

Abstract
Host-microbiome co-evolution drives homeostasis and disease susceptibility, yet regulatory principles governing the integrated intestinal host-commensal microenvironment remain obscure. While inflammasome signaling participates in these interactions, its activators and microbiome-modulating mechanisms are unknown. Here, we demonstrate that the microbiota-associated metabolites taurine, histamine, and spermine shape the host-microbiome interface by co-modulating NLRP6 inflammasome signaling, epithelial IL-18 secretion, and downstream anti-microbial peptide (AMP) profiles. Distortion of this balanced AMP landscape by inflammasome deficiency drives dysbiosis development. Upon fecal transfer, colitis-inducing microbiota hijacks this microenvironment-orchestrating machinery through metabolite-mediated inflammasome suppression, leading to distorted AMP balance favoring its preferential colonization. Restoration of the metabolite-inflammasome-AMP axis reinstates a normal microbiota and ameliorates colitis. Together, we identify microbial modulators of the NLRP6 inflammasome and highlight mechanisms by which microbiome-host interactions cooperatively drive microbial community stability through metabolite-mediated innate immune modulation. Therefore, targeted ‘postbiotic’ metabolomic intervention may restore a normal microenvironment, as treatment or prevention of dysbiosis-driven diseases.
Keywords: Inflammasome, anti-microbial peptides, microbiota, metabolites
Graphical Abstract
Introduction
Aberrant intestinal microbiota composition and function, termed dysbiosis, is suggested to associate with many diseases (Thaiss and Elinav, 2014; Turnbaugh et al., 2006). A dysbiotic state can be established in multiple ways, including intestinal infection (Lupp et al., 2007; Stecher et al., 2007) auto-inflammation (Devkota et al., 2012), altered host genetics (Goodrich et al., 2014), and dietary modulation (David et al., 2014). Remarkably, disease-promoting dysbiotic microbiota in multiple mouse models harbors dominance, upon fecal transfer, over a previously stable wild-type (WT) microbiota, leading to transferable disease susceptibility (Couturier-Maillard et al., 2013; Elinav et al., 2011; Garrett et al., 2007; Henao-Mejia et al., 2012; Ivanov et al., 2009). The host immune system participates in the organization of the ‘healthy’ host-microbial interface, with mechanisms ranging from IgA and mucus secretion to AMP production (Hooper et al., 2012). However, the means by which the indigenous microbiota contributes to its ‘healthy’ niche construction, and mechanisms driving the formation of a dysbiotic microbiota and its persistence in an invaded host, remain poorly understood.
To study in detail the mutualistic host- and microbiota-regulated mechanisms determining the normal and disease-associated intestinal interface, we employed the NLPR6 inflammasome deficiency model, in which host innate immune genetic deficiency is associated with development of an aberrant microbial configuration driving disease susceptibility (Elinav et al., 2011; Henao-Mejia et al., 2012). Inflammasomes are multi-protein complexes consisting of one of several upstream Nod-like Receptor (NLR) proteins, the adaptor apoptosis-associated speck-like protein containing a CARD (ASC), and the effector caspase-1. Upon receipt of defined sets of transcriptional and post-translational signals, inflammasomes are assembled, become active through auto-cleavage of pro-caspase-1, and process the catalytic activation of IL-1β and IL-18 (Martinon et al., 2009).
The newly proposed NLRP6 inflammasome is a key regulator of colonic homeostasis (Elinav et al., 2011; Hu et al., 2013; Wlodarska et al., 2014). NLRP6 is predominantly expressed in intestinal epithelial cells, including goblet cells (Elinav et al., 2011; Gremel et al., 2014; Wlodarska et al., 2014), where it was found to be essential for mucosal self-renewal, proliferation, and mucus secretion (Chen et al., 2011; Normand et al., 2011; Wlodarska et al., 2014). Likewise, ASC and caspase-1 are co-expressed in intestinal epithelial cells, yet no biochemical evidence of their assembly into an NLRP6 inflammasome has been demonstrated to date. Mice deficient in NLRP6, ASC, or caspase-1 feature a distinct form of dysbiosis that drives a context-specific propensity for intestinal auto-inflammation, inflammation-induced colorectal cancer and features of the ‘metabolic syndrome’ (Anand and Kanneganti, 2012; Elinav et al., 2011; Henao-Mejia et al., 2012; Ikuta et al., 2013). It is postulated that factors secreted by the WT microbiota activate and modulate the NLRP6 inflammasome, but no such factors have been discovered to date. Moreover, the mechanisms by which inflammasome deficiency leads to dysbiosis, and how this disease-causing ecosystem assumes dominance upon transfer into a WT host remain elusive.
In this study, we highlight principles by which the host and its microbiota cooperatively utilize inflammasome signaling in the formation and stabilization of the host-microbiota interface. We identify microbial metabolites that signal to the NLRP6 inflammasome, thereby activating host immune circuits to orchestrate an antimicrobial program, which optimizes commensal colonization and persistence. We demonstrate that alteration of this crosstalk is sufficient to induce a state of dysbiosis, and that the altered microbiota is able to exploit these pathways in conferring a preferential microenvironment for its colonization in an invaded host.
Results
The microbiota modulates the NLRP6 inflammasome and downstream anti-microbial peptide secretion
We began our investigation by studying the effects of commensal bacterial colonization on inflammasome signaling in the WT setting. We identified the gut microbiota to be critically important for inflammasome activation, as its absence in germ-free (GF) mice resulted in abolished caspase-1 auto-cleavage (Figures 1A and 1B). This severely altered inflammasome activity, coupled with reduced Il18 mRNA (Figure S1A), resulted in abrogated levels of colonic IL-18 in WT GF mice (Figure 1C). Similarly, WT mice treated with broad-spectrum antibiotics (see methods) featured decreased mRNA (Figure S1B) and protein levels of IL-18 (Figure 1D). To corroborate the role of the microbiota in inducing mucosal IL-18, we examined the levels of IL-18 in the neonatal period, in which newborn mammals feature progressive microbial colonization, accompanied by intestinal barrier formation and immune system maturation. Indeed, intestinal IL-18 levels progressively increased over the first five weeks of post-natal development, paralleling microbial colonization (Figures 1E and S1C, (Kempster et al., 2011)). Together, these results suggest that WT commensal bacterial colonization regulates intestinal inflammasome assembly by providing a ‘signal I’ (transcription of inflammasome components) and possibly a ‘signal II’ (inducing inflammasome assembly), resulting in steady state mucosal IL-18 secretion.
Figure 1. Microbiota activation of inflammasome signaling results in downstream induction of antimicrobial peptides.

(A, B) Immunoblot analysis (A) and quantification (B) of pro-caspase-1 (p45) and cleaved caspase-1 (p20) in colon tissue from germ-free and SPF mice.
(C–E) IL-18 production by colon explants from germ-free mice (C), antibiotics-treated mice (D), and during early stages of post-natal colonization (E).
(F) Differential expression between wild-type (WT) and Il18−/− mice of antimicrobial peptides (AMPs) versus all other genes. Box = interquartile range (IQR) = 25th to 75th percentile, line - median, star - mean, whiskers - IQR*1.5. Mann-Whitney U-test p<0.0001.
(G–O) Expression levels of the indicated AMPs in WT and Il18−/− colonic tissue (G–I), during early stages of post-natal colonization (J), in the colon of bone marrow chimeras generated from WT and Il18−/− mice (K), in colonic explants from germ-free mice cultured with IL-18 compared to controls (L), in IL-18 injected germ-free mice compared to SPF controls (M, N), and in colonic explants cultured with or without the NF-κB inhibitor Bay 11–7085 (O).
Data are expressed as mean ± SEM. * p<0.05; ** p<0.01; *** p<0.001.
Pairwise comparison was performed using Student’s t test, unless stated otherwise.
Results shown are representative of 3 (panels A–D) or two (panels E–O) independent repeats (n= 3–13 per group).
See also Figure S1.
We next determined the consequences of commensal-mediated IL-18 induction on the host mucosal microenvironment, by performing RNA-seq of colonic tissue from WT and Il18−/− mice. We grouped host transcripts into Gene Ontology functional categories and evaluated differential expression of these categories between WT and Il18−/− mice (Figure S1D). One of the most differentially represented categories included anti-microbial pathways (Figure 1F), suggesting a role for IL-18 in regulating the anti-microbial program of the colonic mucosa. Among the AMPs induced by IL-18 were ITLN1, RELMβ, and members of the angiogenin family (Figures 1G–1I and S1E), for which a microbicidal activity has been previously reported (Artis et al., 2004; Tsuji et al., 2001). Of these, angiogenin-4 (Ang4) expression has been suggested to be influenced by the intestinal microbiota (Hooper et al., 2003). Indeed, GF mice featured nearly undetectable levels of Ang4 (Figure S1F), as well as ITLN1 and RELMβ (Figures S1G and S1H), while microbial colonization during early development progressively induced AMP expression in newborn WT mice (Figure 1J). Bone marrow chimera experiments, using WT or Il18−/− mice as either donors or recipients of bone marrow transplants, indicated that intestinal IL-18 is primarily produced, under homeostatic conditions, by the non-hematopoietic compartment (Figures S1I and S1J), and that IL-18 originating from this source is necessary for AMP induction (Figure 1K). Administration of IL-18 under sterile conditions to GF colon explants increased the levels of colonic AMPs (Figure 1L). In-vivo administration of IL-18 to WT GF mice partially rescued AMP levels (Figures 1M, 1N, and S1K), correlating with colonic levels of IL-18 (Figure S1L), while transcript levels of IL-18 remained unaffected (Figure S1M). Induction of AMPs by IL-18 was dependent on NF-κB signaling, since NF-κB inhibition prevented AMP induction (Figure 1O). Altogether, these results suggest that microbiota-induced colonic IL-18 is both necessary and sufficient for the regulation of intestinal anti-microbial peptide production.
We next aimed to determine whether IL-18-mediated induction of colonic anti-microbial activity is dependent on epithelial NLRP6 inflammasome activity, previously found to regulate homeostatic IL-18 levels (Elinav et al., 2011). An ASC-dependent NLRP6 inflammasome complex was found in-vitro (upon expression in HEK293T cells, (Grenier et al., 2002)), by NLRP6, ASC and caspase-1 co-immunoprecipitation (Figures 2A and 2B), and suggested in-vivo as colons from Nlrp6−/− mice colons featured decreased steady-state activation of caspase-1 (Figures 2C and S2A). To determine whether the NLRP6 inflammasome drives IL-18-mediated induction of colonic AMP expression, we performed RNA-seq of colons from WT and inflammasome-deficient mice. Similar to Il18−/− mice, Asc−/− and Nlrp6−/− mice featured an abnormal AMP profile, including impaired levels of Retnlb, Ang4, and Itln1 (Figures 2D, 2E, and S2B), suggesting that control of AMP expression requires an intact NLRP6 inflammasome. In fact, these AMPs were among the most differentially expressed genes when comparing the transcriptomes of Il18−/−, Asc−/− and Nlrp6−/− mice to WT controls (Figures 2E and S2C). An aberrant AMP profile of Asc−/− mice was also noted by RNA-seq of colons obtained from genetically identical mice housed in an independent animal facility (Figures S2D–S2G). To validate these results, we further focused on colonic IL-18 and the prototypical AMP Ang4, and compared their levels in WT mice to those in Nlrp6−/−, Asc−/−, Il18−/−, Casp1/11−/− and Nlrp3−/− mice. While mice lacking NLRP3 had normal levels of colonic IL-18 and Ang4, Nlrp6−/−, Asc−/−, Il18−/−, and Casp1/11−/− mice featured a marked reduction in both IL-18 and Ang4, suggesting that the NLRP6 inflammasome is required for IL-18 production upstream of AMP induction (Figures 2F and S2H). Ang4 reduction at the protein level was confirmed by targeted mass-spectrometry for Ang4 peptides (Figures 2G and 2H). In-vivo administration of recombinant IL-18 into mice lacking NLRP6, ASC, or caspase-1/11 restored IL-18 and AMP levels (Figures 2I, 2J, and S2I–S2L), demonstrating that IL-18 is sufficient for AMP expression downstream of inflammasome signaling.
Figure 2. NLRP6 inflammasome signaling is required for IL-18 production upstream of the induction of antimicrobial peptides.

(A, B) Co-immunoprecipitation of FLAG-NLRP6, HA-ASC and MYC-Caspase-1 overexpressed in HEK293T cells. Cell lysates were precipitated using anti-FLAG sepharose beads (A) or anti-HA sepharose beads (B) followed by immunoblotting with anti HA, anti FLAG and anti MYC antibodies.
(C) Immunoblot analysis of pro-caspase-1 (p45) and cleaved caspase-1 (p20) in colon tissue from SPF WT and Nlrp6−/− mice.
(D) Differential expression between WT and Nlrp6−/− mice of AMPs versus all other genes. Box = interquartile range (IQR) = 25th to 75th percentile, line - median, star - mean, whiskers - IQR*1.5. Mann-Whitney U-test p<0.05.
(E) Heatmap of AMPs from transcriptome analysis of colon tissue from WT, Asc−/−, Nlrp6−/− and Il18−/− mice.
(F) Colonic Ang4 expression in WT, Asc−/−, IL18−/−, Casp1/11−/−, Nlrp6−/− and Nlrp3−/− mice.
(G, H) Targeted mass spectrometry of Ang4 peptides identified in fecal samples obtained from WT or Asc−/− mice. Colors indicate different fragment ions.
(I–J) Colonic expression of the indicated AMPs in Nlrp6−/−, Asc−/−, and Casp1/11−/− mice following injection of IL-18.
(K–M) Immunoblot quantification of pro-caspase-1 (p45) and cleaved caspase-1 (p20) (K), IL-18 production (L) and Ang4 expression (M) in colon tissue from germ-free and SPF WT and Nlrp6−/− mice.
(N) Colonic Ang4 expression levels in germ-free Nlrp6−/− mice, with or without injection of IL-18. Data are expressed as mean ± SEM. * p<0.05; ** p<0.01; *** p<0.001.
Pairwise comparison was performed using Student’s t test, unless stated otherwise.
Results shown are representative of two independent repeats (n= 3–14 per group).
See also Figure S2.
Since both the microbiota and NLRP6 inflammasome were required for IL-18 and AMP induction, we hypothesized that the microbiota may activate NLRP6 upstream of IL-18 secretion. Indeed, while induction of IL-18 transcription in response to microbial colonization of GF mice functioned normally in both WT and Nlrp6−/− mice (Figure S2M), activation of caspase-1, secretion of mature IL-18 protein, and concomitant up-regulation of Ang4 mRNA were induced upon colonization of GF WT mice, but were severely impaired upon conventionalization of GF Nlrp6−/− mice (Figures 2K–2M and S2N). Moreover, sterile administration of IL-18 into GF Nlrp6−/− mice (Figure S2O) rescued this defect and induced Ang4 expression (Figure 2N), indicating that the addition of IL-18 bypasses the need for both the presence of a microbiota and intact inflammasome signaling, and suffices to induce a normal anti-microbial program. Together, these data uncover a pathway by which the microbiota induces NLRP6 inflammasome signaling to produce IL-18, which in turn activates an AMP program in the colonic mucosa.
The inflammasome-antimicrobial peptide axis regulates intestinal microbial composition
We next assessed whether the above NLRP6-IL-18-AMP axis is involved in control of microbiota composition. Nlrp6−/− mice were recently shown to harbor a dysbiotic microbiome configuration (Elinav et al., 2011). However, whether impaired inflammasome deficiency directly drives dysbiosis (as opposed to cross-generational or facility-related dysbiosis) remained to be investigated. We therefore analyzed, using 16S rDNA sequencing, the temporal microbial composition of GF Nlrp6−/− or WT mice that were allowed to spontaneously conventionalize at our vivarium (termed ex-GF mice). Notably, ex-GF Nlrp6−/− mice gradually shifted their microbial community composition towards the dysbiotic configuration of Nlrp6−/− mice that had been housed in our specific pathogen-free (SPF) vivarium for multiple generations (Figures 3A and 3B). Two months following colonization, the microbiota composition of ex-GF Nlrp6−/− mice became similar to that of SPF Nlrp6−/− mice. This shift was accompanied by a gradual reduction in alpha-diversity down to the level of SPF Nlrp6−/− mice (Figure 3C). Indeed, as early as three weeks from the start of spontaneous conventionalization, the gut microbiota of newly colonized Nlrp6−/− mice became profoundly different from that of concomitantly conventionalized ex-GF WT mice (Figure 3D). NLRP6 inflammasome-induced dysbiosis was present across vivaria, as exemplified by similar composition and function of Nlrp6−/− and Asc−/− microbiota assessed in two different facilities (Figures S3A–S3D). Interestingly, WT, but not Nlrp6−/− cohorts, significantly differed across facilities (Figures S3A–S3D). Overall, these results demonstrate that the intestinal inflammasome-deficient microenvironment is associated with a microbial ‘signature’ that is independent of housing conditions, and which can be acquired de-novo upon colonization of GF mice.
Figure 3. The inflammasome-antimicrobial peptide axis regulates intestinal microbial community composition.

(A) Principal coordinate analysis (PCoA) of UniFrac distances based on fecal 16S rDNA analysis from ex-germ-free Nlrp6−/− mice at different time points following spontaneous colonization, and Nlrp6−/− mice born and maintained in an SPF vivarium. Trajectory of the colonization time-course is indicated by an arrow.
(B, C) Relative distances (B) and alpha diversity rarefaction (C) between different stages of ex-germ-free Nlrp6−/− mouse colonization and SPF Nlrp6−/− mice.
(D) PCoA of fecal microbiota from ex-germ-free (GF) WT and Nlrp6−/− mice, 3 weeks following spontaneous colonization.
(E–G) PCoA (E), relative distance (F), and alpha diversity (G) of OTU abundance observed in WT and Casp1/11−/− mice with or without injection of IL-18.
(H, I) Relative distance (H) and alpha diversity (I) of microbiota between WT and Asc−/− mice with or without in-vitro addition of Ang4.
Data are expressed as mean ± SEM. * p<0.05; ** p<0.01; *** p<0.001.
Pairwise comparison was performed using Student’s t test, all other comparisons were performed using ANOVA.
Results shown are representative of two independent repeats (n=2–5).
See also Figure S3.
To determine whether the reduction of intestinal IL-18 and AMP levels is responsible for the inability of inflammasome-deficient mice to develop a normal microbial community structure, we administered recombinant IL-18 to WT, Nlrp6−/−, Asc−/−, or Caspase1/11−/− mice and determined the changes in bacterial composition. While the microbiota of WT mice was only mildly affected by the injection of IL-18, the microbiota of Nlrp6−/−, Asc−/−, and Caspase1/11−/− mice underwent marked compositional changes following IL-18 replenishment (Figures 3E, S3E, and S3F). Importantly, addition of IL-18 to Caspase1/11−/− mice partially reverted the aberrant microbiota composition on the levels of both beta- and alpha-diversity (Figures 3F and 3G). Likewise, administration of recombinant Ang4 to anaerobic in-vitro microbiome cultures of Asc−/− mice partially restored beta- and alpha-diversity (Figures 3H and 3I), corroborating Ang4 as one of the microbiome-modulating effectors downstream of IL-18. Altogether, these data indicate that the identified NLRP6-IL-18-AMP pathway is involved in de-novo determination of the normal intestinal community composition and stability, while its absence drives the development of dysbiosis.
Dominant takeover of a dysbiotic microbiota upon cohabitation through suppression of inflammasome activity
One of the hallmarks of the dysbiotic microbiota in inflammasome-deficient mice is its ability to dominantly transfer to genetically intact mice. Since the IL-18-AMP axis drives the establishment of a normal or dysbiotic microbiome, we determined whether this pathway also plays a role in the transmissibility of the aberrant microbiota composition into WT hosts. To this end, colonized WT mice were cohabitated for 4 weeks with either WT mice and designated colonized recipients (crWT(WT)), or cohabitated with Asc−/− mice and designated crWT(Asc−/−) (Figure 4A). Cohousing equilibrated the microbiota between cohabitated partners, leading to the establishment of dysbiosis in crWT(Asc−/−) mice as compared to the genetically identical crWT(WT) mice (Figure S3G). Surprisingly, cohousing with Asc−/− mice reduced colonic IL-18 levels in recipient WT mice (crWT(Asc−/−)) (Figure 4B). This IL-18 suppression was independent of genetic background and similarly developed when outbred Swiss Webster mice were used as cohoused WT partners (Figure 4C). These results suggested that the dysbiotic microbiota from inflammasome-deficient mice suppresses colonic IL-18 levels when transferred into WT recipients, regardless of their strain.
Figure 4. Dominant takeover of the dysbiotic microbiota upon cohabitation is mediated by suppression of inflammasome activity.

Colonized or germ-free WT Swiss Webster or C57Bl/6 mice were cohoused with WT mice or Asc−/− mice for 4 weeks before analysis, designated crWT(WT) and crWT(Asc−/−) when the recipients were colonized, and grWT(WT) and grWT(Asc−/−), when the recipients were germ-free.
(A) Schematic illustration demonstrating cohousing settings. WT or Asc−/− mice served as microbiota donors to either colonized WT recipients or germ-free WT recipients. In this setting, genetically identical mice harbor distinct microbiota configuration.
(B–E) IL-18 production by colon explants from WT and Asc−/− mice, as well as their respective cohousing partners (crWTs in B and C, grWTs in D and E). Recipient mice were either C57Bl/6 (B, D) or Swiss Webster (C, E).
(F, G) Immunoblot analysis (F) and quantification (G) of pro-caspase-1 (p45) and cleaved caspase-1 (p20) in colon tissue.
(H) Differential expression between grWT(WT) and grWT(Asc−/−) mice of AMPs versus all other genes. Box = interquartile range (IQR) = 25th to 75th percentile, line - median, star - mean, whiskers - IQR*1.5. Mann-Whitney U-test p<0.0001.
(I, J) Colonic transcript levels of Ang4 in WT and Asc−/− mice, as well as their respective cohousing partners (grWTs). Recipient mice were either C57Bl/6 (I) or Swiss Webster (J).
(K, L) PCoA (K) and relative distance (L) of fecal microbiota from grWT(Asc−/−) mice that were injected with either PBS or IL-18, followed by a fecal microbiota analysis. WT mice serve as controls.
Data are expressed as mean ± SEM. * p<0.05; ** p<0.01; *** p<0.001.
Pairwise comparison was performed using Student’s t test, unless stated otherwise.
Results shown are representative of 2–4 independent repeats (n=3–6 per group).
See also Figure S3.
To enable a simplified transmissibility system, we cohabitated GF WT mice with either WT mice (designated germ-free recipient, grWT(WT)) or Asc−/− mice (designated grWT(Asc−/−)) (Figure 4A). Expectedly, this led to full transfer of their respective microbiota composition and diversity to the cohoused GF partners (data not shown), so that both the WT and dysbiotic composition stably persisted in the genetically intact, previously GF mice. Strikingly, and similar to the above observation in co-housed SPF WT mice, colonic IL-18 levels in recipient grWT(Asc−/−) mice were significantly reduced as compared to grWT(WT) mice (Figure 4D). As above, this suppressive phenomenon was present regardless of the genetic background of recipient ex-GF mice (Figure 4E). These differences in colonic IL-18 protein levels did not result from alterations in transcript levels of IL-18 or NLRP6 inflammasome components (Figures S3H–S3J), suggesting that the microbiome-mediated effects were influencing inflammasome activation (‘signal II’) but not the transcription of its components (‘signal I’). Indeed, caspase-1 processing was abrogated in grWT(Asc−/−) mice as compared to grWT(WT) mice, while pro-caspase-1 levels remained similar (Figures 4F and 4G), indicating that the dysbiotic microbiota originating from Asc−/− mice suppressed intestinal inflammasome activation in the new, genetically intact host.
Reduction in inflammasome signaling and IL-18 production in grWT(Asc−/−) mice was accompanied by an alteration in the global anti-microbial transcriptional program of the colonic mucosa (Figure 4H), including a reduction in Ang4 and RELMβ production (Figures 4I, 4J, S3K, and S3L). Continuous IL-18 replenishment of recipient WT mice throughout the cohousing period partially prevented dominant transmission of the dysbiotic microbiota configuration from inflammasome-deficient mice (Figure 4K), resulting in an increased compositional similarity between IL-18-injected cohoused mice and WT controls (Figure 4L). Together, these findings demonstrate that the dysbiotic microbiota stemming from inflammasome-deficient mice modulates inflammasome signaling upon transfer to a new WT host. As a consequence, the anti-microbial milieu of the new colonization microenvironment changes to resemble the dysbiotic niche of origin and allows for community persistence of the invading microbiome.
Microbiota metabolites modulate NLRP6 inflammasome signaling and anti-microbial pathways
To determine the mechanism by which the microbiota modulates inflammasome signaling in the WT and dysbiotic states, we performed shotgun metagenomic sequencing of grWT(WT) and grWT(Asc−/−) mice and found their respective microbiota to feature a large number of differentially abundant functional KEGG modules (Figure 5A). Many of the altered pathways involved the generation of small metabolites downstream of energy and nutrient metabolism, including amino acid and polyamine metabolism (Figure 5A). These changes were consistent across vivaria (Figures S4A and S4B).
Figure 5. Microbiota metabolites modulate NLRP6 inflammasome signaling in the healthy and dysbiotic settings.

(A) Heatmap representation of differentially abundant microbial modules between grWT(WT) and grWT(Asc−/−) mice. Displayed are significant FDR-corrected genes, Mann-Whitney U-test p <0.05.
(B) Heatmap representation of metabolite abundance in grWT(WT) and grWT(Asc−/−) mice. Displayed are FDR-corrected metabolites, Mann-Whitney U-test p <0.1.
(C) Metabolite screen for induction of IL-18 production by cultured WT colon explants.
(D) IL-18 production by WT colon explants cultured with increasing doses of taurine.
(E, F) Immunoblot (E) and quantification (F) of pro-caspase-1 (p45) and cleaved caspase-1 (p20) in colon tissue following 12 hours incubation with taurine.
(G) Ang4 expression in WT explants cultured with taurine for 8 hours.
(H) IL-18 production by WT and Nlrp6−/− colon explants cultured with taurine.
(I) Metabolite screen for IL-18 suppression by cultured WT colon explants.
(J, K) IL-18 production by colon explants cultured with increasing doses of histamine (J) or spermine (K).
(L–O) Immunoblot analysis (L, N) and quantification (M, O) of pro-caspase-1 (p45) and cleaved caspase-1 (p20) in colon tissue following 12 hours incubation with either histamine (L, M) or spermine (N, O).
(P) IL-18 production by colon explants cultured with histamine and spermine, with or without addition of taurine.
Data are expressed as mean ± SEM. * p<0.05; ** p<0.01; *** p<0.001; n.s. not significant.
Pairwise comparison was performed using Student’s t test, unless stated otherwise.
Results shown are representative of 2–5 independent repeats (n=4–16 per group).
See also Figures S4 and S5.
Since metabolites are considered pivotal mediators of host-microbiota communication (Shapiro et al., 2014), we hypothesized that microbiota-modulated metabolites may take part in regulation of NLRP6 inflammasome signaling and downstream anti-microbial pathways. To this end, we performed a metabolomic screen of cecal content of grWT(WT) mice as compared to grWT(Asc−/−) mice. We found more than 70 metabolites to feature significantly differential levels between these genetically identical recipients (Figure 5B), among them amino acids and polyamines. Metabolites enriched in the grWT(WT) as compared to grWT(Asc−/−) mice, potentially being microbiota-associated inflammasome activators, included the bile acid conjugate taurine, carbohydrates, and long-chain fatty acids (Figure S4C). These were tested for inflammasome signaling modulation, by culturing with sterile WT colonic explants and measuring their effect on IL-18 induction (Figure 5C), with taurine featuring the strongest dose-dependent inductive activity (Figure 5D). Taurine depletion from the intestinal lumen of grWT(Asc−/−) mice correlated with a higher metagenomic abundance of the taurine transport system that is required for taurine uptake into bacterial cells and subsequent conjugation to secondary bile acids (Figure S4D). In colonic explants, taurine induced IL-18 secretion by triggering intestinal inflammasome activation, and enhanced caspase-1 cleavage (Figures 5E and 5F), while neither influencing IL-18 transcript levels (Figure S4E) nor cell death (Figure S4F). Consequently, taurine treatment also induced upregulation of AMPs (Figure 5G). This induction of IL-18 was dependent on NLRP6, but not on NLRP3 (Figures 5H and S4G). Together, these results suggest that taurine is a microbiota-dependent positive inflammasome modulator responsible for enhanced NLRP6 inflammasome-induced IL-18 secretion upon intestinal microbial colonization.
We next sought to identify metabolites that are involved in the microbiota-induced suppression of inflammasome signaling upon dysbiosis transfer into a WT host. To this end, we focused on metabolites enriched in grWT(Asc−/−) as compared to grWT(WT) mice (Figure S4H), and screened the most differentially abundant metabolites as potential inflammasome suppressors using the colonic explant system. The two strongest suppressors of IL-18 secretion were histamine and spermine (Figure 5I), both found to be over-represented in colons of grWT(Asc−/−) (Figure S4H). The accumulation of spermine in the lumen of grWT(Asc−/−) mice was in line with the metagenomic enrichment of the polyamine biosynthesis and transport pathways that are required for the conversion of ornithine into spermine (Figure S4I). These differences were mainly accounted for by members of the Lactobacillus genus (Figures S4J and S4K). Higher histamine levels in grWT(Asc−/−) mice were in line with both increased histidine biosynthesis and transport pathways, as well as decreased histidine degradation (Figure S5A). A number of bacterial genera contributed to these histamine-related pathways (Figures S5B–S5D). Both histamine and spermine featured concentration-dependent IL-18 suppressive functions in the colonic explant-based validation screen (Figures 5J and 5K), while no change in IL-18 mRNA or cell death was observed (Figures S5E and S5F). The metabolite putrescine, a metabolite structurally similar to spermine, equally suppressed IL-18 in colon explants system (Figure S5G).
Intestinal IL-18 suppression mediated by spermine and histamine resulted from a reduction in NLRP6 inflammasome assembly, as indicated by reduced caspase-1 processing (Figures 5L–5O) and decreased IL-18 protein levels seen in Nlrp3−/−, but not Nlrp6−/− explants (Figure S5H). Of note, metabolite-induced inflammasome suppression by either spermine or histamine could be rescued by concomitant administration of taurine (Figure 5P), suggesting that relative in-vitro contribution of metabolites determine the overall activation of the NLRP6 inflammasome and downstream cytokine production. To validate the ex-vivo colonic explant results, we employed colonic spheroids, an organ-like system amenable to long-term culture (Miyoshi and Stappenbeck, 2013). As in explants, taurine administration to WT but not to Nlrp6−/− organoids induced IL-18 secretion (Figures S5I and S5J), while not affecting organoid growth or morphology (Figure S5K). Additional supplementation with histamine and spermine diminished the taurine-mediated increase in IL-18 production (Figures S5I and S5J), while likewise not affecting spheroid growth or morphology (Figure S5K).
To provide in-vivo validation to the above metabolite activities and to further characterize their effects on the host-microbiome interface, we administered taurine to mice in the drinking water. This led to a marked activation of colonic caspase-1 (Figures 6A and 6B), IL-18 secretion (Figure 6C), and epithelial Ang4 production (Figure 6D), while not affecting IL-18 mRNA levels (Figure S6A). These metabolite-induced epithelial changes were not accompanied by alterations in major colonic lamina propria hematopoietic cell populations (Figure S6B). These and our previous results (Figure 1) suggested that taurine may constitute a microbial ‘signal II’ for the activation of the colonic NLRP6 inflammasome. To test this notion directly, we administered to GF WT mice either LPS (a known ‘signal I’), taurine, or both. Indeed, LPS, but not taurine, potently induced colonic mRNA of Il18 (Figure 6E). However, caspase-1 activation, IL-18 secretion, and subsequent Ang4 production were only induced when both LPS and taurine were co-administered (Figures 6F–6H and S6C). Taurine’s ‘signal II’ function required an intact NLRP6 inflammasome, since no IL-18 induction was observed in taurine-administered Asc−/− or Nlrp6−/− mice (Figure 6I). In contrast to taurine, in-vivo administration of histamine and spermine reduced the levels of activated caspase-1 (Figures 6J and 6K), while not altering the composition of hematopoietic cells in the lamina propria (Figure S6D). These data verify the in-vivo effects of the identified metabolites and suggest that the microbial ‘signal II’ for NLRP6 inflammasome activation has distinct molecular identities in the form of microbiota-related metabolites.
Figure 6. Microbiota metabolites are functionally involved in inflammasome modulation.

(A, B) Immunoblot analysis (A) and quantification (B) of pro-caspase-1 (p45) and cleaved caspase-1 (p20) in colon tissue obtained from WT mice drinking taurine for 14 days.
(C) IL-18 production by colon explants obtained from WT mice drinking taurine for 14 days.
(D) Ang4 expression in sorted epithelial cells (CD45−EpCAM+) obtained from WT mice drinking taurine for 14 days.
(E–H) WT germ-free mice were drinking taurine and on day 7 administered with LPS. IL-18 expression in the colon (E), immunoblot quantifications of pro-caspase-1 (p45) and cleaved caspase-1 (p20) (F), IL-18 production by colon explants (G), colonic Ang4 expression (H).
(I) IL-18 production by colon explants from WT, Asc−/− or Nlrp6−/− mice drinking taurine for 14 days.
(J, K) Immunoblot analysis (J) and quantification (K) of pro-caspase-1 (p45) and cleaved caspase-1 (p20) in colon tissue obtained from WT mice drinking histamine with or without spermine for 14 days.
(L–O) PCoA (M, N) and relative distance (L, O) of fecal microbiota from WT, Nlrp6−/− and Asc−/− mice administered with taurine.
Data are expressed as mean ± SEM. * p<0.05; ** p<0.01; *** p<0.001; n.s. not significant.
Pairwise comparison was performed using Student’s t test, unless stated otherwise.
Results shown are representative of 2–5 independent repeats (n=4–16 per group). A–D represent a single experiment.
See also Figure S6.
Administration of mice with taurine, histamine, or spermine in drinking water induced compositional changes in the intestinal microbiota (Figures 6L, 6M, S6E and S6F), which did not occur upon taurine administration to Asc−/− or Nlrp6−/− mice (Figures 6N, 6O, and S6G). Anaerobic microbiota cultures supplemented with taurine, histamine, or spermine did not feature significant compositional alterations (Figure S6H), further indicating that the metabolites do not act directly on commensal bacteria, but required signaling through the host to alter microbial ecology. Metabolite treatment also induced pronounced compositional changes in the epithelial-adherent microbiota, as determined by 16S sequencing and electron microscopy (Figures 7A, 7B, and S6I–S6K). Together, these results identify distinct microbiome-modulated metabolites as in-vitro and in-vivo regulators of the NLRP6 inflammasome and downstream control of microbiota composition.
Figure 7. Restoration of the inflammasome-antimicrobial peptide axis ameliorates colitis.

(A) PCoA of colonic mucosal-adherent microbiota from WT mice drinking taurine.
(B) Electron microscopy images of epithelial associated bacteria from WT mice drinking taurine.
(C–F) Acute DSS colitis (1.5% DSS) was induced in antibiotics-treated WT mice with or without administration of 1% taurine in the drinking water. Weight loss (C), colonoscopy severity score on day 7 (D), representative histology images on day 11 (E), pathology scoring on day 11 (F).
(G) Mortality following 2% DSS in WT mice administered with taurine (n=10 mice in each group).
(H, I) Acute DSS colitis (1.5% DSS) was induced in Asc−/− mice with or without administration of 1% taurine in the drinking water. Weight loss (H), colonoscopy severity score on day 7 (I).
(J–L) Acute DSS colitis (1.5% DSS) was induced in Asc−/− mice with or without daily administration of IL-18 for 5 days before induction of colitis. Weight loss (J), representative colonoscopy images (K), colonoscopy severity score on day 7 (L).
Data are expressed as mean ± SEM. * p<0.05; ** p<0.01; *** p<0.001; n.s. not significant.
Pairwise comparison was performed using Student’s t test, unless stated otherwise.
Results shown in panels A, C–L represent 2–5 independent repeats (n=5–10 per group).
Images shown in panel B are representative of 24 randomly taken electron micrographs.
See also Figures S6 and S7.
Restoration of the inflammasome-antimicrobial peptide axis ameliorates colitis
Finally, we sought to determine whether the identification of the metabolite-IL-18-AMP axis has functional significance in disease settings, with a focus on inflammatory bowel disease (IBD). This auto-inflammatory disorder is driven by an impaired host-microbiota microenvironment, and modeled by intestinal auto-inflammation mediated by a dysbiotic microbiota configuration in NLRP6 inflammasome-deficient mice (Elinav et al., 2011). To determine the potential of the identified metabolites to ameliorate colonic auto-inflammation, taurine was administered in the drinking water to naïve WT mice for two weeks and dextran sodium sulphate (DSS) colitis was induced. Taurine-treated mice featured improved weight loss (Figure 7C), reduced colitis severity (Figures 7D–7F and S7A (Shimizu et al., 2009; Zhao et al., 2008)), enhanced survival (Figure 7G), and improved mucosal barrier integrity as indicated by a reduced systemic FITC-dextran influx, decreased hepatic bacterial load, and sustained epithelial tight junction integrity (Figures S7B–S7E). Importantly, taurine’s beneficial effects were also observed when taurine administration was stopped before induction of DSS colitis (Figures S7F–S7I), suggesting that the microbial changes, rather than any direct anti-inflammatory effects, were responsible for the amelioration of auto-inflammation. Taurine failed to have beneficial effects when administered to WT mice treated with broad-spectrum antibiotics (Figures 7C–7F), GF WT mice (Figures S7J and S7K), or mice lacking either ASC or NLRP6 (Figures 7H, 7I, S7L, and S7M), suggesting that its activity requires an intact NLRP6 inflammasome and presence of the microbiota. In contrast to taurine, histamine treatment exacerbated DSS colitis in WT mice, an effect that was prevented when mice were concomitantly treated with antibiotics (Figures S7N and S7O), further underlining the microbiome-dependency of the metabolite-mediated modulation of inflammation.
As inflammasome-deficient mice were resistant to taurine-mediated improvement of colitis, we tested whether an intervention targeting host signaling downstream of the NLRP6 inflammasome would improve disease severity in these mice. To this aim, we systemically (i.p) administered twice-daily IL-18 or vehicle control to Asc−/− mice for two weeks, followed (after cessation of IL-18 treatment) by induction of DSS colitis. Indeed, administration of IL-18 prior to induction of colonic inflammation diminished colitis severity in Asc−/− mice, as assessed by reduced weight loss (Figure 7J) and an improved colonoscopy score (Figures 7K and 7L). Together, these results suggest that metabolite administration can modulate inflammasome signaling and downstream microbial composition, host physiology, and disease susceptibility.
Discussion
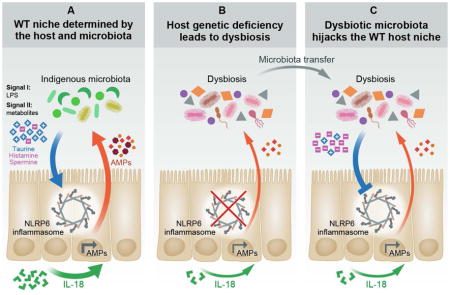
In this study, we characterize a molecular mechanism contributing to the construction of a symbiotic intestinal microenvironment. We identify a set of microbiota-modulated metabolites whose integrative activity drives NLRP6 inflammasome activation and downstream epithelial IL-18-induced AMP secretion, thereby providing favorable conditions for WT microbiota colonization. Inflammasome deficiency leads to development of an aberrant AMP program, leading to formation of a dysbiotic microbiota that acquires inflammasome-suppressive capabilities through an altered metabolite profile. Upon invasion into a WT host, hijacking and suppression of NLRP6 inflammasome signaling leads to modification of its anti-microbial landscape towards one that resembles the invading microbiota environment of origin, thereby ensuring its persistent colonization and conferring competitive advantage over the invaded host’s microbiota.
Of note, in addition to IL-18’s roles in steady state intestinal niche regulation, it features distinct and equally important functions during an inflammatory response. As demonstrated by Nowarski et al. in this issue, IL-18 drives intestinal auto-inflammation and breakdown of mucosal barrier integrity, through direct transcriptional effects exerted on goblet cells, impacting their maturation and function. Temporal and spatial integration of these IL-18-driven processes in orchestrating the mucosal immune response merit further studies.
Using an integrated metabolomics-metagenomics approach, we identify the organic acid taurine as a mucosal inflammasome activator, and the metabolites histamine and spermine as inflammasome inhibitors. Taurine is a bile acid component modulated by commensal bacteria, which plays multiple roles in regulation of immune and metabolic processes (Brestoff and Artis, 2013). Histamine and the polyamine spermine are metabolites produced by both the microbiota and the host. Inflammasome deficiency creates a microenvironment favoring the outgrowth of commensals that are capable of polyamine synthesis (Figure S4I–K), as well as differential expression of histamine production and degradation pathways (Figure S5A–D), thereby leading to an accumulation of intestinal spermine and histamine.
The downstream consequence of these shifts in intestinal metabolites is the generation of a set of integrative signals to the host epithelium. Signal I recognizes microbial presence, primarily by sensing of Toll-like recpetor ligands. Signal II may assess microbial function, by sensing of metabolite levels, indicative of commensal activity. Combinatorial metabolite levels may thus serve as a ‘rheostat’ of microbiota function in distinct environmental scenarios. A combination of high taurine and low spermine/histamine levels provides an inflammasome-activating signal, resulting in formation of a supportive host microenvironment for the WT microbiota. In contrast, dysbiotic microbiota-induced low taurine and high spermine/histamine levels provides an inflammasome suppressive signal, driving an altered host-microbial interface that supports the dysbiotic microbiota (graphical abstract). The involvement of more metabolites in these complex interactions, and the molecular mechanisms by which they influence innate immune signaling will constitute an exciting area of future research.
Equally interesting is the ability of certain dysbiotic microbiome configurations to invade a stable microbiota. ‘Inflammasome hijacking’ highlights one such mechanism, in which the microbiome is able to modulate host signaling to enable optimal colonization conditions. Microbiota-mediated intestinal secretory IgA modulation represents another such ‘host hijacking’ example (Moon et al., 2015). Our findings, demonstrating that a given microbial configuration may persist and ‘synchronize’ colonization conditions across genetically different hosts, provide the functional counterpart to the recent finding that host genetics influence microbial composition (Goodrich et al., 2014). Deciphering these and other unrecognized molecular mechanisms driving microbiota resilience may enable to elucidate the ability of certain microbiota to transmit disease susceptibility between individuals.
Finally, our study suggests that microbiota composition and function may be therapeutically exploited by methods different from, and complementary to probiotic and prebiotic approaches. As such, rather than attempting to change the microbiota itself, such ‘postbiotic’ treatment employing or manipulating downstream microbiota-produced or -modulated metabolites, may enable to control the host-microbiota interface. Harnessing the endogenous physiological forces shaping the microbiota to drive it from disease-prone towards a healthy configuration may circumvent the strong inter-individual variability in microbiota composition that severely limits effective pre- and pro-biotic treatment. Such novel therapeutic approaches may potentially present opportunities for rational design of microbiota-modulating interventions in a variety of multi factorial disorders.
Experimental Procedures
A detailed description of experimental procedures can be found in the Supplemental Information.
Mice
C57Bl/6 mice were purchased from Harlan and allowed to acclimatize for two weeks before experimentation. GF mice were bred at the Weizmann Institute GF facility. In experiments where both C57Bl/6 and outbred Swiss Webster mice were used, the respective strain is indicated. In all experiments, age- and gender-matched 8–9 week old mice were used. In cohousing experiments, age matched female mice were cohoused at 1:1 ratios for 4 weeks. Fresh stool samples were collected in tubes, snap frozen in liquid nitrogen, and stored at −80°C until DNA isolation. Broad-spectrum antibiotics included a combination of vancomycin (1 g/l), ampicillin (1 g/l), kanamycin (1 g/l), and metronidazole (1 g/l) in drinking water. In spontaneous colonization experiments, GF WT mice and GF Nlrp6−/− mice (both C57Bl/6) were removed from sterile isolators and housed in SPF conditions. Inter-facility analysis refers to: Weizmann Institute of Science (facility A) and University of Massachusetts Medical School (facility B). All experimental procedures were approved by the local IACUC.
Taxonomic Microbiota Analysis
Frozen fecal samples were processed for DNA isolation using the MoBio PowerSoil kit according to the manufacturer’s instructions and sequenced using the Illumina MiSeq platform. Data was processed using the QIIME analysis pipeline (Caporaso et al., 2010).
Statistical Analysis
Data are expressed as mean ± SEM. P-values < 0.05 were considered significant. * p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001. Pairwise comparison was performed using Student’s t test, unless specified otherwise. Comparison between multiple groups was performed using ANOVA, and Mann-Whitney U-test was used to correct for multiple comparisons.
Accession numbers
Microbial shotgun and 16S sequences have been deposited at The European Nucleotide Archive (ENA) under accession number PRJEB11300.
Supplementary Material
Acknowledgments
We thank the members of the Elinav and Segal labs, Ilana Kolodkin-Gal, and Yifat Merbel for fruitful discussions, and Ayelet Erez and Atan Gross for valuable support. We acknowledge Elena Kartvelishvily for help with electron microscopy, and Raya Eilam for assistance with immunofluorescence. We thank Millenium Pharmaceuticals for kindly providing transgenic mice. We thank Carmit Bar-Nathan for dedicated GF mouse care taking. C.A.T. is the recipient of a Boehringer Ingelheim Fonds PhD Fellowship. E.E. is supported by Yael and Rami Ungar, Israel; Leona M. and Harry B. Helmsley Charitable Trust; the Gurwin Family Fund for Scientific Research; Crown Endowment Fund for Immunological Research; estate of Jack Gitlitz; estate of Lydia Hershkovich; the Benoziyo Endowment Fund for the Advancement of Science; Adelis Foundation; John L. and Vera Schwartz, Pacific Palisades; Alan Markovitz, Canada; Cynthia Adelson, Canada; CNRS; estate of Samuel and Alwyn J. Weber; Mr. and Mrs. Donald L. Schwarz, Sherman Oaks; grants funded by the European Research Council; the German-Israel Binational foundation; the Israel Science Foundation; the Minerva Foundation; the Rising Tide foundation; and the Alon Foundation scholar award. E.E. is the incumbent of the Rina Gudinski Career Development Chair.
Footnotes
Author Contributions
M.L. and C.A.T. designed, performed, and analyzed all experiments, interpreted the results, and wrote the manuscript. D.Z. performed bioinformatics analysis and interpreted the results. L.D., J.A.M., Y.H., M.P-F., H.S., and A.C. helped with experiments. G.Z.-S., E.D., and T.K. helped with bioinformatics. A.S. performed the proteomic experiments. A.H. supervised the germ-free mouse experiments. Z.H. provided critical insights and suggestions. E.L. and R.A.F. provided critical tools and valuable insights. I.A. supervised the transcriptomic analysis and provided critical insights. E.S. co-directed the project, supervised the computational analysis, interpreted the results, and provided critical insights. E.E. conceived and directed the project, designed experiments, interpreted the results, and wrote the manuscript.
References
- Anand PK, Kanneganti TD. Targeting NLRP6 to enhance immunity against bacterial infections. Future microbiology. 2012;7:1239–1242. doi: 10.2217/fmb.12.94. [DOI] [PubMed] [Google Scholar]
- Artis D, Wang ML, Keilbaugh SA, He W, Brenes M, Swain GP, Knight PA, Donaldson DD, Lazar MA, Miller HR, et al. RELMbeta/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:13596–13600. doi: 10.1073/pnas.0404034101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brestoff JR, Artis D. Commensal bacteria at the interface of host metabolism and the immune system. Nature immunology. 2013;14:676–684. doi: 10.1038/ni.2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Liu M, Wang F, Bertin J, Nunez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. Journal of immunology. 2011;186:7187–7194. doi: 10.4049/jimmunol.1100412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, Huot L, Grandjean T, Bressenot A, Delanoye-Crespin A, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. The Journal of clinical investigation. 2013;123:700–711. doi: 10.1172/JCI62236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature. 2012;487:104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, Glickman JN, Glimcher LH. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131:33–45. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, et al. Human genetics shape the gut microbiome. Cell. 2014;159:789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremel G, Wanders A, Cedernaes J, Fagerberg L, Hallstrom B, Edlund K, Sjostedt E, Uhlen M, Ponten F. The human gastrointestinal tract-specific transcriptome and proteome as defined by RNA sequencing and antibody-based profiling. Journal of gastroenterology. 2014 doi: 10.1007/s00535-014-0958-7. [DOI] [PubMed] [Google Scholar]
- Grenier JM, Wang L, Manji GA, Huang WJ, Al-Garawi A, Kelly R, Carlson A, Merriam S, Lora JM, Briskin M, et al. Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-kappaB and caspase-1. FEBS letters. 2002;530:73–78. doi: 10.1016/s0014-5793(02)03416-6. [DOI] [PubMed] [Google Scholar]
- Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV, Stappenbeck TS, Hong CV, Gordon JI. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nature immunology. 2003;4:269–273. doi: 10.1038/ni888. [DOI] [PubMed] [Google Scholar]
- Hu B, Elinav E, Huber S, Strowig T, Hao L, Hafemann A, Jin C, Wunderlich C, Wunderlich T, Eisenbarth SC, et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:9862–9867. doi: 10.1073/pnas.1307575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikuta T, Kobayashi Y, Kitazawa M, Shiizaki K, Itano N, Noda T, Pettersson S, Poellinger L, Fujii-Kuriyama Y, Taniguchi S, et al. ASC-associated inflammation promotes cecal tumorigenesis in aryl hydrocarbon receptor-deficient mice. Carcinogenesis. 2013;34:1620–1627. doi: 10.1093/carcin/bgt083. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempster SL, Belteki G, Forhead AJ, Fowden AL, Catalano RD, Lam BY, McFarlane I, Charnock-Jones DS, Smith GC. Developmental control of the Nlrp6 inflammasome and a substrate, IL-18, in mammalian intestine. American journal of physiology Gastrointestinal and liver physiology. 2011;300:G253–263. doi: 10.1152/ajpgi.00397.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell host & microbe. 2007;2:204. doi: 10.1016/j.chom.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annual review of immunology. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, Stappenbeck TS. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nature protocols. 2013;8:2471–2482. doi: 10.1038/nprot.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon C, Baldridge MT, Wallace MA, Burnham CA, Virgin HW, Stappenbeck TS. Vertically transmitted faecal IgA levels determine extra-chromosomal phenotypic variation. Nature. 2015 doi: 10.1038/nature14139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L, Lemoine Y, Hot D, Chamaillard M. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:9601–9606. doi: 10.1073/pnas.1100981108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro H, Thaiss CA, Levy M, Elinav E. The cross talk between microbiota and the immune system: metabolites take center stage. Current opinion in immunology. 2014;30C:54–62. doi: 10.1016/j.coi.2014.07.003. [DOI] [PubMed] [Google Scholar]
- Shimizu M, Zhao Z, Ishimoto Y, Satsu H. Dietary taurine attenuates dextran sulfate sodium (DSS)-induced experimental colitis in mice. Advances in experimental medicine and biology. 2009;643:265–271. doi: 10.1007/978-0-387-75681-3_27. [DOI] [PubMed] [Google Scholar]
- Stecher B, Robbiani R, Walker AW, Westendorf AM, Barthel M, Kremer M, Chaffron S, Macpherson AJ, Buer J, Parkhill J, et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS biology. 2007;5:2177–2189. doi: 10.1371/journal.pbio.0050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaiss CA, Elinav E. Exploring new horizons in microbiome research. Cell host & microbe. 2014;15:662–667. doi: 10.1016/j.chom.2014.05.016. [DOI] [PubMed] [Google Scholar]
- Tsuji S, Uehori J, Matsumoto M, Suzuki Y, Matsuhisa A, Toyoshima K, Seya T. Human intelectin is a novel soluble lectin that recognizes galactofuranose in carbohydrate chains of bacterial cell wall. The Journal of biological chemistry. 2001;276:23456–23463. doi: 10.1074/jbc.M103162200. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM, Frankel G, Levy M, Katz MN, Philbrick WM, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell. 2014;156:1045–1059. doi: 10.1016/j.cell.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Satsu H, Fujisawa M, Hori M, Ishimoto Y, Totsuka M, Nambu A, Kakuta S, Ozaki H, Shimizu M. Attenuation by dietary taurine of dextran sulfate sodium-induced colitis in mice and of THP-1-induced damage to intestinal Caco-2 cell monolayers. Amino acids. 2008;35:217–224. doi: 10.1007/s00726-007-0562-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.