

Abstract
CENP‐R is a component of the CENP‐O complex, including CENP‐O, CENP‐P, CENP‐Q, CENP‐R, and CENP‐U and is constitutively localized to kinetochores throughout the cell cycle in vertebrates. CENP‐R‐deficient chicken DT40 cells are viable and show a very minor effect on mitosis. To investigate the functional roles of CENP‐R in vivo, we generated CENP‐R‐deficient mice (Cenp‐r −/−). Mice heterozygous or homozygous for Cenp‐r null mutation are viable and healthy, with no apparent defect in growth and morphology, indicating Cenp‐r is not essential for normal development. Accordingly, to investigate the role of the Cenp‐r gene in skin carcinogenesis, we subjected Cenp‐r −/− mice to the 7,12‐dimethylbenz(a)anthracene (DMBA)/TPA chemical carcinogenesis protocol and monitored tumor development. As a result, Cenp‐r −/− mice initially developed significantly more papillomas than control wild‐type mice. However, papillomas in Cenp‐r −/− mice showed a decrease of proliferative cells and an increase of apoptotic cells. As a result, they did not grow bigger and some papillomas showed substantial regression. Furthermore, papillomas in Cenp‐r −/− mice showed lower frequency of malignant conversion to squamous cell carcinomas. These results indicate Cenp‐r functions bilaterally in cancer development: during early developmental stages, Cenp‐r functions as a tumor suppressor, but during the expansion and progression of papillomas it functions as a tumor‐promoting factor.
Keywords: CENP, malignant conversion, mouse model, papilloma, two‐stage skin carcinogenesis
The centromere is a critical chromosomal region for faithful chromosome segregation. The mitotic/meiotic kinetochore structure is assembled on the centromere to capture microtubules correctly for equal segregation of sister chromatids.1, 2 The kinetochore is a large protein complex and contains more than 100 proteins. The kinetochore protein complex is roughly divided into two groups. One group constitutively localizes to centromeres throughout the cell cycle, and is known as the constitutive centromere‐associated network (CCAN). CCAN contains at least 16 proteins and is further divided into subcomplexes.1, 2 The other group, called the KMN (KNL1‐Mis12 complex‐Ndc80 complex) network, is targeted into CCAN during late G2 to establish the complete kinetochore structure in mitosis. CCAN associates with centromere‐specific chromatin, which is marked by centromere‐specific histone H3 variant CENP‐A. CENP‐A gene disruption results in severe mitotic defects in various organisms3, 4, 5, 6 and Cenp‐a (symbol for mouse CENP‐A) null mutation causes early embryonic cell death,7 indicating that CENP‐A plays an important role in chromosome segregation and cell survival. Recently, overexpression of CENP‐A was found in several human malignancies, including hepatocellular carcinoma,8, 9 breast cancer,10 and ovarian cancer.11 Prognostic significance of CENP‐A was described for various cancers. For instance, Qiu et al.11 reported that CENP‐A is upregulated in epithelial ovarian cancer and predicts poor outcomes in patients with this disease. CENP‐A also shows a poor prognostic impact in estrogen receptor‐positive breast cancer. In addition to CENP‐A, CCAN proteins are also associated with tumor promotion, including CENP‐M, CENP‐U/50, and CENP‐W. Like CENP‐A, CENP‐H was found to be overexpressed in colorectal cancers.12, 13
CCAN proteins are distributed in several functional groups as follows: CENP‐C, CENP‐H/I/K/M, CENP‐L/M, CENP‐O/P/Q/R/U, CENP‐T/W, CENP‐S/X.14, 15, 16 Among CCAN proteins, we focused on CENP‐R in the present study, which forms a complex with CENP‐O/P/Q/U (CENP‐O complex).17, 18, 19 We previously demonstrated that CENP‐O/P/Q/U‐deficient chicken DT40 cells are viable, but show defects during the process of recovery from spindle damage.17 However, CENP‐R‐deficient DT40 cells showed only subtle defects in the recovery process from spindle damage.17 In addition, CENP‐O/P/Q/U localization is not changed in CENP‐R‐deficient cells, whereas CENP‐R localization was abolished in CENP‐O/P/Q/U‐deficient DT40 cells, suggesting that CENP‐R functions downstream of all other proteins in the CENP‐O complex.17 Therefore, the functional role of CENP‐R seems distinct from other members of the CENP‐O complex and CENP‐R may have other roles from other CCAN (CENP‐O complex members) in tumor promotion/suppression.
To gain insight into the role of Cenp‐r in vivo, especially in the process of carcinogenesis, we generated Cenp‐r‐deficient mice and analyzed its function in normal development and skin carcinogenesis, using a two‐stage carcinogenesis mouse model.
Pathology of the two‐stage chemically induced skin carcinogenesis mouse model is almost identical to the development of human skin cancers and thus offers an ideal model to study skin cancer initiation and growth.20, 21 In the first step of the chemically induced carcinogenesis protocol, mice are treated with a low dose of the mutagen 7,12‐dimethylbenz(a)anthracene (DMBA) to start tumor development. This first chemical treatment step leads to “tumor initiation”. In the second step, mice are treated continuously with TPA to stimulate epidermal tumor proliferation. This second chemical treatment step influences “tumor promotion”. During tumor promotion, benign tumors, known as papillomas, are thought to arise by additional mutations caused by the TPA chemical treatment. After prolonged treatment (~20 weeks), some of the papillomas will progress into carcinogenic tumors, such as squamous cell carcinomas (SCC). The role of various genes and cell‐signaling pathways involved in skin tumor development can be explored in this two‐stage skin carcinogenesis model by the use of genetically engineered mouse models.22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32
In the present study, we show, for the first time, the crucial function of Cenp‐r in the two‐stage carcinogenesis model. We present findings that demonstrate the tumor suppressive role of Cenp‐r in papilloma development and an oncogenic role in progression and malignant conversion.
Materials and Methods
Mouse strains
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Ministry of Education, Culture, Sports, Science, and Technology of Japan. All mice were maintained under the guidelines for animal experiments at the National Institute of Genetics and the carcinogenesis protocol was approved by the Committee on the Ethics of Animal Experiments of Chiba Cancer Center (Permit Number: 13‐18). All efforts were made to minimize suffering. The C57BL/6 strain was used as recipients for targeted ES cells and as the background strain in this study. After establishing Cenp‐r +/− heterozygous mice by crossing with C57BL/6, Cenp‐r −/− mice were generated by Cenp‐r +/− heterozygous intercross.
Skin carcinogenesis
7,12‐Dimethylbenz(a)anthracene (DMBA) was purchased from Sigma‐Aldrich, Merck Milipore, Billerica, MA, USA, and TPA was purchased from Calbiochem, Merck Milipore, Billerica, MA, USA. DMBA is used as a carcinogen and TPA as a promoter. A total of 30 Cenp‐r −/− mice and 11 Cenp‐r +/+ mice were treated according to the modified two‐stage carcinogenesis protocol. At 8–10 weeks of age, the backs of the mice were carefully shaved with an electric clipper. Two days after shaving, DMBA (25 μg per mouse in 200 μL acetone) was applied to shaved dorsal back skin. Three days after the first DMBA treatment, TPA (10 μg per mouse in 200 μL acetone) was applied. After four rounds of this single DMBA and TPA treatment, the mice were treated with TPA twice weekly for 20 weeks. Papilloma number and size (mm in diameter) of each papilloma were recorded from 7 weeks up to 20 weeks, and carcinoma development was monitored up to 38 weeks post‐TPA treatment.
Cell culture
Mouse ES cells were cultured in Dulbecco's modified medium supplemented with 15% fetal calf serum, 0.1 mM non‐essential amino acids solution (Gibco, Thermofisher, Waltham, MA, USA), 500 or 1000 U/mL Leukemia inhibitory factor (ESGRO) (Merck Milipore, Billerica, MA, USA), 100 μM beta‐mercaptoethanol, and penicillin–streptomycin (Gibco). ES cells were cultured on mitotically inactivated embryonic feeder cells in gelatin‐coated dishes. The ES cells (129/terSV/J1ES) and G418‐resistant feeder cells were used to generate CENP‐U‐deficient mice. Mouse embryonic fibroblast (MEF) cells were prepared from day E14.5 embryos from Cenp‐r +/+, Cenp‐r +/− and Cenp‐r −/− mice. The MEF cells were cultured in MEF medium and were used for growth rate analysis or immunocytochemistry after the third passage.
Immunofluorescence
Dorsal back skin or tumors were fixed in 4% paraformaldehyde at 4°C overnight. Frozen skin and tumor embedded in optimum cutting temperature compound (Sakura Finetek, Tokyo, Japan) were cut into 10‐μm sections. In contrast, dehydrated samples were embedded in paraffin and sectioned as 10‐μm slices, which were stained with H&E. Endogenous peroxidase activity in the specimens was blocked by treatment with 0.3% H2O2 and samples were then rinsed with PBS. Sections were incubated with primary antibodies diluted in blocking buffer overnight at 4°C. Rabbit anti‐keratin 14 (1:500; Covance Research, Denver, PA, USA) and rat anti‐Ki‐67 (1:200; DakoCytomation, Agilent, Santa Clara, CA, USA) were used as primary antibodies. Secondary antibodies were Alexa Fluor 488‐conjugated anti‐rat antibody (1:100; Molecular Probes, Invitrogen, Thermofisher, Waltham, MA, USA) and Alexa Fluor 568‐conjugated anti‐rabbit antibody (1:100; Molecular Probes, Invitrogen). Nuclei were counterstained with Hard Set Mounting Medium with DAPI (Vector laboratories, Burlingame, CA, USA). All fluorescence images were obtained with a Leica TCS SPE confocal microscope equipped with a DMI4000B (Leica Microsystems, Wetzlar, Germany) (10 × /0.40, 20 × /0.70, and 40 × /1.25 oil immersion objective).
MEF cells were placed on slides using a cytocentrifuge and fixed in 3% paraformaldehyde in 250 mM HEPES at room temperature for 15 min or with chilled methanol at −20°C for 20 min. Then, samples were permeabilized with 0.5% NP‐40 in PBS at room temperature for 15 min and incubated with an appropriate primary antibody diluted with 0.5% BSA at 37°C for 1 h or at 4°C overnight. After washing, FITC‐conjugated secondary antibodies diluted with 0.5% BSA/PBS were used. Nuclei or chromosomes were counterstained with DAPI. Affinity‐purified rabbit polyclonal antibodies were generated against recombinant chicken CENP‐R as described previously.33
TUNEL staining
Apoptotic scores were obtained with a TUNEL assay using an in situ Apoptosis Detection Kit (Takara, Kusatsu, Japan). Briefly, skin tumor sections were deparaffinized, rehydrated, and digested with protein K and labeled with TUNEL reaction mixture for 90 min at 37°C. Sections were screened for positive nuclei under a light microscope, and 10 random tumor cell fields were counted for every papilloma under 400× magnification. Data from all fields and all papillomas were pooled to obtain the apoptotic index, which is the percentage of TUNEL‐positive cells in total cells manually counted in five randomly selected fields and compared between Cenp‐r −/− and Cenp‐r +/+ mice.
Classification between papillomas and carcinomas
The majority of papillomas were determined by visual inspection. Papillomas appeared as outgrowths on the dorsal skin of mice. Some of the papillomas began to convert to carcinomas by becoming flatter on the skin and penetrating deeper into the dermis. They were normally easily distinguishable from one another. However, in some intermediate‐type tumors, we prepared a paraffin section and confirmed the histology.
Statistical analysis
Statistical significance was calculated by the unpaired two‐tailed Student's t‐test or 2 × 2 chi squared test (Fisher's test). P‐value <0.05 was considered statistically significant and a P‐value <0.01 was considered highly statistically significant.
Results
Generation of Cenp‐r knockout mice
We first generated Cenp‐r null mice (Fig. 1). We prepared a gene‐targeting construct, which disrupted exon 4 of the Cenp‐r gene (Fig. 1a) and introduced it into mouse ES cells. This construct contains lox‐p sequences in the flanking region of Cenp‐r exon 4 (Fig. 1a). We picked up clones in which one allele was targeted by the construct (Fig. 1a) and produced mice having this allele (Fig. 1b; Fig. S1a). Then, exon 4 was deleted by crossing with mice expressing Cre recombinase. We isolated mice with Cenp‐r +/− alleles and finally generated Cenp‐r −/− alleles by intercross. We confirmed exon 4 disruption by Southern blot analysis and observed that the CENP‐R protein was not detected in these cells based on immunofluorescence and Western blot analyses with anti‐CENP‐R antibody (Fig. 1c; Fig. S1b).
Figure 1.
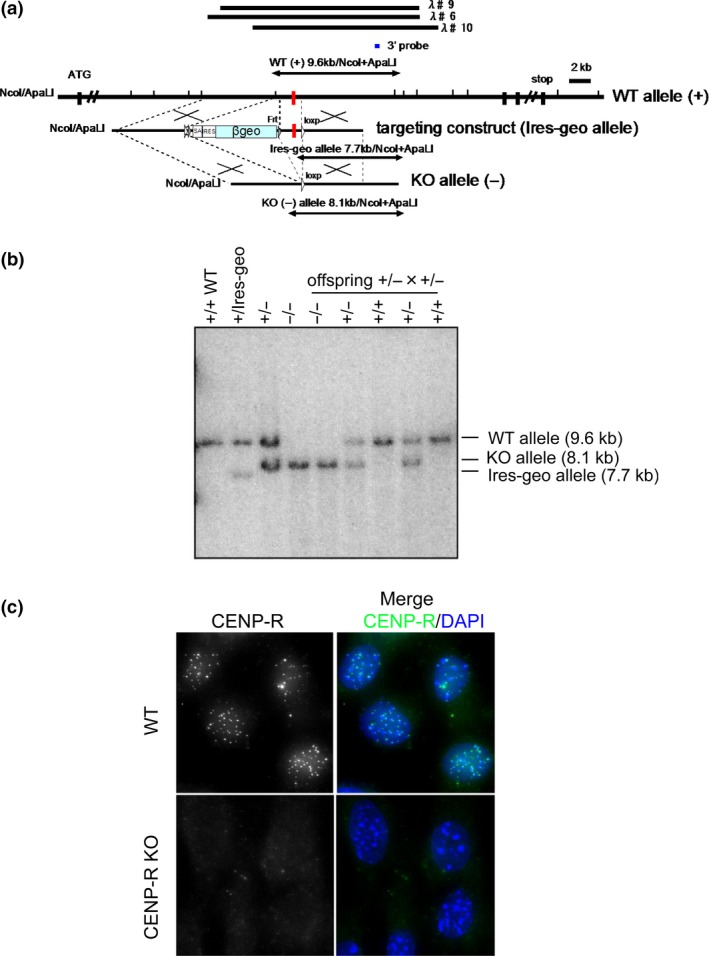
Generation of Cenp‐r −/− mice. (a) Targeting strategy to generate mice lacking CENP‐R. λ♯6, 9, 10 indicate three genomic clones used as starting materials. Black boxes indicate the positions of exons. Short vertical lines indicate NcoI and Apa LI sites. Red boxes indicate exon 4. Position of the probe used for Southern hybridization is indicated. CENP‐R allele was targeted using the vector to disrupt exon 4. Targeting construct shows the selection marker βgeo flanked by loxP is inserted into the genomic region in the 5′ end of exon4. (b) Southern blot analysis of NcoI/Apa LI‐digested genomic DNA from mice at each step using 3′ probe shown in part (a). (c) Immunofluorescence analysis of mouse embryonic fibroblast (MEF) cells (Cenp‐r +/+ and Cenp‐r −/−) using anti CENP‐R antibody.
Disruption of Cenp‐r is not essential for embryogenesis
Mice with the heterozygous Cenp‐r allele: Cenp‐r +/− were mated and the offspring were genotyped (Table1). Animals derived from these crosses displayed all three expected genotypes, indicating that homozygous null animals are viable (Table 2). The observed distribution of genotypes was not significantly different from the expected values for crosses between heterozygous animals. In addition, crosses between Cenp‐r null males and females demonstrated that they have normal fertility and produce the expected proportion of male and female progeny. From 40 independent crosses between null males and females, a total of 100% progeny was obtained of which 45% were male and 55% were female (Table2).
Table 1.
Frequency of possible genotyping from crosses between CENP‐R heterozygous animals
| WT | +/– | –/– | Total | |
|---|---|---|---|---|
| Offspring | ||||
| Male | 13 | 23 | 15 | 51 |
| Female | 16 | 26 | 13 | 55 |
| Total (%) | 29 (27) | 49 (46) | 28 (26) | 106 (100) |
Table 2.
Sex ratio of offspring from crosses between CENP‐R null male and female
| Male | Female | Total | |
|---|---|---|---|
| Offspring (%) | 18 (45) | 22 (55) | 40 (100) |
Cenp‐r functions as a tumor suppressor in papilloma development
Although Cenp‐r deficiency has no effect on normal development, it may have a critical role in carcinogenesis. To investigate the role of the Cenp‐r gene in skin carcinogenesis, we subjected 30 Cenp‐r −/− mice and 15 wild‐type control mice to the DMBA/TPA chemical carcinogenesis protocol and monitored their tumor development for a period of 38 weeks. In the standard DMBA/TPA protocol, mice were treated once with DMBA, followed by repeated application of TPA. However, as the Cenp‐r −/− mice were in a C57BL6/J background, which is known to be resistant to tumor induction by DMBA/TPA chemical treatment,34 we modified the standard DMBA/TPA protocol and included four additional rounds of DMBA‐TPA treatment to overcome the chemical resistance of C57BL6/J mice.35
Papillomas are visually identified by their cauliflower‐like projections that may appear white or normal‐colored and may be pedunculated or sessile. Carcinomas are distinguished from papillomas by their flattened appearance. For each mouse, we documented the number of papillomas present for 24 weeks after tumor initiation (Fig. 2a) as well as the number of carcinomas present for 38 weeks after tumor promotion. In addition, the size of each papilloma was measured and recorded (Fig. 2b–d). Between 8 and 20 weeks after the initial DMBA treatment, we found that Cenp‐r −/− mice developed significantly more papillomas than control mice (Fig. 2a, P = 0.0002 at 12 weeks). We found that within those 20 weeks, some of the papillomas formed in Cenp‐r −/− mice regressed (Fig. 2a,b,e,f). Regression of papillomas was much more notable after termination of the 20th week of TPA treatments (Fig. 2a,b,e,f). Cenp‐r +/+ mice developed more papillomas than Cenp‐r −/− mice at 24 weeks after initiation, although the different was not statistically significant (Fig. 2a, P = 0.2907). In addition, Cenp‐r +/+ mice developed 1.09 ± 1.22 papillomas >6 mm in diameter at 24 weeks after initiation (Fig. 2d). In contrast, Cenp‐r −/− mice developed 0.20 ± 0.41 papillomas >6 mm (Fig. 2d), indicating that Cenp‐r functions as a tumor suppressor in papilloma development. However, as an important note, Cenp‐r supports tumor growth during the expansions of papillomas.
Figure 2.
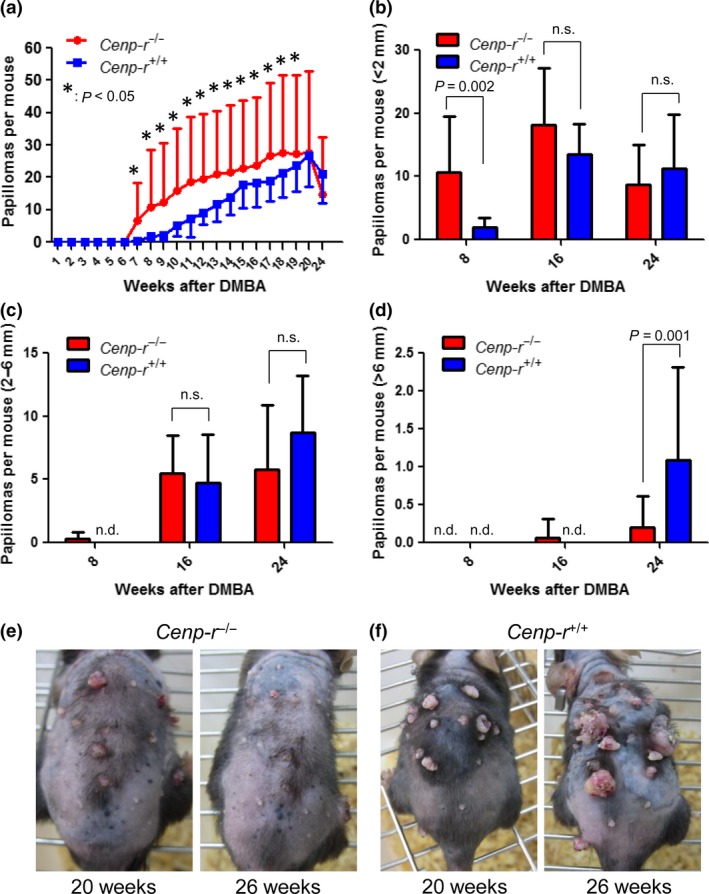
7,12‐Dimethylbenz(a)anthracene (DMBA)/TPA‐induced papilloma development in Cenp‐r‐deficient mice. (a) Comparison of DMBA/TPA‐induced papilloma numbers per mouse between Cenp‐r −/− mice (n = 30) (shown in red) and Cenp‐r +/+ mice (n = 11) (shown in blue). Error bars are SD. P‐value was calculated for papilloma number at 7 weeks up to 20 weeks by t‐test. (b) Number of papillomas <2 mm per mouse. Red bars represent Cenp‐r −/− mice. Blue bars represent Cenp‐r +/+ mice. (c) Number of papillomas 2–6 mm per mouse. Red bars represent Cenp‐r −/− mice. Blue bars represent Cenp‐r +/+ mice. (d) Number of papillomas >6 mm per mouse. Red bars represent Cenp‐r −/− mice. Blue bars represent Cenp‐r +/+ mice. (e) Photos of representative mice on TPA treatment. Dorsal back skin of a Cenp‐r −/− mouse at 20 weeks, a Cenp‐r −/− mouse at 26 weeks after initiation from left to right. (f) Photos of representative mice on TPA treatment. Dorsal back skin of a Cenp‐r +/+ mouse at 20 weeks, a Cenp‐r +/+ mouse at 26 weeks after initiation from left to right. n.s., not significant.
Cenp‐r deficiency decreases proliferative cells and increases apoptotic cells in papillomas
To investigate the mechanism by which Cenp‐r supports papilloma development, we analyzed papillomas in Cenp‐r −/− and Cenp‐r +/+ by standard histology methods. H&E staining of papillomas showed no significant morphological differences (Fig. S2). We then carried out immunostaining with the cell proliferation marker Ki‐67 to investigate the cell proliferation capacities of papillomas. We observed fewer Ki‐67‐positive cells in papillomas of Cenpr −/− mice (n = 9) than in papillomas of control mice (n = 10) (P = 0.0269, t‐test) (Fig. 3a–c). These observations showed that papillomas from Cenp‐r −/− mice grow more slowly, indicating that Cenp‐r supports the growth of papillomas by maintaining cell proliferation. We then conducted TUNEL staining to investigate apoptotic cells in papillomas. As a result, we observed more apoptotic cells in papillomas of Cenp‐r −/− mice (n = 5) than in papillomas of the Cenp‐r +/+ mice (n = 5) (P = 0.0316, t‐test) (Fig. 3d–f). These findings are consistent with previous reports, which showed that knockdown of some CCAN proteins caused apoptosis through long mitotic arrest.36
Figure 3.
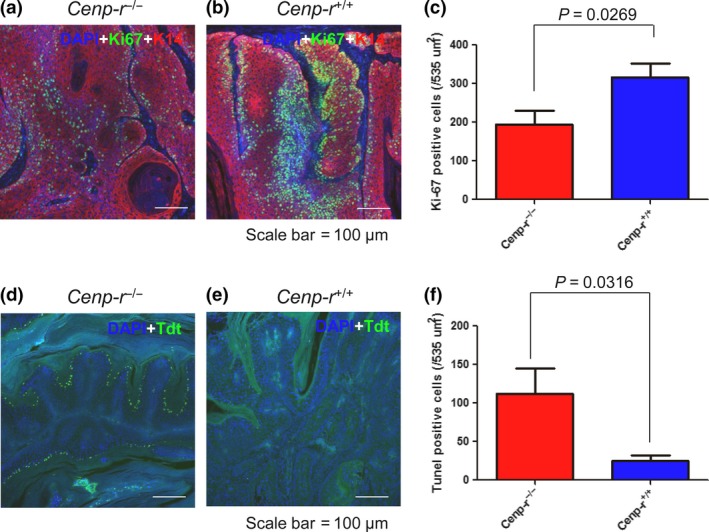
Cenp‐r‐deficient mice show a decrease of proliferative cells and an increase of apoptotic cells in papillomas. (a) Representative double‐immunostaining pattern of Ki‐67 (green) and K14 (red) in a papilloma from a Cenp‐r −/− mouse. Cells were counterstained with DAPI (blue). (b) Representative double‐immunostaining pattern of Ki‐67 (green) and K14 (red) in a papilloma from a control Cenp‐r +/+ mouse. Cells were counterstained with DAPI (blue). (c) Numbers of Ki67‐positive cells per 535 μm2 in papillomas of control Cenp‐r −/− (n = 9) and Cenp‐r +/+ (n = 10) mice. Error bars are SD. Scale bars, 100 μm. P‐value was calculated for Ki‐67‐positive cell number by t‐test. (d) Representative TUNEL staining pattern of Tdt (green) in a papilloma from a Cenp‐r −/− mouse. Cells were counterstained with DAPI (blue). (e) Representative TUNEL staining pattern of Tdt (green) in a papilloma from a control Cenp‐r +/+ mouse. Cells were counterstained with DAPI (blue). (f) Numbers of TUNEL‐positive cells per 535 μm2 in papillomas of control Cenp‐r −/− (n = 5) and Cenp‐r +/+ (n = 5) mice. Error bars are SD. Scale bars, 100 μm. P‐value was calculated for TUNEL‐positive cell number by t‐test.
Cenp‐r is required for malignant conversion
To investigate the effect of Cenp‐r deletion on carcinoma development, we monitored 30 chemically treated Cenp‐r −/− mice and 15 chemically treated control mice up to 35 weeks after tumor initiation. We observed that Cenp‐r −/− mice have a lower incidence of carcinoma development as well as a later onset of carcinoma development compared with control mice (Fig. 4).
Figure 4.
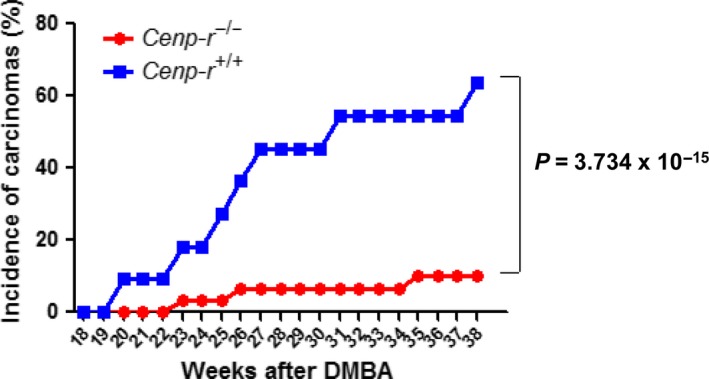
Disruption of Cenp‐r inhibits malignant conversion. Comparison of 7,12‐dimethylbenz(a)anthracene (DMBA)/TPA‐induced carcinoma incidence between Cenp‐r −/− (n = 30) (shown in red) and Cenp‐r +/+ (n = 11) (shown in blue). P‐value was calculated for carcinoma incidence at 38 weeks by Fisher's test.
We observed that the Cenp‐r −/− mice developed the first carcinoma at 23 weeks after initiation, whereas control Cenp‐r +/+ mice developed the first carcinoma at 20 weeks. In addition, we observed that less than 10 percent of Cenp‐r −/− mice had developed carcinoma at 38 weeks, whereas about 60 percent of control mice had done so. This difference between Cenp‐r −/− and Cenp‐r +/+ was statistically significant (P = 3.734 × 10−15, Fisher's test) (Fig. 4). Our investigations of Cenp‐r in tumorigenesis indicate that, in addition to the role in papilloma development and growth, Cenp‐r also functions to support conversion of benign papillomas into malignant tumors (Fig. 5).
Figure 5.
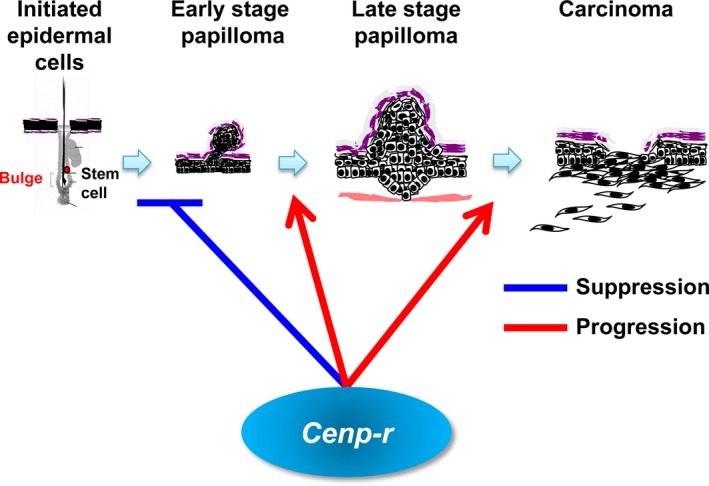
Schematic drawing of the functions of Cenp‐r in the process of skin carcinogenesis. Cenp‐r negatively regulates papilloma development. In contrast, Cenp‐r positively regulates papilloma growth as well as malignant conversion from papilloma into carcinoma.
Discussion
In the present study, we demonstrated that Cenp‐r −/− mice were viable and developed more papillomas than control mice in the two‐stage skin carcinogenesis experiments. However, those papillomas in Cenp‐r −/− mice did not grow larger and showed a lower frequency of malignant conversion to squamous cell carcinomas. This indicates that Cenp‐r functions bilaterally in cancer development: during early developmental stages, Cenp‐r functions as a tumor suppressor, but during the expansion and progression of papillomas it functions as a tumor‐promoting factor (Fig. 5).
Complete kinetochore structure generally ensures faithful chromosome segregation and normal cell proliferation. Therefore, Cenp‐r must function as a tumor suppressor in the early stage of papilloma development, because we postulate Cenp‐r‐deficient cells do not form a complete kinetochore structure. We have previously demonstrated that CENP‐R‐deficient DT40 cells only showed subtle defects in the process of recovery from spindle damage.18 However, compared with wild‐type cells, CENP‐R‐deficient cells had defects, suggesting that the kinetochore is not fully functional in these cells.
Interestingly, once the tumor was created, Cenp‐r promotes malignant conversion. We speculate that once the cell environment adapts to cancer, the complete kinetochore must promote cell proliferation. The full kinetochore structure may change its role depending on the cell environment. It usually suppresses transformation to cancer cells, but once cells adapt to cancer environments, it promotes growth. Consistent with our idea, CENP‐E deficiency showed similar results.37 Cells and mice with reduced levels of CENP‐E were shown to develop aneuploidy and chromosomal instability in vitro and in vivo, indicating that full kinetochore structure with CENP‐E suppresses aneuploidy and chromosomal instability, which cause cancer formation.38 However, in cells with an increased rate of aneuploidy, CENP‐E induced tumors,37 indicating that Cenp‐e functions both oncogenically and as a tumor suppressor in cancer development like Cenp‐r.
The phenotype observed in Cenp‐e and Cenp‐r‐deficient mice is also found in transforming growth factor alpha (TGF‐α) transgenic mice. Mice that overexpress TGF‐α in their epidermis develop papillomas on wounding or TPA treatment.39, 40 However, the papillomas from the TGF‐α transgenic mice also have the tendency to regress and were never observed to progress to malignancy.41 TGF‐α is the ligand for the epidermal growth factor (EGF) receptor, which activates Ras through the guanine nucleotide exchange factor SOS.42 Hras is the most important molecule in the two‐stage DMBA/TPA model.21 A specific mutation in codon 61 of Hras (Q61L) was identified as the initiating mutation in the two‐stage mouse skin model.21 However, the papillomas that arise in TGF‐α transgenic mice do not have a mutation in Hras, suggesting that activation of the Ras pathway through TGF‐α overexpression is sufficient to induce skin tumor initiation in the absence of Hras mutational activation, but is not sufficient to progress to malignancy. It might be expected that Cenp‐r deficiency affects the expression of TGF‐α and Hras indirectly in the initiated cells.
Although genotoxic and non‐genotoxic carcinogens alone can cause aneuploidy, tumor promoters such as TPA are especially potent inducers of chromosomal damage, in part because of the release of reactive oxygen species (ROS) such as H2O2.43 Damaged cells can be removed by apoptosis.44 During the promotion phase, the papillomas that arise frequently show trisomy of chromosome 7,45 where the Hras gene is located. Further, the duplicated chromosome is invariably that which bears the mutant Hras allele, which suggests a strong preference for amplification of the mutant Hras allele.46, 47 Cenp‐r is not essential in normal cells. However, it is obviously required for papilloma growth, indicating that papilloma cells in Cenp‐r −/− mice do not tolerate the stress exposed to tumor cells. The papilloma cells in Cenp‐r −/− mice showed mitotic defects followed by apoptosis (Fig. 3), indicating that Cenp‐r is involved in tumor cell‐specific chromosome separation, although the precise molecular mechanisms are yet to be elucidated.
We have uncovered a novel role of Cenp‐r in the two‐stage carcinogenesis model. Cenp‐r functions as a tumor suppressor during the early papillomagenesis stage, but during the expansion and the progression stage it functions as an oncogene. Cenp‐r is not essential in normal development and tissues, and it functions specifically in tumors. This indicates that inhibition of CENP‐R has potential as a novel therapeutic strategy for cancer without side‐effects.
Disclosure Statement
Authors declare no conflicts of interest for this article.
Supporting information
Fig. S1. Genotyping and western blot analysis. (a) PCR primers used for genotyping Cenp‐r +/+, Cenp‐r +/− and Cenp‐r −/− mice. (b) PCR genotyping of mice displaying the three potential genotypes using the three primer sets shown in part (a). (c) Western blot analysis of MEF cells (Cenp‐r +/+, Cenp‐r +/− and Cenp‐r −/−) using anti CENP‐R antibody.
Fig. S2. Histology of tumors. H&E staining of papillomas and carcinomas from Cenp‐r +/+ and Cenp‐r −/− mice. Scale bar, 100 μm.
Cancer Sci 108 (2017) 2142–2148
References
- 1. Fukagawa T, Earnshaw WC. The centromere: chromatin foundation for the kinetochore machinery. Dev Cell 2014; 30: 498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McKinley KL, Cheeseman IM. The molecular basis for centromere identity and function. Nat Rev Mol Cell Biol 2016; 17: 16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mendiburo MJ, Padeken J, Fülöp S, Schepers A, Heun P. Drosophila CENH3 is sufficient for centromere formation. Science 2011; 334: 686–90. [DOI] [PubMed] [Google Scholar]
- 4. Guse A, Carroll CW, Moree B, Fuller CJ, Straight AF. In vitro centromere and kinetochore assembly on defined chromatin templates. Nature 2011; 477: 354–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Foltz DR1, Jansen LE, Black BE et al The human CENP‐A centromeric nucleosome‐associated complex. Nat Cell Biol 2006; 8: 458–69. [DOI] [PubMed] [Google Scholar]
- 6. Blower MD, Daigle T, Kaufman T, Karpen GH. Drosophila CENP‐A mutations cause a BubR1‐dependent early mitotic delay without normal localization of kinetochore components. PLoS Genet 2006; 2: e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Howman EV, Fowler KJ, Newson AJ et al Early disruption of centromeric chromatin organization in centromere protein A (Cenpa) null mice. Proc Natl Acad Sci USA 2000; 97: 1148–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wonsey DR, Follettie MT. Loss of the forkhead transcription factor FoxM1 causes centrosome amplification and mitotic catastrophe. Cancer Res 2005; 65: 5181–9. [DOI] [PubMed] [Google Scholar]
- 9. Li Y, Zhu Z, Zhang S et al ShRNA‐targeted centromere protein A inhibits hepatocellular carcinoma growth. PLoS One 2011; 6: e17794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McGovern SL, Qi Y, Pusztai L, Symmans WF, Buchholz TA. Centromere protein‐A, an essential centromere protein, is a prognostic marker for relapse in estrogen receptor‐positive breast cancer. Breast Cancer Res 2012; 14: R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qiu JJ, Guo JJ, Lv TJ et al Prognostic value of centromere protein‐A expression in patients with epithelial ovarian cancer. Tumour Biol 2013; 34: 2791–5. [DOI] [PubMed] [Google Scholar]
- 12. Tomonaga T, Matsushita K, Yamaguchi S et al Overexpression and mistargeting of centromere protein‐A in human primary colorectal cancer. Cancer Res 2003; 63: 3511–6. [PubMed] [Google Scholar]
- 13. Tomonaga T, Matsushita K, Ishibashi M et al Centromere protein H is up‐regulated in primary human colorectal cancer and its overexpression induces aneuploidy. Cancer Res 2005; 65: 4683–9. [DOI] [PubMed] [Google Scholar]
- 14. Perpelescu M, Fukagawa T. The ABCs of CENPs. Chromosoma 2011; 120: 425–46. [DOI] [PubMed] [Google Scholar]
- 15. Okada M, Cheeseman IM, Hori T et al The CENP‐H‐I complex is required for the efficient incorporation of newly synthesized CENP‐A into centromeres. Nat Cell Biol 2006; 8: 446–57. [DOI] [PubMed] [Google Scholar]
- 16. Weir JR, Faesen AC, Klare K et al Insights from biochemical reconstitution into the architecture of human kinetochores. Nature 2016; 537: 249–53. [DOI] [PubMed] [Google Scholar]
- 17. Hori T, Okada M, Maenaka K, Fukagawa T. CENP‐O class proteins form a stable complex and are required for proper kinetochore function. Mol Biol Cell 2008; 19: 843–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kagawa N, Hori T, Hoki Y et al The CENP‐O complex requirement varies among different cell types. Chromosome Res 2014; 22: 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Samejima I, Spanos C, Alves Fde L et al Whole‐proteome genetic analysis of dependencies in assembly of a vertebrate kinetochore. J Cell Biol 2015; 211: 1141–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kemp CJ. Multistep skin cancer in mice as a model to study the evolution of cancer cells. Semin Cancer Biol 2005; 15: 460–73. [DOI] [PubMed] [Google Scholar]
- 21. Abel EL, Angel JM, Kiguchi K, DiGiovanni J. Multi‐stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc 2009; 4: 1350–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wilker E, Lu J, Rho O et al Role of PI3K/Akt signaling in insulin‐like growth factor‐1 (IGF‐1) skin tumor promotion. Mol Carcinog 2005; 44: 137–45. [DOI] [PubMed] [Google Scholar]
- 23. Amornphimoltham P, Leelahavanichkul K, Molinolo A, Patel V, Gutkind JS. Inhibition of Mammalian target of rapamycin by rapamycin causes the regression of carcinogen‐induced skin tumor lesions. Clin Cancer Res 2008; 14: 8094–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brown K, Strathdee D, Bryson S, Lambie W, Balmain A. The malignant capacity of skin tumours induced by expression of a mutant H‐ras transgene depends on the cell type targeted. Curr Biol 1998; 8: 516–24. [DOI] [PubMed] [Google Scholar]
- 25. Kemp CJ, Donehower LA, Bradley A, Balmain A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell 1993; 74: 813–22. [DOI] [PubMed] [Google Scholar]
- 26. Glick AB, Lee MM, Darwiche N et al Targeted deletion of the TGF‐beta 1 gene causes rapid progression to squamous cell carcinoma. Genes Dev 1994; 8: 2429–40. [DOI] [PubMed] [Google Scholar]
- 27. Han G, Lu SL, Li AG et al Distinct mechanisms of TGF‐beta1‐mediated epithelial‐to‐mesenchymal transition and metastasis during skin carcinogenesis. J Clin Invest 2005; 115: 1714–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matsumoto T, Jiang J, Kiguchi K et al Targeted expression of c‐Src in epidermal basal cells leads to enhanced skin tumor promotion, malignant progression, and metastasis. Cancer Res 2003; 63: 4819–28. [PubMed] [Google Scholar]
- 29. Chan KS, Sano S, Kiguchi K et al Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J Clin Invest 2004; 114: 720–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rundhaug JE, Pavone A, Kim E, Fischer SM. The effect of cyclooxygenase‐2 overexpression on skin carcinogenesis is context dependent. Mol Carcinog 2007; 46: 981–92. [DOI] [PubMed] [Google Scholar]
- 31. Segrelles C, Lu J, Hammann B et al Deregulated activity of Akt in epithelial basal cells induces spontaneous tumors and heightened sensitivity to skin carcinogenesis. Cancer Res 2007; 67: 10879–88. [DOI] [PubMed] [Google Scholar]
- 32. Rundhaug JE, Gimenez‐Conti I, Stern MC et al Changes in protein expression during multistage mouse skin carcinogenesis. Mol Carcinog 1997; 20: 125–36. [PubMed] [Google Scholar]
- 33. Fukagawa T, Pendon C, Morris J, Brown W. CENP‐C is necessary but not sufficient to induce formation of functional centromere. EMBO J 1999; 18: 4196–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sundberg JP, Sundberg BA, Beamer WG. Comparison of chemical carcinogen skin tumor induction efficacy in inbred, mutant, and hybrid strains of mice: morphologic variations of induced tumors and absence of a papillomavirus cocarcinogen. Mol Carcinog 1997; 20: 19–32. [DOI] [PubMed] [Google Scholar]
- 35. Okumura K, Saito M, Isogai E et al Meis1 regulates epidermal stem cells and is required for skin tumorigenesis. PLoS One 2014; 9: e102111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Amano M1, Suzuki A, Hori T, Backer C, Okawa K. The CENP‐S complex is essential for the stable assembly of outer kinetochore structure. J Cell Biol 2009; 186: 172–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weaver BA, Silk AD, Montagna C, Verdier‐Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 2007; 11: 25–36. [DOI] [PubMed] [Google Scholar]
- 38. Weaver BA, Cleveland DW. Aneuploidy: instigator and inhibitor of tumorigenesis. Cancer Res 2007; 67: 10103–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dominey AM, Wang XJ, King LE Jr et al Targeted overexpression of transforming growth factor alpha in the epidermis of transgenic mice elicits hyperplasia, hyperkeratosis, and spontaneous, squamous papillomas. Cell Growth Differ 1993; 12: 1071–81. [PubMed] [Google Scholar]
- 40. DiGiovanni J, Rho O, Xian W, Beltràn L. Role of the epidermal growth factor receptor and transforming growth factor alpha in mouse skin carcinogenesis. Prog Clin Biol Res 1994; 387: 113–38. [PubMed] [Google Scholar]
- 41. Jhappan C1, Takayama H, Dickson RB, Merlino G. Transgenic mice provide genetic evidence that transforming growth factor alpha promotes skin tumorigenesis via H‐ras‐dependent and H‐ras‐independent pathways. Cell Growth Differ 1994; 5: 385–94. [PubMed] [Google Scholar]
- 42. Damjanov N, Meropol NJ. Epidermal growth factor receptor inhibitors for the treatment of colorectal cancer: a promise fulfilled? Oncology (Williston Park) 2004; 18: 479–88. [PubMed] [Google Scholar]
- 43. Wang CY, Liu LN, Zhao ZB. The role of ROS toxicity in spontaneous aneuploidy in cultured cells. Tissue Cell 2013; 45: 47–53. [DOI] [PubMed] [Google Scholar]
- 44. Beltrami E, Valtorta S, Moresco R et al The p53‐p66Shc apoptotic pathway is dispensable for tumor suppression whereas the p66Shc‐generated oxidative stress initiates tumorigenesis. Curr Pharm Des 2013; 19: 2708–14. [DOI] [PubMed] [Google Scholar]
- 45. Schvartzman JM, Sotillo R, Benezra R. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer 2010; 10: 102–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bremner R, Kemp CJ, Balmain A. Induction of different genetic changes by different classes of chemical carcinogens during progression of mouse skin tumors. Mol Carcinog 1994; 11: 90–7. [DOI] [PubMed] [Google Scholar]
- 47. Nagase H, Mao JH, Balmain A. Allele‐specific Hras mutations and genetic alterations at tumor susceptibility loci in skin carcinomas from interspecific hybrid mice. Cancer Res 2003; 63: 4849–53. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Genotyping and western blot analysis. (a) PCR primers used for genotyping Cenp‐r +/+, Cenp‐r +/− and Cenp‐r −/− mice. (b) PCR genotyping of mice displaying the three potential genotypes using the three primer sets shown in part (a). (c) Western blot analysis of MEF cells (Cenp‐r +/+, Cenp‐r +/− and Cenp‐r −/−) using anti CENP‐R antibody.
Fig. S2. Histology of tumors. H&E staining of papillomas and carcinomas from Cenp‐r +/+ and Cenp‐r −/− mice. Scale bar, 100 μm.