

SUMMARY
Importance
KRAS mutations are very common in pancreatic cancer, but directly targeting the KRAS protein has thus far been unsuccessful. The aim of this trial was to block the MEK and PI3K/AKT pathways downstream of the KRAS protein as an alternate treatment strategy to slow cancer growth and prolong survival. This was the first cooperative group trial to evaluate this strategy using molecularly targeted oral combination therapy for the treatment of chemotherapy refractory pancreatic cancer.
Objective
SWOG S1115 was a randomized phase 2 study of selumetinib and MK-2206 versus modified FOLFOX in patients who failed gemcitabine-based therapy.
Design, Setting, and Participants
Between September 2012 and May 2014, 137 patients with metastatic pancreatic adenocarcinoma who failed gemcitabine-based chemotherapy were randomized to selumetinib plus MK-2206 or mFOLFOX. Patients were randomized in a 1:1 fashion and stratified according to duration of prior systemic therapy and presence of liver metastases.
Interventions
Patients received selumetinib 100 mg orally per day plus MK-2206 135 mg orally once per week or mFOLFOX (oxaliplatin 85 mg/m2 intravenous and 5-fluorouracil 2,400 mg/m2 intravenous infusion over 46–48 hours) on days 1 and 15 with each cycle being 28 days.
Main Outcomes and Measures
The primary endpoint of the study was overall survival. Secondary objectives included evaluating toxicities, objective tumor response and progression free survival (PFS).
Results
Median OS was shorter in the experimental arm (3.9 vs 6.7 months, HR=1.37, p=0.15). PFS was also inferior in the experimental arm (HR=1.61, p=0.02). One vs five patients had a partial response and 12 vs 14 patients had stable disease in the selumetinib plus MK-2206 arm versus (vs) mFOLFOX. Grade 3 or higher toxicities were observed in 39 patients treated with selumetinib and MK-2206 vs 23 patients treated with mFOLFOX. More patients on the experimental arm discontinued therapy due to adverse events, as well.
Conclusions and Relevance
Although, dual targeting of MEK and PI3K/AKT pathways downstream of KRAS by selumetinib plus MK-2206 did not improve overall survival in patients who failed gemcitabine-based chemotherapy, this was the first randomized prospective evaluation of mFOLFOX in the U.S. population which showed comparable results to CONKO-003 and PANCREOX.
Trial Registration
ClinicalTrials.gov, number NCT01658943
INTRODUCTION
Metastatic pancreatic cancer remains resistant to conventional systemic treatments with median overall survival being less than a year. Due to toxicities of combination cytotoxics, rational approaches with targeted therapies have been attempted in hope of minimizing toxicities. Erlotinib is the only FDA approved molecularly targeted treatment for pancreatic cancer; however, the combination with gemcitabine improved median overall survival by only 2 weeks compared to gemcitabine alone.1 Therefore, traditional cytotoxics have been the mainstay of treatment. FOLFIRINOX has the highest reported objective response rate with a median overall survival just under 1 year but at the cost of increased toxicities.2 The applicability of FOLFIRINOX is therefore limited to younger patients with a good performance status, near normal liver function and a willingness to undergo aggressive therapy for metastatic disease. Gemcitabine and nab-paclitaxel provides another treatment option albeit with a shorter median overall survival.3
KRAS protein is a GTPase that regulates cell growth, angiogenesis and survival. Over 90% of pancreatic ductal adenocarcinomas have activating mutations in this protein which is also one of the earliest genetic alterations resulting in neoplastic transformation.4, 5 Many attempts have been made to target mutant KRAS.6 Earlier studies targeting RAS were directed against its farnesylation, a critical step in its activation. Unfortunately there was no evidence of benefit to patients, partly because of the alternate activation of RAS by geranylgeranylation.7 Since there are currently no drugs that directly target mutant RAS, inhibiting its downstream canonical RAF/MEK/ERK and PI3K/AKT signaling pathways was a rational alternate treatment strategy.8, 9 An earlier trial of tremetinib, an orally bioavailable, reversible inhibitor of MEK 1/2, in combination with gemcitabine showed a trend for improved survival suggesting that single pathway blockade was not sufficient for a clinically worthwhile benefit that was suggested by preclinical studies.10 Subsequent preclinical studies explored the cross-talk between pathways downstream of RAS to determine mechanisms to overcome resistance.11, 12 Indeed, there was evidence that inhibition of MEK led to upregulation of AKT phosphorylation that in turn allowed continued cell survival and proliferation.13, 14 Furthermore, enhanced cytotoxicity in pancreatic cancer cell lines was demonstrated by blocking both MEK and AKT.15, 16
Selumetinib is a potent, selective, ATP-uncompetitive inhibitor of MEK 1/2 with a maximum tolerated dose (MTD) of 200 mg twice per day as a single agent. The phase 1 trial showed significant rash at this dose and it was therefore decreased to the well-tolerated 100 mg twice per day. The median half-life was 8 hours and treatment tumor biopsies demonstrated inhibition of ERK phosphorylation.17, 18 MK-2206 was the first allosteric AKT inhibitor and at nanomolar concentrations, inhibited all three isoforms of AKT. In a phase 1 study, the MTD was determined to be 60 mg every other day with a mean half-life of 63–76 hrs.Due to its dose limiting toxicities of rash and mucositis,19 when combined with selumetinib, the dosing schedule had to be modified because of overlapping toxicities. Selumetinib could only be given at a dose of 100 mg per day rather than twice per day and MK-2206 was given at a dose of 135 mg per week rather than every other day. Despite the decreased dose density, there were two pancreatic cancer patients that had stable disease with one of the patients having a KRAS mutated tumor.15
KRAS mutation is the most frequent genomic alteration in pancreatic cancer and considered essential in the biology of this disease.4 Previous attempts at targeting this mutant protein have been unsuccessful; therefore, we embarked on this novel trial utilizing the combination of selumetinib and MK2206 to target downstream effectors. Our hypothesis was that blockade of signaling downstream of KRAS by dual targeting of MEK and AKT pathways would slow tumor growth and prolong survival of patients with metastatic pancreatic cancer. This was the first second-line pancreas cancer trial exploring a non-cytotoxic combination regimen conducted by SWOG. This was also the first study to prospectively evaluate mFOLFOX in the United States population.
METHODS
Study Design and Participants
S1115 was an open-label randomized phase 2 study completed within the National Cancer Institutes’s National Clinical Trials Network groups SWOG, ECOG-ACRIN and Alliance (Figure 1). Sixty-one sites participated with SWOG being the coordinating group. The participating sites obtained institutional review board approval and informed, written consent was obtained from all patients prior to enrollment. The study was registered on ClinicalTrials.gov, number NCT01658943.
Figure 1.
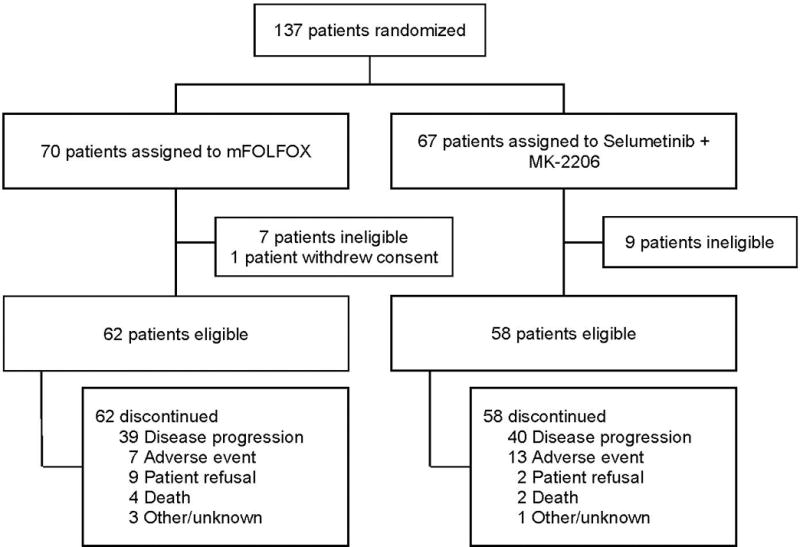
Trial profile
Patients age 18 years and older with a Zubrod performance status of 0–1were eligible if they had histologically or cytologically confirmed diagnosis of pancreatic adenocarcinoma that was metastatic. Patients with neuroendocrine tumors, lymphoma or ampullary adenocarcinoma were excluded. Prior gemcitabine-based chemotherapy must have been completed at least 14 days prior to registration and toxicities from therapy must have recovered to CTCAE grade ≤ 1. If prior treatment included FOLFIRINOX, FOLFOX, other oxaliplatin-based chemotherapy, MEK, PI3K or AKT inhibitors, the patient was deemed ineligible. Normal cardiac and renal functions were required. In patients with hepatic metastases, the total bilirubin was required to be ≤ Institutional Upper Limit of Normal (IULN); AST and ALT both to be ≤ 2.5 × IULN and serum alkaline phosphatase to be ≤ 3 × IULN. Patients were required to have an albumin level ≥ 2.5 g/dL and uncontrolled diarrhea was an exclusion criterion. Patients with any visual abnormalities except myopia, hyperopia, and presbyopia were excluded.
Randomization
Patients were randomized 1:1 to selumetinib plus MK-2206 or mFOLFOX by the SWOG Statistical Center using a dynamic balancing algorithm (Pocock and Simon, 1975) with stratification based on duration of prior systemic chemotherapy (≤ or > 4 months) and presence or absence of liver metastases.
Procedures
Baseline evaluation including history and physical examination, laboratory evaluations and imaging by computed tomography (CT) or magnetic resonance imaging (MRI) were completed prior to registration. Laboratory tests were within 14 days while imaging was within 28 days of registration. Patients received selumetinib 100 mg orally per day plus MK-2206 135 mg orally once per week or mFOLFOX (oxaliplatin 85 mg/m2 intravenous and 5-fluorouracil 2,400 mg/m2 intravenous infusion over 46–48 hours) on days 1 and 15 with each cycle being 28 days. Hematopoietic growth factors were allowed per ASCO guidelines and antiemetic medications were prescribed per institutional guidelines. Tumor assessments by CT or MRI scan were performed every 2 cycles until disease progression.
Study Endpoints
The primary objective of this study was to estimate the overall survival in patients receiving selumetinib plus MK-2206 or mFOLFOX after failing gemcitabine-based chemotherapy. Secondary objectives included evaluating toxicities per National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) v4.0, objective tumor response and progression free survival (PFS) according to RECIST 1.0. OS and PFS endpoints were measured from the time of randomization and censoring time was defined as the date of last contact. Patients were followed until death or 3 years after registration, whichever occurred first.
Statistical Analysis
Median OS was assumed to be 6 months for the mFOLFOX arm based upon previously published results of the CONKO-003 and PANCREOX trials.20, 21 Assuming a one-sided type 1 error of 10%, approximately 2 years of accrual, and 1.5 years of follow-up, 120 eligible patients provided 80% power to detect a 0.66 hazard ratio22. An interim analysis of OS was planned once 34% of the expected events in the mFOLFOX arm were observed. The study was to close early if the alternative hypothesis was rejected at a one-sided 0.05 level. According to the intent-to-treat principle, all eligible patients were included in the analyses according to randomized treatment assignment, regardless of treatments received.
Probabilities of OS and PFS were estimated using the Kaplan-Meier method. Statistical differences in event rates between treatment arms were assessed via Cox proportional hazards model. Rates of objective tumor response (confirmed and unconfirmed complete and partial response), were compared via Fisher’s exact test, in the subset of patients with measurable disease. Heterogeneity between treatment arms was tested using a two-sample t-test for age and chi-square tests for sex, race, prior systemic therapy duration, and presence of liver metastases.
We instituted careful adverse event monitoring for the first 20 patients randomized to the experimental arm because of limited pre-existing clinical data on the safety of selumetinib combined with MK-2206. Toxicities were closely monitored by the Study Coordinator, Study Statistician, Disease Committee Chair, and SWOG GI Executive Officer, in conjunction with the SWOG DSMC and CTEP.
RESULTS
Between September 2012 and May 2014, 137 patients with metastatic pancreatic cancer failing gemcitabine-based chemotherapy were randomized to selumetinib plus MK-2206 or mFOLFOX. (Figure 1). Sixteen patients were ineligible after not meeting protocol specified eligibility criteria. One additional patient withdrew consent prior to protocol treatment. Thus, 58 patients in the selumetinib plus MK-2206 arm and 62 patients in the mFOLFOX arm were available for toxicity and efficacy analyses. eTable 1 in the supplement provides the patient characteristics. In the experimental arm, patients were older with more males compared to the mFOLFOX arm (p=0.001 and p=0.01, respectively). Overall, 50% of patients (n=60) had received combination, rather than single agent, gemcitabine-based chemotherapy with 48% (n=28) in the experimental arm and 52% (n=32) in the mFOLFOX arm. Since the approval of nab-paclitaxel for pancreatic cancer in 2013, this was most commonly used combination with gemcitabine, Prior systemic therapy duration and percentage of patients with liver metastases was not significantly different between treatment arms (p = 0.99 and p=0.24).
Adverse Events
eTable 2 in the supplement compares the frequency of treatment related adverse events occurring in at least 10% of the patients. The most common toxicities observed in selumetinib plus MK-2206 treated patients were nausea and vomiting occurring in 41.4% and 31.0% of patients, respectively. The frequency of nausea and vomiting for mFOLFOX was higher at 59.7% and 30.6%, with a higher incidence of grade 3 toxicity compared to the oral therapy arm. In the experimental arm, fatigue and anorexia occurred in 41.4% and 32.8% of patients, compared to 56.5% and 32.3% in the mFOLFOX arm. Overall, more patients in the experimental arm vs the mFOLFOX arm experienced grade 3 or higher toxicities (67% vs 37%). Rash and mucositis, which are common side effects for this class of drugs, occurred in 51.7% and 22.4% of patients. The experimental arm had 45% of the patients require dose modifications or delays in the first cycle compared to 10% for mFOLFOX (eTable 3). In addition more patients on the experimental arm discontinued treatment compared to mFOLFOX 22% versus 10% (eTable 4).
Efficacy
Based upon the assumptions made at the beginning of the study, the interim analysis was expected that to occur at 15 months with approximately 62% of accrual met. Due to very robust accrual and a lower than expected event rate, we completed accrual to the trial prior to the pre-specified interim analysis. After the data was analyzed, the futility boundary was crossed and the results were released early to the public. The median PFS was 1.9 months in the selumetinib plus MK-2206 arm and 2.0 months in the mFOLFOX arm with a hazard ratio (HR) of 1.61 (95% confidence interval (CI) 1.07–2.43, p=0.02) (Figure 2). The median OS was 3.9 months in the selumetinib plus MK-2206 arm and 6.7 months in the mFOLFOX arm (Figure 3). The estimated hazard ratio for the comparison of the selumetinib plus MK-2206 arm to the mFOLFOX arm was 1.37 (95% CI 0.90–2.08, p=0.15). Of the 55 and 57 evaluable patients with measurable disease in the selumetinib plus MK-2206 and mFOLFOX arms, no complete responses were seen. In the selumetinib plus MK- 2206 arm vs mFOLFOX, there were 1 vs 4 patients with a partial response (p=0.21) and 12 vs 14 patients with stable disease, respectively.
Figure 2.
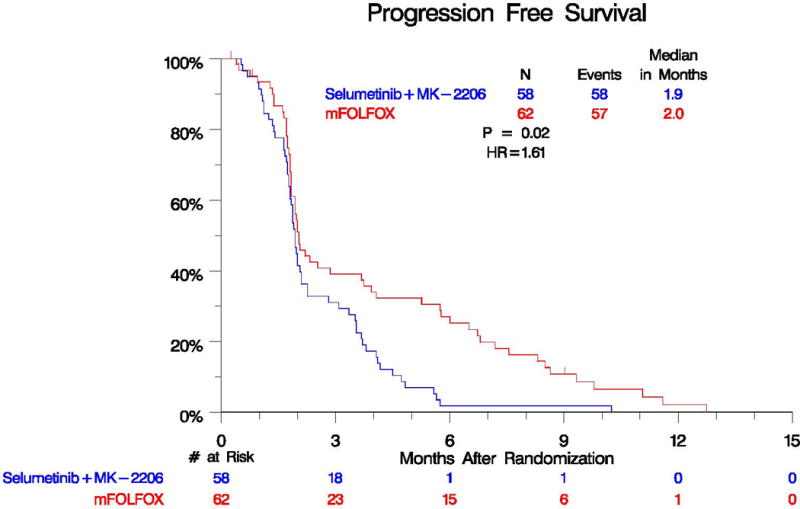
Progression Free Survival by Treatment Arm
Figure 3.
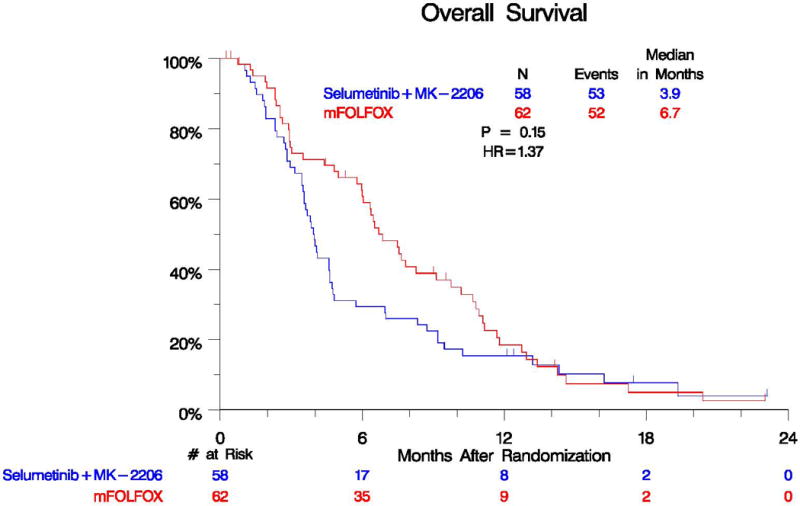
Overall Survival by Treatment Arm
DISCUSSION
The very modest advances in the treatment of pancreatic cancer were made exclusively with conventional cytotoxic drugs. Targeted agents inhibiting a single pathway such as IGF-1R or MEK, though promising in the preclinical setting, failed to improve survival in a variety of cancers.23, 24 This study addresses mutant RAS signaling specifically targeting downstream survival and proliferation pathways utilizing a dual inhibitory strategy with two oral molecularly targeted drugs, selumetinib and MK-2206. The results of this trial did not meet its primary endpoint of improving survival over a standard treatment with modified FOLFOX. However, our trial showed that in an American population, efficacy of mFOLFOX was similar to those obtained in the CONKO-003 and PANCREOX trials. With OFF and mFOLFOX6 chemotherapy, the median overall survival was 5.9 mos and 6.1 mos respectively which is comparable to our survival of 6.7 mos. This was the first trial to prospectively evaluate and show efficacy of this regimen in the US population.
The lack of clinical activity of this combined targeted strategy in advanced pancreatic cancer may be attributed to a number of factors related to sustained blockage of signaling pathways in vivo. In single agent phase 1 studies of selumetinib and MK-2206, target inhibition was achieved with twice daily dosing of selumetinib and alternate day dosing of MK-2206.18, 19 However, the maximum tolerated dose of the phase 1 study using the combination of the two drugs was only 100 mg daily for selumetinib and 135 mg once per week for MK2206. The lower dose intensity was because of their overlapping toxicities.15 It is reasonable to assume that a non-cytotoxic regimen must be given at an optimal dose intensity for sustained inhibition of signaling pathways that are necessary for an effective clinical outcome. This was demonstrated in biomarker trial studying this drug combination in colorectal cancer patients.25 Pre- and post-treatment biopsies were obtained to evaluate pAKT and pERK inhibition. In their trial, target inhibition of 70% was pre-specified and considered necessary for clinical activity based upon preclinical data. Dual target inhibition at the specified levels was not seen using selumetinib 75 mg daily and MK-2206 90 mg weekly. Escalation to doses similar to our study also did not produce worthwhile dual target inhibition and there was no clinical responses in this population of patients with advanced colorectal cancer.25 This indicates that the strategy of utilizing two or more kinase inhibitors, though scientifically justified, is challenged by the overlapping toxicities that would significantly influence the delivery of effective inhibitory doses of both drugs in vivo.
A major contributing factor to the lack of benefit was the frequency of toxicity related treatment delays and dose reductions in the experimental arm undermining sustained signaling inhibition. Dose delays or dose reductions occurred in 45% of patients on the experimental arms compared to only 10% in the mFOLFOX arm. Mucositis and rash were more frequently observed in the experimental arm and although managed symptomatically, ultimately dose modifications were required. The self-limiting nature of these toxicities in the mFOLFOX arm with every 2 week infusions made it more tolerable to patients.
Results of this trial can also be explained by alternate signaling pathways that drive pancreatic cell growth and survival. As an example, effectors such as Ral protein signaling may be relevant to tumor progression. Recently, Lauffenburger’s group reported on posttranslational modification of the cell surface as a mechanism of resistance to targeting MAPK signaling.26 Normally, proteolytic shedding of cell surface receptors can provide negative feedback on signaling activity; however, MAPK inhibition decreases this post-translational event by enhancing the signaling of sheddase substrates. Decreased proteolysis leads to surface accumulation of receptor tyrosine kinases (RTK) and increased signaling through other pathways such a JNK to promote cellular proliferation. The RTK shedding also impacts the tumor microenvironment amplifying prosurvival and prometastatic tumor-stroma interactions.26 This illustrates the complexity of the signaling pathways necessitating the need to block multiple ones for clinical benefit. However, the challenge lies in combining multiple inhibitors that will be tolerable to the patient
This study is informative for the design of future trials with targeted agents especially for clinical testing of multi-targeted strategies. Despite preclinical models which demonstrated synergy and target inhibition of MEK and AKT signaling, this did not translate into a clinical benefit in patients with refractory metastatic pancreatic cancer.15, 27, 28 Whether this was due to inadequate target inhibition because of suboptimal dosing, additional preclinical studies need to be performed. An alternative treatment schedule with intermittent (higher intensity) dosing of the targeted agents may be considered. Inclusion of a pro-apoptotic cytotoxic agent may also be necessary because the selumetinib and MK-2206 combination is not synthetically lethal. Even as newer formulations of drugs blocking the PI3K/AKT and RAF/MEK/ERK pathways with improved therapeutic index are developed, targeting these pathways downstream of KRAS may lead to additional resistant phenotypes. For example in melanoma cell lines, inhibition of MEK and BRAF induced STAT3 signaling potentially leading to increased invasion and metastasis.13, 29 STAT3 has been associated with a poor prognosis in pancreatic cancer and preclinical studies have shown that the loss of p53 function activates JAK2-STAT3 signaling.30
KRAS mutations differ across tumor types and understanding which pathways are specifically activated may be predictive of response to targeted therapies. For example, PI3K/Pdk1 signaling is required for tumor initiation in KRAS driven pancreatic cancer but not in non-small cell lung cancer models. In murine lung cancer models, inhibition of the MEK/PI3K pathways showed pronounced and sustained responses.28 However, pancreatic cancer models showed transient benefit to MEK/PI3K inhibition potentially due to the differing genetic and epigenetic changes.8, 27 Pancreatic cancer can also be subdivided into epithelial and mesenchymal subtypes and a synergistic effect of MEK and EGFR inhibition was only seen in the epithelial subtype with HER3 knockdown.31 It is clear that a better understanding of the underlying signaling networks driving pancreatic cancer progression and potential escape mechanisms is required. Also, the role of preclinical models of pancreatic cancer and the optimal translation of preclinical successes into trial design must be improved. Moreover, clinical testing of targeted agents must include validation of target modulation in treated patients. Obtaining tissue before and after treatment is challenging for this population of patients but the recent advances in liquid biopsies may help to remove the hurdles of serial tissue samples.32
Supplementary Material
Acknowledgments
The authors wish to thank he Hope Foundation for sponsoring the SWOG Young Investigators Course; the Susan E. Riley Foundation; and AstraZeneca for provision of selumetinib and; Merck & Co., Inc. for provision of MK2206.
Funding/Support: This work was supported by National Institutes of Health/National Cancer Institute/National Clinical Trials Network [grant numbers CA180888, CA180819, CA180820, CA180847, CA180821, CA180835, CA 180846, CA180818, CA180801, CA180828, CA189804, CA180798]; National Institutes of Health/National Cancer Institute Community Oncology Research Program [grant numbers CA189821, CA189830, CA189960, CA189971, CA189808, CA189954, CA189822, CA189809, CA189853, CA189953, CA189858]; and National Institutes of Health/National Cancer Institute legacy [grant numbers CA11083, CA46368, CA58723, CA04919 and CA46113]
Role of the Funder/Sponsor: The funders of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. The corresponding author had full access to all of the data and the final responsibility to submit for publication.
Footnotes
Author Contributions:
VC, LAD, AML, and PAP participated in study conception and design. DC, VC, IAD, KAG, AEH, TES, and MAT were responsible for collection and assembly of data. TAB, CDB, VC, KAG, HSH, SM, and AW-G were responsible for data analysis and interpretation. TAB, CDB, DC, VC, LAD, IAD, KAG, AEH, HSH, AML, SD, PAP, TES, and MAT, participated in manuscript writing. TAB, CDB, DC, VC, LAD, IAD, KAG, AEH, HSH, LH, AML, SM, PAP, TES, and AW-G participated in final approval of the manuscript. PAP and AW-G participated in provision of study materials or patients. CDB, LAD and HSH were responsible for administrative support.
Declaration of interests
Stock or other ownership: Dr. Al Baghdadi (Exelixis, Array biopharma, Tracon pharmaceuticals, Cerulean and Spectrum pharmaceuticals/self); Honoraria: Drs. Chung (Celgene/self); Lowy (Pfizer/self); Philip (Celgene/self); Wang-Gillam (Axis/self); Consulting or advisory role: Drs. Al Baghdadi (Seattle genetics and Incyte/self); Chung (Celgene/self); Lowy (Merck, Halozyme/self); Philip (Celgene, Merrimack, Halozyme/self); Seery (Bayer, Halozyme/self); Wang-Gillam (Newlink, Merrimack, Pfizer/self); Speakers’ bureau: Drs. Chung (Celgene/self); Research funding: Drs. Chung (Novartis/self); Philip (Celgene, Merck/self); Tejani (Bayer/self and institution); Wang-Gilliam (Aduro, Chemocentry, CTI Biopharma, Millennium, Pfizer, Merrimack, Newlink, EMD Precision, AstraZeneca; Patent or intellectual property interest: Drs. Doyle (patent as co-discoverer of the ABCG2 (BCRP) transporter gene. The patent and the function of this gene have no bearing on the S1115 trial, and there was no conflict in participation in the trial; Lowy (Amgen/self)
Expert testimony: Dr. Lowy (Merck/self); Travel, accommodations, expenses: Dr. Al Baghdadi (Celgene and Cardinal health/self). All remaining authors have declared no conflicts of interest.
References
- 1.Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2007;25(15):1960–6. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 2.Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. The New England journal of medicine. 2011;364(19):1817–25. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 3.Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. The New England journal of medicine. 2013;369(18):1691–703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53(4):549–54. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 5.Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. British journal of cancer. 2014;111(5):817–22. doi: 10.1038/bjc.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zagouri F, Sergentanis TN, Chrysikos D, et al. Molecularly targeted therapies in metastatic pancreatic cancer: a systematic review. Pancreas. 2013;42(5):760–73. doi: 10.1097/MPA.0b013e31827aedef. [DOI] [PubMed] [Google Scholar]
- 7.Macdonald JS, McCoy S, Whitehead RP, et al. A phase II study of farnesyl transferase inhibitor R115777 in pancreatic cancer: a Southwest oncology group (SWOG 9924) study. Investigational new drugs. 2005;23(5):485–7. doi: 10.1007/s10637-005-2908-y. [DOI] [PubMed] [Google Scholar]
- 8.Junttila MR, Devasthali V, Cheng JH, et al. Modeling targeted inhibition of MEK and PI3 kinase in human pancreatic cancer. Molecular cancer therapeutics. 2015;14(1):40–7. doi: 10.1158/1535-7163.MCT-14-0030. [DOI] [PubMed] [Google Scholar]
- 9.Hofmann I, Weiss A, Elain G, et al. K-RAS mutant pancreatic tumors show higher sensitivity to MEK than to PI3K inhibition in vivo. PloS one. 2012;7(8):e44146. doi: 10.1371/journal.pone.0044146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Infante JR, Somer BG, Park JO, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. European journal of cancer (Oxford, England: 1990) 2014;50(12):2072–81. doi: 10.1016/j.ejca.2014.04.024. [DOI] [PubMed] [Google Scholar]
- 11.Zhong H, Sanchez C, Spitzer D, et al. Synergistic effects of concurrent blockade of PI3K and MEK pathways in pancreatic cancer preclinical models. PloS one. 2013;8(10):e77243. doi: 10.1371/journal.pone.0077243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janku F, Lee JJ, Tsimberidou AM, et al. PIK3CA mutations frequently coexist with RAS and BRAF mutations in patients with advanced cancers. PloS one. 2011;6(7):e22769. doi: 10.1371/journal.pone.0022769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagle N, Van Allen EM, Treacy DJ, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer discovery. 2014;4(1):61–8. doi: 10.1158/2159-8290.CD-13-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wee S, Jagani Z, Xiang KX, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer research. 2009;69(10):4286–93. doi: 10.1158/0008-5472.CAN-08-4765. [DOI] [PubMed] [Google Scholar]
- 15.Tolcher AW, Khan K, Ong M, et al. Antitumor activity in RAS-driven tumors by blocking AKT and MEK. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21(4):739–48. doi: 10.1158/1078-0432.CCR-14-1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turke AB, Song Y, Costa C, et al. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer research. 2012;72(13):3228–37. doi: 10.1158/0008-5472.CAN-11-3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adjei AA, Cohen RB, Franklin W, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26(13):2139–46. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banerji U, Camidge DR, Verheul HM, et al. The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): a phase I open-label multicenter trial in patients with advanced cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16(5):1613–23. doi: 10.1158/1078-0432.CCR-09-2483. [DOI] [PubMed] [Google Scholar]
- 19.Yap TA, Yan L, Patnaik A, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29(35):4688–95. doi: 10.1200/JCO.2011.35.5263. [DOI] [PubMed] [Google Scholar]
- 20.Oettle H, Riess H, Stieler JM, et al. Second-line oxaliplatin, folinic acid, and fluorouracil versus folinic acid and fluorouracil alone for gemcitabine-refractory pancreatic cancer: outcomes from the CONKO-003 trial. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2014;32(23):2423–9. doi: 10.1200/JCO.2013.53.6995. [DOI] [PubMed] [Google Scholar]
- 21.Gill SKY-J, Cripps C, et al. PANCREOX: A randomized phase 3 study of 5FU/LV with or without oxaliplatin for second-line advanced pancreatic cancer (APC) in patients (pts) who have received gemcitabine (GEM)-based chemotherapy (CT) Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2014;32(5s) doi: 10.1200/JCO.2016.68.5776. [DOI] [PubMed] [Google Scholar]
- 22.Bernstein D, Lagakos SW. Sample size and power determination for stratified clinical trials. Journal of Statistical Computation and Simulation. 1978;8(1):65–73. [Google Scholar]
- 23.Philip PA, Goldman B, Ramanathan RK, et al. Dual blockade of epidermal growth factor receptor and insulin-like growth factor receptor-1 signaling in metastatic pancreatic cancer: phase Ib and randomized phase II trial of gemcitabine, erlotinib, and cixutumumab versus gemcitabine plus erlotinib (SWOG S0727) Cancer. 2014;120(19):2980–5. doi: 10.1002/cncr.28744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2004;22(22):4456–62. doi: 10.1200/JCO.2004.01.185. [DOI] [PubMed] [Google Scholar]
- 25.Do K, Speranza G, Bishop R, et al. Biomarker-driven phase 2 study of MK-2206 and selumetinib (AZD6244, ARRY-142886) in patients with colorectal cancer. Investigational new drugs. 2015;33(3):720–8. doi: 10.1007/s10637-015-0212-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller MA, Oudin MJ, Sullivan RJ, et al. Reduced Proteolytic Shedding of Receptor Tyrosine Kinases Is a Post-Translational Mechanism of Kinase Inhibitor Resistance. Cancer discovery. 2016 doi: 10.1158/2159-8290.CD-15-0933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alagesan B, Contino G, Guimaraes AR, et al. Combined MEK and PI3K inhibition in a mouse model of pancreatic cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21(2):396–404. doi: 10.1158/1078-0432.CCR-14-1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meng J, Dai B, Fang B, et al. Combination treatment with MEK and AKT inhibitors is more effective than each drug alone in human non-small cell lung cancer in vitro and in vivo. PloS one. 2010;5(11):e14124. doi: 10.1371/journal.pone.0014124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vultur A, Villanueva J, Krepler C, et al. MEK inhibition affects STAT3 signaling and invasion in human melanoma cell lines. Oncogene. 2014;33(14):1850–61. doi: 10.1038/onc.2013.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wormann SM, Song L, Ai J, et al. Loss of P53 Function Activates JAK2-STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and is Associated With Patient Survival. Gastroenterology. 2016 doi: 10.1053/j.gastro.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 31.Mirzoeva OK, Collisson EA, Schaefer PM, et al. Subtype-specific MEK-PI3 kinase feedback as a therapeutic target in pancreatic adenocarcinoma. Molecular cancer therapeutics. 2013;12(10):2213–25. doi: 10.1158/1535-7163.MCT-13-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwaederle M, Husain H, Fanta PT, et al. Use of Liquid Biopsies in Clinical Oncology: Pilot Experience in 168 Patients. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016 doi: 10.1158/1078-0432.CCR-16-0318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.