

Abstract
Antimicrobial peptides (AMPs) are a diverse class of well-studied membrane-permeating peptides with important functions in innate host defense. In this short review, we provide a historical overview of AMPs, summarize previous applications of machine learning to AMPs, and discuss the results of our studies in the context of the latest AMP literature. Much work has been recently done in leveraging computational tools to design new AMP candidates with high therapeutic efficacies for drug-resistant infections. We show that machine learning on AMPs can be used to identify essential physico-chemical determinants of AMP functionality, and identify and design peptide sequences to generate membrane curvature. In a broader scope, we discuss the implications of our findings for the discovery of membrane-active peptides in general, and uncovering membrane activity in new and existing peptide taxonomies.
Keywords: machine learning, antimicrobial peptides, membrane curvature, amphiphilic peptides
1. Organization of review
In recent work, we have established a computational model that can predict and detect membrane-permeating activity in arbitrary peptide sequences by learning from a dataset of antimicrobial peptides (AMPs) [1]. We use the results of this study to examine critically the nature of AMPs and the process of machine learning. In this review, we explore the impact of these experimental studies in the context of prior applications of machine learning to the synthesis and design of novel AMPs; another recent invited mini-review contextualizes the computational aspects of the study [2]. We begin with a brief historical overview of AMPs and a summary of previously developed machine learning tools for AMP discovery, and show how our work fits into and contrasts with this body of work. In particular, machine learning is proficient at discovering ‘known unknowns’ by extrapolating from known AMP sequences to unknown ones. We show that it is possible to use machine learning as an engine to discover ‘unknown unknowns’, by using it reflexively to identify limitations in existing assumptions or classifications. Since machine learning allows us to quantify the key properties of peptides that enable them to permeate membranes, in principle, it contains information about the physico-chemical mechanism of membrane permeation. In practice, it can prove difficult to extract this information due to inherent difficulties in translating machine learning results into governing physical principles. We suggest an approach to circumvent these difficulties by using machine learning to guide calibrating experiments to reveal the physico-chemical determinants and mechanisms of membrane permeabilization. Results of these findings are critically compared with general trends and principles identified in the current literature on the biophysics of membrane curvature, amphiphilicity and AMP mechanisms. Finally, we describe the explanatory potential of this tool and conclude with an outlook on future applications. We stress that this review is not meant to be a comprehensive review on the details of machine learning techniques, but rather highlights a recent surprising application of machine learning to understand how the physical chemistry of peptide sequences relates to the geometry of membrane permeation.
2. Introduction to antimicrobial peptides
AMPs are essential components of innate host defense [3–8]. As of today, over 2000 natural and synthetic AMPs have been discovered and characterized [9–12]. While these heterogeneous peptide-based broad-spectrum antibiotics span an enormous diversity of sequences and secondary structures [3,8], early studies have identified common characteristics among AMPs. They tend to have short amino acid sequences (<50 amino acids), net cationic charge (+2 to +9) and amphiphilicity [3–8]. AMPs are often divided into three classes: α-helical AMPs [13,14], β-sheet AMPs [15] and extended linear peptides enriched with specific amino acids [16,17]. While AMPs in general are amphiphilic with segregated groups of polar and hydrophobic residues, a large class of AMPs can form α-helical structures that have polar (charged) and hydrophobic residues arranged onto opposite faces along the helical axis, thus creating facial amphiphilicity (figure 1a). This unique presentation of residues is often described as being ‘amphipathic’.
Figure 1.
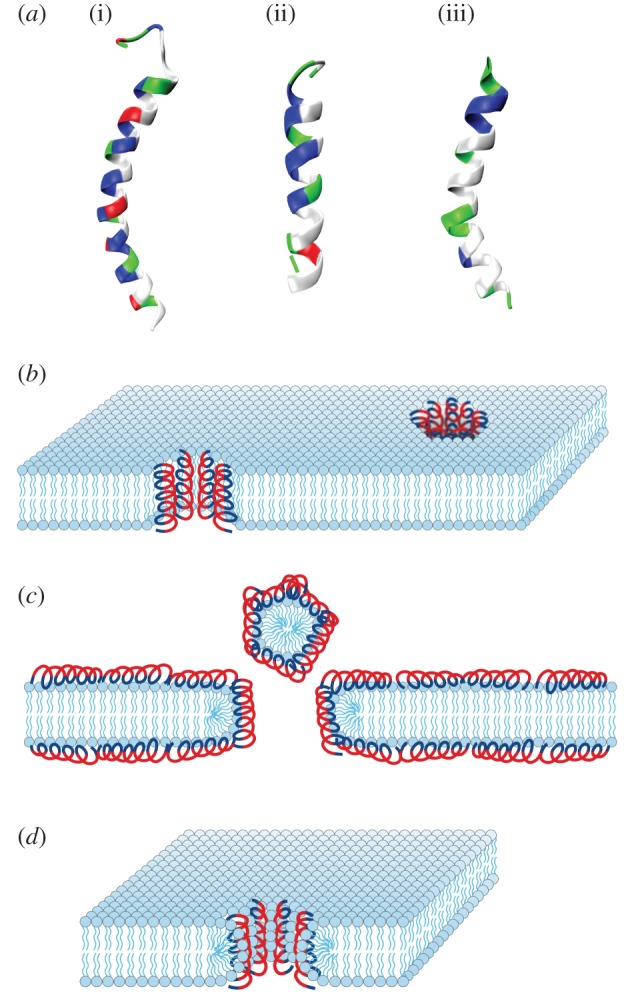
AMPs and their mechanisms of action. (a) Examples of cationic AMPs: LL-37 [18] (i, PDB ID: 2K6O), magainin [19] (ii, PDB ID: 2MAG) and melittin [20] (iii, PDB ID: 1MLT, 2MLT). Cationic residues are coloured blue and hydrophobic residues are coloured white. Structures were taken from the Protein Data Bank and visualized in VMD. Proposed mechanisms of AMP antimicrobial activity include the ‘barrel-stave’ model (b), the ‘carpet’ model (c) and the ‘toroidal-pore’ model (d). Reproduced with permission from [5,14,21].
In vitro experiments suggest that AMPs generally function by selectively disrupting microbial membranes. In theory, this leads to cell death due to the loss of electrochemical gradients, reduction in resistance to osmotic stress, leakage of cellular contents and disruption of metabolic processes [4,8]. This bactericidal activity typically is dependent on interactions between AMPs and bacterial membranes [22], which has been demonstrated using a variety of experimental techniques, including X-ray scattering, nuclear magnetic resonance (NMR), dye leakage assays, electron microscopy and circular dichroism [8]. Different models describing membrane permeation have been proposed, including the ‘barrel-stave’ model, the ‘carpet’ model and the ‘toroidal-pore’ model among others (figure 1b–d). In the ‘barrel-stave’ model, amphipathic α-helical AMPs self-assemble into cylindrical bundles that are embedded perpendicularly into the cell membrane to form pores [23–27]. Within the membrane, the hydrophobic faces of the individual AMPs are oriented towards the hydrophobic interior of the bilayer, and the hydrophilic faces are oriented towards one another to form the lumen of the aqueous pore (figure 1b) [5,14]. In the ‘carpet’ model, AMPs adsorb onto the cell membrane in a parallel orientation. Once a critical local concentration is reached, the peptides disintegrate the membrane via micellization (figure 1c) [5,28–32]. Here, pore formation does not occur, unlike the ‘barrel-stave’ model. In the ‘toroidal-pore’ model, AMPs insert perpendicularly into the membrane to form a pore [21,33] but differs from the ‘barrel-stave’ model in that the membrane is integrated into the pore lining, forming a continuous interface with the peptides (figure 1d) [5,34,35].
The selectivity of AMPs for bacterial membranes over eukaryotic membranes is generally believed to result from the compositional differences between their membranes [3,4,8]. More specifically, bacteria membranes contain large amounts of anionic lipids (e.g. phosphatidylglycerol and cardiolipin), while eukaryotic membranes contain mostly zwitterionic lipids (e.g. phosphatidylcholine and sphingomyelin) [36–38]. In human cells, cholesterol is particularly important [39]. Indeed, in vitro experiments have shown that the presence of anionic lipids results in increased membrane disruption and permeabilization by cationic membrane-active antimicrobials [3]. However, while the existence of anionic lipids is a necessary condition for permeation by AMPs, it is not a sufficient one. Bacterial membranes also contain high amounts of negative intrinsic curvature lipids, such as phosphatidylethanolamine and cardiolipin, which predispose their membranes to poration [39–44]. In the simplest models, AMPs first interact with bacterial cells by binding electrostatically to their membrane surfaces, during which the cationic residues of the peptide bind to the anionic lipid head groups and other anionic surface components. After adsorbing onto the membrane with its helical axis parallel to the surface, the AMP partitions into the lipid bilayer, driven primarily by hydrophobic interactions between its hydrophobic residues and the membrane core. The amphipathic nature of AMPs allows for this direct interaction with the cell membrane, which can then lead to membrane permeation and cell death [28]. In fact, the majority of early experimental studies conclude that membrane permeation underlies the primary mechanism of action of AMPs. We will revisit this question when we discuss our machine learning results.
3. A. brief history of machine learning on AMPs
3.1. Machine learning fundamentals
Machine learning leverages expertise from mathematics, statistics and computer science to learn from data. Machine learning models can be divided into supervised and unsupervised methods. Supervised learning builds a prediction model based on existing ‘ground truth’ data, which consists of actual measured outcomes for each object of interest. In turn, each object can be characterized by any number of input variables or ‘features’. The presence of the outcome variable guides the learning process. Supervised models will take the ‘features’ as input and output a prediction, which can be a binary, categorical or continuous variable. By contrast, unsupervised learning approaches do not rely on measurements of outcome and rely purely on the input ‘features’ of the objects. Unsupervised approaches must infer a function to discover hidden trends in the data. The majority of established learning methods revolve around supervised learning but the literature on unsupervised methods is growing quickly. Most of the learning methods discussed in this review centre upon supervised learning on validated datasets of AMPs.
3.2. Simultaneous maturation of machine learning methods and AMP studies
The mathematical and statistical framework for machine learning has existed for centuries, far before computers were invented. Ideas and elements most recognizable today in machine learning, including Bayes' Theorem, principal component analysis, multiple linear regression, least-squares fitting and Markov chains, were established by mathematicians before 1950 [45]. Alan Turing's pioneering work on the Turing machine in 1950 [46] led to the development of the first artificial neural network (ANN) [36]. After the invention of the modern computer, research in machine learning exploded with the development of many of the modern methods used today, including partial least-squares regression, recurrent neural networks, hidden Markov models (HMMs), support vector machines (SVMs) and random forests (RFs) [45]. Most recently, advances in scalable training algorithms and the availability of large datasets has spurred a resurgence of interest in ANNs with deep network architectures capable of tasks including handwriting recognition [47], cancer diagnosis [48], HIV classification [49], facial recognition [50], spam filtering [51] and quantum chemistry [52]. In 2016 the ‘AlphaGo’ ANN rose to prominence by beating a 9-dan human grand master at the ancient board game Go [53]. As machine learning methods matured in the latter half of the century, the same was occurring to the field of AMPs. As described in §2, decades of biophysical studies centred upon understanding AMP mechanisms of action and sequence rules gradually filled the literature. The advent of high-throughput screening coupled with decades of experimental data allowed for curation of large annotated datasets [11]. In the last 10–15 years, the focus of machine learning has shifted to an intensely data-driven approach. Significant advancements in computational power and easy-to-use statistical learning tools has made supervised machine learning a viable strategy for leveraging large datasets for the high-throughput and high-accuracy classification of AMPs. Typical readouts from biophysical assays on AMPs include calculations of minimum inhibitory concentrations, minimum bactericidal concentrations and binding affinities. These quantities, coupled with sequence information about AMPs, allow for the training of various supervised learning models using peptide sequence information as an input. Before this era, methods for de novo AMP discovery relied on long-standing bioinformatics methods, including sequence alignment and homology modelling for prediction of biological activity. Now, the convergence of innovations in machine learning models, the presence of modern computational tools, and the availability of high-quality datasets has enabled the machine learning-aided design of AMP candidates.
3.3. Recent applications of machine learning to AMP classification and discovery
The diversity of AMP sequences and structures coupled with the time and expense associated with experimental design, production and testing of AMP candidates precludes comprehensive experimental screening of peptide sequence space. Thus, the earliest machine learning models were quantitative structure–active relationship (QSAR) models that proved useful in efficient screening and optimization of a small number of promising sequences for experimental evaluation. QSAR models seek to use physico-chemical descriptors to predict biological activity of a molecule that is typically expensive and/or time consuming to measure or calculate. In contrast, many physico-chemical properties of a peptide can be inexpensively computed directly from its amino acid sequence [54,55]. Models are trained and validated over experimentally characterized databases, then employed in high-throughput in silico screening to identify novel candidates with the desired biological activity. This approach relies on statistical learning to infer empirical relationships between physico-chemical properties and biological activity, and is, therefore, contingent on an underlying relationship between (some subset of) the descriptors, the capacity of the machine learning model to discover and encode this relationship in a mathematical expression, and sufficiently large and diverse training databases to produce robust predictive models [45,54,56].
QSAR models for computational AMP design have been developed using a variety of statistical learning approaches. The majority of prior AMP machine learning studies have tended to focus on either optimizing classification accuracy of AMPs or identifying potent AMP candidates with low minimum inhibitory concentrations. In one of the first applications of machine learning to AMPs, Lata et al. [57] developed a QSAR AMP classification tool based on ANN, SVM and quantitative matrix models based on unique motifs found in the C- and N-terminal residues of known AMPs. In 2008, Chersakov et al. used high-throughput screening methods to train an ANN model on the measured antimicrobial efficacies of thousands of nine-residue peptides to discover potent antimicrobials that were potent against multi-drug-resistant bacteria [58]. Fjell et al. [59] published a 2008 study using HMMs to screen for AMPs in the bovine genome, which led to the discovery of a previously unknown AMP and confirmed the absence of α-defensins. In a similar vein, this group later developed an ANN model in 2009 to screen a larger number of synthetic AMP candidates, characterizing 18 sequences with high antimicrobial efficacy against multi-drug-resistant bacteria [54]. In 2011, Wang et al. [60] used a combination of sequence alignment and feature selection methods to design a computational model to more accurately classify AMPs. Similarly, Torrent et al. [61] trained an eight-descriptor SVM to classify AMPs with 75–90% accuracy while taking into account new factors like peptide aggregation. In 2013, Maccari et al. [62] used RF models to design and validate the antimicrobial activity of two natural peptides and one peptide with non-natural amino acids, and Xiao et al. [63] designed a two-level classifier to first classify peptide sequences as an AMP, and then sub-classify them into 10 functional AMP categories. In 2015, Giguère et al. [64] used a kernel method based on graph theory to train a 100 peptide dataset based on multiple measures of bioactivity to predict novel candidates. Most recently in 2017, Schneider et al. [65] reported the first application of unsupervised–supervised two-step models to classify AMPs. They used self-organizing maps to apply nonlinear dimensionality reduction to the training data, which were then used as an input for a supervised neural network model. Together, these studies highlight a diversity of methods and approaches that have been used to classify and design AMPs with great success.
4. A machine learning model that detects membrane activity
In our recent work, we aimed to use machine learning not to directly discover and design AMPs with enhanced potency and antimicrobial efficacy, but rather to help glean understanding about the relationship between AMP sequence and function. Rather than optimizing for antimicrobial efficacy, we trained and interrogated a machine learning model to determine which physico-chemical characteristics were most defining of ‘antimicrobial-ness’. Furthermore, we focused on α-helical AMPs, which have structures common to many peptides and proteins. To do this, we collated a dataset of known, experimentally characterized AMPs from the antimicrobial peptide database [11], and a separate dataset of α-helical non-antimicrobial decoy peptides in the same length range drawn from the Protein Data Bank of Transmembrane Proteins (PDBTM) [66]. The selection of the decoy dataset is consistent with the methodology used in prior machine learning studies [61,67]. The peptides were collated without regard to their organism of origin. From these datasets, we developed a QSAR model to differentiate between antimicrobial and non-antimicrobial α-helical peptide sequences. Using this model as a search tool, we carried out a directed search of the unknown peptide sequence space to find new antimicrobial candidates evolutionarily distant from known AMP sequences, and combined our computational modelling with calibrating experiments to elucidate the physico-chemical basis for AMP function. Surprisingly, we found that our classification metric did not, as we had anticipated, correlate with antimicrobial efficacy, but instead correlated with the capacity to permeate cell membranes [1]. While antimicrobial activity is often associated with membrane permeation ability, these two properties are inherently distinct from one another and are not coextensive. The results here imply that membrane activity, while a common feature for AMP action, may not be the main mode of antimicrobial action. Interestingly, membrane permeation has been previously demonstrated to be highly dependent on the generation of membrane curvature [68–71]. Thus, our finding dovetails with recent work [72,73] showing how diverse sequences from drastically different peptide families can generate similar magnitudes of membrane curvature. These findings also agree nicely with studies on the phase behaviour of AMP–lipid complexes, and biophysical mechanisms of membrane curvature generation. More generally, our model not only enables identification of new AMP sequences, but also facilitates recognition of previously undetected membrane activity in existing protein families.
4.1. Computational model
We constructed and trained a SVM classifier on a dataset of 286 antimicrobial and 286 decoy α-helical peptides. We used the Python package propy [74] to generate physico-chemical descriptors from the peptide sequences, and the package scikit-learn [75] to create the SVM classifier. We began with an initial broad panel of 1588 physico-chemical descriptors [56,67,74,76,77], including simple peptide metrics of length, charge, hydrophobicity, residue composition and more complicated metrics such as autocorrelation, physico-chemical compositions and sequence order. These descriptors can be quickly calculated directly from the peptide sequence independent of experimental measurement. We rationally selected the k = 12 most highly predictive descriptors of the original 1588 descriptors identified using the L1-norm sparse variable selection approach of Bi et al. [78] (table 1). The linear SVM trained over this subset of variables possesses as good or better predictive performance and more intuitive interpretability than those employing entire descriptor ensemble and/or implementing nonlinear kernels. The trained linear SVM based on a training set of 486 peptides and a validation set of 86 peptides had a prediction accuracy of 91.9%, a specificity of 93.0% and a sensitivity of 90.7%. The SVM takes an arbitrary peptide sequence as an input, and outputs σ, the distance of the peptide to the (k − 1)-dimensional SVM hyperplane that separates the antimicrobial and non-antimicrobial sequences. By construction, σ can also be converted into a probability of positive classification, in which a larger positive value of σ denotes higher confidence of antimicrobial activity, and a large negative value denotes a higher confidence of lack of antimicrobial activity. Using the SVM as a screening tool, we conducted a directed search of two regions of the unknown peptide sequence space: peptides close in homology to known AMPs, and those far in homology to known AMPs. For all candidates, we also calculated their helicity in addition to σ and homology to known AMPs (minHomologyAMP). Because an exhaustive screen of all possible peptides of length n is not feasible, we decided to screen peptides with lengths of 20–25 amino acids, which corresponds to the most common lengths of membrane-spanning AMPs [4,8]. We also directed our search towards ideal candidates with large positive values of σ. To that end, using Monte Carlo sampling, we randomly mutated AMPs in the size range of interest, and accepted or rejected sequences based on the Metropolis criterion 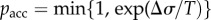 , where
, where  and T = 0.8 is an effective temperature [81–83]. Then, to identify optimal candidates, we applied multi-objective optimization to our sequence map. Specifically, employing helicity, σ and minHomologyAMP as optimization criteria, we constructed a ‘Pareto frontier’ from our candidate sequences defined as the subset of sequences not strictly dominated by any other in all three of these metrics [84,85]. In other words, sequences away from the frontier are non-optimal in the sense that better sequences exist with simultaneously higher helicity, larger σ and smaller minHomologyAMP, whereas for those on the frontier no other sequences exist for which all three of these metrics can be improved simultaneously—improvements in any one are necessarily accompanied by diminishment in at least one other. Full details of the computational model are found in Lee et al. [1].
and T = 0.8 is an effective temperature [81–83]. Then, to identify optimal candidates, we applied multi-objective optimization to our sequence map. Specifically, employing helicity, σ and minHomologyAMP as optimization criteria, we constructed a ‘Pareto frontier’ from our candidate sequences defined as the subset of sequences not strictly dominated by any other in all three of these metrics [84,85]. In other words, sequences away from the frontier are non-optimal in the sense that better sequences exist with simultaneously higher helicity, larger σ and smaller minHomologyAMP, whereas for those on the frontier no other sequences exist for which all three of these metrics can be improved simultaneously—improvements in any one are necessarily accompanied by diminishment in at least one other. Full details of the computational model are found in Lee et al. [1].
Table 1.
Subset of 12 bagged descriptors identified by the L1-SVM variable selection procedure. For each descriptor we provide a description of its physical interpretation [77], and its weight in the linear SVM trained over the training data employing only these 12 descriptors. Positive (negative) weights correspond to a positive (negative) association of the descriptor with antimicrobial activity, and the magnitude of the weight indicates the relative importance of the Z-scored descriptor in the classification prediction.
| rank | descriptor | physical interpretation | SVC weight |
|---|---|---|---|
| 1 | netCharge | net charge of peptide | 0.80 |
| 2 | 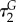 |
length normalized sequence order coupling number measuring physico-chemical correlations between residues separated by two positions (i, i + 2) measured by the Grantham chemical distance matrix [79,80] | 0.48 |
| 3 | 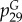 |
PseAAC generalization at tier k = 9 measuring pairwise correlations of the physico-chemical properties of residues separated by nine positions (i, i + 9) measured by the Grantham chemical distance matrix [79,80] | 0.36 |
| 4 | SolventAccessD1025 | fraction of the peptide length containing 25% of the buried residues {A,L,F,C,G,I,V,W} | −0.24 |
| 5 | pc(M,K) | relative fraction of M residues to K residues | −0.21 |
| 6 | 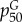 |
PseAAC generalization at tier k = 30 measuring pairwise correlations of the physico-chemical properties of residues separated by 30 positions (i, i + 30) measured by the Grantham chemical distance matrix [79,80] | 0.20 |
| 7 | AE | fraction of contiguous AE residue pairs | 0.18 |
| 8 | 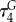 |
length normalized sequence order coupling number measuring physico-chemical correlations between residues separated by four positions (i, i + 4) measured by the Grantham chemical distance matrix [79,80] | −0.17 |
| 9 | LW | fraction of contiguous LW residue pairs | 0.17 |
| 10 | NK | fraction of contiguous NK residue pairs | 0.13 |
| 11 | DP | fraction of contiguous DP residue pairs | −0.12 |
| 12 | FC | fraction of contiguous FC residue pairs | −0.04 |
4.2. The support vector machine model detects membrane activity rather than antimicrobial efficacy
Informed by our computational model, we continued to explore the relationship between AMPs and their purported modes of activity. Traditionally, the assessment of antimicrobial activity is accomplished in vitro by using the minimum inhibitory concentration (MIC) assay, which determines the lowest concentration of an agent needed to inhibit growth of a specific bacteria, and correspondingly, its efficacy against that bacteria. Using a database of MIC values for 478 AMPs against Staphylococcus aureus, we calculated the σ values for each peptide and plotted them against their reported MIC values. We found no correlation between σ and MIC (RSpearman = −0.060 [−0.154, 0.034], p = 0.187) (figure 2a) [1]. This lack of correlation of antimicrobial efficacy with distance to the hyperplane of known AMPs can be explained by a superposition of membrane activity with other multiplexed functions. Analysis of the literature suggests that the majority of the known AMPs we tested are compounds that have other bactericidal activities in addition to membrane penetration. For example, buforin is known to bind intracellularly to DNA [86], and indolicidin enters bacteria and inhibits DNA synthesis [87]. In addition to membrane permeation, pleurocidin inhibits bacterial macromolecular synthesis [88], and mersacidin inhibits peptidoglycan synthesis [89]. Several AMPs like the cathelicidin LL-37 also have immunomodulatory activities alongside the ability to kill bacteria via membrane permeation [90,91]. Upregulation of the immune system can enhance the killing ability of a peptide, thereby leading to increased antimicrobial potency. Here, we find several cases of peptides that have extremely low values of MIC (high potency) and have low or negative values of σ assigned by our classifier (figure 2a). Thus, one might imagine that this deviant antimicrobial potency of an AMP results from a superposition of multiple contributing effects. The above limitation in AMP analysis also highlights a generic problem with machine learning approaches: classifier accuracy does not always straightforwardly translate to human understanding of the underlying mechanisms of action. Although it is known that AMPs can have immunologically relevant activity outside of membrane activity, there is currently no general way to identify AMPs with additional functions. This has been a salient problem in the field of AMPs. Thus, we proposed a way to identify candidate sequences with multiplexed functions, but doing so requires some understanding of the SVM parameter σ. Since membrane activity is a common mode of AMP activity, we hypothesized that the SVM has learned to recognize not antimicrobial activity directly, but membrane activity as the discriminant between AMPs and decoys in the training data.
Figure 2.

The SVM detects the ability of peptides to generate NGC. (a) We observe a lack of correlation between distance to margin σ and antimicrobial potency (MIC) of known AMPs against S. aureus (RSpearman = −0.060 [−0.154, 0.034], p = 0.187). (b) Using SAXS, we observe that test peptides derived from machine learning generate NGC. (i) Shows the 3D topology of a Pn3m cubic phase induced by test peptides in model membranes. (ii) Illustrates the concept of NGC with positive curvature (+) in one principal direction and negative curvature (−) in the orthogonal direction. (c) We find that σ correlates strongly with the ability to generate membrane curvature (RSpearman = 0.653 [0.234, 0.891], p = 0.006). Adapted from data in [1].
Using the trained classifier as a guide, we designed calibrating experiments to test this hypothesis. From the ensemble of peptide sequences screened by the classifier, we selected and synthesized 16 peptide candidates with varying homologies to known AMPs that were proximal to the Pareto frontier. We characterized their activity using antimicrobial assays and small-angle X-ray scattering (SAXS) with artificial membranes. When candidate peptides were incubated with artificial membranes in the form of small unilamellar vesicles (SUVs), we observed a topological transition from lipid vesicles to liquid-crystalline cubic phases rich in negative Gaussian curvature (NGC) (figure 2b). Most importantly, we found that σ correlated strongly with a peptide's ability to generate negative Gaussian membrane curvature (RSpearman = 0.653 [0.234, 0.891], p = 0.006) (figure 2c) [1]. This provides experimental support for our assertion that the machine learning classifier has learned to discriminate peptides based on their membrane activity, discovering and encoding a relationship between the 12 physico-chemical descriptors used to train the classifier (table 1) and an emergent structural capacity to induce NGC.
4.3. Negative Gaussian curvature
To understand how peptides can generate negative Gaussian membrane curvature, we provide a brief overview of the physics of membrane curvature and deformation processes. Cell membranes are fluid bilayers containing two leaflets that can stretch and deform. Although cell membranes exist in three dimensions, they are better visualized as a curved two-dimensional surface. At any given point on the surface, the curvature can be defined by a plane tangent to that point. Any plane orthogonal to this tangent plane can intersect the surface with some curvature value c = 1/R defined by the radius of curvature R. Given all possible orthogonal planes, the two that give the maximal and minimal curvatures are defined as c1 = 1/Rmin and c2 = 1/Rmax, respectively. These are called the two principal curvatures, and in combination, can be used to describe the shape of the surface at that point. Mathematically, the arithmetic mean of these two principal curvatures is the mean curvature H = ½ (c1 + c2), and the product of the two principal curvatures is the Gaussian curvature K = c1c2 [92]. Curvature can be positive or negative, however, when describing a membrane monolayer, the sign of curvature is conventionally defined by the direction of bending. Positive curvature is defined by bending of the monolayer to form a convex hydrophilic surface (such as the outside of a spherical cell), while negative curvature is defined as the bending of the monolayer to form a concave hydrophilic surface (such as on the neck of a budding vesicle) [69,93,94] (figure 2b). Furthermore, positive Gaussian (‘dome-like’) curvature, K > 0, results from principal curvatures of the same sign, while negative Gaussian (‘saddle-like’) curvature, K < 0, results from principal curvatures of the opposite sign (figure 2b). Membrane curvature can also be influenced by the geometry and packing behaviour of the various phospholipids present in the bilayer, which together define the equilibrium shape of the membrane. Thus, deformation of the membrane from equilibrium exacts an energetic penalty that can be calculated from curvature changes and mechanical properties of the membrane.
Peptides and proteins can generate membrane curvature through several mechanisms, including membrane partitioning and insertion [95], membrane scaffolding [39,96], curvature sensing [97], molecular crowding [98,99] and membrane wrapping [100,101]. Often, the mechanism of membrane deforming peptides and proteins involve one or more of the above. The specific structure of amphipathic α-helical AMPs, with separate polar and hydrophobic faces, facilitates the interactions associated with these mechanisms. The hydrophobic AMP domains interact with the hydrocarbon chains in the lipid core, driving positive curvature generation via steric impingement in the membrane. Simultaneously, electrostatic interactions between the cationic groups of the peptide and anionic lipid head groups drive negative curvature by inducing wrapping of the membrane.
The variation in membrane disruption and curvature effects among amphipathic cationic α-helical AMPs has been attributed to differences in the charge distribution and sizes of the polar and hydrophobic faces of their helices [102,103]. A large number of studies have explored correlations between the relative sizes of the polar and hydrophobic faces with biological effects, such as cell lysis [102–107]. Specifically, lytic antibacterial helical peptides that are known to destabilize membranes can often be described as having an inverted wedge-shaped cross-section, with a narrow polar face to form the apex, and a wide hydrophobic face to form the base [4,102,104,105]. A large hydrophobic face allows the peptide to deeply penetrate into the membrane, perturbing the packing of the bilayer core. This can lead to membrane thinning and cause the membrane to become more susceptible to curvature deformations as a result of reduced bending moduli [21,108–110]. Moreover, previous work has found that increasing the bulkiness or angle subtended by the hydrophobic face further increases the ability of the peptide to destabilize membranes and cause lysis [102,104,111]. For peptides with narrow polar faces and wide hydrophobic faces, we expect the hydrophobic insertion effects to dominate, resulting in strong positive curvature. Conversely, for peptides with wide polar faces and narrow hydrophobic faces, electrostatic interactions between the cationic residues and anionic lipid head groups cause membrane wrapping (negative curvature) to dominate over the positive curvature from hydrophobic insertion.
This ability of a cationic amphipathic AMP to induce both positive and negative curvature in mutually orthogonal directions at a single location results in NGC, which is the type of curvature that is topologically necessary for membrane destabilization processes. In fact, AMPs have been shown to destabilize membranes through a variety of modes, including pore formation [23,112,113], blebbing [114,115], budding [116] and vesicularization [117,118], all of which require NGC. As AMPs vary in relative amounts and distributions of cationic charge and hydrophobicity, and thus, can yield different levels of negative and positive curvature, an optimal balance among these properties would enable efficient generation of NGC. For instance, for many AMPs, the angle subtended by the polar face tends to be ≤100° and the ratio of cationic/anionic residues is approximately 4–5 [104].
While the actual form of membrane destabilization is dependent on the physical chemistry of the specific AMP and the target membrane, AMP-induced disruption of bacterial membranes has been found to correlate with the ability of the peptides to generate NGC [70]. Synchrotron SAXS is an efficient way for measuring NGC in lipid membranes. Recent work has used SAXS to map out the phase behaviour of lipids complexed with membrane deforming peptides and proteins. For example, three families of defensins, a class of AMPs, were assayed for their induction of membrane curvature deformations by incubating the peptides with SUVs composed of binary and ternary mixtures of phospholipids. By tuning the phospholipid compositions of the SUVs, one can model the lipid compositions of prokaryotic and eukaryotic membranes. For compositions mimicking bacterial membranes, defensins were found to restructure SUVs to liquid-crystalline cubic phases, which are characterized by a minimal surface with NGC at every point. [70,119]. The ability to generate NGC has also been observed for cell-penetrating peptides [71], natural and synthetic AMPs [120,121] and viral fusion proteins [72,122]. Taken together, these results suggest that the generation of NGC is not only a feature of AMPs, but a common root mechanism for membrane destabilizing processes in general.
4.4. Metrics that define membrane-permeating peptides
Having summarized different modes of NGC generation, we now examine the details of the machine learning model in the context of prior work on AMP amphiphilicity, cationicity, hydrophobicity and NGC. This analysis is facilitated by our use of sparse variable selection techniques to reduce the number of descriptors to just 12, and our selection of a linear SVM model in which the separating hyperplane exists in this 12-dimensional space. Deriving interpretability from classifiers containing large numbers of descriptors and/or performing nonlinear classification can be very challenging. Remarkably, our analysis shows that the machine learning model efficiently learned a long-observed but poorly quantified characteristic of AMPs: facial amphiphilicity (often termed amphipathicity for helical AMPs). The training dataset for our SVM contains many amphipathic AMPs, as extensively summarized in the literature [28,123]. Our SVM, using physico-chemical descriptors independent of geometry, was able to illuminate the importance of amphipathicity in predicting whether peptides are membrane active and is implicitly defined in our classification scheme. From the original set of 1588 descriptors, we obtained a subset of 12 descriptors for our SVM that were most predictive for antimicrobial activity based on sparse variable selection approaches (table 1), and four out of these 12 descriptors directly enforce amphiphilicity. Three of the four descriptors (nos. 2, 3, 6 in table 1) have positive weights in the SVM model indicating that any two residues radially spaced apart by two, nine and 30 positions along the helical wheel likely have opposite amino acid character (e.g. polar and hydrophobic) in candidates predicted to be highly membrane active, while the fourth (no. 8) has negative weight indicating that residues spaced apart by four positions along the helical wheel likely have similar character (e.g. polar and polar or hydrophobic and hydrophobic). The four descriptors are illustrated in the helical wheel diagram in figure 3a. Accordingly, our SVM model discovers, detects and enforces amphipathicity in its classification process, which results in membrane-active candidates with inherent amphipathicity. Using a quantitative metric, the mean hydrophobic moment (magnitude of the vector hydrophobic moment), we can calculate the amphipathicity of our test peptides that are near the Pareto frontier. Using the Eisenberg consensus scale [124], we calculate the mean hydrophobic moment of the 16 test peptides from machine learning. We find they fall in agreement with the expected amphipathicities of known AMPs as we show in figure 3b. To explore correlations with membrane activity, we compared the hydrophobic moment and the magnitude of NGC produced by the peptides from SAXS. The Spearman rank correlation between mean hydrophobic moment and NGC is RSpearman = 0.680 [0.259, 0.856], p = 0.0038, indicating that amphipathicity is indeed an important determining factor in the ability to generate NGC (figure 3c). This makes sense because the amphiphilic nature of the molecule is one of the structural features that enables its interaction with membranes, as discussed in §4.3. Taken together with our finding that σ also correlates well with NGC (figure 2c), we confirm that our SVM classifier has learned to pick out amphipathic helices that generate the requisite topological criteria for membrane permeation.
Figure 3.

Machine learning model learns amphipathicity of membrane-permeating helices. (a) Helical wheel plot demonstrating four physico-chemical descriptors that describe amphipathicity (see nos. 2, 3, 6 and 8 in table 1). Residues spaced apart by two, nine and 30 positions along the helix likely have opposite character (red lines), while residues spaced apart by four positions likely have similar character (green line). (b) Mean hydrophobic moments (measure of amphipathicity) of test peptides derived from machine learning are similar to those of known AMPs. (c) Positive, statistically significant correlation between NGC generated and amphipathicity of test peptides (RSpearman = 0.680 [0.259, 0.856], p = 0.0038). (d) Tested peptides were classified into two groups based on the angle (θ) subtended by the hydrophobic face. Simplified diagrams of helix cross-sections depict the different widths of the polar (upper) and hydrophobic (lower) faces for each of the two groups, θ > 120° and θ ≤ 120°. For a subset of the tested peptides, the membrane activity in terms of |〈K〉| (units of 10−4 Å−2) and σ are listed alongside the mean hydrophobic moment 〈μH〉 (units of kcal mol−1), mean hydrophobicity 〈H〉 (units of kcal/mol) and charge z at pH 7.4. Adapted from data in [1].
The membrane-permeating activity and the bactericidal activity of a given peptide are distinct and both depend on a variety of factors. Examples of factors that impact membrane activity include charge, hydrophobicity, amphipathicity and the ability to generate NGC. Together, these relations point towards the idea of optimal balance between electrostatic and hydrophobic peptide–membrane interactions that overall determines AMP activity [125], which is often also used for synthetic mimics of AMPs [126]. Thus, it is not surprising that no simple correlations exist between the individual parameters and activity. However, we find that our SVM classifier captures the collective effects of the various peptide properties as a multi-dimensional profile, as σ correlates with the ability to generate NGC, which in turn correlates with various mechanisms of peptide-induced membrane permeabilization [70].
That we have 16 test peptides that are ranked by machine learning and calibrated by direct synchrotron X-ray measurements of induced membrane NGC generation affords us the opportunity to qualitatively examine some of the trends and principles in membrane permeation identified experimentally in biophysical studies. We observe that membrane activity, as described by σ and NGC, is dependent upon the relationships between several intrinsic physico-chemical properties, specifically the mean hydrophobic moment μH, mean hydrophobicity H and charge z. Here, we examine where our present observations agree with those from previous biophysical studies, where we deviate, and propose potential explanations. Previous work has shown that a larger hydrophobic face tends to result in a more lytic peptide [102,104,111], a trend that is generally consistent among our 16 peptides. We categorized the peptides into two groups based on the size of the angle (θ) subtended by the hydrophobic face and found that those having larger angles (greater than 120°) typically are associated with greater membrane activity, and therefore, more likely to be antimicrobial (figure 3d). Interestingly, peptide 12 is characterized by a narrower hydrophobic face (θ ≤ 120°), yet it has considerably high membrane activity. This finding may appear contrary to the trend, however, it is in some ways ‘the exception that proves the rule’. Previous work has shown that a peptide with a low mean hydrophobicity can achieve membrane activity when combined with a large mean hydrophobic moment [111,125,127–129]. Conversely, a peptide with a small mean hydrophobic moment may exhibit antimicrobial activity if it is has a high mean hydrophobicity [111,125,130,131]. Indeed, a comparison between peptides 10 and 14 further illustrates precisely these relations. Furthermore, with lower peptide charge, activity has been found to experience more influence from the mean hydrophobic moment, mean hydrophobicity and size of the hydrophobic face [111], as is shown accordingly by peptides 7 and 8. As we can see, our data are also in good agreement with previous findings.
To better quantify sequence trends of AMPs, we observe that it is well known that AMPs and membrane-permeating peptides are abundant in the cationic residues arginine, lysine and histidine, and the presence of hydrophobic residues (greater than 30%) is common. Based on bioinformatics analysis of the AMP database [9], we previously established the ‘saddle-splay selection rule’, a tool for predicting whether a peptide sequence could generate NGC [70] from its amino acid content. This rule predicts that there will be a compositional trade-off between the number of arginine and lysine residues, and the hydrophobicity of an AMP. Biophysical studies and quantum mechanical simulations show that arginine alone can generate NGC due to bidentate hydrogen bonding with phosphate head groups, while lysine and hydrophobicity must work in concert together to generate NGC [132]. This is because hydrophobicity generates positive curvature while lysine generates a negative mean curvature due to membrane wrapping from charge compensation, which can combine to form NGC. This observation is consistent with both experimental studies and molecular simulations [132–134]. Our prior analyses have shown all peptides in the AMP database to adhere to this saddle-splay curvature selection rule [70]. Furthermore, this relation also explains why cell-penetrating peptides rich in arginines like HIV-TAT can generate the same kind of membrane curvature as traditional AMPs rich in lysines and hydrophobicity [135]. Given that our computational model learned the physico-chemical characteristics underlying charge (#1 in table 1) and amphiphilicity of AMPs (nos. 2, 3, 6 and 8 in table 1), we were curious to see whether our screening tool could identify peptides that followed the saddle-splay selection rule. We previously established that our classification metric σ correlates with the induction of NGC, and showed that the SVM classifier can be used to predict NGC magnitudes for arbitrary peptides [1]. We plotted the Pareto-optimal peptides on the saddle-splay selection rule. Remarkably, the physico-chemically restricted Pareto sequences (i.e. those with physico-chemical descriptors no more than 10% outside the range observed in the training data) were found to closely follow the rule governing amino acid content previously shown for AMPs [70] (figure 4a, green diamonds). This result also demonstrates the surprising result that our QSAR approach and SVM classifier trained solely on physico-chemical descriptors learned a rule that describes the geometry and topology of membrane deformation. For the physico-chemically unrestricted Pareto-optimal sequences (i.e. those without any restriction on the values of the physico-chemical descriptors), the sequences tend to segregate into two clusters, ones that follow the saddle-splay selection rule (figure 4b, orange diamonds, no border), and ones that fall below the curve (figure 4b, orange diamonds, black borders). These off-trend sequences suggest a greater plasticity in the peptide sequence and imply that these candidates may contain additional multiplexed functions in addition to membrane activity. We also compared the positions of our synthesized test peptides along the saddle-splay selection rule with their ability to generate NGC. We observe that the magnitude of NGC generated by a peptide does not seem to depend on where the sequences fall on the saddle-splay selection rule (figure 4c). In other words, there is no significant correlation (p < 0.05) between position on this saddle-splay curve and degree of induced NGC in membranes, consistent with the idea that this curve represents a trade-off between using different combinations of amino acids to enable NGC generation (RSpearman = −0.433, p = 0.094 for NGC and K/K + R, RSpearman = −0.292, p = 0.273 for NGC and mean hydrophobicity).
Figure 4.

Pareto-optimal sequences and newly discovered curvature-generating sequences generally follow the saddle-splay selection rule. Physico-chemically restricted (a) and physico-chemically unrestricted (b) Pareto-optimal sequences follow the saddle-splay selection rule denoted by the AMP database (black). (c) Tested peptides (stems) demonstrate NGC generating ability regardless of their placement along the saddle-splay selection rule (black circles). |〈K〉| has units of units of 10−4 Å−2. Adapted from data in [1].
It is tempting here to ask what the contribution of machine learning is, if it seems to re-learn previously identified trends or rules. When confronted by a phenomenon governed by a complex set of rules, more traditional modes of inquiry would typically ask which of these rules are the most dominant, and perhaps design experiments to highlight that in a specific context (e.g. is hydrophobic moment more important than the size of the hydrophobic face?) Here machine learning is more subtle. In our context of AMPs, machine learning does not use rules or define hierarchies, but is rather able to implicitly subsume all of these hierarchies into a single data-driven metric and tease out these complex relationships directly from the data with little human intervention. Whether a physico-chemical parameter is important or how two such parameters compare depends on how it contributes to this metric, which our calibrating experiments have shown to correlate strongly with NGC generation. Indeed, our classifier remains equally effective whether or not we interrogate it post hoc to ascertain how it performs discrimination, but doing so is valuable in revealing the connection between physico-chemical peptide properties and activity, and illuminating the capacity of the classifier to recapitulate understanding gained over a large body of prior ‘human learning’.
4.5. Discovery of unanticipated membrane-active peptide taxonomies
To further examine the sequence space of peptides that are dissimilar to known AMPs by sequence homology, we turned our classifier backwards to reflexively search for known and unknown peptide taxonomies that encode membrane activity. We pulled several peptides and proteins of interest with known crystal or NMR structures from the PDB and ran them through our classifier. At first blush, one might expect that these peptide sequences far in homology from known AMPs should have lower or negative values of σ. Surprisingly, a diverse number of existing peptide families were predicted by our classifier to be membrane active [1]. Within these families, we identified several candidate peptides that have not been previously described as having antimicrobial or membrane activity (figure 5a). We also found several proteins that have known membrane interactions, but had not been characterized to generate NGC. These include neuropeptides, amyloids, viral fusion proteins and many other membrane-associated proteins.
Figure 5.

Diverse families of membrane curvature-generating peptides discovered from a directed search of the unknown peptide sequence space. Plot of discovered peptides and proteins from the Protein Data Bank on a sequence map obtained from a Monte Carlo search of unknown peptide sequence space (a) and on the saddle-splay selection rule (b). Adapted from data in [1].
In fact, the positions along the saddle-splay rule of the newly discovered families show a similar distribution to that of the Pareto sequences (figure 5b). The majority fall along the trend (figure 5b, stars without borders) while a subset deviates, having less lysine and more arginine content relative to their hydrophobic content (figure 5b, stars with black borders). This behaviour makes sense because the purported functions of these proteins are more complicated than simply pore formation. For example, the members of the membrane anchor proteins family should likely persist in the lipid bilayer, which is reflected by their higher average hydrophobicity and closer correlation to the selection rule. In contrast, the topogenic peptides must penetrate through bilayers and deliver cargo rather than reside within membranes. Thus, possessing increased arginine content makes sense, because cell-penetrating peptides tend to have less hydrophobicity, and thereby deviate below the saddle-splay curve for AMPs. Using the directed search of the sequence space enabled by the SVM classifier, we can now efficiently identify and discover curvature-generating sequences in existing peptide families and also predict novel membrane-active peptides evolutionarily distant from any known AMPs.
5. Outlook
In this review, we discussed an unusual way to apply machine learning to understand membrane-active peptides. In doing so, we simultaneously highlight the potentialities and limitations of machine learning—we do not always ‘learn’ what we think we are learning. The present AMP system illustrates this limitation. By combining computational modelling with targeted experimentation, we were able to identify precisely what our SVM model learned about ‘AMP-ness’ (i.e. membrane activity rather than antimicrobial activity). Permeation of membranes can lead to cell death by disrupting the barrier function, so antimicrobial activity can be directly caused by membrane activity. However, antimicrobial activity of a peptide can also result from other functions, such as the binding of intracellular targets. Therefore, membrane activity and antimicrobial activity are not coextensive with one another. What is most surprising to us is not the sometimes unintended and exasperatingly precise literalness to machine learning results (with an uncanny resemblance to the Delphic Oracle in mythology), but rather the fact that machine learning came to essentially the same conclusions regarding membrane activity that researchers did in early work on AMPs. In learning how to distinguish antimicrobial from non-antimicrobial sequences, our classifier has foregrounded the most recognizable feature common to known AMPs, which is membrane activity. This is encoded within the SVM algorithm as the distance from hyperplane σ, which we show using experimental data to be a quantitative measure that correlates strongly with NGC generation. In this process, we showed the SVM model based its predictions on ‘rules’ it had learned regarding structure, amphiphilicity and sequence content, contextualizing findings from prior studies of peptides that generate NGC. By combining our classifier with Monte Carlo tools, we discovered multiple taxonomies of peptides and proteins with predicted membrane activity, some of which have never been previously described to possess that function. Based on these initial studies, there likely exist other undiscovered peptide and protein taxonomies with diverse primary functions and unanticipated membrane activity.
There are many potential applications of this screening tool. In addition to designing new AMPs for multi-drug-resistant infections, it also enables us to borrow inspiration from nature's unique ability to encode multiple functions into the same amino acid sequence. The screening tool identified several peptides and proteins with known ability to generate NGC, but also many others with diverse annotated functions, including receptor signalling, homeostasis and cellular transport. When combined with other protein design techniques, we can construct multi-functional molecules to have membrane permeating and/or antimicrobial activity. As a genomic tool, the classifier can enable rapid screening of new genomes for membrane-active sequences, and contribute to our knowledge of how membrane activity is used with other functions in prokaryotes, eukaryotes and viruses.
Authors' contributions
E.Y.L., A.L.F. and G.C.L.W. conceived and designed the studies and analysed the data. E.Y.L., B.M.F. and A.L.F. developed the computational tools. E.Y.L. carried out the laboratory experiments and performed bioinformatics and statistical analyses. E.Y.L., M.W.L., A.L.F. and G.C.L.W. drafted the manuscript. All authors gave final approval for publication.
Competing interests
We have no competing interests.
Funding
E.Y.L. acknowledges support from the T32 Systems and Integrative Biology Training Grant at University of California, Los Angeles (UCLA) (T32GM008185) and the T32 Medical Scientist Training Program at UCLA (T32GM008042). B.M.F. acknowledges support from National Science Foundation (NSF) Grant DMS 1345032 ‘MCTP: PI4: Program for Interdisciplinary and Industrial Internships at Illinois.’ G.C.L.W. acknowledges support from NIH grant no. 1R21AI122212. X-ray research was conducted at Stanford Synchrotron Radiation Lightsource, SLAC National Laboratory, supported by the US DOE Office of Basic Energy Sciences under contract no. DE-AC02-76SF00515.
References
- 1.Lee EY, Fulan BM, Wong GCL, Ferguson AL. 2016. Mapping membrane activity in undiscovered peptide sequence space using machine learning. Proc. Natl Acad. Sci. USA 113, 13 588–13 593. ( 10.1073/pnas.1609893113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee EY, Wong GCL, Ferguson AL. 2017. Machine learning-enabled discovery and design of membrane-active peptides. Bioorg. Med. Chem. ( 10.1016/j.bmc.2017.07.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zasloff M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415, 389–395. ( 10.1038/415389a) [DOI] [PubMed] [Google Scholar]
- 4.Shai Y. 1999. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta 1462, 55–70. ( 10.1016/S0005-2736(99)00200-X) [DOI] [PubMed] [Google Scholar]
- 5.Brogden KA. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3, 238–250. ( 10.1038/nrmicro1098) [DOI] [PubMed] [Google Scholar]
- 6.Hancock REW, Lehrer R. 1998. Cationic peptides: a new source of antibiotics. Trends Biotechnol. 16, 82–88. ( 10.1016/S0167-7799(97)01156-6) [DOI] [PubMed] [Google Scholar]
- 7.Hancock REW, Sahl H-G. 2006. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24, 1551–1557. ( 10.1038/nbt1267) [DOI] [PubMed] [Google Scholar]
- 8.Yeaman MR, Yount NY. 2003. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 55, 27–55. ( 10.1124/pr.55.1.2) [DOI] [PubMed] [Google Scholar]
- 9.Wang Z, Wang G. 2004. APD: the antimicrobial peptide database. Nucleic Acids Res. 32, D590–D592. ( 10.1093/nar/gkh025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang G, Li X, Wang Z. 2009. APD2: the updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 37, D933–D937. ( 10.1093/nar/gkn823) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang G, Li X, Wang Z. 2016. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 44, D1087–D1093. ( 10.1093/nar/gkv1278) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skarnes RC, Watson DW. 1957. Antimicrobial factors of normal tissues and fluids. Bacteriol. Rev. 21, 273–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zasloff M. 1987. Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl Acad. Sci. USA 84, 5449–5453. ( 10.1073/pnas.84.15.5449) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oren Z, Shai Y. 1998. Mode of action of linear amphipathic α-helical antimicrobial peptides. Biopolymers 47, 451–463. ( 10.1002/(SICI)1097-0282(1998)47:6%3C451::AID-BIP4%3E3.0.CO;2-F) [DOI] [PubMed] [Google Scholar]
- 15.Ganz T. 2003. Defensins: antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 3, 710–720. ( 10.1038/nri1180) [DOI] [PubMed] [Google Scholar]
- 16.Selsted ME, Novotny MJ, Morris WL, Tang YQ, Smith W, Cullor JS. 1992. Indolicidin, a novel bactericidal tridecapeptide amide from neutrophils. J. Biol. Chem. 267, 4292–4295. [PubMed] [Google Scholar]
- 17.Agerberth B, Lee JY, Bergman T, Carlquist M, Boman HG, Mutt V, Jörnvall H. 1991. Amino acid sequence of PR-39. Isolation from pig intestine of a new member of the family of proline-arginine-rich antibacterial peptides. Eur. J. Biochem. 202, 849–854. ( 10.1111/j.1432-1033.1991.tb16442.x) [DOI] [PubMed] [Google Scholar]
- 18.Wang G. 2008. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J. Biol. Chem. 283, 32637–32643. ( 10.1074/jbc.M805533200) [DOI] [PubMed] [Google Scholar]
- 19.Gesell J, Zasloff M, Opella SJ. 1997. Two-dimensional 1H NMR experiments show that the 23-residue magainin antibiotic peptide is an α-helix in dodecylphosphocholine micelles, sodium dodecylsulfate micelles, and trifluoroethanol/water solution. J. Biomol. NMR 9, 127–135. ( 10.1023/A:1018698002314) [DOI] [PubMed] [Google Scholar]
- 20.Terwilliger TC, Eisenberg D. 1982. The structure of melittin. I. Structure determination and partial refinement. J. Biol. Chem. 257, 6010–6015. [DOI] [PubMed] [Google Scholar]
- 21.Huang HW, Chen F-Y, Lee M-T. 2004. Molecular mechanism of peptide-induced pores in membranes. Phys. Rev. Lett. 92, 198304 ( 10.1103/PhysRevLett.92.198304) [DOI] [PubMed] [Google Scholar]
- 22.Hancock RE. W, Rozek A. 2002. Role of membranes in the activities of antimicrobial cationic peptides. FEMS Microbiol. Lett. 206, 143–149. ( 10.1111/j.1574-6968.2002.tb11000.x) [DOI] [PubMed] [Google Scholar]
- 23.Yang L, Harroun TA, Weiss TM, Ding L, Huang HW. 2001. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 81, 1475–1485. ( 10.1016/S0006-3495(01)75802-X) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ehrenstein G, Lecar H. 1977. Electrically gated ionic channels in lipid bilayers. Q. Rev. Biophys. 10, 1–34. ( 10.1017/S0033583500000123) [DOI] [PubMed] [Google Scholar]
- 25.Lee M-T, Chen F-Y, Huang HW. 2004. Energetics of pore formation induced by membrane active peptides. Biochemistry 43, 3590–3599. ( 10.1021/bi036153r) [DOI] [PubMed] [Google Scholar]
- 26.Spaar A, Münster C, Salditt T. 2004. Conformation of peptides in lipid membranes studied by X-ray grazing incidence scattering. Biophys J. 87, 396–407. ( 10.1529/biophysj.104.040667) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He K, Ludtke SJ, Worcester DL, Huang HW. 1996. Neutron scattering in the plane of membranes: structure of alamethicin pores. Biophys J. 70, 2659–2666. ( 10.1016/S0006-3495(96)79835-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bechinger B, Kim Y, Chirlian LE, Gesell J, Neumann JM, Montal M, Tomich J, Zasloff M, Opella SJ. 1991. Orientations of amphipathic helical peptides in membrane bilayers determined by solid-state NMR spectroscopy. J. Biomol. NMR 1, 167–173. ( 10.1007/BF01877228) [DOI] [PubMed] [Google Scholar]
- 29.Pouny Y, Rapaport D, Mor A, Nicolas P, Shai Y. 1992. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogs with phospholipid membranes. Biochemistry 31, 12 416–12 423. ( 10.1021/bi00164a017) [DOI] [PubMed] [Google Scholar]
- 30.Yamaguchi S, Huster D, Waring A, Lehrer RI, Kearney W, Tack BF, Hong M. 2001. Orientation and dynamics of an antimicrobial peptide in the lipid bilayer by solid-state NMR spectroscopy. Biophys J. 81, 2203–2214. ( 10.1016/S0006-3495(01)75868-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bechinger B. 1999. The structure, dynamics and orientation of antimicrobial peptides in membranes by multidimensional solid-state NMR spectroscopy. Biochim. Biophys. Acta 1462, 157–183. ( 10.1016/S0005-2736(99)00205-9) [DOI] [PubMed] [Google Scholar]
- 32.Ladokhin AS, White SH. 2001. ‘Detergent-like’ permeabilization of anionic lipid vesicles by melittin. Biochim. Biophys. Acta 1514, 253–260. ( 10.1016/S0005-2736(01)00382-0) [DOI] [PubMed] [Google Scholar]
- 33.Matsuzaki K, Murase O, Fujii N, Miyajima K. 1996. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry 35, 11 361–11 368. ( 10.1021/bi960016v) [DOI] [PubMed] [Google Scholar]
- 34.Hallock KJ, Lee D-K, Ramamoorthy A. 2003. MSI-78, an analogue of the magainin antimicrobial peptides, disrupts lipid bilayer structure via positive curvature strain. Biophys J. 84, 3052–3060. ( 10.1016/S0006-3495(03)70031-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamaguchi S, Hong T, Waring A, Lehrer RI, Hong M. 2002. Solid-state NMR investigations of peptide–lipid interaction and orientation of a β-sheet antimicrobial peptide, protegrin. Biochemistry 41, 9852–9862. ( 10.1021/bi0257991) [DOI] [PubMed] [Google Scholar]
- 36.Epand RM, Epand RF. 2011. Bacterial membrane lipids in the action of antimicrobial agents. J. Pept. Sci. 17, 298–305. ( 10.1002/psc.1319) [DOI] [PubMed] [Google Scholar]
- 37.Zachowski A. 1993. Phospholipids in animal eukaryotic membranes: transverse asymmetry and movement. Biochem. J. 294, 1–14. ( 10.1042/bj2940001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Meer G, Voelker DR, Feigenson GW. 2008. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124. ( 10.1038/nrm2330) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zimmerberg J, Kozlov MM. 2006. How proteins produce cellular membrane curvature. Nat. Rev. Mol. Cell Biol. 7, 9–19. ( 10.1038/nrm1784) [DOI] [PubMed] [Google Scholar]
- 40.Epand RF, Savage PB, Epand RM. 2007. Bacterial lipid composition and the antimicrobial efficacy of cationic steroid compounds (ceragenins). Biochim. Biophys. Acta 1768, 2500–2509. ( 10.1016/j.bbamem.2007.05.023) [DOI] [PubMed] [Google Scholar]
- 41.Siegel DP, Kozlov MM. 2004. The Gaussian curvature elastic modulus of N-monomethylated dioleoylphosphatidylethanolamine: relevance to membrane fusion and lipid phase behavior. Biophys J. 87, 366–374. ( 10.1529/biophysj.104.040782) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Som A, Yang L, Wong GCL, Tew GN. 2009. Divalent metal ion triggered activity of a synthetic antimicrobial in cardiolipin membranes. J. Am. Chem. Soc. 131, 15 102–15 103. ( 10.1021/ja9067063) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang L, et al. 2008. Mechanism of a prototypical synthetic membrane-active antimicrobial: efficient hole-punching via interaction with negative intrinsic curvature lipids. Proc. Natl. Acad. Sci. USA 105, 20 595–20 600. ( 10.1073/pnas.0806456105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang L, Gordon VD, Mishra A, Som A, Purdy KR, Davis MA, Tew GN, Wong GC. L. 2007. Synthetic antimicrobial oligomers induce a composition-dependent topological transition in membranes. J. Am. Chem. Soc. 129, 12 141–12 147. ( 10.1021/ja072310o) [DOI] [PubMed] [Google Scholar]
- 45.Hastie T, Tibshirani R, Friedman J. 2009. The elements of statistical learning. New York, NY: Springer Science & Business Media. [Google Scholar]
- 46.Turing AM. 1950. Computing machinery and intelligence. Mind 49, 433–460. ( 10.1093/mind/LIX.236.433) [DOI] [Google Scholar]
- 47.Graves A, Liwicki M, Fernández S, Bertolami R, Bunke H, Schmidhuber J. 2009. A novel connectionist system for unconstrained handwriting recognition. IEEE Trans. Pattern Anal. Mach. Intell. 31, 855–868. ( 10.1109/TPAMI.2008.137) [DOI] [PubMed] [Google Scholar]
- 48.Ganesan N, Venkatesh K, Rama MA. 2010. Application of neural networks in diagnosing cancer disease using demographic data. Inter. J. Comput. Theory Eng. 1, 81–97. ( 10.5120/476-783) [DOI] [Google Scholar]
- 49.Betechuoh BL, Marwala T, Tettey T. 2006. Autoencoder networks for HIV classification. Curr. Sci. 91, 1467–1473. [Google Scholar]
- 50.Agarwal M, Jain N, Kumar MM. 2010. Face recognition using eigen faces and artificial neural network. Inter. J. Comput. Theory Eng. 2, 624–629. ( 10.7763/IJCTE.2010.V2.213) [DOI] [Google Scholar]
- 51.Rothwell AC, Jagger LD, Dennis WR, Clarke DR. 2004. Intelligent spam detection system using an updateable neural analysis engine. Patent no. WO/2003/010680. [Google Scholar]
- 52.Balabin RM, Lomakina EI. 2009. Neural network approach to quantum-chemistry data: accurate prediction of density functional theory energies. J. Chem. Phys. 131, 074104 ( 10.1063/1.3206326) [DOI] [PubMed] [Google Scholar]
- 53.Silver D, et al. 2016. Mastering the game of Go with deep neural networks and tree search. Nature 529, 484–489. ( 10.1038/nature16961) [DOI] [PubMed] [Google Scholar]
- 54.Fjell CD, Jenssen H, Hilpert K, Cheung WA, Panté N, Hancock REW, Cherkasov A. 2009. Identification of novel antibacterial peptides by chemoinformatics and machine learning. J. Med. Chem. 52, 2006–2015. ( 10.1021/jm8015365) [DOI] [PubMed] [Google Scholar]
- 55.Mitchell JBO. 2014. Machine learning methods in chemoinformatics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 4, 468–481. ( 10.1002/wcms.1183) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hilpert K, Fjell CD, Cherkasov A. 2008. Short linear cationic antimicrobial peptides: screening, optimizing, and prediction. Methods Mol. Biol. 494, 127–159. ( 10.1007/978-1-59745-419-3_8) [DOI] [PubMed] [Google Scholar]
- 57.Lata S, Sharma BK, Raghava G. 2007. Analysis and prediction of antibacterial peptides. BMC Bioinformatics 8, 1 ( 10.1186/1471-2105-8-263) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cherkasov A, Hilpert K, Jenssen H, Fjell CD, Waldbrook M, Mullaly SC, Volkmer R, Hancock REW. 2008. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chem. Biol. 4, 65–74. ( 10.1021/cb800240j) [DOI] [PubMed] [Google Scholar]
- 59.Fjell CD, Jenssen H, Fries P, Aich P, Griebel P, Hilpert K, Hancock RE. W, Cherkasov A. 2008. Identification of novel host defense peptides and the absence of α-defensins in the bovine genome. Proteins Struct. Funct. Bioinform. 73, 420–430. ( 10.1002/prot.22059) [DOI] [PubMed] [Google Scholar]
- 60.Wang P, et al. 2011. Prediction of antimicrobial peptides based on sequence alignment and feature selection methods. PLoS ONE 6, e18476 ( 10.1371/journal.pone.0018476) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Torrent M, Andreu D, Nogués VM, Boix E. 2011. Connecting peptide physicochemical and antimicrobial properties by a rational prediction model. PLoS ONE 6, e16968 ( 10.1371/journal.pone.0016968) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maccari G, Di Luca M, Nifosí R, Cardarelli F, Signore G, Boccardi C, Bifone A. 2013. Antimicrobial peptides design by evolutionary multiobjective optimization. PLoS Comput. Biol. 9, e1003212 ( 10.1371/journal.pcbi.1003212) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiao X, Wang P, Lin W-Z, Jia J-H, Chou K-C. 2013. iAMP-2 L: a two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 436, 168–177. ( 10.1016/j.ab.2013.01.019) [DOI] [PubMed] [Google Scholar]
- 64.Giguère S, Laviolette F, Marchand M, Tremblay D, Moineau S, Liang X, Biron É, Corbeil J. 2015. Machine learning assisted design of highly active peptides for drug discovery. PLoS Comput. Biol. 11, e1004074 ( 10.1371/journal.pcbi.1004074) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schneider P, Müller AT, Gabernet G, Button AL, Posselt G, Wessler S, Hiss JA, Schneider G. 2017. Hybrid network model for ‘deep learning’ of chemical data: application to antimicrobial peptides. Mol. Inform. 36, 1600011 ( 10.1002/minf.201600011) [DOI] [PubMed] [Google Scholar]
- 66.Kozma D, Simon I, Tusnády GE. 2012. PDBTM: protein data bank of transmembrane proteins after 8 years. Nucleic Acids Res. 41, 1169 ( 10.1093/nar/gks1169) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Porto WF, Pires ÁS, Franco OL. 2012. CS-AMPPred: an updated SVM model for antimicrobial activity prediction in cysteine-stabilized peptides. PLoS ONE 7, e51444 ( 10.1371/journal.pone.0051444) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gelbart WM, Ben-Shaul A, Roux DD. 1994. Micelles, membranes, microemulsions, and monolayers. New York, NY: Springer. [Google Scholar]
- 69.Schmidt NW, Wong GCL. 2013. Antimicrobial peptides and induced membrane curvature: geometry, coordination chemistry, and molecular engineering. Curr. Opin. Solid State Mat. Sci. 17, 151–163. ( 10.1016/j.cossms.2013.09.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schmidt NW, et al. 2011. Criterion for amino acid composition of defensins and antimicrobial peptides based on geometry of membrane destabilization. J. Am. Chem. Soc. 133, 6720–6727. ( 10.1021/ja200079a) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schmidt NW, Mishra A, Lai GH, Wong GCL. 2010. Arginine-rich cell-penetrating peptides. FEBS Lett. 584, 1806–1813. ( 10.1016/j.febslet.2009.11.046) [DOI] [PubMed] [Google Scholar]
- 72.Schmidt NW, Mishra A, Wang J, DeGrado WF, Wong GC. L. 2013. Influenza virus A M2 protein generates negative gaussian membrane curvature necessary for budding and scission. J. Am. Chem. Soc. 135, 13710–13719. ( 10.1021/ja400146z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Braun AR, Sevcsik E, Chin P, Rhoades E, Tristram-Nagle S, Sachs JN. 2012. α-Synuclein induces both positive mean curvature and negative Gaussian curvature in membranes. J. Am. Chem. Soc. 134, 2613–2620. ( 10.1021/ja208316h) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cao D-S, Xu Q.-S, Liang Y-Z. 2013. propy: A tool to generate various modes of Chou's PseAAC. Bioinformatics 29, 960–962. ( 10.1093/bioinformatics/btt072) [DOI] [PubMed] [Google Scholar]
- 75.Pedregosa F, et al. 2011. Scikit-learn: machine learning in python. J. Mach. Learn. Res. 12, 2825–2830. [Google Scholar]
- 76.Mauri A, Ballabio D, Consonni V, Manganaro A, Todeschini R. 2008. Peptides multivariate characterisation using a molecular descriptor based approach. Match Commun. Math. Comput. Chem. 60, 671–690. [Google Scholar]
- 77.Li ZR, Lin HH, Han LY, Jiang L, Chen X, Chen YZ. 2006. PROFEAT: a web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res. 34, W32–W37. ( 10.1093/nar/gkl305) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bi J, Bennett K, Embrechts M, Breneman C, Song M. 2003. Dimensionality reduction via sparse support vector machines. J. Mach. Learn. Res. 3, 1229–1243. [Google Scholar]
- 79.Chou K-C. 2009. Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology. Curr. Proteomics 6, 262–274. ( 10.2174/157016409789973707) [DOI] [Google Scholar]
- 80.Grantham R. 1974. Amino acid difference formula to help explain protein evolution. Science 185, 862–864. ( 10.1126/science.185.4154.862) [DOI] [PubMed] [Google Scholar]
- 81.Gilks WR. 2005. Markov chain Monte Carlo. Chichester, UK: John Wiley & Sons, Ltd. [Google Scholar]
- 82.Gilks WR, Richardson S, Spiegelhalter D. 1995. Markov chain Monte Carlo in practice. Boca Raton, FL: CRC Press. [Google Scholar]
- 83.Geyer CJ. 1992. Practical Markov chain Monte Carlo. Stat. Sci. 7, 473–483. ( 10.2307/2246094) [DOI] [Google Scholar]
- 84.Arora JS. 2011. Introduction to optimum design. New York, NY: Academic Press. [Google Scholar]
- 85.Shoval O, Sheftel H, Shinar G, Hart Y, Ramote O, Mayo A, Dekel E, Kavanagh K, Alon U. 2012. Evolutionary trade-offs, Pareto optimality, and the geometry of phenotype space. Science 336, 1157–1160. ( 10.1126/science.1217405) [DOI] [PubMed] [Google Scholar]
- 86.Park CB, Kim HS, Kim SC. 1998. Mechanism of action of the antimicrobial peptide buforin II: buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 244, 253–257. ( 10.1006/bbrc.1998.8159) [DOI] [PubMed] [Google Scholar]
- 87.Subbalakshmi C, Sitaram N. 1998. Mechanism of antimicrobial action of indolicidin. FEMS Microbiol. Lett. 160, 91–96. ( 10.1111/j.1574-6968.1998.tb12896.x) [DOI] [PubMed] [Google Scholar]
- 88.Patrzykat A, Friedrich CL, Zhang L, Mendoza V, Hancock REW. 2002. Sublethal concentrations of pleurocidin-derived antimicrobial peptides inhibit macromolecular synthesis in Escherichia coli. Antimicrob. Agents Chemother. 46, 605–614. ( 10.1128/AAC.46.3.605-614.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brötz H, Bierbaum G, Leopold K, Reynolds PE, Sahl HG. 1998. The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrob. Agents Chemother. 42, 154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lande R, et al. 2007. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449, 564–569. ( 10.1038/nature06116) [DOI] [PubMed] [Google Scholar]
- 91.Zhang L.-J, Gallo RL. 2016. Antimicrobial peptides. Curr. Biol. 26, R14–R19. ( 10.1016/j.cub.2015.11.017) [DOI] [PubMed] [Google Scholar]
- 92.Kreyszig E. 1991. Differential geometry. New York, NY: Dover Publications. [Google Scholar]
- 93.Koller D, Lohner K. 2014. The role of spontaneous lipid curvature in the interaction of interfacially active peptides with membranes. Biochim. Biophys. Acta 1838, 2250–2259. ( 10.1016/j.bbamem.2014.05.013) [DOI] [PubMed] [Google Scholar]
- 94.Deserno M. 2015. Fluid lipid membranes: from differential geometry to curvature stresses. Chem. Phys. Lipids 185, 11–45. ( 10.1016/j.chemphyslip.2014.05.001) [DOI] [PubMed] [Google Scholar]
- 95.Bechinger B. 2009. Rationalizing the membrane interactions of cationic amphipathic antimicrobial peptides by their molecular shape. Curr. Opin. Colloid Interface Sci. 14, 349–355. ( 10.1016/j.cocis.2009.02.004) [DOI] [Google Scholar]
- 96.Baumgart T, Capraro BR, Zhu C, Das SL. 2011. Thermodynamics and mechanics of membrane curvature generation and sensing by proteins and lipids. Annu. Rev. Phys. Chem. 62, 483–506. ( 10.1146/annurev.physchem.012809.103450) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Antonny B. 2011. Mechanisms of membrane curvature sensing. Annu. Rev. Biochem. 80, 101–123. ( 10.1146/annurev-biochem-052809-155121) [DOI] [PubMed] [Google Scholar]
- 98.Stachowiak JC, Hayden CC, Sasaki DY. 2010. Steric confinement of proteins on lipid membranes can drive curvature and tubulation. Proc. Natl Acad. Sci. USA 107, 7781–7786. ( 10.1073/pnas.0913306107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stachowiak JC, Schmid EM, Ryan CJ, Ann HS, Sasaki DY, Sherman MB, Geissler PL, Fletcher DA, Hayden CC. 2012. Membrane bending by protein–protein crowding. Nat. Cell Biol. 14, 944–949. ( 10.1038/ncb2561) [DOI] [PubMed] [Google Scholar]
- 100.Koltover I, Salditt T, Rädler JO, Safinya CR. 1998. An inverted hexagonal phase of cationic liposome–DNA complexes related to DNA release and delivery. Science 281, 78–81. ( 10.1126/science.281.5373.78) [DOI] [PubMed] [Google Scholar]
- 101.Raviv U, Needleman DJ, Li Y, Miller HP, Wilson L, Safinya CR. 2005. Cationic liposome-microtubule complexes: pathways to the formation of two-state lipid–protein nanotubes with open or closed ends. Proc. Natl Acad. Sci. USA 102, 11 167–11 172. ( 10.1073/pnas.0502183102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tytler EM, Segrest JP, Epand RM, Nie SQ, Epand RF, Mishra VK, Venkatachalapathi YV, Anantharamaiah GM. 1993. Reciprocal effects of apolipoprotein and lytic peptide analogs on membranes. Cross-sectional molecular shapes of amphipathic alpha helixes control membrane stability. J. Biol. Chem. 268, 22 112–22 118. [PubMed] [Google Scholar]
- 103.Epand RM, Shai Y, Segrest JP, Anantharamiah GM. 1995. Mechanisms for the modulation of membrane bilayer properties by amphipathic helical peptides. Pept. Sci. 37, 319–338. ( 10.1002/bip.360370504) [DOI] [PubMed] [Google Scholar]
- 104.Segrest JP, De Loof H, Dohlman JG, Brouillette CG, Anantharamaiah GM. 1990. Amphipathic helix motif: classes and properties. Proteins Struct. Funct. Bioinform. 8, 103–117. ( 10.1002/prot.340080202) [DOI] [PubMed] [Google Scholar]
- 105.Zemel A, Ben-Shaul A, May S. 2008. Modulation of the spontaneous curvature and bending rigidity of lipid membranes by interfacially adsorbed amphipathic peptides. J. Phys. Chem. B 112, 6988–6996. ( 10.1021/jp711107y) [DOI] [PubMed] [Google Scholar]
- 106.Wieprecht T, Dathe M, Epand RM, Beyermann M, Krause E, Maloy WL, MacDonald DL, Bienert M. 1997. Influence of the angle subtended by the positively charged helix face on the membrane activity of amphipathic, antibacterial peptides. Biochemistry 36, 12 869–12 880. ( 10.1021/bi971398n) [DOI] [PubMed] [Google Scholar]
- 107.Drin G, Antonny B. 2009. Amphipathic helices and membrane curvature. FEBS Lett. 584, 1840–1847. ( 10.1016/j.febslet.2009.10.022) [DOI] [PubMed] [Google Scholar]
- 108.Chen F-Y, Lee M-T, Huang HW. 2003. Evidence for membrane thinning effect as the mechanism for peptide-induced pore formation. Biophys J. 84, 3751–3758. ( 10.1016/S0006-3495(03)75103-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tristram-Nagle S, Chan R, Kooijman E, Uppamoochikkal P, Qiang W, Weliky DP, Nagle JF. 2010. HIV fusion peptide penetrates, disorders, and softens T-cell membrane mimics. J. Mol. Biol. 402, 139–153. ( 10.1016/j.jmb.2010.07.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Szleifer I, Kramer D, Ben-Shaul A, Roux D, Gelbart WM. 1988. Curvature elasticity of pure and mixed surfactant films. Phys. Rev. Lett. 60, 1966–1969. ( 10.1103/PhysRevLett.60.1966) [DOI] [PubMed] [Google Scholar]
- 111.Dathe M, Wieprecht T. 1999. Structural features of helical antimicrobial peptides: their potential to modulate activity on model membranes and biological cells. Biochim. Biophys. Acta 1462, 71–87. ( 10.1016/S0005-2736(99)00201-1) [DOI] [PubMed] [Google Scholar]
- 112.Yang L, Weiss TM, Lehrer RI, Huang HW. 2000. Crystallization of antimicrobial pores in membranes: magainin and protegrin. Biophys J. 79, 2002–2009. ( 10.1016/S0006-3495(00)76448-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ludtke SJ, He K, Heller WT, Harroun TA, Yang L, Huang HW. 1996. Membrane pores induced by magainin. Biochemistry 35,13 723–13 728. ( 10.1021/bi9620621) [DOI] [PubMed] [Google Scholar]
- 114.Saiman L, Tabibi S, Starner TD, San Gabriel P, Winokur PL, Jia HP, McCray PB, Tack BF. 2001. Cathelicidin peptides inhibit multiply antibiotic-resistant pathogens from patients with cystic fibrosis. Antimicrob. Agents Chemother. 45, 2838–2844. ( 10.1128/AAC.45.10.2838-2844.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kalfa VC, Jia HP, Kunkle RA, McCray PB, Tack BF, Brogden KA. 2001. Congeners of SMAP29 kill ovine pathogens and induce ultrastructural damage in bacterial cells. Antimicrob. Agents Chemother. 45, 3256–3261. ( 10.1128/AAC.45.11.3256-3261.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yu Y, Vroman JA, Bae SC, Granick S. 2010. Vesicle budding induced by a pore-forming peptide. J. Am. Chem. Soc. 132, 195–201. ( 10.1021/ja9059014) [DOI] [PubMed] [Google Scholar]
- 117.Falagas ME, Kasiakou SK. 2005. Colistin: the revival of polymyxins for the management of multidrug-resistant Gram-negative bacterial infections. Clin. Infect. Dis. 40, 1333–1341. ( 10.1086/429323) [DOI] [PubMed] [Google Scholar]
- 118.Gidalevitz D, Ishitsuka Y, Muresan AS, Konovalov O, Waring AJ, Lehrer RI, Lee KYC. 2003. Interaction of antimicrobial peptide protegrin with biomembranes. Proc. Natl. Acad. Sci. USA 100, 6302–6307. ( 10.1073/pnas.0934731100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schmidt NW, Tai KP, Kamdar K, Mishra A, Lai GH, Zhao K, Ouellette AJ, Wong GCL. 2012. Arginine in α-defensins: differential effects on bactericidal activity correspond to geometry of membrane curvature generation and peptide–lipid phase behavior. J. Biol. Chem. 287, 21 866–21 872. ( 10.1074/jbc.M112.358721) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lee MW, Chakraborty S, Schmidt NW, Murgai R, Gellman SH, Wong GCL. 2014. Two interdependent mechanisms of antimicrobial activity allow for efficient killing in nylon-3-based polymeric mimics of innate immunity peptides. Biochim. Biophys. Acta 1838, 2269–2279. ( 10.1016/j.bbamem.2014.04.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Xiong M, et al. 2015. Helical antimicrobial polypeptides with radial amphiphilicity. Proc. Natl Acad. Sci. USA 112, 13 155–13 160. ( 10.1073/pnas.1507893112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yao H, Lee MW, Waring AJ, Wong GC. L, Hong M. 2015. Viral fusion protein transmembrane domain adopts β-strand structure to facilitate membrane topological changes for virus–cell fusion. Proc. Natl. Acad. Sci. USA 112, 10 926–10 931. ( 10.1073/pnas.1501430112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Epand RM. 1993. The amphipathic helix. Boca Raton, FL: CRC Press. [Google Scholar]
- 124.Eisenberg D, Weiss RM, Terwilliger TC, Wilcox W. 1982. Hydrophobic moments and protein structure. Faraday Symp. Chem. Soc. 17, 109 ( 10.1039/fs9821700109) [DOI] [Google Scholar]
- 125.Dathe M, Wieprecht T, Nikolenko H, Handel L, Maloy WL, MacDonald DL, Beyermann M, Bienert M. 1997. Hydrophobicity, hydrophobic moment and angle subtended by charged residues modulate antibacterial and haemolytic activity of amphipathic helical peptides. FEBS Lett. 403, 208–212. ( 10.1016/S0014-5793(97)00055-0) [DOI] [PubMed] [Google Scholar]
- 126.Hu K, et al. 2013. A critical evaluation of random copolymer mimesis of homogeneous antimicrobial peptides. Macromolecules 46, 1908–1915. ( 10.1021/ma302577e) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pérez-Payá E, Houghten RA, Blondelle SE. 1995. The role of amphipathicity in the folding, self-association and biological activity of multiple subunit small proteins. J. Biol. Chem. 270, 1048–1056. ( 10.1074/jbc.270.3.1048) [DOI] [PubMed] [Google Scholar]
- 128.Subbalakshmi C, Nagaraj R, Sitaram N. 1999. Biological activities of C-terminal 15-residue synthetic fragment of melittin: design of an analog with improved antibacterial activity. FEBS Lett. 448, 62–66. ( 10.1016/S0014-5793(99)00328-2) [DOI] [PubMed] [Google Scholar]
- 129.Wieprecht T, Dathe M, Krause E, Beyermann M, Maloy WL, MacDonald DL, Bienert M. 1997. Modulation of membrane activity of amphipathic, antibacterial peptides by slight modifications of the hydrophobic moment. FEBS Lett. 417, 135–140. ( 10.1016/S0014-5793(97)01266-0) [DOI] [PubMed] [Google Scholar]
- 130.Dathe M, Schümann M, Wieprecht T, Winkler A, Beyermann M, Krause E, Matsuzaki K, Murase O, Bienert M. 1996. Peptide helicity and membrane surface charge modulate the balance of electrostatic and hydrophobic interactions with lipid bilayers and biological membranes. Biochemistry 35, 12 612–12 622. ( 10.1021/bi960835f) [DOI] [PubMed] [Google Scholar]
- 131.Wieprecht T, Dathe M, Beyermann M, Krause E, Maloy WL, MacDonald DL, Bienert M. 1997. Peptide hydrophobicity controls the activity and selectivity of magainin 2 amide in interaction with membranes. Biochemistry 36, 6124–6132. ( 10.1021/bi9619987) [DOI] [PubMed] [Google Scholar]
- 132.Schmidt NW, Lis M, Zhao K, Lai GH, Alexandrova AN, Tew GN, Wong GCL. 2012. Molecular basis for nanoscopic membrane curvature generation from quantum mechanical models and synthetic transporter sequences. J. Am. Chem. Soc. 134, 19 207–19 216. ( 10.1021/ja308459j) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wu Z, Cui Q, Yethiraj A. 2013. Why do arginine and lysine organize lipids differently? Insights from coarse-grained and atomistic simulations. J. Phys. Chem. B 117, 12 145–12 156. ( 10.1021/jp4068729) [DOI] [PubMed] [Google Scholar]
- 134.Cui Q, Zhang L, Wu Z, Yethiraj A. 2013. Generation and sensing of membrane curvature: where materials science and biophysics meet. Curr. Opin. Solid State Mat. Sci. 17, 164–174. ( 10.1016/j.cossms.2013.06.002) [DOI] [Google Scholar]
- 135.Mishra A, Gordon VD, Yang L, Coridan R, Wong GCL. 2008. HIV TAT forms pores in membranes by inducing saddle-splay curvature: potential role of bidentate hydrogen bonding. Angew. Chem. Int. Ed. Engl. 47, 2986–2989. ( 10.1002/anie.200704444) [DOI] [PubMed] [Google Scholar]