

Abstract
Primary carnitine deficiency is caused by a defect in the OCTN2 carnitine transporter encoded by the SLC22A5 gene. It can cause hypoketotic hypoglycemia or cardiomyopathy in children, and sudden death in children and adults. Fibroblasts from affected patients have reduced carnitine transport. We evaluated carnitine transport in fibroblasts from 358 subjects referred for possible carnitine deficiency. Carnitine transport was reduced to 20% or less of normal in fibroblasts of 140/358 subjects. Sequencing of the 10 exons and flanking regions of the SLC22A5 gene in 95/140 subjects identified causative variants in 84% of the alleles. The missense variants identified in our patients and others previously reported (n=92) were expressed in CHO cells. Carnitine transport was impaired by 73/92 variants expressed. Prediction algorithms (Polyphen-2, SIFT) correctly predicted the functional effects of expressed variants in about 80% of cases. These results indicate that mutations in the coding region of the SLC22A5 gene cannot be identified in about 16% of the alleles causing primary carnitine deficiency. Prediction algorithms failed to determine the functional effects of amino acid substitutions in this transmembrane proteins in about 20% of cases. Therefore, functional studies in fibroblasts remain the best strategy to confirm or exclude a diagnosis of primary carnitine deficiency.
Keywords: carnitine deficiency, carnitine uptake defect, fatty acid oxidation, carnitine transport, SLC22A5, OCTN2, mutations, newborn screening
INTRODUCTION
Primary carnitine deficiency (MIM# 212140) is an autosomal recessive disorder of the carnitine cycle resulting in defective fatty acid oxidation (Longo, et al., 2016). The incidence of primary carnitine deficiency varies with a frequency of approximately 1:40,000 newborns in Japan, 1:37,000–1:100,000 newborns in Australia, and 1:142,000 in the USA (Koizumi, et al., 1999; Longo, et al., 2016; Therrell, et al., 2014; Wilcken, et al., 2003). The highest incidence of primary carnitine deficiency (1:300) is in the Faroe Islands, an archipelago in the North Atlantic that remained geographically isolated for many centuries, where about 5% of the population is carrier for an abnormal allele (Longo, et al., 2016; Rasmussen, et al., 2014a; Rasmussen, et al., 2014b; Steuerwald, et al., 2017).
The gene for primary carnitine deficiency, SLC22A5 (MIM# 603377), spans about 30 kb on chromosome 5q31 (chr5:132,369,752–132,395,614, hg38) and encodes for the organic cation transporter novel 2 (OCTN2) carnitine transporter (Longo, et al., 2016; Tamai, et al., 1998; Wu, et al., 1998). OCTN2 was identified in 1998 for its homology to the organic cation transport novel 1 (OCTN1) (Tamai, et al., 1998; Wu, et al., 1999; Wu, et al., 1998). OCTN1 and OCTN2 are part of the SLC22 family of membrane transporters, along with organic anion and cation transporters (Burckhardt and Wolff, 2000; Frigeni, et al., 2016; Longo, et al., 2016). OCTN2 operates as a Na+-independent organic cation transporter as well as a high affinity (Km = 2.9 ± 0.7 μM) Na+-dependent carnitine transporter (Longo, et al., 2016; Tamai, et al., 1998; Wang, et al., 2000b; Wu, et al., 1999). Over 150 genetic variants are described in the SLC22A5 mutation database (http://www.arup.utah.edu/database/OCTN2/OCTN2_display.php), of which 86 are pathogenic (including missense, nonsense, indels, frame-shifting, splice-site altering mutations and one large gene deletion).
Defective carnitine transport results in urinary carnitine wasting, low serum carnitine levels (< 9 μM, normal 25–50 μM), and decreased intracellular carnitine accumulation (Longo, et al., 2006; Longo, et al., 2016). Patients with primary carnitine deficiency lose most (10–95%) of the filtered carnitine in urine, and their heterozygous parents lose 2 to 3 times the normal amount, explaining their mildly reduced plasma carnitine levels (Longo, et al., 2016). Since carnitine is required for the transfer of long-chain fatty acids from the cytoplasm to the mitochondrial matrix for subsequent β-oxidation, the lack of carnitine impairs the ability to use fat as energy source during periods of metabolic stress (Longo, et al., 2016). If carnitine supplements are not promptly started, patients with primary carnitine deficiency can present with an acute metabolic decompensation early in life, or later in life with skeletal and cardiac myopathy or sudden death from arrhythmia (Longo, et al., 2016; Rose, et al., 2012; Wang, et al., 2000a). Siblings of affected children had only mild developmental delays or were asymptomatic (Longo, et al., 2016; Spiekerkoetter, et al., 2003; Wang, et al., 2001).
With the advent of expanded newborn screening, infants with primary carnitine deficiency or their affected mothers can be identified by very low levels of free carnitine (Longo, et al., 2016). The diagnosis of primary carnitine deficiency can be biochemically confirmed by demonstration of low free carnitine levels in plasma with reduced renal reabsorption (less than 90%), and normal renal function with no abnormalities in the urine organic acids (although a non-specific dicarboxylic aciduria has been reported during acute attacks) (Longo, et al., 2016; Scaglia, et al., 1998). Given the possibility of a maternal disorder causing primary or secondary carnitine deficiency, plasma and urine carnitine, plasma acylcarnitine profile, and urine organic acids should be evaluated in the mother as well when carnitine levels are low in the newborn screening (Longo, et al., 2016). The diagnosis can be definitively confirmed by verifying reduced carnitine transport activity (< 20% of normal controls) in skin fibroblasts from the patient or by mutational analysis of the SLC22A5 gene (Longo, et al., 2016).
The measurement of carnitine transport in cultured fibroblasts is a very reliable method to confirm the diagnosis of primary carnitine deficiency, but is time demanding and requires a skin biopsy (Longo, et al., 2016). For this reason, molecular analysis (sequencing and deletion/duplication analysis) of the 10 exons of the SLC22A5 gene and exon/intron boundaries in DNA obtained from blood is now the first-line test for diagnostic confirmation (Longo, et al., 2016).
Here the reliability of DNA testing is compared to functional studies in fibroblasts to confirm the diagnosis of primary carnitine deficiency in 95 subjects. In addition, several novel missense mutations are expressed in heterologous cells to confirm their pathogenicity.
MATERIALS AND METHODS
Patients and Membrane Transport
All studies were approved by the IRB of the University of Utah. Patients were referred for diagnostic confirmation either after symptomatic presentation or after identification by newborn screening programs. Some of the patients reported in this study were previously individually described (Amat di San Filippo and Longo, 2004; Amat di San Filippo, et al., 2006; Amat di San Filippo, et al., 2008; Amat di San Filippo, et al., 2003; De Biase, et al., 2012; Filippo, et al., 2011; Rose, et al., 2012; Schimmenti, et al., 2007; Wang, et al., 2000a; Wang, et al., 2001; Wang, et al., 2000b; Wang, et al., 2000c; Wang, et al., 1999). Fibroblasts from patients with primary carnitine deficiency were obtained by skin biopsy for diagnostic purposes. They were grown in Dulbecco’s Modified Eagle Medium (Thermo Fisher Scientific Inc., Waltham, MA) supplemented with 12% fetal bovine serum (Sigma-Aldrich Corp., St. Louis, MO), 2 mM L-Glutamine (Thermo Fisher Scientific Inc., Waltham, MA), 100 U/ml Penicillin-Streptomycin (GE Healthcare Life Sciences HyClone Laboratories, South Logan, UT), 2.5 μg/mL Amphotericin B solution (GE Healthcare Life Sciences HyClone Laboratories, South Logan, UT). 3H-Carnitine (0.5 μM) transport was measured for 4 hours at 37 ˚C in adherent cells using the cluster-tray method (Scaglia, et al., 1999; Wang, et al., 2000b). Non-saturable transport was measured in the presence of 2 mM cold carnitine and was subtracted from total transport to obtain saturable transport (Scaglia, et al., 1999; Wang, et al., 2000b). Values are reported as means ± SE of 6 independent determinations obtained in two separate experiments.
DNA Analysis and Molecular Techniques
Genomic DNA was extracted from fibroblasts or peripheral blood by standard methods. GenBank sequence AB016625.1 was used as reference for the gene, NM_003060.2 was used as the reference sequence for the cDNA, and NP_001295051.1 for the protein. Nucleotide numbering uses the A of the ATG translation initiation start site as nucleotide +1. The 10 exons of the SLC22A5 gene including exon/intron boundaries were amplified by PCR using a total of 10 primer pairs tailed with universal M-13 forward and reverse sequences. Amplification was verified by automated gel electrophoresis in the HDA-GT12™ Genetic Analyzer. PCR fragments were purified with ExoSap-IT (USB) and bi-directionally sequenced using BigDye-Terminator v.1.1 cycle sequencing kit and capillary electrophoresis in the ABI3730. Generated sequences were aligned to reference sequence (GenBank AB016625.1) using Mutation Surveyor v.4.0 (Softgenetics) to detect mutations (Wang, et al., 2001). Multiplex ligation-dependent probe amplification (MLPA) analysis was performed to detect large deletions within the SLC22A5 gene. Each DNA sample (400 ng) was analyzed using the SALSA MLPA P076 ACADVL-SLC22A5 Probemix for SLC22A5 (MRC-Holland, Amsterdam, The Netherlands) following the manufacturer’s recommendations (http://mlpa.com/WebForms/WebFormMain.aspx).
Missense variants identified in patients with primary carnitine deficiency were re-created in the OCTN2-EGFP expression vector by site-directed mutagenesis using the Quick Change system (Agilent Technologies Inc., Santa Clara, CA) following the manufacturer’s instructions (Amat di San Filippo, et al., 2003; Wang, et al., 2000b). The final plasmids were sequenced to confirm the presence of the mutation and the absence of PCR artifacts and stably transfected into Chinese Hamster Ovary (CHO) cells. The plasmid contained the neomycin-resistance gene and cells were selected for resistance to G418 (Geneticin, 0.8 mg/ml) for 3 weeks before testing. A mass culture including different clones was then used for functional studies. Confocal microscopy was used to confirm protein production, since in our expression vector the OCTN2 transporter is tagged with the green fluorescent protein (Wang, et al., 2000b). CHO cells were grown in Ham’s F12 Nutrient Mixture (Thermo Fisher Scientific Inc., Waltham, MA) supplemented with 6% fetal bovine serum (Sigma-Aldrich Corp., St. Louis, MO), 2 mM L-Glutamine (Thermo Fisher Scientific Inc., Waltham, MA), 100 U/ml Penicillin-Streptomycin (GE Healthcare Life Sciences HyClone Laboratories, South Logan, UT), 2.5 μg/ml Amphotericin B solution (GE Healthcare Life Sciences HyClone Laboratories, South Logan, UT). Carnitine (0.5 μM) transport was measured at 37°C with the cluster-tray method as previously described (Amat di San Filippo, et al., 2003; Wang, et al., 2000b).
In silico analysis of missense mutations
The missense variants identified and expressed in CHO cells were evaluated using Sorting Intolerant From Tolerant (SIFT, http://sift.jcvi.org/www/SIFT_enst_submit.html) and Polymorphism Phenotyping Version v2.2.2r398 (POLYPHEN-2, http://genetics.bwh.harvard.edu/pph2/index.shtml) software programs for variant evaluation.
Estimate of allele frequency
The Exome Aggregation Consortium Browser Beta (ExAC, http://exac.broadinstitute.org/) and the Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/) were used to determine the allele frequency of the variants identified in the SLC22A5 gene (Lek, et al., 2016).
RESULTS
Functional and molecular characterization of fibroblasts from patients with primary carnitine deficiency
Carnitine transport was measured in skin fibroblasts from 358 subjects referred for possible primary carnitine deficiency (Figure 1). A wide range of carnitine transport activity was observed in these cells with a nearly continuous distribution of activity from 0% of control to values above that measured in matched controls. A reduction of carnitine transport to 20% or less than paired normal control fibroblasts was used as cut-off for the diagnosis of primary carnitine deficiency, since all of our patients whose fibroblasts had carnitine transport below 20% of normal controls were not able to maintain free carnitine level ≥9 μmol/L (normal 25–50 μM) without supplements (Longo, et al., 2016). Cells from 140/358 subjects (39%) had carnitine transport activity below 20% of paired normal controls.
Figure 1. Carnitine transport by fibroblasts obtained from 358 individuals referred for possible primary carnitine deficiency.

3H-Carnitine (0.5 μM) transport was measured for 4 hours at 37°C. Nonsaturable transport, measured in the presence of 2 mM cold carnitine, was subtracted from total transport to obtain saturable carnitine transport. Transport activity was normalized to the one of simultaneous normal controls (100% activity). Each bar represents the average of 6 observations in two separate experiments with the sample numbered from 1 (lowest activity) to 358 (highest activity) indicated on the X-axis. Carnitine transport activity of 20% or less of normal controls was indicative of primary carnitine deficiency (continuous line). Fibroblasts from some of the subjects had carnitine transport higher than their paired normal controls (100% activity, dashed line).
Molecular studies (sequencing of the 10 exons and flanking regions of the SLC22A5 gene encoding the OCTN2 carnitine transporter and deletion/duplication analysis) were performed in 95/140 of the affected subjects. Table 1 shows carnitine transport activity in fibroblasts of the 95 patients diagnosed with primary carnitine deficiency along with the variants identified in their SLC22A5 gene. Among the 72 variants identified, 48 were missense, 9 caused a frameshift, 8 were nonsense, 2 were in-frame deletions, 1 was a synonymous variant, and 4 were localized in flanking regions of the SLC22A5 gene. Overall, the most frequent mutation was c.136C>T (p.Pro46Ser), which was identified in 13/190 alleles. This mutation has been identified in asymptomatic or minimally symptomatic (easy fatigability, muscle pain with exercise, fasting intolerance) adult women (Longo, et al., 2016; Schimmenti, et al., 2007) and in a mother (compound heterozygous p.Pro46Ser/p.Asn32Ser) who suffered repeated episodes of cardiac arrest requiring placement of a defibrillator, episodes that resolved after her diagnosis and initiation of carnitine therapy (De Biase, et al., 2012). This mutation (p.Pro46Ser) reduces, but does not abolish carnitine transport and affects glycosylation and maturation to the plasma membrane of the OCTN2 carnitine transporter (Filippo, et al., 2011; Longo, et al., 2016). This and other similar missense mutations seem protective against early clinical manifestations of primary carnitine deficiency, but can still be associated with fatal cardiac arrhythmia leading to sudden death (De Biase, et al., 2012; Longo, et al., 2016; Rose, et al., 2012). Molecular analysis identified causative variants in 84% of the affected alleles, but failed to identify significant variations in 6/95 patients, and a second mutation in 19/95 patients (Table 2), despite a functional diagnosis of primary carnitine deficiency.
Table 1. Functional and molecular analysis of cells from 95 patients with primary carnitine deficiency.
Carnitine transport activity was measured in fibroblasts from subjects referred for possible primary carnitine deficiency (Figure 1) and expressed as percent of normal activity. The 10 exons and flanking regions of the SLC22A5 gene encoding the OCTN2 carnitine transporter were sequenced and subjected to deletion/duplication analysis in patients whose fibroblasts had <20% of normal transport activity (transport activity is reported in the second column). Variations from the standard sequence (NM_003060.2) are reported in columns 3–10. No causative variants were identified in 16% of the alleles (NOT FOUND).
| Pt # | Transport activity % of control | ALLELE 1 cDNA | PROTEIN | ALLELE 2 cDNA | PROTEIN | ADDITIONAL VARIANT | |||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.0 | c.12C>G | p.Tyr4* | c.12C>G | p.Tyr4* | ||||
| 2 | 0.1 | c.865C>T | p.Arg289* | c.865C>T | p.Arg289* | ||||
| 3 | 0.1 | c.458_459del | p.Val153Alafs*41 | c.844C>T | p.Arg282* | ||||
| 4 | 0.2 | c.1202_1203insA | p.Tyr401* | c.1302del | p.Gly435fs*24 | ||||
| 5 | 0.2 | c.844C>T | p.Arg282* | c.844C>T | p.Arg282* | ||||
| 6 | 0.3 | c.740C>G | p.Pro247Arg | c.740C>G | p.Pro247Arg | ||||
| 7 | 0.3 | c.64_66del | p.Phe22del | c.64_66del | p.Phe22del | ||||
| 8 | 0.3 | c.254_264dup | p.Ile89Glyfs*45 | c.254_264dup | p.Ile89Glyfs*45 | ||||
| 9 | 0.3 | c.1319C>T | p.Thr440Met | c.1319C>T | p.Thr440Met | ||||
| 10 | 0.4 | c.248G>T | p.Arg83Leu | c.248G>T | p.Arg83Leu | ||||
| 11 | 0.5 | c.760C>T | p.Arg254* | c.760C>T | p.Arg254* | ||||
| 12 | 0.5 | c.844C>T | p.Arg282* | c.3G>T | p.Met1Leu | ||||
| 13 | 0.5 | c.12C>G | p.Tyr4* | c.12C>G | p.Tyr4* | ||||
| 14 | 0.5 | c.839C>T | p.Ser280Phe | c.1463G>A | p.Arg488His | c.424G>T | p.Ala142Ser | ||
| 15 | 0.6 | c.506G>A | p.Arg169Gln | c.506G>A | p.Arg169Gln | ||||
| 16 | 0.7 | c.1392_1409delinsCA | p.Val465Thrfs*29 | c.1392_1409delinsCA | p.Val465Thrfs*29 | ||||
| 17 | 0.9 | c.458_459del | p.Val153Alafs*41 | c.865C>T | p.Arg289* | ||||
| 18 | 0.9 | c.760C>T | p.Arg254* | c.760C>T | p.Arg254* | ||||
| 19 | 0.9 | c.760C>T | p.Arg254* | c.760C>T | p.Arg254* | ||||
| 20 | 1.0 | c.565_568del | p.Phe189Argfs*14 | c.1316T>G | p.Val439Gly | ||||
| 21 | 1.0 | c.1340A>G | p.Tyr447C | c.1340A>G | p.Tyr447Cys | ||||
| 22 | 1.0 | c.768G>A | p.Trp256* | c.1403C>G | p.Thr468Arg | ||||
| 23 | 1.0 | c.825G>A | p.Trp275* | c.1267+3_1267+23del | |||||
| 24 | 1.1 | c.505C>T | p.Arg169Trp | c.1051T>C | p.Trp351Arg | ||||
| 25 | 1.1 | c.1193C>T | p.Pro398Leu | c.839C>T | p.Ser280Phe | ||||
| 26 | 1.2 | c.1319C>T | p.Thr440Met | c.1463G>A | p.Arg488H | c.424G>T | p.Ala142Ser | ||
| 27 | 1.3 | c.344A>G | p.Asp115Gly | c.344A>G | p.Asp115Gly | c.283C>G | p.Leu95Val | c.283C>G | p.Leu95Val |
| 28 | 1.4 | c.844C>T | p.Arg282* | c.1463G>A | p.Arg488His | c.424G>T | p.Ala142Ser | ||
| 29 | 1.5 | c.1193C>T | p.Pro398Leu | c.557T>C | p.Leu186Pro | c.1645C>T | p.Pro549Ser | ||
| 30 | 1.5 | c.506G>C | p.Arg169Pro | c.1088T>C | p.Leu363Pro | ||||
| 31 | 2.0 | c.902C>A | p.Ala301Asp | c.902C>A | p.Ala301Asp | ||||
| 32 | 2.0 | c.760C>T | p.Arg254* | c.1400C>G | p.Ser467Cys | ||||
| 33 | 2.1 | c.248G>T | p.Arg83Leu | c.248G>T | p.Arg83Leu | ||||
| 34 | 2.2 | c.629A>G | p.Asn210Ser | c.1463G>A | p.Arg488His | c.424G>T | p.Ala142Ser | ||
| 35 | 2.2 | c.56G>C | p.Arg19Pro | c.254_264dup | p.Ile89Glyfs*45 | ||||
| 36 | 2.5 | c.136C>T | p.Pro46Ser | c.760C>T | p.Arg254* | ||||
| 37 | 2.8 | c.1196G>A | p.Arg399Gln | c.1463G>A | p.Arg488His | c.424G>T | p.Ala142Ser | ||
| 38 | 2.8 | c.725G>T | p.Gly242Val | c.725G>T | p.Gly242Val | ||||
| 39 | 2.8 | c.844C>T | p.Arg282* | c.136C>G | p.Pro46Ser | ||||
| 40 | 2.9 | c.847T>A | p.Trp283Arg | c. 847T>A | p.Trp283Arg | ||||
| 41 | 2.9 | c.43G>T | p.Gly15Trp | c.248G>T | p.Arg83Leu | ||||
| 42 | 3.0 | c.760C>T | p.Arg254* | c.760C>T | p.Arg254* | ||||
| 43 | 3.2 | c.505C>T | p.Arg169Trp | c.505C>T | p.Arg169Trp | ||||
| 44 | 3.3 | c.1196G>A | p.Arg399Gln | c.1196G>A | p.Arg399Gln | ||||
| 45 | 3.3 | c.695C>T | p.Thr232Met | c.1403C>G | p.Thr468Arg | ||||
| 46 | 3.4 | c.136C>T | p.Pro46Ser | c.1556_1559dup | p.Ile521Hisfs*3 | ||||
| 47 | 3.4 | c.232delC | p.His79Thrfs*51 | c.232delC | p.His79Thrfs*51 | ||||
| 48 | 3.5 | c.248G>T | p.Arg83Leu | c.641C>T | p.Ala214Val | ||||
| 49 | 3.8 | c.254_264dup | p.Ile89Glyfs*45 | c.1385G>T | p.Gly462Val | ||||
| 50 | 3.8 | c.845G>A | p.Arg282Gln | c.845G>A | p.Arg282Gln | ||||
| 51 | 4.0 | c.695C>T | p.Thr232Met | NOT FOUND | |||||
| 52 | 4.0 | c.136C>G | p.Pro46Ser | c.1463G>A | p.Arg488His | c.424G>T | p.Ala142Ser | ||
| 53 | 4.2 | c.136C>G | p.Pro46Ser | c.1556_1559dup | p.Ile521Hisfs*3 | ||||
| 54 | 4.2 | c.1354G>A | p.Glu452Lys | c.1354G>A | p.Glu452Lys | ||||
| 55 | 4.3 | c.77G>A | p.Ser26Asn | c.845G>A | p.Arg282Gln | ||||
| 56 | 4.3 | c.232delC | p.His79Thrfs*51 | c.1342G>T | p.Val448Leu | c.641C>T | p.Ala214Val | ||
| 57 | 4.8 | c.95A>G | p.Asn32Ser | c.136C>G | p.Pro46Ser | ||||
| 58 | 4.9 | c.1324_1325GC>AT | p.Ala442Ile | c.1324_1325GC>AT | p.Ala442Ile | ||||
| 59 | 5.0 | c.1462C>T | p.Arg488Cys | c.1462C>T | p.Arg488Cys | ||||
| 60 | 5.3 | Exon 3 deletion | p.Phe167Aspfs*61 | NOT FOUND | c.394-16T>A | ||||
| 61 | 5.5 | c.695C>T | p.Thr232Met | c.695C>T | p.Thr232Met | ||||
| 62 | 5.6 | c.136C>T | p.Pro46Ser | c.695C>T | p.Thr232Met | ||||
| 63 | 5.6 | c.136C>T | p.Pro46Ser | c.1463G>A | p.Arg488His | c.424G>T | p.Ala142Ser | ||
| 64 | 5.7 | c.844C>T | p.Arg282* | NOT FOUND | c.824+13T>C | ||||
| 65 | 5.7 | c.136C>T | p.Pro46Ser | c.1193C>T | p.Pro398Leu | ||||
| 66 | 5.8 | c.1319C>T | p.Thr440Met | c.453G>A | p.Val151Val | ||||
| 67 | 5.9 | c.1319C>T | p.Thr440Met | NOT FOUND | |||||
| 68 | 6.3 | c.136C>T | p.Pro46Ser | c.523G>A | p.Val175Met | ||||
| 69 | 7.0 | c.458_459del | p.Val153Alafs*41 | NOT FOUND | |||||
| 70 | 7.2 | c.137C>T | p.Pro46Leu | c.1175_1177del | p.Leu394del | ||||
| 71 | 7.4 | c.1195C>T | p.Arg399Trp | c.653-2A>C | c.653-2A>C | ||||
| 72 | 7.5 | c.865C>T | p.Arg289* | NOT FOUND | |||||
| 73 | 8.5 | c.248G>T | p.Arg83Leu | c.641C>T | p.Ala214Val | ||||
| 74 | 8.9 | c.43G>T | p.Gly15Trp | NOT FOUND | |||||
| 75 | 9.0 | c.453G>A | p.Val151Val | Exon 3 deletion | p.Phe167Aspfs*61 | ||||
| 76 | 9.1 | c.136C>T | p.Pro46Ser | NOT FOUND | |||||
| 77 | 9.6 | NOT FOUND | NOT FOUND | c.1441G>T | p.Val481Phe | ||||
| 78 | 11.1 | c.136C>T | p.Pro46Ser | NOT FOUND | |||||
| 79 | 11.1 | NOT FOUND | NOT FOUND | c.1451G>T | p.Gly484Val | ||||
| 80 | 11.7 | NOT FOUND | NOT FOUND | ||||||
| 81 | 12.4 | c.806delT | p.Leu269Argfs*26 | c.1345T>G | p.Tyr449Asp | ||||
| 82 | 13.7 | c.839C>T | p.Ser280Phe | NOT FOUND | |||||
| 83 | 13.9 | NOT FOUND | NOT FOUND | ||||||
| 84 | 14.0 | c.505C>T | p.Arg169Trp | NOT FOUND | |||||
| 85 | 14.0 | c.419G>A | p.Trp140* | NOT FOUND | |||||
| 86 | 14.3 | c.51C>G | p.Phe17Leu | NOT FOUND | |||||
| 87 | 15.0 | NOT FOUND | NOT FOUND | ||||||
| 88 | 16.2 | c.641C>T | p.Ala214Val | c.641C>T | p.Ala214Val | ||||
| 89 | 16.8 | c.393G>C | p.Glu131Asp | NOT FOUND | |||||
| 90 | 16.9 | NOT FOUND | NOT FOUND | ||||||
| 91 | 17.7 | c.1202_1203insA | p.Tyr401* | NOT FOUND | |||||
| 92 | 17.8 | c.136C>T | p.Pro46Ser | NOT FOUND | |||||
| 93 | 17.9 | c.1193C>T | p.Pro398Leu | NOT FOUND | |||||
| 94 | 18.7 | c.458_459del | p.Val153Alafs*41 | NOT FOUND | |||||
| 95 | 19.4 | c.1193C>T | p.Pro398Leu | NOT FOUND | |||||
Table 2. Molecular studies in patients with primary carnitine deficiency.
Sequencing and deletion/duplication analysis of all 10 exons of the SLC22A5 gene and flanking regions was performed in 95 patients diagnosed with primary carnitine deficiency because of a low level of carnitine transport in fibroblasts (activity <20% of matching normal controls). No variants were identified in 31/190 (16%) of the alleles sequenced. For 6/95 (6%) affected patients no variants were identified in either alleles, while for 19/95 (20%) affected patients no variant was identified in one allele.
| NUMBER of PATIENTS | 95 |
|---|---|
| NUMBER of ALLELES | 190 |
| Alleles with unknown mutations | 31/190 (16%) |
| Patients with no identified variant in one allele | 19/95 (20%) |
| Patients with no identified variant in either alleles | 6/95 (6%) |
Expression of SLC22A5 variants in CHO cells
Molecular analysis is becoming the preferred method for diagnostic confirmation in patients with suspected primary carnitine deficiency since it does not require a skin biopsy and is relatively rapid. Among samples received for molecular analysis (including the 95 patients who had carnitine transport analyzed in fibroblasts), 133 different variants were identified, with 33 being novel (Table 3A). Of the 133 variants identified, 90 were missense, 16 caused a frameshift, 14 were nonsense, 8 affected splicing, 2 were in-frame deletions of single amino acids, 1 was a ~1.6 Mb deletion encompassing the entire OCTN2 gene (Li, et al., 2010), 1 was a 113 bp deletion encompassing the initiation codon of exon 1 (Nezu, et al., 1999), and 1 was a synonymous variant that created a splice site (c.453 G>A, p.Val151Val). An additional missense variant (c.393G>C, p.Glu131Asp) in the last nucleotide abolished the donor site of exon 1 and is predicted to affect splicing. Twenty-six new missense variants and 1 new in-frame deletion detected in our patients, as well as 64 missense variants and 1 in-frame deletion reported in the literature (most never previously expressed in heterologous cells, see Figure 2 for their location on the OCTN2 transporter), were expressed in CHO cells to determine their effect on carnitine transport. Carnitine transport was expressed as percent of the value measured in CHO cells expressing the wild-type OCTN2 carnitine transporter, assumed to be 100% (Table 3A). Carnitine transport was reduced to less than 20% of control in 73/92 variants, indicating that these amino acid substitutions significantly impaired carnitine transport activity.
Table 3. Variants in the SLC22A5 gene in patients referred for possible primary carnitine.
(A) Molecular analysis of the SLC22A5 gene in our patients identified 133 variants, with 33 being novel (*). Of the 133 variants identified, 90 missense variants and 2 in-frame deletions were expressed in CHO cells to evaluate carnitine transport (functional activity). Carnitine transport was expressed as percent of the activity in CHO cells expressing the wild-type OCTN2 carnitine transporter (100% activity). Carnitine transport was reduced to less than 20% of wild-type OCTN2 by 73/92 variants. (B) Table 3B shows the frequency in normal individuals in the ExAC Browser Beta and in the gnomAD of the alleles found in our patients along with the number of normal individual homozygous for a specific allele. 1:221 alleles contained either one of the missense mutations expressed in this study or a mutation causing the premature insertion of a stop codon or affecting splicing in the SLC22A5 gene. (C) The 90 missense variants and 2 in-frame deletions whose carnitine transport was tested in CHO cells were evaluated for pathogenicity using the PolyPhen-2 v2.2.2r398 (http://genetics.bwh.harvard.edu/pph2/index.shtml, using default parameters, defining damaging any variant with a score >0.15) and SIFT (http://sift.jcvi.org/www/SIFT_enst_submit.html, using default parameters and assuming as damaging any change with a score of <0.05) software with the predicted effects indicated. Actual values for each amino acid substitution are reported in Supplemental Table S2. Variants were submitted to ClinVar https://www.ncbi.nlm.nih.gov/clinvar/?term=SLC22A5%5Bgene%5D.
| A | B | C | ||||||
|---|---|---|---|---|---|---|---|---|
| Exon | cDNA | PROTEIN | Transport activity % wild-type OCTN2 | Reference(s) | Allele Frequency (ExAC) | Homozygous (ExAC) | Polyphen-2 | SIFT |
| OCTN2 deletion | (Li, et al., 2010) | |||||||
| 1 | c.-91_22del | (Nezu, et al., 1999) | ||||||
| 1 | c.3G>T | p.Met1Leu | 0.16 | (Dobrowolski, et al., 2005) | 3.23E-05 | 0 | damaging | damaging |
| 1 | c.4_5insC | p.Arg2Profs*136 | (Nezu, et al., 1999) | |||||
| 1 | c.12C>G | p.Tyr4* | (Wang, et al., 2001) | 0.0000173 | 0 | |||
| 1 | c.34G>A | p.Gly12Ser | 51.7 | (Li, et al., 2010) | 0.0007152 | 1 | damaging | damaging |
| 1 | c.43G>T | p.Gly15Trp | 11.05 | (El-Hattab, et al., 2010) | 0.0001028 | 0 | damaging | damaging |
| 1 | c.47C>T | p.Pro16Leu | 0.00 | * | benign | benign | ||
| 1 | c.51C>G | p.Phe17Leu | 10.74 | (Lee, et al., 2010; Urban, et al., 2006) | 0.0000601 | 0 | damaging | damaging |
| 1 | c.56G>C | p.Arg19Pro | 2.03 | (Wang, et al., 2001) | damaging | benign | ||
| 1 | c.59T>A | p.Leu20H | 46.77 | * | 0.0013610 | 1 | damaging | benign |
| 1 | c.64_66del | p.Phe23del | 0.46 | (Lamhonwah, et al., 2002) | 0.0000259 | 0 | / | damaging |
| 1 | c.77G>A | p.Ser26Asn | 5.51 | (Rose, et al., 2012) | 0.0000262 | 0 | damaging | benign |
| 1 | c.83G>T | p.Ser28Ile | 1.35 | (Rahbeeni, et al., 2002) | damaging | benign | ||
| 1 | c.95A>G | p.Asn32Ser | 0.88 | (Lamhonwah, et al., 2002; Rasmussen, et al., 2014b) | 0.0000268 | 0 | damaging | benign |
| 1 | c.131C>T | p.Ala44Val | 8.22 | * | 0.0000197 | 0 | damaging | damaging |
| 1 | c.136C>T | p.Pro46Ser | 2.47 | (Schimmenti, et al., 2007) | 0.0005515 | 0 | damaging | damaging |
| 1 | c.137C>T | p.Pro46Leu | 4.10 | * | 0.0000202 | 0 | damaging | damaging |
| 1 | c.149G>A | p.Cys50Tyr | 0.00 | * | damaging | damaging | ||
| 1 | c.196A>C | p.Thr66Pro | 1.80 | (Li, et al., 2010) | damaging | damaging | ||
| 1 | c.224G>C | p.Arg75Pro | 1.93 | (Li, et al., 2010) | 0.0000177 | 0 | benign | benign |
| 1 | c.232delC | p.His79Thrfs*51 | (Amat di San Filippo, et al., 2006) | 0.0000181 | 0 | |||
| 1 | c.248G>T | p.Arg83Leu | 0.14 | (Makhseed, et al., 2004) | 0.0004323 | 0 | damaging | damaging |
| 1 | c.254_264dup | p.Ile89Glyfs*45 | (Lamhonwah, et al., 2002; Wang, et al., 2001) | 0.0000385 | 0 | |||
| 1 | c.278C>G | p.Ser93Trp | 0.00 | * | damaging | damaging | ||
| 1 | c.283C>G | p.Leu95Val | 28.92 | * | benign | benign | ||
| 1 | c.287G>C | p.Gly96Ala | 19.65 | (Li, et al., 2010) | 0.0003927 | 0 | damaging | damaging |
| 1 | c.344A>G | p.Asp115Gly | 4.04 | * | damaging | damaging | ||
| 1 | c.350G>A | p.Trp117* | (Li, et al., 2010) | 0.0000042 | 0 | |||
| 1 | c.368T>G | p.Val123Gly | 13.96 | (Li, et al., 2010) | damaging | damaging | ||
| 1 | c.393G>C | p.Glu131Asp | 32.84 | * | damaging | damaging | ||
| IVS1 | c.393+5G>A | (Han, et al., 2014) | ||||||
| IVS1 | c.394-16T>A | (Rose, et al., 2012) | ||||||
| 2 | c.396G>A | p.Trp132* | (Koizumi, et al., 1999; Nezu, et al., 1999; Tang, et al., 1999) | 0.0000082 | 0 | |||
| 2 | c.419G>A | p.Trp140* | * | |||||
| 2 | c.424G>T | p.Ala142Ser | 25.68 | (Amat di San Filippo, et al., 2006) | 0.0000494 | 0 | benign | benign |
| 2 | c.428C>T | p.Pro143Leu | 1.18 | (Li, et al., 2010) | 0.0000081 | 0 | damaging | damaging |
| 2 | c.433dupA | p. Thr145Asnfs*50 | (Han, et al., 2014) | |||||
| 2 | c.451G>A | p.Val151Met | 65.93 | * | 0.0000330 | 0 | damaging | benign |
| c.453G>A | p.Val151Val | * | 0.0000433 | 0 | ||||
| 2 | c.458_459del | p.Val153fs*41 | (Dobrowolski, et al., 2005) | |||||
| IVS2 | c.497+1G>T | (Han, et al., 2014) | ||||||
| Exon 3 deletion | p.Phe167Aspfs*61 | * | ||||||
| 3 | c.505C>T | p.Arg169Trp | 0.01 | (Lamhonwah, et al., 2002; Wang, et al., 2000c) | 0.0000082 | 0 | damaging | damaging |
| 3 | c.506G>C | p.Arg169Pro | 0.24 | * | 0.0000577 | 0 | damaging | damaging |
| 3 | c.506G>A | p.Arg169Gln | 0.22 | (Burwinkel, et al., 1999) | 0.0000447 | 0 | damaging | damaging |
| 3 | c.517delC | p.Leu173Cysfs*3 | (Han, et al., 2014) | |||||
| 3 | c.523G>A | p.Val175Met | 7.03 | * | 0.0000330 | 0 | damaging | benign |
| 3 | c.529A>G | p.Met177Val | 14.40 | (Li, et al., 2010) | 0.0001071 | 0 | damaging | damaging |
| 3 | c.535A>T | p.Met179Leu | 48.13 | (Koizumi, et al., 1999) | benign | benign | ||
| 3 | c.557T>C | p.Leu186Pro | 0.00 | (Li, et al., 2010) | 0.0000041 | 0 | damaging | damaging |
| 3 | c.565_568del | p.Phe189Argfs*13 | * | |||||
| 3 | c.573delG | p.Asn192Ilefs*12 | (Li, et al., 2010) | 0.0000494 | 0 | |||
| 3 | c.614T>G | p.Met205Arg | 0.00 | * | damaging | damaging | ||
| 3 | c.629A>G | p.Asn210Ser | 0.13 | * | 0.0000203 | 0 | damaging | benign |
| 3 | c.632A>G | p.Tyr211Cys | 0.00 | (Vaz, et al., 1999) | 0.0000081 | 0 | damaging | damaging |
| 3 | c.641C>T | p.Ala214Val | 32.56 | * | 0.0007989 | 2 | damaging | damaging |
| IVS3 | c.652+1G>A | (Lamhonwah, et al., 2002) | 0.0000122 | 0 | ||||
| IVS3 | c.653-2A>C | * | ||||||
| 4 | c.656C>A | p.Thr219Lys | 28.25 | * | damaging | damaging | ||
| 4 | c.674C>T | p.Ser225Leu | 11.34 | * | damaging | benign | ||
| 4 | c.680G>A | p.Arg227His | 7.25 | (Li, et al., 2010) | 0.0000577 | 0 | damaging | damaging |
| 4 | c.688T>C | p.Phe230Leu | 0.80 | (Li, et al., 2010) | 0.0000108 | 0 | damaging | damaging |
| 4 | c.692C>T | p.Ser231Phe | 0.00 | * | damaging | damaging | ||
| 4 | c.695C>T | p.Thr232Met | 19.14 | (Dobrowolski, et al., 2005) | 0.0001318 | 0 | damaging | damaging |
| 4 | c.718G>A | p.Ala240Thr | 2.06 | (Li, et al., 2010) | damaging | benign | ||
| 4 | c.725G>T | p.Gly242Val | 0.00 | (Wang, et al., 2000c) | damaging | damaging | ||
| 4 | c.740C>G | p.Pro247Arg | 0.07 | * | damaging | damaging | ||
| 4 | c.745_748del | p.Phe249Leufs*13 | (Han, et al., 2014) | |||||
| 4 | c.760C>T | p.Arg254* | (Tang, et al., 2002) | 0.0001071 | 0 | |||
| 4 | c.761G>A | p.Arg254Gln | 34.24 | * | 0.0001236 | 0 | damaging | damaging |
| 4 | c.768G>A | p.Trp256* | (Amat di San Filippo, et al., 2006) | |||||
| 4 | c.769C>T | p.Arg257Trp | 7.65 | (Li, et al., 2010) | 0.0000906 | 0 | damaging | damaging |
| 4 | c.791C>T | p.Thr264Met | 46.00 | (Amat di San Filippo, et al., 2008) | 0.0000330 | 0 | damaging | damaging |
| 4 | c.791C>G | p.Thr264Arg | 2.48 | (Li, et al., 2010) | 0.0000406 | 0 | damaging | damaging |
| 4 | c.806delT | p.Leu269Argfs*27 | (Cederbaum, et al., 2002) | |||||
| 4 | c.806T>C | p.Leu269Pro | 44.86 | * | damaging | damaging | ||
| IVS4 | c.824+13T>C | (Mutlu-Albayrak, et al., 2015) | 0.4346000 | 27668 | ||||
| 5 | c.825G>A | p.Trp275* | (Dobrowolski, et al., 2005) | 0.0000082 | 0 | |||
| 5 | c.839delC | p.Arg282Aspfs*14 | (Lamhonwah, et al., 2002) | 0.0000082 | 0 | |||
| 5 | c.839C>T | p.Ser280Phe | 0.53 | (Amat di San Filippo, et al., 2006) | 0.0000081 | 0 | damaging | damaging |
| 5 | c.844C>T | p.Arg282* | (Burwinkel, et al., 1999; Vaz, et al., 1999);(Wang, et al., 1999) | 0.0000494 | 0 | |||
| 5 | c.844delC | p.Val295* | (Komlosi, et al., 2009) | |||||
| 5 | c.845G>A | p.Arg282Gln | 5.60 | (Amat di San Filippo, et al., 2006) | damaging | benign | ||
| 5 | c.847T>C; T>A | p.Trp283Arg | 0.45 | (Mayatepek, et al., 2000) | damaging | damaging | ||
| 5 | c.849G>T | p.Trp283Cys | 0.00 | (Koizumi, et al., 1999) | damaging | damaging | ||
| 5 | c.865C>T | p.Arg289* | (Dobrowolski, et al., 2005) | 0.0000082 | 0 | |||
| 5 | c.902C>A | p.Ala301Asp | 0.33 | (Wang, et al., 2000c) | damaging | damaging | ||
| 5 | c.934A>G | p.Ile312Val | 67.14 | (Amat di San Filippo, et al., 2008; Li, et al., 2010) | 0.0008072 | 0 | benign | benign |
| 5 | c.949G>A | p.Glu317Lys | 100.00 | (Amat di San Filippo, et al., 2008) | 0.0000082 | 0 | damaging | benign |
| 5 | c.955C>T | p.Gln319* | (Li, et al., 2010) | 0.0000041 | 0 | |||
| 6 | c.1008delA | p.Thr337Profs*12 | (Lamhonwah, et al., 2002) | |||||
| 6 | c.1043T>C | p.Ile348Thr | 57.69 | * | 0.0000496 | 0 | damaging | benign |
| 6 | c.1051T>C | p.Trp351Arg | 0.00 | (Wang, et al., 2000c) | damaging | damaging | ||
| 7 | c.1064C>T | p.Ser355Leu | 1.55 | (Li, et al., 2010) | 0.0000081 | 0 | benign | damaging |
| 7 | c.1072T>A | p.Tyr358Asn | 0.08 | (Li, et al., 2010) | 0.0000082 | 0 | damaging | damaging |
| 7 | c.1088T>C | p.Leu363Pro | 0.00 | (Akpinar, et al., 2010) | 0.0000041 | 0 | damaging | damaging |
| 7 | c.1161T>G | p.Tyr387* | (Tang, et al., 2002) | 0.0000323 | 0 | |||
| 7 | c.1175_1177del | p.Leu394del | 5.29 | * | 0.0000082 | 0 | / | damaging |
| 7 | c.1193C>T | p.Pro398Leu | 0.46 | (Amat di San Filippo, et al., 2006) | 0.0000989 | 0 | damaging | damaging |
| 7 | c.1195C>T | p.Arg399Try | 4.54 | (El-Hattab, et al., 2010) | 0.0000741 | 0 | damaging | damaging |
| 7 | c.1196G>A | p.Arg399Gln | 0.45 | (Wang, et al., 2001) | 0.0000162 | 0 | damaging | damaging |
| 7 | c.1202_1203insA | p.Tyr401* | (Lamhonwah, et al., 2002; Wang, et al., 1999) | 0.0000082 | 0 | |||
| 7 | c.1234A>G | p.Ser412Gly | 107.72 | * | benign | benign | ||
| IVS7 | c.1267+3_1267+23del | (Dobrowolski, et al., 2005) | ||||||
| 8 | c.1302delG | p.Gly435Alafs*24 | (Wang, et al., 1999) | |||||
| 8 | c.1316T>G | p.Val439Gly | 0.52 | * | damaging | damaging | ||
| 8 | c.1319C>T | p.Thr440Met | 0.00 | (Lamhonwah, et al., 2002) | 0.0000165 | 0 | damaging | damaging |
| 8 | c.1324_1325delGCinsAT | p.Ala442Ile | 14.83 | (El-Hattab, et al., 2010) | damaging | damaging | ||
| 8 | c.1327T>G | p.Phe443Val | 0.34 | (Li, et al., 2010) | damaging | benign | ||
| 8 | c.1336G>T | p.Val446Phe | 0.58 | (Mayatepek, et al., 2000) | 0.0000041 | 0 | damaging | damaging |
| 8 | c.1340A>G | p.Tyr447Cys | 0.00 | (Amat di San Filippo and Longo, 2004; Rahbeeni, et al., 2002) | damaging | damaging | ||
| 8 | c.1342G>T | p.Val448Leu | 10.62 | * | damaging | benign | ||
| 8 | c.1345T>G | p.Tyr449Asp | 16.56 | (Amat di San Filippo and Longo, 2004) | 0.0003247 | 1 | damaging | damaging |
| 8 | c.1354G>A | p.Glu452Lys | 2.14 | (Wang, et al., 2000a) | 0.0000494 | 0 | damaging | damaging |
| 8 | c.1364C>G | p.Pro455Arg | 0.18 | (Li, et al., 2010) | 0.0000072 | 0 | damaging | damaging |
| 8 | c.1372delG | p. Val458* | (Han, et al., 2014) | |||||
| 8 | c.1385G>T | p.Gly462Val | 2.80 | * | damaging | damaging | ||
| 8 | c.1392_1409delinsCA | p.Val465Thrfs*29 | * | |||||
| 8 | c.1400C>G | p.Ser467Cys | 16.56 | (Koizumi, et al., 1999) | 0.0001565 | 0 | damaging | damaging |
| 8 | c.1403C>G | p.Thr468Arg | 0.64 | (Lamhonwah, et al., 2002) | 0.0000081 | 0 | damaging | damaging |
| 8 | c.1409C>T | p.Ser470Phe | 0.00 | (Lamhonwah, et al., 2002) | 0.0000165 | 0 | damaging | damaging |
| 8 | c.1412G>C | p.Arg471Pro | 0.00 | (Filippo, et al., 2011) | damaging | damaging | ||
| 8 | c.1412G>A | p.Arg471His | 1.40 | (Spiekerkoetter, et al., 2003) | damaging | damaging | ||
| 8 | c.1427T>G | p.Leu476Arg | 0.00 | (Mutlu-Albayrak, et al., 2015) | damaging | damaging | ||
| 8 | c.1433C>T | p.Pro478Leu | 0.28 | (Tang, et al., 1999) | damaging | damaging | ||
| 8 | c.1441G>T | p.Val481Phe | 63.00 | (Amat di San Filippo, et al., 2008) | benign | benign | ||
| IVS8 | c.1451-1G>A | (Nezu, et al., 1999) | 0.0004777 | 0 | ||||
| 9 | c.1462C>T | p.Arg488Cys | 9.50 | (Schimmenti, et al., 2007) | 0.0000412 | 0 | damaging | damaging |
| 9 | c.1463G>A | p.Arg488His | 40.19 | (Amat di San Filippo, et al., 2006) | 0.0032620 | 4 | damaging | damaging |
| 9 | c.1520T>C | p.Leu507Ser | 4.85 | (Li, et al., 2010) | 0.0000323 | 0 | damaging | damaging |
| 9 | c.1556_1559dup | p.Ile521Hisfs*3 | (Schimmenti, et al., 2007) | |||||
| 10 | c.1645C>T | p.Pro549Ser | 23.79 | (Amat di San Filippo, et al., 2008; Urban, et al., 2006) | 0.0064990 | 28 | benign | benign |
Figure 2. Schematic of the OCTN2 carnitine transporter with location of the 92 variants expressed in CHO cells.
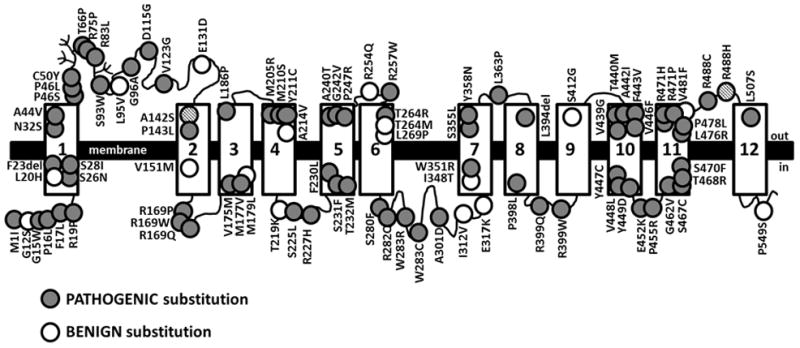
The OCTN2 carnitine transporter is composed of 557 amino acids. Hydropathy analysis and conservation with other organic cation transporters indicates that the transporter forms 12 transmembrane spanning domains (rectangles) with both the N- and C-termini facing the cytoplasm. Glycosylation sites are indicated by branching (Filippo, et al., 2011). The 92 OCTN2 variants identified and expressed in CHO cells are indicated by circles. Mutations reducing carnitine transport to 20% or less than wild-type when expressed in CHO cells are indicated by full circles; variants retaining more that 20% residual activity are indicated by empty circles. The A142S and R488H mutations (indicated by black stripes circles) impair carnitine transport when in cis on the same allele, but not alone (Amat di San Filippo, et al., 2006).
Frequency of tested variants in the general population
Of the 92 variants tested, 19 retained significant residual carnitine transport activity (>20% of normal), and several of them (p.Gly12Ser; p.Leu20His; p.Ala142Ser; p.Val151Met; p.Ala214Val; p.Arg254Gln; p.Thr264Met; p.Ile312Val; p.Glu317Lys; p.Ile348Thr; p.Arg488H; p.Pro549Ser) had frequency in the ExAC Browser Beta and in the gnomAD much higher than those impairing carnitine transport activity, with some individuals being homozygous for some of these milder variants (Table 3B). These variants affected different domains of the OCTN2 carnitine transporter (open circles in Figure 2), including transmembrane and hydrophilic regions in a manner similar to pathogenic missense variants (filled circles in Figure 2). While we cannot exclude that some of these changes might represent mutations (they could affect splicing which was not directly tested here, or they could be in combination with other variants impairing carnitine transport such when p.Ala142Ser is in cis with p.Arg488His (Amat di San Filippo, et al., 2006)), we can conclude that they do not affect the function of the protein if synthesized.
Functional studies allowed us to calculate the minimal estimated frequency of people carrying two abnormal alleles from the frequency in normal individuals reported in the ExAC Browser Beta and in the gnomAD (Table 3B). From these data, 1:221 (0.0045289) alleles contained either one of the missense mutations expressed in this study or a mutation causing the premature insertion of a stop codon or affecting splicing in the SLC22A5 gene (Tables 3B and Supplemental Table 1). Note that one normal subject reported in the gnomAD browser was homozygous for the p.Tyr449Asp variant (Table 3B). This variant reduced carnitine transport to about 17% of normal OCTN2 and has only been identified in one heterozygous patient whose fibroblasts had about 50% of normal carnitine transport (carrier of primary carnitine deficiency) (Amat di San Filippo and Longo, 2004). We cannot exclude that the subject homozygous for this variant has a late-onset form of primary carnitine deficiency.
Given the frequency of pathogenic variants in the normal population (p), the predicted frequency of individuals with two abnormal alleles (p2) in a general population would be 1.6817*10−5 or 1:59,465.
Variant analysis software
Polyphen-2 and SIFT are software programs commonly used to predict the pathogenicity of missense variants. The 90 missense variants and 2 in-frame deletions whose carnitine transport was tested in CHO cells were evaluated using these software programs and the results compared (Table 3C). Both programs were effective in separating benign from damaging variants (p=0.0005 for Polyphen-2 and p=0.0009 for SIFT using analysis of variance, Table 4). However, about 20% of the variants were incorrectly assigned by either program, with a more marked error in the prediction of benign variants (Table 4). When only variants in which the two programs provided a concordant interpretation (74/92) were analyzed, the correct prediction rate increased to 86% (Table 4).
Table 4. Correlation between functional studies and predictions of variant analysis software programs.
Polyphen-2 and SIFT are software programs commonly used to predict the pathogenicity of missense variants. Functional studies of carnitine transport in CHO cells expressing 90 missense variants and 2 in-frame deletions identified in our patients were compared to the predictions of Polyphen-2 and SIFT. Carnitine transport activity of 20% or less that the wild-type transporter was considered pathogenic. Both programs were effective in separating benign from damaging variants (p=0.0005 for Polyphen-2 and p=0.0009 for SIFT using Fisher exact text). However, about 20% of the variants were incorrectly predicted by either program, with a more marked error in the prediction of benign variants. Polyphen-2 and SIFT provided a concordant prediction of the effect of a variant in 80% of the cases (74 of 92 variants). In 86% of the cases the results provided by the two programs were concordant with the results obtained analyzing carnitine transport in CHO cells, with 57/59 (97%) variants correctly identified as pathogenic, but only 7/15 (47%) correctly identified as benign.
| POLYPHEN-2 VARIANT ANALYSIS SOFTWARE | ||
|---|---|---|
| 90 variants analyzed | benign | damaging |
| transport <20% of WT-OCTN2 | 3 | 68 |
| transport >20% of WT-OCTN2 | 7 | 12 |
| 75/90 (83%) correctly identified. Two-tailed p value 0.0005 | ||
| SIFT VARIANT ANALYSIS SOFTWARE | ||
|---|---|---|
| 92 variants analyzed | benign | damaging |
| transport <20% of WT-OCTN2 | 13 | 60 |
| transport >20% of WT-OCTN2 | 11 | 8 |
| 71/92 (77%) correctly identified. Two-tailed p value 0.0009 | ||
| POLYPHEN-2 AND SIFT CONCORDANT PREDICTIONS | ||
|---|---|---|
| benign | damaging | |
| transport <20% of WT-OCTN2 | 2 | 57 |
| transport >20% of WT-OCTN2 | 7 | 8 |
| 64/74 (86%) correctly identified. | Two-tailed | p value 0.0001 |
DISCUSSION
Primary carnitine deficiency is a recessive condition caused by mutations in the OCTN2 carnitine transporter encoded by the SLC22A5 gene (Longo, et al., 2016). The defective OCTN2 carnitine transporter is expressed in fibroblasts and these cells can be used for diagnostic confirmation (Longo, et al., 2016). By looking at a large number of diagnostic samples on which functional studies were performed, there was a continuum of distribution of carnitine transport activity, from 0% in some patients up to 1.5 times normal in certain subjects (Figure 1). While some of this variability might be in part related to variability of the assay, the measurements reported in individual cells were reproducible in repeated experiments, indicating the likely contribution of genetic factors. It is interesting that 218 of 358 fibroblast strains had no evidence of a carnitine uptake defect and that carnitine transport activity was above 60% of normal (the highest levels we have seen in a carrier) in about 70 patients. This suggests that in many cases confirmatory testing might have been requested immediately at the time of initial presentation, without following over time plasma carnitine levels to remove cases of secondary carnitine deficiency (in most cases nutritional).
Genetic analysis identified a number of missense variants, some of which decreased substantially transport activity, others that caused only a modest or no impairment (Table 3). The variability reported here for a limited number of subjects is only a portion of that observed in normal individuals, with about 354 missense variants reported in the ExAC Browser Beta (http://exac.broadinstitute.org/gene/ENSG00000197375) and in the gnomAD browser (http://http://gnomad.broadinstitute.org/gene/ENSG00000197375). It is conceivable that missense variations, in addition to variations in regulatory sequences of the gene, are responsible for this continuous, rather that discontinuous distribution of carnitine transport activity in fibroblasts.
In our experience, patients whose fibroblasts had less than 20% carnitine transport activity were unable to maintain free carnitine level ≥9 μmol/L and required carnitine supplementation. For this reason, we established 20% of normal transport as the cut-off below which we diagnose primary carnitine deficiency and provide carnitine supplementation. Fibroblasts from 95 patients had carnitine transport activity <20% (Table 1). Molecular analysis identified causative mutations in 159/190 alleles, while no variants were identified in 31/190 alleles (Table 2), indicating that some mutations lie outside the coding region or flanking sequence of the SLC22A5 gene. It must be noted that we still know very little of the promoter region of this gene and other regulatory sequences that are not routinely sequenced. For this reason, the diagnosis of primary carnitine deficiency can be missed in some patients if only DNA studies are performed. For example, 6 out of 95 of our patients would have been missed completely by molecular analysis (no significant sequence variations identified in either allele), if functional studies in fibroblasts had not been performed. This has profound implications since these patients could have escaped diagnosis and could have been left untreated without functional studies.
It is crucial to correctly identify all the patients affected by primary carnitine deficiency and promptly start carnitine supplements. If left untreated, primary carnitine deficiency can lead to hypoketotic hypoglycemia early in life, or manifest with skeletal and cardiac myopathy, and in some cases sudden death at any age (Longo, et al., 2016). At the same time, a subject wrongly diagnosed with the disease will have to face an unnecessary lifelong therapy with carnitine supplements (Longo, et al., 2016).
Unlike mutations causing the premature insertion of a stop codon or affecting splicing, the interpretation of missense variants found by molecular analysis can be difficult and requires additional studies. Here we expressed 90 missense variants and 2 in-frame deletions identified in our patients in CHO cells to determine their carnitine transport activity (Table 3A). A carnitine transport activity of 20% or less than CHO expressing the wild-type OCTN2 carnitine transporter (transport activity of 100%) was considered pathogenic. Of the variants tested, 73/92 were pathogenic, and 19/92 were benign (Table 3A).
Polyphen-2 and SIFT are software programs commonly used to predict the effect of missense variants on protein function. We compared the predictions of Polyphen-2 and SIFT against functional studies of carnitine transport in CHO cells (Table 3C). Our results indicate that while Polyphen-2 and SIFT were effective in separating benign from damaging variants, they incorrectly identified the effect of variants in about 20% of the cases (Table 4). For variants in which Polyphen-2 and SIFT provided a concordant interpretation, the effect of benign variants was wrongly predicted in 53% of the cases, while the error was reduced to 3% when predicting the effect of damaging variants (Table 4). For these reasons, these programs seem skewed in predicting more damaging effects of missense mutations than experimentally observed (false positive results) in the OCTN2 carnitine transporter. We do not know whether this could extend to the interpretation of functional effects in other membrane transporters.
The incidence of primary carnitine deficiency is approximately 1:142,000 in the USA as ascertained by newborn screening (Therrell, et al., 2014). From the frequency of pathogenic alleles reported in the ExAC Browser Beta and in the gnomAD Browser in about 120,000 healthy (heterozygotes) individuals (Table 3B and supplemental table 1) (Lek, et al., 2016), the calculated frequency of mutant alleles is 1:221, with a minimal estimated frequency of people carrying two abnormal alleles in the general population of 1.6817*10−5 or 1:59,465 individuals. This frequency is significantly higher than the one reported by newborn screening, indicating that some affected individuals might be missed by the current neonatal screening protocols. We do not know whether this is due to specific mutations (Chen, et al., 2013) and/or to problems related to the timing of collection of the newborn screening sample, in which the blood samples are collected at 1–3 days of age and carnitine levels might reflect those of the mother (Pasquali and Longo, 2013; Rasmussen, et al., 2017; Schimmenti, et al., 2007). Carnitine is transferred from the mother to the child via the placenta, and immediately after birth levels of free carnitine (C0) are usually lower in infants of mothers with primary carnitine deficiency as compared to infants with the disease themselves (Pasquali and Longo, 2013). With time, levels decrease in infants with primary carnitine deficiency, but remain stable or slightly increase in infants of mothers with primary carnitine deficiency (Pasquali and Longo, 2013). The diagnosis can therefore be missed if screening is performed too close to birth and no second screening is obtained. In the Faroe Islands, post-neonatal screening (age >2 months) was able to identify additional patients who had unrevealing newborn screening, confirming that patients with primary carnitine deficiency are missed by current newborn screening methods (Steuerwald, et al., 2017). For these reasons, primary carnitine deficiency should be considered in children and adults with a suitable clinical presentation (hypoglycemia, cardiomyopathy, arrhythmia, sudden death) even in the presence of a normal newborn screening test.
Supplementary Material
Acknowledgments
Grant support: Supported in part by NIH grant R01-DK53824.
This work was supported in part by NIH grant DK 53824. We thank Dr. Vadivel Ganapathy for providing the cDNAs for the OCTN2 carnitine transporter.
The Authors have no conflict of interest to declare for this manuscript.
Footnotes
Study approved by IRB of the University of Utah
BIBLIOGRAPHY
- Akpinar B, Coteli C, Yucel D, Ozgul RK, Dursun A, Demirkol M, Baykal T, Gokcay G. A novel mutation (L363P) in the OCTN2 gene and its effect on secondary protein structure. Clinical Genetics. 2010;78(Suppl 1):89. (Abs J06) [Google Scholar]
- Amat di San Filippo C, Longo N. Tyrosine residues affecting sodium stimulation of carnitine transport in the OCTN2 carnitine/organic cation transporter. J Biol Chem. 2004;279(8):7247–53. doi: 10.1074/jbc.M309171200. [DOI] [PubMed] [Google Scholar]
- Amat di San Filippo C, Pasquali M, Longo N. Pharmacological rescue of carnitine transport in primary carnitine deficiency. Hum Mutat. 2006;27(6):513–23. doi: 10.1002/humu.20314. [DOI] [PubMed] [Google Scholar]
- Amat di San Filippo C, Taylor MR, Mestroni L, Botto LD, Longo N. Cardiomyopathy and carnitine deficiency. Mol Genet Metab. 2008;94(2):162–6. doi: 10.1016/j.ymgme.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amat di San Filippo C, Wang Y, Longo N. Functional domains in the carnitine transporter OCTN2, defective in primary carnitine deficiency. J Biol Chem. 2003;278(48):47776–84. doi: 10.1074/jbc.M307911200. [DOI] [PubMed] [Google Scholar]
- Burckhardt G, Wolff NA. Structure of renal organic anion and cation transporters. Am J Physiol Renal Physiol. 2000;278(6):F853–66. doi: 10.1152/ajprenal.2000.278.6.F853. [DOI] [PubMed] [Google Scholar]
- Burwinkel B, Kreuder J, Schweitzer S, Vorgerd M, Gempel K, Gerbitz KD, Kilimann MW. Carnitine transporter OCTN2 mutations in systemic primary carnitine deficiency: a novel Arg169Gln mutation and a recurrent Arg282ter mutation associated with an unconventional splicing abnormality. Biochem Biophys Res Commun. 1999;261(2):484–7. doi: 10.1006/bbrc.1999.1060. [DOI] [PubMed] [Google Scholar]
- Cederbaum SD, Koo-McCoy S, Tein I, Hsu BY, Ganguly A, Vilain E, Dipple K, Cvitanovic-Sojat L, Stanley C. Carnitine membrane transporter deficiency: a long-term follow up and OCTN2 mutation in the first documented case of primary carnitine deficiency. Mol Genet Metab. 2002;77(3):195–201. doi: 10.1016/s1096-7192(02)00169-5. [DOI] [PubMed] [Google Scholar]
- Chen YC, Chien YH, Chen PW, Leung-Sang Tang N, Chiu PC, Hwu WL, Lee NC. Carnitine uptake defect (primary carnitine deficiency): risk in genotype-phenotype correlation. Hum Mutat. 2013;34(4):655. doi: 10.1002/humu.22286. [DOI] [PubMed] [Google Scholar]
- De Biase I, Champaigne NL, Schroer R, Pollard LM, Longo N, Wood T. Primary Carnitine Deficiency Presents Atypically with Long QT Syndrome: A Case Report. JIMD Rep. 2012;2:87–90. doi: 10.1007/8904_2011_52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolski SF, McKinney JT, Amat di San Filippo C, Giak Sim K, Wilcken B, Longo N. Validation of dye-binding/high-resolution thermal denaturation for the identification of mutations in the SLC22A5 gene. Hum Mutat. 2005;25(3):306–13. doi: 10.1002/humu.20137. [DOI] [PubMed] [Google Scholar]
- El-Hattab AW, Li FY, Shen J, Powell BR, Bawle EV, Adams DJ, Wahl E, Kobori JA, Graham B, Scaglia F, et al. Maternal systemic primary carnitine deficiency uncovered by newborn screening: clinical, biochemical, and molecular aspects. Genet Med. 2010;12(1):19–24. doi: 10.1097/GIM.0b013e3181c5e6f7. [DOI] [PubMed] [Google Scholar]
- Filippo CA, Ardon O, Longo N. Glycosylation of the OCTN2 carnitine transporter: study of natural mutations identified in patients with primary carnitine deficiency. Biochim Biophys Acta. 2011;1812(3):312–20. doi: 10.1016/j.bbadis.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigeni M, Iacobazzi F, Yin X, Longo N. Wide tolerance to amino acids substitutions in the OCTN1 ergothioneine transporter. Biochim Biophys Acta. 2016;1860(6):1334–1342. doi: 10.1016/j.bbagen.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Wang F, Wang Y, Ye J, Qiu W, Zhang H, Gao X, Gong Z, Gu X. Analysis of genetic mutations in Chinese patients with systemic primary carnitine deficiency. Eur J Med Genet. 2014;57(10):571–5. doi: 10.1016/j.ejmg.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Koizumi A, Nozaki J, Ohura T, Kayo T, Wada Y, Nezu J, Ohashi R, Tamai I, Shoji Y, Takada G, et al. Genetic epidemiology of the carnitine transporter OCTN2 gene in a Japanese population and phenotypic characterization in Japanese pedigrees with primary systemic carnitine deficiency. Hum Mol Genet. 1999;8(12):2247–54. doi: 10.1093/hmg/8.12.2247. [DOI] [PubMed] [Google Scholar]
- Komlosi K, Magyari L, Talian GC, Nemes E, Kaposzta R, Mogyorosy G, Mehes K, Melegh B. Plasma carnitine ester profile in homozygous and heterozygous OCTN2 deficiency. J Inherit Metab Dis. 2009;32(Suppl 1):S15–9. doi: 10.1007/s10545-009-0926-1. [DOI] [PubMed] [Google Scholar]
- Lamhonwah AM, Olpin SE, Pollitt RJ, Vianey-Saban C, Divry P, Guffon N, Besley GT, Onizuka R, De Meirleir LJ, Cvitanovic-Sojat L, et al. Novel OCTN2 mutations: no genotype-phenotype correlations: early carnitine therapy prevents cardiomyopathy. Am J Med Genet. 2002;111(3):271–84. doi: 10.1002/ajmg.10585. [DOI] [PubMed] [Google Scholar]
- Lee NC, Tang NL, Chien YH, Chen CA, Lin SJ, Chiu PC, Huang AC, Hwu WL. Diagnoses of newborns and mothers with carnitine uptake defects through newborn screening. Mol Genet Metab. 2010;100(1):46–50. doi: 10.1016/j.ymgme.2009.12.015. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FY, El-Hattab AW, Bawle EV, Boles RG, Schmitt ES, Scaglia F, Wong LJ. Molecular spectrum of SLC22A5 (OCTN2) gene mutations detected in 143 subjects evaluated for systemic carnitine deficiency. Hum Mutat. 2010;31(8):E1632–51. doi: 10.1002/humu.21311. [DOI] [PubMed] [Google Scholar]
- Longo N, Amat di San Filippo C, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006;142C(2):77–85. doi: 10.1002/ajmg.c.30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo N, Frigeni M, Pasquali M. Carnitine transport and fatty acid oxidation. Biochim Biophys Acta. 2016;1863(10):2422–35. doi: 10.1016/j.bbamcr.2016.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhseed N, Vallance HD, Potter M, Waters PJ, Wong LT, Lillquist Y, Pasquali M, Amat di San Filippo C, Longo N. Carnitine transporter defect due to a novel mutation in the SLC22A5 gene presenting with peripheral neuropathy. J Inherit Metab Dis. 2004;27(6):778–80. doi: 10.1023/b:boli.0000045837.23328.f4. [DOI] [PubMed] [Google Scholar]
- Mayatepek E, Nezu J, Tamai I, Oku A, Katsura M, Shimane M, Tsuji A. Two novel missense mutations of the OCTN2 gene (W283R and V446F) in a patient with primary systemic carnitine deficiency. Hum Mutat. 2000;15(1):118. doi: 10.1002/(SICI)1098-1004(200001)15:1<118::AID-HUMU28>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Mutlu-Albayrak H, Bene J, Oflaz MB, Tanyalcin T, Caksen H, Melegh B. Identification of SLC22A5 Gene Mutation in a Family with Carnitine Uptake Defect. Case Rep Genet. 2015;2015:259627. doi: 10.1155/2015/259627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nezu J, Tamai I, Oku A, Ohashi R, Yabuuchi H, Hashimoto N, Nikaido H, Sai Y, Koizumi A, Shoji Y, et al. Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat Genet. 1999;21(1):91–4. doi: 10.1038/5030. [DOI] [PubMed] [Google Scholar]
- Pasquali M, Longo N. Response to chen et Al.: carnitine uptake defect (primary carnitine deficiency): risk in genotype-phenotype correlation. Hum Mutat. 2013;34(4):656. doi: 10.1002/humu.22285. [DOI] [PubMed] [Google Scholar]
- Rahbeeni Z, Vaz FM, Al-Hussein K, Bucknall MP, Ruiter J, Wanders RJ, Rashed MS. Identification of two novel mutations in OCTN2 from two Saudi patients with systemic carnitine deficiency. J Inherit Metab Dis. 2002;25(5):363–9. doi: 10.1023/a:1020143632011. [DOI] [PubMed] [Google Scholar]
- Rasmussen J, Hougaard DM, Sandhu N, Fjaellegaard K, Petersen PR, Steuerwald U, Lund AM. Primary Carnitine Deficiency: Is Foetal Development Affected and Can Newborn Screening Be Improved? JIMD Rep. 2017 doi: 10.1007/8904_2016_30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen J, Kober L, Lund AM, Nielsen OW. Primary Carnitine deficiency in the Faroe Islands: health and cardiac status in 76 adult patients diagnosed by screening. J Inherit Metab Dis. 2014a;37(2):223–30. doi: 10.1007/s10545-013-9640-0. [DOI] [PubMed] [Google Scholar]
- Rasmussen J, Nielsen OW, Janzen N, Duno M, Gislason H, Kober L, Steuerwald U, Lund AM. Carnitine levels in 26,462 individuals from the nationwide screening program for primary carnitine deficiency in the Faroe Islands. J Inherit Metab Dis. 2014b;37(2):215–22. doi: 10.1007/s10545-013-9606-2. [DOI] [PubMed] [Google Scholar]
- Rose EC, di San Filippo CA, Ndukwe Erlingsson UC, Ardon O, Pasquali M, Longo N. Genotype-phenotype correlation in primary carnitine deficiency. Hum Mutat. 2012;33(1):118–23. doi: 10.1002/humu.21607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaglia F, Wang Y, Longo N. Functional characterization of the carnitine transporter defective in primary carnitine deficiency. Arch Biochem Biophys. 1999;364(1):99–106. doi: 10.1006/abbi.1999.1118. [DOI] [PubMed] [Google Scholar]
- Scaglia F, Wang Y, Singh RH, Dembure PP, Pasquali M, Fernhoff PM, Longo N. Defective urinary carnitine transport in heterozygotes for primary carnitine deficiency. Genet Med. 1998;1(1):34–9. doi: 10.1097/00125817-199811000-00008. [DOI] [PubMed] [Google Scholar]
- Schimmenti LA, Crombez EA, Schwahn BC, Heese BA, Wood TC, Schroer RJ, Bentler K, Cederbaum S, Sarafoglou K, McCann M, et al. Expanded newborn screening identifies maternal primary carnitine deficiency. Mol Genet Metab. 2007;90(4):441–5. doi: 10.1016/j.ymgme.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Spiekerkoetter U, Huener G, Baykal T, Demirkol M, Duran M, Wanders R, Nezu J, Mayatepek E. Silent and symptomatic primary carnitine deficiency within the same family due to identical mutations in the organic cation/carnitine transporter OCTN2. J Inherit Metab Dis. 2003;26(6):613–5. doi: 10.1023/a:1025968502527. [DOI] [PubMed] [Google Scholar]
- Steuerwald U, Lund AM, Rasmussen J, Janzen N, Hougaard DM, Longo N. Neonatal Screening for Primary Carnitine Deficiency: Lessons Learned from the Faroe Islands. Int J Neonatal Screen. 2017;3(1):1. [Google Scholar]
- Tamai I, Ohashi R, Nezu J, Yabuuchi H, Oku A, Shimane M, Sai Y, Tsuji A. Molecular and functional identification of sodium ion-dependent, high affinity human carnitine transporter OCTN2. J Biol Chem. 1998;273(32):20378–82. doi: 10.1074/jbc.273.32.20378. [DOI] [PubMed] [Google Scholar]
- Tang NL, Ganapathy V, Wu X, Hui J, Seth P, Yuen PM, Wanders RJ, Fok TF, Hjelm NM. Mutations of OCTN2, an organic cation/carnitine transporter, lead to deficient cellular carnitine uptake in primary carnitine deficiency. Hum Mol Genet. 1999;8(4):655–60. doi: 10.1093/hmg/8.4.655. [DOI] [PubMed] [Google Scholar]
- Tang NL, Hwu WL, Chan RT, Law LK, Fung LM, Zhang WM. A founder mutation (R254X) of SLC22A5 (OCTN2) in Chinese primary carnitine deficiency patients. Hum Mutat. 2002;20(3):232. doi: 10.1002/humu.9053. [DOI] [PubMed] [Google Scholar]
- Therrell BL, Jr, Lloyd-Puryear MA, Camp KM, Mann MY. Inborn errors of metabolism identified via newborn screening: Ten-year incidence data and costs of nutritional interventions for research agenda planning. Mol Genet Metab. 2014;113(1–2):14–26. doi: 10.1016/j.ymgme.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban TJ, Gallagher RC, Brown C, Castro RA, Lagpacan LL, Brett CM, Taylor TR, Carlson EJ, Ferrin TE, Burchard EG, et al. Functional genetic diversity in the high-affinity carnitine transporter OCTN2 (SLC22A5) Mol Pharmacol. 2006;70(5):1602–11. doi: 10.1124/mol.106.028126. [DOI] [PubMed] [Google Scholar]
- Vaz FM, Scholte HR, Ruiter J, Hussaarts-Odijk LM, Pereira RR, Schweitzer S, de Klerk JB, Waterham HR, Wanders RJ. Identification of two novel mutations in OCTN2 of three patients with systemic carnitine deficiency. Hum Genet. 1999;105(1–2):157–61. doi: 10.1007/s004399900105. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kelly MA, Cowan TM, Longo N. A missense mutation in the OCTN2 gene associated with residual carnitine transport activity. Hum Mutat. 2000a;15(3):238–45. doi: 10.1002/(SICI)1098-1004(200003)15:3<238::AID-HUMU4>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Wang Y, Korman SH, Ye J, Gargus JJ, Gutman A, Taroni F, Garavaglia B, Longo N. Phenotype and genotype variation in primary carnitine deficiency. Genet Med. 2001;3(6):387–92. doi: 10.1097/00125817-200111000-00002. [DOI] [PubMed] [Google Scholar]
- Wang Y, Meadows TA, Longo N. Abnormal sodium stimulation of carnitine transport in primary carnitine deficiency. J Biol Chem. 2000b;275(27):20782–6. doi: 10.1074/jbc.M000194200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Taroni F, Garavaglia B, Longo N. Functional analysis of mutations in the OCTN2 transporter causing primary carnitine deficiency: lack of genotype-phenotype correlation. Hum Mutat. 2000c;16(5):401–7. doi: 10.1002/1098-1004(200011)16:5<401::AID-HUMU4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ye J, Ganapathy V, Longo N. Mutations in the organic cation/carnitine transporter OCTN2 in primary carnitine deficiency. Proc Natl Acad Sci U S A. 1999;96(5):2356–60. doi: 10.1073/pnas.96.5.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcken B, Wiley V, Hammond J, Carpenter K. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N Engl J Med. 2003;348(23):2304–12. doi: 10.1056/NEJMoa025225. [DOI] [PubMed] [Google Scholar]
- Wu X, Huang W, Prasad PD, Seth P, Rajan DP, Leibach FH, Chen J, Conway SJ, Ganapathy V. Functional characteristics and tissue distribution pattern of organic cation transporter 2 (OCTN2), an organic cation/carnitine transporter. J Pharmacol Exp Ther. 1999;290(3):1482–92. [PubMed] [Google Scholar]
- Wu X, Prasad PD, Leibach FH, Ganapathy V. cDNA sequence, transport function, and genomic organization of human OCTN2, a new member of the organic cation transporter family. Biochem Biophys Res Commun. 1998;246(3):589–95. doi: 10.1006/bbrc.1998.8669. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.