMA), and whole exome sequencing (WES). Karyotype is the most basic and detects abnormalities of chromosome number (aneuploidies). CMA is a more advanced technique used to analyze the entire coding and non-coding regions of a patient’s genome for CNV mutations. Currently, CMA analysis detects approximately 70% of CNV mutations (62). WES is the most comprehensive test currently available. It evaluates a patient’s entire exome for point mutations and sometimes CNV mutations (not all testing protocols include CNV analysis). WES may miss up to 5% of specific gene mutations, but its sensitivity continues to improve with time (62).
Targeted testing modalities include fluorescence in-situ hybridization (FISH) and targeted sequencing, they are best suited to identify a mutation known to cause a specific genetic syndrome with a clear phenotype. FISH is used to identify aneuploidies and CNV mutations by using a fluorescent probe to visualize a specific gene or portion of a gene. FISH does not detect abnormalities other than the one the probe is designed to look for. Targeted sequencing of one or more genes evaluates specific genes for point mutations. One example of a syndrome in which targeted sequencing of a single gene is recommended is Marfan syndrome. Marfan syndrome is a connective tissue disorder that results from mutations in FBN1 (Fibrillin 1). Patients have an increased risk of arterial aneurysms and dissections and often have a number of distinctive musculoskeletal features including tall stature, scoliosis, long fingers and flat feet. Targeted sequencing that identifies pathologic mutation of FBN1 is diagnostic and identifies patients that would benefit from annual surveillance for arterial aneurysms.
B. Clinical Approach to Genetic Testing in CHD
Figure 2 presents an algorithm for clinicians to use when beginning a genetics evaluation of a CHD patient (11). The key to efficient and appropriate testing of CHD patients is the clinical context of the patient. Testing patients with either mild isolated CHD, without a family history of CHD, or with no significant comorbidities, is unlikely to provide a genetic cause of the CHD and is not currently recommended. Testing patients with complex CHD, a positive family history of CHD, comorbid congenital malformations, or other significant comorbidities, such as NDD, increases the likelihood of a genetic cause of the CHD and genetic testing is strongly recommended. (To determine whether a patient’s CHD is considered “complex”, the reader is referred to the list of lesions included in the AHA neurodevelopmental screening guidelines that place a CHD patient at risk for NDD shown in Table 2) (4).
Figure 2. Genetics screening algorithm for CHD patients (Previously Published Figure).
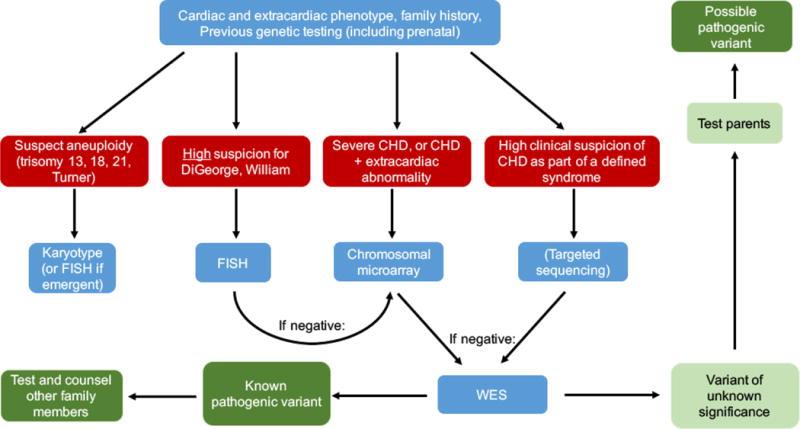
[11] Zaidi S, Brueckner M. Genetics and Genomics of Congenital Heart Disease. Circulation research. 2017;120(6):923-40.
Once the clinician decides to pursue genetic testing of a CHD patient, he or she must then decide whether targeted testing or general testing is most appropriate. If the clinical presentation, family history, extra-cardiac comorbidities, or prenatal testing is strongly suggestive of a specific syndrome, targeted testing for that syndrome should be pursued. For suspected aneuploidy, karyotype (or FISH if the result is required within a short time to make major clinical decisions) are the recommended first line tests. If the clinician strongly suspects a specific syndrome caused by CNV mutations, FISH for the specific deletion or duplication is recommended. When a patient’s clinical phenotype does not strongly suggest a specific syndrome, general genetic testing is the most efficient means to identify a genetic cause. CMA is the first recommended test due to its shorter time to result and lower cost. If this is negative, the next step is WES.
C. The Role of Genetics Consultation and Genetic Counselling
Genetics consultation can assist patients and clinicians in obtaining and understanding the results of genetic testing as it pertains to CHD. Clinical geneticists provide rigorous patient and family phenotyping and assist clinicians in selecting appropriate genetic testing modalities. They also assist in the interpretation of genetic test results, which can be particularly helpful when testing reveals a variant of unknown significance (VUS). These mutations are rare or novel variants that have not been previously reported in association with clinical disease. Methods to determine whether a VUS is pathogenic and the likely cause of a patient’s CHD include: rigorous family phenotyping with targeted genetic sequencing, evaluation of the evolutionary conservation of the mutated allele, and the effect of the mutation on gene transcription and protein function. Mutations that are de novo or segregate with affected family members are more likely to be pathogenic than mutations that are seen with equal frequency in affected and non-affected family members. Mutations that either occur in highly conserved regions of a gene, cause the introduction of a stop codon, or cause a loss of function mutation are all more likely to be pathogenic (63).
Genetic counselling is another service that clinicians may consider as part of the genetic evaluation of a CHD patient. Genetic counsellors are trained in counselling patients and families about genetic disease, the risk of disease recurrence in future offspring, and decisions about genetic testing both in the pre- and post-natal time periods. Their service is complementary to that of the clinical geneticist. By utilizing both services, CHD patients, families and the clinicians caring for them receive cost effective testing designed to meet the clinical needs of CHD patients and their families (64).
Conclusion
Care of CHD patients in the 21st century demands a comprehensive approach to patient care that includes not only surgical palliation of anatomical defects but also proactive strategies to address the comorbidities that cause morbidity and mortality in CHD patients. The timely and efficient use of genetics evaluation of CHD patients continues to gain importance in the long term care of CHD patients through the identification of patients at high risk for specific comorbidities. Clinicians are increasingly being asked to obtain basic genetics evaluation of CHD patients and incorporate the results of this testing in their management strategies. This review has provided an introduction to the genetics of CHD and its associated comorbidities as well as a clinical algorithm that clinicians can use to efficiently incorporate a genetics evaluation into the care of CHD patients.
Key Points.
Genetic mutations can be identified in 35% of patients with congenital heart disease. These mutations include aneuploidy, copy number variant mutations, and point mutations. They may be inherited through Mendelian inheritance of occur de novo.
Genetic causes of congenital heart disease also account for many of the comorbidities seen with increased frequency in congenital heart disease patients, including neurodevelopmental disability, pulmonary disease, arrhythmia, renal disease, heart failure and an increased incidence of malignancy.
Several testing modalities are available to evaluate for genetic causes of congenital heart disease. These include general tests such as karyotype, chromosomal microarray, and whole exome sequencing and targeted testing that identifies only specific mutations such as fluorescent in-situ hybridization and targeted genetic sequencing.
A patient’s clinical findings and family history are the most important elements to consider when deciding which genetic tests are most appropriate.
Clinicians are encouraged to utilize consultation with clinical geneticists and genetic counsellors in order to ensure efficient genetic testing and appropriate counselling of patients about the results of genetic testing.
Acknowledgments
None.
Financial support and sponsorship
M. Brueckner is supported by NIHLBI UM1HL098162-07
Footnotes
Conflicts of Interest
None.
References
- 1.Šamánek M. Children with congenital heart disease: probability of natural survival. Pediatric cardiology. 1992;13(3):152–8. doi: 10.1007/BF00793947. [DOI] [PubMed] [Google Scholar]
- 2•.van der Bom T, Mulder BJ, Meijboom FJ, van Dijk AP, Pieper PG, Vliegen HW, et al. Contemporary survival of adults with congenital heart disease. Heart. 2015 doi: 10.1136/heartjnl-2015-308144. heartjnl-2015-308144. This is a large, registry based epidemiological report on mortality outcomes of 14,327 patients with CHD. The study also includes limited data on the incidence of arrhythmia, heart failure and pulmonary hypertension in the adult CHD population. [DOI] [PubMed] [Google Scholar]
- 3.Alonso-Gonzalez R, Borgia F, Diller G-P, Inuzuka R, Kempny A, Martinez-Naharro A, et al. Abnormal Lung Function in Adults With Congenital Heart Disease: Prevalence, Relation to Cardiac Anatomy, and Association With SurvivalClinical Perspective. Circulation. 2013;127(8):882–90. doi: 10.1161/CIRCULATIONAHA.112.126755. [DOI] [PubMed] [Google Scholar]
- 4•.Marino BS, Lipkin PH, Newburger JW, Peacock G, Gerdes M, Gaynor JW, et al. Neurodevelopmental outcomes in children with congenital heart disease: evaluation and management. Circulation. 2012;126(9):1143–72. doi: 10.1161/CIR.0b013e318265ee8a. This is the current consensus statement on the incidence and management of neurodevelopmental abnormalities associated with CHD. [DOI] [PubMed] [Google Scholar]
- 5.Dimopoulos K, Diller G-P, Koltsida E, Pijuan-Domenech A, Papadopoulou SA, Babu-Narayan SV, et al. Prevalence, predictors, and prognostic value of renal dysfunction in adults with congenital heart disease. Circulation. 2008;117(18):2320–8. doi: 10.1161/CIRCULATIONAHA.107.734921. [DOI] [PubMed] [Google Scholar]
- 6•.Fahed AC, Roberts AE, Mital S, Lakdawala NK. Heart failure in congenital heart disease: a confluence of acquired and congenital. Heart failure clinics. 2014;10(1):219–27. doi: 10.1016/j.hfc.2013.09.017. This is a comprehensive review of the current understanding of the genetics that underlie heart failure in congenital heart disease. The studies reviewed in the summary provide an excellent bibliography for those looking for more indepth knowledge of the genetics of heart failure in CHD patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7•.Gurvitz M, Ionescu-Ittu R, Guo L, Eisenberg MJ, Abrahamowicz M, Pilote L, et al. Prevalence of Cancer in Adults With Congenital Heart Disease Compared With the General Population. The American Journal of Cardiology. 2016;118(11):1742–50. doi: 10.1016/j.amjcard.2016.08.057. This is an intriguing study of 34,965 adults with CHD patients enrolled in the Quebec CHD database that showed that CHD patients have a 1.6-2 times higher prevalence of cancer than the general population. [DOI] [PubMed] [Google Scholar]
- 8.Herskind AM, Pedersen DA, Christensen K. Increased prevalence of congenital heart defects in monozygotic and dizygotic twins. Circulation. 2013;113:002453. doi: 10.1161/CIRCULATIONAHA.113.002453. CIRCULATIONAHA. [DOI] [PubMed] [Google Scholar]
- 9.Øyen N, Poulsen G, Boyd HA, Wohlfahrt J, Jensen PK, Melbye M. Recurrence of congenital heart defects in families. Circulation. 2009;120(4):295–301. doi: 10.1161/CIRCULATIONAHA.109.857987. [DOI] [PubMed] [Google Scholar]
- 10.Øyen N, Poulsen G, Wohlfahrt J, Boyd HA, Jensen PK, Melbye M. Recurrence of discordant congenital heart defects in families. Circulation: Cardiovascular Genetics. 2010;109:890103. doi: 10.1161/CIRCGENETICS.109.890103. CIRCGENETICS. [DOI] [PubMed] [Google Scholar]
- 11•.Zaidi S, Brueckner M. Genetics and Genomics of Congenital Heart Disease. Circulation research. 2017;120(6):923–40. doi: 10.1161/CIRCRESAHA.116.309140. This is a comprehensive review of the current advances in understanding the genetics of CHD. The review requires a basic understanding of genetics and includes significant amount of translational research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartman RJ, Rasmussen SA, Botto LD, Riehle-Colarusso T, Martin CL, Cragan JD, et al. The contribution of chromosomal abnormalities to congenital heart defects: a population-based study. Pediatric cardiology. 2011;32(8):1147–57. doi: 10.1007/s00246-011-0034-5. [DOI] [PubMed] [Google Scholar]
- 13.Ferencz C, Neill CA, Boughman JA, Rubin JD, Brenner JI, Perry LW. Congenital cardiovascular malformations associated with chromosome abnormalities: an epidemiologic study. The Journal of pediatrics. 1989;114(1):79–86. doi: 10.1016/s0022-3476(89)80605-5. [DOI] [PubMed] [Google Scholar]
- 14.Korbel JO, Tirosh-Wagner T, Urban AE, Chen X-N, Kasowski M, Dai L, et al. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proceedings of the National Academy of Sciences. 2009;106(29):12031–6. doi: 10.1073/pnas.0813248106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riccardi V. Trisomy 8: an international study of 70 patients. Birth defects original article series. 1976;13(3C):171–84. [PubMed] [Google Scholar]
- 16.Arnold GL, Kirby RS, Stern TP, Sawyer JR. Trisomy 9: review and report of two new cases. American Journal of Medical Genetics Part A. 1995;56(3):252–7. doi: 10.1002/ajmg.1320560303. [DOI] [PubMed] [Google Scholar]
- 17.Pont SJ, Robbins JM, Bird T, Gibson JB, Cleves MA, Tilford JM, et al. Congenital malformations among liveborn infants with trisomies 18 and 13. American Journal of medical Genetics Part A. 2006;140(16):1749–56. doi: 10.1002/ajmg.a.31382. [DOI] [PubMed] [Google Scholar]
- 18.Jones KL, Jones MC, Del Campo M. Smith’s recognizable patterns of human malformation: Elsevier Health Sciences. 2013 [Google Scholar]
- 19.Bruns D, Campbell E. Twenty‐two survivors over the age of 1 year with full trisomy 18: Presenting and current medical conditions. American Journal of Medical Genetics Part A. 2014;164(3):610–9. doi: 10.1002/ajmg.a.36318. [DOI] [PubMed] [Google Scholar]
- 20.Freeman SB, Taft LF, Dooley KJ, Allran K, Sherman SL, Hassold TJ, et al. Population-based study of congenital heart defects in Down syndrome. American journal of medical genetics. 1998;80(3):213–7. [PubMed] [Google Scholar]
- 21.Gøtzsche C, Krag-Olsen B, Nielsen J, Sørensen K, Kristensen BO. Prevalence of cardiovascular malformations and association with karyotypes in Turner’s syndrome. Archives of disease in childhood. 1994;71(5):433–6. doi: 10.1136/adc.71.5.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Battaglia A, Filippi T, Carey JC, editors. American Journal of Medical Genetics Part C: Seminars in Medical Genetics. Wiley Online Library; 2008. Update on the clinical features and natural history of Wolf–Hirschhorn (4p‐) syndrome: Experience with 87 patients and recommendations for routine health supervision. [DOI] [PubMed] [Google Scholar]
- 23.Hills C, Moller JH, Finkelstein M, Lohr J, Schimmenti L. Cri du chat syndrome and congenital heart disease: a review of previously reported cases and presentation of an additional 21 cases from the Pediatric Cardiac Care Consortium. Pediatrics. 2006;117(5):e924–e7. doi: 10.1542/peds.2005-1012. [DOI] [PubMed] [Google Scholar]
- 24.Kobrynski L, Sullivan K. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. The Lancet. 2007;370(9596):1443–1452. doi: 10.1016/S0140-6736(07)61601-8. [DOI] [PubMed] [Google Scholar]
- 25.Soemedi R, Topf A, Wilson IJ, Darlay R, Rahman T, Glen E, et al. Phenotype-specific effect of chromosome 1q21. 1 rearrangements and GJA5 duplications in 2436 congenital heart disease patients and 6760 controls. Human molecular genetics. 2011:ddr589. doi: 10.1093/hmg/ddr589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, et al. Recurrent reciprocal 1q21. 1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nature genetics. 2008;40(12):1466–71. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, et al. Recurrent rearrangements of chromosome 1q21. 1 and variable pediatric phenotypes. New England Journal of Medicine. 2008;359(16):1685–99. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Battaglia A. 1p36 deletion syndrome. Gene Reviews 2013 [Google Scholar]
- 29.Green EK, Priestley MD, Waters J, Maliszewska C, Latif F, Maher ER. Detailed mapping of a congenital heart disease gene in chromosome 3p25. Journal of medical genetics. 2000;37(8):581–7. doi: 10.1136/jmg.37.8.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Digilio MC, Versacci P, Lepri F, Baban A, Dallapiccola B, Marino B. Atrioventricular canal defect and associated genetic disorders: new insights into polydactyly syndromes. Cardiogenetics. 2011;1(1):7. [Google Scholar]
- 31.Figueroa JDR, Rodríguez LMO, Hach JLP, Ruíz VDC, Martínez HO. Cardiovascular spectrum in Williams-Beuren syndrome: the Mexican experience in 40 patients. Texas Heart Institute Journal. 2008;35(3):279. [PMC free article] [PubMed] [Google Scholar]
- 32.Pehlivan T, Pober BR, Brueckner M, Garrett S, Slaugh R, Van Rheeden R, et al. GATA4 haploinsufficiency in patients with interstitial deletion of chromosome region 8p23. 1 and congenital heart disease. American Journal of Medical Genetics Part A. 1999;83(3):201–6. [PubMed] [Google Scholar]
- 33.Mattina T, Perrotta CS, Grossfeld P. Jacobsen syndrome. Orphanet journal of rare diseases. 2009;4(1):9. doi: 10.1186/1750-1172-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouquillon S, Boute O, Holder-Espinasse M, Delobel B, Duban B, Vallee L, et al. 15q11. 2 microdeletion (BP1eBP2) and developmental delay, behaviour issues, epilepsy and congenital heart disease: A series of 52 patients. European Journal of Medical Genetics. 2015;30:1e8. doi: 10.1016/j.ejmg.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 35.Thienpont B, Béna F, Breckpot J, Philip N, Menten B, Van Esch H, et al. Duplications of the critical Rubinstein–Taybi deletion region on chromosome 16p13. 3 cause a novel recognisable syndrome. Journal of medical genetics. 2010;47(3):155–61. doi: 10.1136/jmg.2009.070573. [DOI] [PubMed] [Google Scholar]
- 36•.Kim DS, Kim JH, Burt AA, Crosslin DR, Burnham N, Kim CE, et al. Burden of potentially pathologic copy number variants is higher in children with isolated congenital heart disease and significantly impairs covariate-adjusted transplant-free survival. The Journal of thoracic and cardiovascular surgery. 2016;151(4):1147–51. e4. doi: 10.1016/j.jtcvs.2015.09.136. This study demonstrated the effect of CNV mutations on long term outcomes in CHD patients. The study demonstrated a decreased transplant-free survival of CHD patients with CNV mutations as compared to CHD patients without CNV mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nature genetics. 2001;27(3):286–91. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- 38.Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104(4):619–29. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 39.Goodship J, Cross I, Scambler P, Burn J. Monozygotic twins with chromosome 22q11 deletion and discordant phenotype. Journal of medical genetics. 1995;32(9):746–8. doi: 10.1136/jmg.32.9.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437(7056):270–4. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 41.Vetrini F, D’Alessandro LC, Akdemir ZC, Braxton A, Azamian MS, Eldomery MK, et al. Bi-allelic Mutations in PKD1L1 Are Associated with Laterality Defects in Humans. The American Journal of Human Genetics. 2016;99(4):886–93. doi: 10.1016/j.ajhg.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, et al. Mutations in the cardiac transcription factor NKX2. 5 affect diverse cardiac developmental pathways. The Journal of clinical investigation. 1999;104(11):1567–73. doi: 10.1172/JCI8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43•.Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350(6265):1262–6. doi: 10.1126/science.aac9396. This is a translational research study suggesting a genetic link between neurodevelopmental outcomes and CHD. The study demonstrated that there are overlapping genetic mutations between CHD patients with neurodevelopmental disability and patients with isolated neurodevelopmental disorders. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44•.Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498(7453):220–3. doi: 10.1038/nature12141. This is the first study linking de-novo mutation to CHD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sifrim A, Hitz M-P, Wilsdon A, Breckpot J, Al Turki SH, Thienpont B, et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nature genetics. 2016 doi: 10.1038/ng.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nature genetics. 1997;16(3):243–51. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- 47.Eldadah ZA, Hamosh A, Biery NJ, Montgomery RA, Duke M, Elkins R, et al. Familial Tetralogy of Fallot caused by mutation in the jagged1 gene. Human molecular genetics. 2001;10(2):163–9. doi: 10.1093/hmg/10.2.163. [DOI] [PubMed] [Google Scholar]
- 48.Kennedy MP, Omran H, Leigh MW, Dell S, Morgan L, Molina PL, et al. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation. 2007;115(22):2814–21. doi: 10.1161/CIRCULATIONAHA.106.649038. [DOI] [PubMed] [Google Scholar]
- 49.Limperopoulos C, Majnemer A, Shevell MI, Rohlicek C, Rosenblatt B, Tchervenkov C, et al. Predictors of developmental disabilities after open heart surgery in young children with congenital heart defects. The Journal of pediatrics. 2002;141(1):51–8. doi: 10.1067/mpd.2002.125227. [DOI] [PubMed] [Google Scholar]
- 50.Diamond A, Barnett WS, Thomas J, Munro S. Preschool program improves cognitive control. Science (New York, NY) 2007;318(5855):1387. doi: 10.1126/science.1151148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51•.Raissadati A, Nieminen H, Haukka J, Sairanen H, Jokinen E. Late causes of death after pediatric cardiac surgery: a 60-year population-based study. Journal of the American College of Cardiology. 2016;68(5):487–98. doi: 10.1016/j.jacc.2016.05.038. This is a large-scale epidemiological study of causes of death in CHD patients undergoing CHD surgery in Finland. It provides details a breakdown of cause and age of death. The study identifies the significant causes of cardiac and non-cardiac morbidity in CHD patients. [DOI] [PubMed] [Google Scholar]
- 52.Lucas JS, Burgess A, Mitchison HM, Moya E, Williamson M, Hogg C, et al. Diagnosis and management of primary ciliary dyskinesia. Archives of disease in childhood. 2014 doi: 10.1136/archdischild-2013-304831. archdischild-2013-304831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knowles MR, Daniels LA, Davis SD, Zariwala MA, Leigh MW. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. American journal of respiratory and critical care medicine. 2013;188(8):913–22. doi: 10.1164/rccm.201301-0059CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harden B, Tian X, Giese R, Nakhleh N, Kureshi S, Francis R, et al. Increased postoperative respiratory complications in heterotaxy congenital heart disease patients with respiratory ciliary dysfunction. The Journal of thoracic and cardiovascular surgery. 2014;147(4):1291–8. e2. doi: 10.1016/j.jtcvs.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 55.Norozi K, Wessel A, Alpers V, Arnhold JO, Geyer S, Zoege M, et al. Incidence and risk distribution of heart failure in adolescents and adults with congenital heart disease after cardiac surgery. The American journal of cardiology. 2006;97(8):1238–43. doi: 10.1016/j.amjcard.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 56.Book WM, Gerardin J, Saraf A, Marie Valente A, Rodriguez F. Clinical Phenotypes of Fontan Failure: Implications for Management. Congenital heart disease. 2016;11(4):296–308. doi: 10.1111/chd.12368. [DOI] [PubMed] [Google Scholar]
- 57.Hsu DT, Zak V, Mahony L, Sleeper LA, Atz AM, Levine JC, et al. Enalapril in infants with single ventricle. Circulation. 2010;122(4):333–40. doi: 10.1161/CIRCULATIONAHA.109.927988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kouatli AA, Garcia JA, Zellers TM, Weinstein EM, Mahony L. Enalapril does not enhance exercise capacity in patients after Fontan procedure. Circulation. 1997;96(5):1507–12. doi: 10.1161/01.cir.96.5.1507. [DOI] [PubMed] [Google Scholar]
- 59.Ching Y-H, Ghosh TK, Cross SJ, Packham EA, Honeyman L, Loughna S, et al. Mutation in myosin heavy chain 6 causes atrial septal defect. Nature genetics. 2005;37(4):423–8. doi: 10.1038/ng1526. [DOI] [PubMed] [Google Scholar]
- 60.Norton N, Robertson PD, Rieder MJ, Zuchner S, Rampersaud E, Martin E, et al. Evaluating pathogenicity of rare variants from dilated cardiomyopathy in the exome era. Circulation: Cardiovascular Genetics. 2012:111.961805. doi: 10.1161/CIRCGENETICS.111.961805. CIRCGENETICS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Theis JL, Zimmermann MT, Evans JM, Eckloff BW, Wieben ED, Qureshi MY, et al. Recessive MYH6 mutations in hypoplastic left heart with reduced ejection fraction. Circulation: Cardiovascular Genetics. 2015:115.001070. doi: 10.1161/CIRCGENETICS.115.001070. CIRCGENETICS. [DOI] [PubMed] [Google Scholar]
- 62.Glessner J, Bick AG, Ito K, Homsy J, Rodriguez-Murillo L, Fromer M, et al. Increased frequency of de novo copy number variations in congenital heart disease by integrative analysis of SNP array and exome sequence data. Circulation research. 2014:114.304458. doi: 10.1161/CIRCRESAHA.115.304458. CIRCRESAHA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine. 2015;17(5):405–23. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ladouceur VB, Gelb BD. Training in Cardiovascular Genetics. Journal of the American College of Cardiology. 2015 doi: 10.1016/j.jacc.2015.01.003. [DOI] [PubMed] [Google Scholar]