

Abstract
Emerging preclinical evidence suggests the critical role of androgen-mediated androgen receptor (AR) signals in the development of bladder cancer. However, little is known about the efficacy of enzalutamide, an AR signaling inhibitor, in androgen-induced urothelial tumorigenesis. We therefore aimed to assess the effects of enzalutamide on neoplastic transformation of urothelial cells. An immortalized normal urothelial cell line SVHUC stably expressing wild-type AR (SVHUC-AR) was exposed to a chemical carcinogen 3-methylcholanthrene (MCA) to induce neoplastic transformation, and subsequently cultured for 6 weeks in the presence of anti-androgens, including enzalutamide, hydroxyflutamide, and bicalutamide. Tumorigenesis was then monitored, using plate and soft agar colony formation assays as well as mouse xenograft models. In SVHUC-AR cells exposed to MCA, each anti-androgen inhibited AR-mediated transcriptional activity, but only enzalutamide prevented AR nuclear translocation. In vitro transformation showed that treatment with each anti-androgen during the process of neoplastic transformation reduced the efficiency of colony formation in vitro. Compared with mock treatment, culture with enzalutamide (P = 0.028), hydroxyflutamide (P = 0.033), or bicalutamide (P = 0.038) also resulted in prevention/retardation of tumor formation in male NOD-SCID mice. In addition, anti-androgens up-regulated the expression of several molecules that play a protective role in bladder tumorigenesis, including p53, p21, and PTEN, and down-regulated that of several oncogenic genes, such as c-myc, cyclin D1, and cyclin E, in MCA-exposed SVHUC-AR cells. Thus, enzalutamide, flutamide, and bicalutamide were found to similarly prevent neoplastic transformation of urothelial cells. These findings offer a potential chemopreventive approach for urothelial tumors using AR antagonists.
Keywords: Bicalutamide, enzalutamide, flutamide, neoplastic transformation, urothelial cancer
Introduction
Urinary bladder cancer has been one of commonly diagnosed malignancies predominantly affecting males throughout the world [1]. The imbalance in the risk of bladder cancer between the genders has thus been observed for many years. In the United States, it is estimated in 2017 that 60,490 men and 18,540 women will newly develop bladder cancer and that 12,240 men and 4,630 women will die of the disease [2]. Cigarette smoking and exposure to industrial chemicals were thought to be major causes that might have contributed to male dominance in bladder cancer. However, the male-to-female ratio of the tumor remains virtually unchanged before and after controlling for these carcinogenic factors [1-3]. In the meantime, recent advances in bladder cancer research have indicated that sex hormones and their receptor signals, in addition to lifestyle or environmental risk factors, play an important role in urothelial carcinogenesis [4-7]. Specifically, androgen deprivation therapy or androgen receptor (AR) knockout resulted in inhibition of tumor development in male rodents treated with a bladder carcinogen N-butyl-N-(4-hydroxybutyl)nitrosamine [8-10]. AR signals have also been found to down-regulate the expression of P450 CYP4B1 [11], UDP-glucuronosyltransferase-1A (UGT1A) [12], and GATA3 [13], all of which are known to inhibit urothelial tumorigenesis. These findings may at least partially explain the gender-specific difference in the incidence of bladder cancer. In addition, recent retrospective clinical studies have suggested that androgen deprivation therapy for prostate cancer prevents bladder cancer development [14] and recurrence [15].
Patients with superficial bladder tumor are at high risk of tumor recurrence after transurethral surgery and currently available intravesical pharmacotherapy. Importantly, some of these patients ultimately develop more invasive disease for which aggressive treatment modalities, such as radical cystectomy and systemic chemotherapy, are often required. Therefore, novel therapeutic options that more effectively prevent the recurrence of superficial bladder tumor need to be urgently developed.
Enzalutamide is a synthetic AR signaling inhibitor that not only blocks androgen binding to the AR but is also shown to prevent AR nuclear translocation, DNA binding, and co-activator recruitment in prostate cancer cells [16-19]. Due to its mechanisms of action, enzalutamide likely provides a more significant and clinically meaningful benefit, compared with the first generation non-steroidal anti-androgenic drugs including flutamide and bicalutamide, in improving the prognosis especially in men with castration-resistant prostate cancer. Meanwhile, some of the above studies [8,9,12,13] have indicated the inhibition of bladder carcinogenesis by hydroxyflutamide and/or bicalutamide. In contrast, the efficacy of enzalutamide in bladder cancer development remains largely unknown. In the current study, we assessed the inhibitory effects of enzalutamide on neoplastic transformation of urothelial cells by comparing with those of hydroxyflutamide and bicalutamide.
Materials and methods
Cell culture and chemicals
The SV40-immortalized human urothelial cell line (SVHUC) was originally obtained from the American Type Culture Collection and was recently authenticated using GenePrint 10 System (Promega). A stable cell line expressing a full-length wild-type human AR (i.e. SVHUC-AR) and a control line expressing only the vector (i.e. SVHUC-control) were described in our previous studies [12,13]. These cells were maintained in Kaighn’s Modification of Ham’s F-12K (Mediatech) supplemented with 10% fetal bovine serum (FBS) and penicillin (100 units/mL)/streptomycin (100 units/mL) at 37°C in a humidified atmosphere of 5% CO2. We obtained hydroxyflutamide from Sigma and bicalutamide from Santa Cruz Biotechnology. Enzalutamide was provided by Astellas Pharma Global Development and Medivation, Inc.
Western blot
Protein extraction and western blotting were performed, as described previously [19-21] with minor modifications. Equal amounts of protein (30 µg) obtained from cell extracts were separated in 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membrane (Immun-Blot PVDF Membrane, Bio-Rad) by electroblotting. Specific antibody binding was detected, using an anti-AR antibody (clone N20; dilution 1:1000; Santa Cruz Biotechnology) or an anti-GAPDH antibody (clone 6C5; dilution 1:5000; Santa Cruz Biotechnology), and a secondary antibody (rabbit IRDye 680LT or mouse IRDye 800CW, LI-COR), followed by scanning with an infrared imaging system (Odyssey, LI-COR).
Reverse transcription (RT) and real-time polymerase chain reaction (PCR)
Total RNA (0.5 μg) was isolated from cultured cells, using TRIzol (Invitrogen), and reverse transcribed with 1 μM oligo (dT) primers (Qiagen) and 4 units of Omniscript reverse transcriptase (Qiagen) in a total volume of 20 μL. Real-time PCR was then performed, using iQ SYBR Green Supermix (Bio-Rad), as described previously [19-21]. The primer sequences are given in Table 1. Human GAPDH was used as an internal control.
Table 1.
Sequences of PCR primers
| Gene | Sense | Anti-sense | Product size (bp) |
|---|---|---|---|
| p53 | 5’-GGAGGGGCGATAAATACC-3’ | 5’-AACTGTAACTCCTCAGGCAGGC-3’ | 132 |
| p27 | 5’-CGAGTGGCAAGAGGTGGAGA-3 | 5’-GGAGCCCCAATTAAAGGCG-3’ | 144 |
| PTEN | 5’-GTTTACCGGCAGCATCAAAT-3’ | 5’-CCCCCACTTTAGTGCACAGT-3’ | 197 |
| c-myc | 5’-ACCAGATCCCGGAGTTGGAA-3’ | 5’-CGTCGTTTCCGCAACAAGTC-3’ | 136 |
| Cyclin D1 | 5’-GTGCTGCGAAGTGGAAACC-3’ | 5’-ATCCAGGTGGCGACGATCT-3’ | 174 |
| Cyclin E | 5’-GCCAGCCTTGGGACAATAATG-3’ | 5’-CTTGCACGTTGAGTTTGGGT-3’ | 104 |
| GAPDH | 5’-AAGGTGAAGGTCGGAGTCAAC-3’ | 5’-GGGGTCATTGATGGCAACAATA-3’ | 102 |
Reporter gene assay
Cells at a density of 50-70% confluence in 24-well plates were co-transfected with 250 ng of mouse mammary tumor virus (MMTV)-Luc reporter plasmid DNA [22] and 2.5 ng of pRL-TK plasmid DNA, using GeneJuice (Novagen), as described previously [19,21]. After transfection, the cells were cultured in the presence or absence of AR antagonists for 24 hours. Cell lysates were then assayed for luciferase activity determined using a Dual-Glo™ Luciferase Assay System (Promega) and luminometer (FLUOstar Omega, BMG Labtech).
Immunofluorescent staining
Cells plated onto 8-well chamber slides (Nunc™ Lab-Tek™, Thermo Fisher Scientific) were cultured in the presence or absence of AR antagonists for 24 hours. At the end of the drug treatment, the adherent cells were fixed by 4% paraformaldehyde. The cells were blocked with 1% bovine serum albumin for 1 hour at 37°C, and an anti-AR antibody (clone N20; dilution 1:50) was added and incubated overnight at 4°C. 4’,6’-diamidino-2-phenylindole (DAPI) was used to visualize nuclei. Fluorescence images were acquired with a fluorescence microscope (Olympus BX41) and a digital camera (Olympus DP70). The number of nuclear staining was quantitated in 6 randomly selected visual fields per chamber by a single observer who was unaware of the treatment group for the cells.
In vitro transformation
We used a method for neoplastic transformation in SVHUC with exposure to a carcinogen 3-methylcholanthrene (MCA), as described in a previous study [23], with minor modifications. Briefly, cells (2 × 106/10-cm culture dish incubated for 48 hours) were cultured in serum-free F-12K containing 5 μg/mL MCA (Sigma). After the first 24 hours of MCA exposure, FBS (1%) was added to the medium. After additional 24 hours, the cells were cultured in medium containing 5% FBS without MCA until near confluence. Subcultured cells (1/3 split) were again cultured in the presence of 5 μg/mL MCA for a 48-hour exposure period, using the above protocol. MCA exposure was repeated one more time. These cells were then subcultured for 6 weeks in the presence or absence of each AR antagonist and thereafter used for further assays.
Plate colony formation and soft agar colony formation assays
The SVHUC sublines exposed to MCA were used for colony formation assays, as described previously [13,21], with minor modifications. For plate colony formation assay, 5 × 102 cells per well seeded in 12-well plates were allowed to grow until colonies in the control well were easily distinguishable. Fresh medium was replaced every other day. The cells were fixed with methanol, stained with 0.1% crystal violet, and photographed. For soft agar colony formation assay, after being dispersed to single cells, they were suspended at 2 × 104 cells per well in 6-well plates in a solution of 0.25% agarose and F-12K with 1% FBS. The cell suspension was layered into wells with a solidified 0.5% agarose and 1% FBS-containing medium sublayer. F-12K medium containing 10% FBS was then added and changed every three days. The cells were cultured until colonies were visible and stained with 0.05% crystal violet. Colonies (≥20 cells) were counted by adjusting the depth of the field and changing the focus under the dissecting microscope. The numbers of colonies and their areas were quantitated, using ImageJ software (National Institutes of Health).
Tumor formation in vivo
The animal protocol in accordance with National Institutes of Health Guidelines for the Care and Use of Experimental Animals was approved by the Institutional Animal Care and Use Committee. SVHUC cells (1 × 106) exposed to MCA, as described above, were suspended, mixed with 100 μL Matrigel (BD Biosciences), and subcutaneously injected into the flank of 6-week-old male NOD-SCID mice (Johns Hopkins University Research Animal Resources), as described previously [19,21]. Tumor formation was then monitored every day.
Statistical analysis
Student’s t-test or Mann-Whitney U test was used to compare the numerical data. P values less than 0.05 were considered to be statistically significant.
Results
Suppression of AR expression and its activity by enzalutamide in urothelial cells undergoing neoplastic transformation
We first assessed the effects of short-term treatment with enzalutamide on AR expression and its activity in immortalized human normal urothelial SVHUC cells with the carcinogen challenge. We compared the expression levels of AR protein in MCA-exposed SVHUC sublines treated with hydroxyflutamide, bicalutamide, or enzalutamide. Compared with mock treatment, treatment with each AR antagonist resulted in down-regulation of AR expression in SVHUC-AR cells cultured in medium containing normal FBS (Figure 1A). Consistent with our previous observations [12,13], AR was undetectable in SVHUC-control cells. In addition, MCA exposure did not alter the levels of AR expression in SVHUC-AR cells. We also compared the transcriptional activities of AR in the extracts of MCA-exposed SVHUC sublines with transfection of an AR luciferase reporter plasmid and subsequent treatment with AR antagonists. Treatment with hydroxyflutamide, bicalutamide, or enzalutamide similarly inhibited AR-mediated transactivation in SVHUC-AR cells cultured in medium containing normal FBS (Figure 1B). We further performed immunofluorescence to investigate whether enzalutamide could affect nuclear translocation of AR in urothelial cells (Figure 1C). Hydroxyflutamide or bicalutamide treatment in MCA-SVHUC-AR cells cultured in medium containing normal FBS did not significantly alter AR expression in their nuclei. In contrast, enzalutamide treatment significantly reduced nuclear AR expression, compared with mock treatment.
Figure 1.
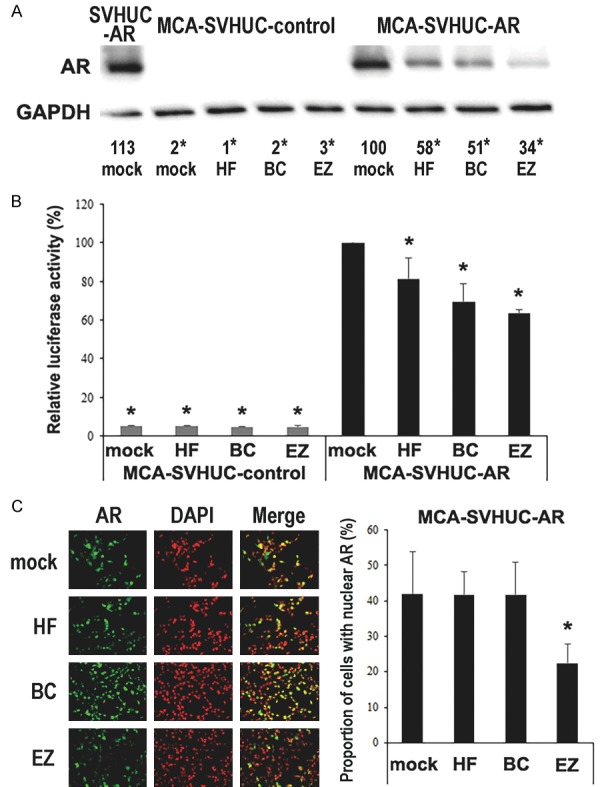
Effects of enzalutamide on AR in MCA-exposed urothelial cells. A. Cell extracts from SVHUC-AR without MCA exposure or SVHUC-control/SVHUC-AR exposed to MCA and subsequently treated with ethanol (mock), hydroxyflutamide (HF; 5 μM), bicalutamide (BC; 5 μM), or enzalutamide (EZ; 5 μM) for 48 hours were analyzed on western blotting, using an antibody to AR (110 kDa). GAPDH (37 kDa) served as an internal control. The means of densitometry values for AR standardized by GAPDH that are relative to the value of mock treatment (6th lane; set as 100%) from three independent experiments are included below the lanes. B. Luciferase reporter gene assay was performed in SVHUC-control or SVHUC-AR exposed to MCA, transfected with MMTV-Luc and pRL-TK, and subsequently treated with ethanol (mock), hydroxyflutamide (HF; 5 μM), bicalutamide (BC; 5 μM), or enzalutamide (EZ; 5 μM) for 24 hours. Luciferase activity is presented relative to that of mock treatment in SVHUC-AR cells. Each value represents the mean + standard deviation from three independent experiments. C. Immunofluorescence of AR in SVHUC-AR cells exposed to MCA and subsequently treated with ethanol (mock), hydroxyflutamide (HF; 5 μM), bicalutamide (BC; 5 μM), or enzalutamide (EZ; 5 μM) for 24 hours. We merged the images between AR and DAPI that was used to visualize nuclei, and the proportion of cells with AR expression only in their nuclei was quantitated. Each value represents the mean + standard deviation of 6 determinants. *P < 0.05 (vs. mock treatment in SVHUC-AR).
Suppression of neoplastic transformation of urothelial cells by enzalutamide
We next assessed the efficacy of long-term treatment with enzalutamide in urothelial tumorigenesis via an in vitro transformation system in SVHUC cells where stepwise transformation could be observed upon exposure to a chemical carcinogen MCA [23]. MCA-exposed SVHUC sublines were treated with hydroxyflutamide, bicalutamide, or enzalutamide during the process of neoplastic transformation. Tumorigenic activity was then monitored, using colony formation assays and mouse xenograft models without further anti-androgen treatment that could directly affect cell/tumor growth.
Plate and soft agar colony formation assays that determine the ability of anchorage-dependent and anchorage-independent growth, respectively, were performed in MCA-exposed SVHUC sublines subsequently treated with hydroxyflutamide, bicalutamide, or enzalutamide for 6 weeks. In the plate colony formation assay, all 3 anti-androgens significantly reduced the area of SVHUC-AR colonies, compared with mock treatment, after 2-week culture without anti-androgens (Figure 2). Only enzalutamide treatment also resulted in a significant decrease in the number of SVHUC-AR colonies. However, there were no significant effects of AR antagonists on the number or area of AR-negative SVHUC-control colonies. Similarly, in the soft agar colony formation assay assessing colony number at 3 weeks, hydroxyflutamide, bicalutamide, or enzalutamide significantly reduced the efficiency of colony formation in SVHUC-AR, but not in SVHUC-control (Figure 3). In these assays, higher numbers/areas of colonies were observed in mock-treated SVHUC-AR cells than in mock-treated SVHUC-control cells.
Figure 2.
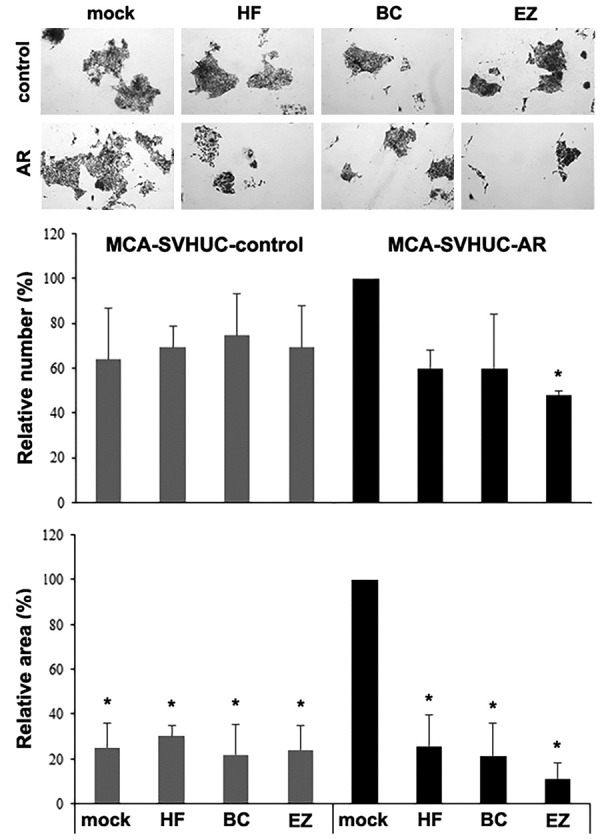
Effects of enzalutamide on anchorage-dependent growth of MCA-exposed urothelial cells. SVHUC-control or SVHUC-AR exposed to MCA and subsequently cultured for 6 weeks in the presence of ethanol (mock), hydroxyflutamide (HF; 5 μM), bicalutamide (BC; 5 μM), or enzalutamide (EZ; 5 μM) was seeded for plate colony formation assay (cultured for additional 2 weeks). The number and area of the colonies were counted and are presented relative to those in mock-treated SVHUC-AR cells. Each value represents the mean + standard deviation from three independent experiments. *P < 0.05 (vs. mock treatment in SVHUC-AR).
Figure 3.
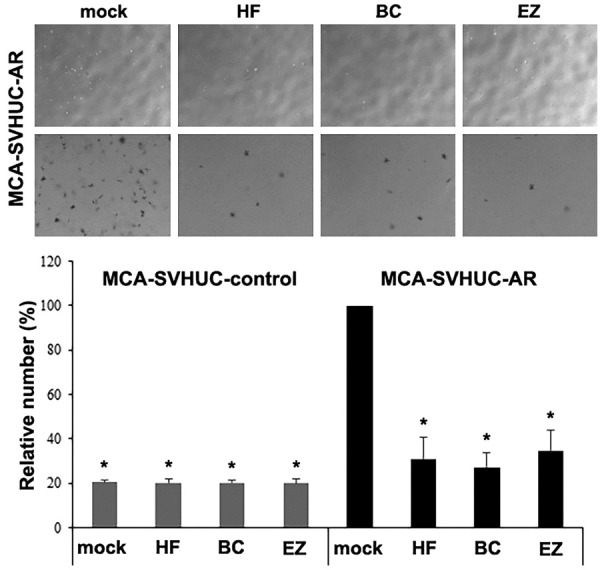
Effects of enzalutamide on anchorage-independent growth of MCA-exposed urothelial cells. SVHUC-control or SVHUC-AR exposed to MCA and subsequently cultured for 6 weeks in the presence of ethanol (mock), hydroxyflutamide (HF; 5 μM), bicalutamide (BC; 5 μM), or enzalutamide (EZ; 5 μM) was seeded for soft agar colony formation assay (cultured for additional 3 weeks). The number of the colonies was counted and is presented relative to that in mock-treated SVHUC-AR cells. Each value represents the mean + standard deviation from three independent experiments. *P < 0.05 (vs. mock treatment in SVHUC-AR).
Mouse xenograft models were also employed to assess urothelial tumorigenesis in vivo. SVHUC cells with the carcinogen challenge and subsequent 6-week hydroxyflutamide/bicalutamide/enzalutamide treatment were inoculated subcutaneously into immunocompromised mice, and tumor formation was monitored as an endpoint. In accordance with our previous in vitro transformation data [13], AR overexpression significantly induced tumor formation (mock-treated SVHUC-control vs. mock-treated SVHUC-AR, P = 0.032). In addition, hydroxyflutamide (P = 0.033), bicalutamide (P = 0.038), or enzalutamide (P = 0.028) treatment strikingly delayed or prevented the formation of SVHUC-AR xenograft tumors compared with mock treatment (Figure 4). In the SVHUC-AR xenografts, there was no statistically significant difference in tumor formation between treatments with hydroxyflutamide vs. bicalutamide (P = 0.707), hydroxyflutamide vs. enzalutamide (P = 0.713), or bicalutamide vs. enzalutamide (P = 0.974). These 3 anti-androgens showed no significant effects on the formation of SVHUC-control xenograft tumors.
Figure 4.
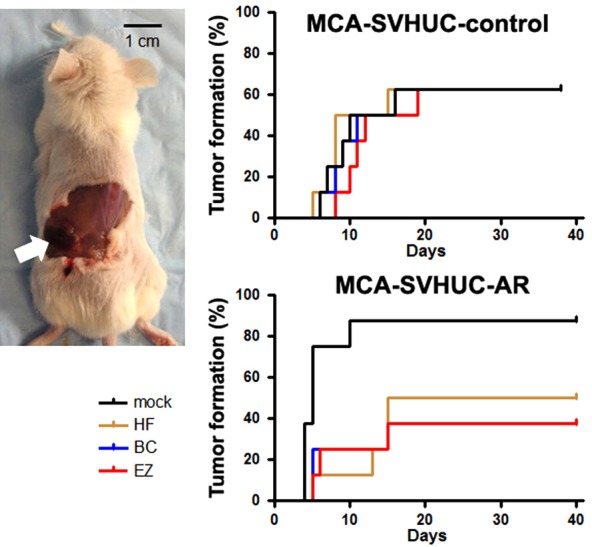
Effects of enzalutamide on urothelial tumor formation in mouse xenograft models. SVHUC-control or SVHUC-AR exposed to MCA and subsequently cultured for 6 weeks in the presence of ethanol (mock), hydroxyflutamide (HF; 5 μM), bicalutamide (BC; 5 μM), or enzalutamide (EZ; 5 μM) was suspended, mixed with Matrigel (1 × 106 cells/100 μL), and subcutaneously implanted into the flank of 6-week-old male NOD-SCID mice (n = 8 in each group). The endpoint for this study was tumor formation (exceeding 10 mm3 in its estimated volume [by the following formula: (short diameter)2 × (longest diameter) × 0.5] or 5 mm in greatest dimension). Comparisons were made by log-rank test.
Up/down-regulation of the expression of tumor suppressor genes/oncogenes by enzalutamide in urothelial cells undergoing neoplastic transformation
By using a quantitative RT-PCR method, we finally compared the expression levels of various molecules that are known to play a protective role in bladder tumorigenesis, such as p53, p27, and PTEN (Figure 5A), and oncogenic molecules, including c-myc, cyclin D1, and cyclin E (Figure 5B), in SVHUC cells with MCA exposure and 6-week hydroxyflutamide/bicalutamide/enzalutamide treatment. As expected, significantly lower and higher expression levels of tumor suppressor genes and oncogenes, respectively, were detected in mock-treated SVHUC-AR cells than in mock-treated SVHUC-control cells. Moreover, 3 anti-androgens up-regulated the expression of these tumor suppressors while down-regulated the expression of these oncogenes in the resultant SVHUC-AR cells, but not in the SVHUC-control cells. No significant effects of AR antagonists on the expression of these genes were observed in SVHUC-AR cells without the carcinogen challenge (data not shown).
Figure 5.
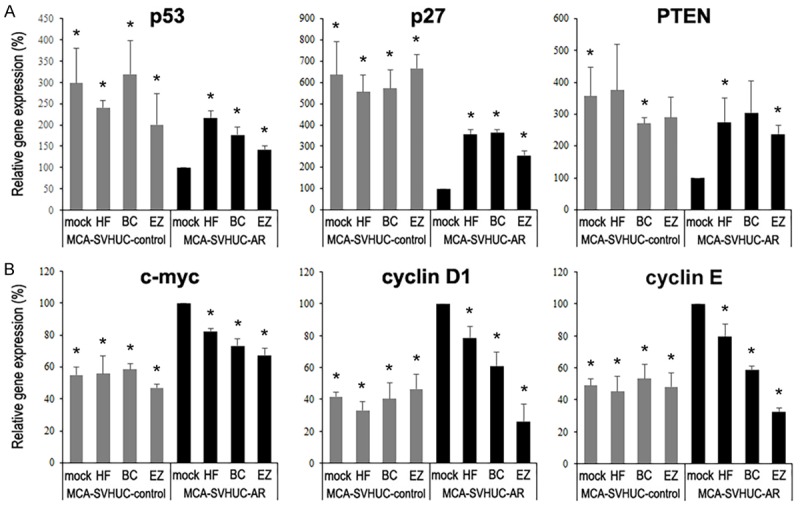
Effects of enzalutamide on the expression of tumor suppressors and oncogenes in MCA-exposed urothelial cells. SVHUC-control or SVHUC-AR exposed to MCA and subsequently cultured for 6 weeks in the presence of ethanol (mock), hydroxyflutamide (HF; 5 μM), bicalutamide (BC; 5 μM), or enzalutamide (EZ; 5 μM) was subjected to a quantitative RT-PCR for p53, p27, and PTEN (A) as well as c-myc, cyclin D1, and cyclin E (B). Expression of each gene was normalized to that of GAPDH. Transcription amount is presented relative to that in mock-treated SVHUC-AR cells. Each value represents the mean + standard deviation from three independent experiments. *P < 0.05 (vs. mock treatment in SVHUC-AR).
Discussion
Using cell line and animal models, activation of the AR pathway has been suggested to promote urothelial carcinogenesis [4,7,8-13], which may be a critical reason why men have a substantially higher risk of bladder cancer than women. Specifically, androgen deprivation via treatment with the first generation non-steroidal anti-androgenic drugs, such as flutamide and bicalutamide, results in inhibition of bladder cancer development [8,9,12,13]. In the current study, we confirmed that AR signals play a critical role in inducing urothelial carcinogenesis by showing differences in tumorigenic activity between AR-negative SVHUC control cells versus SVHUC-AR cells. More importantly, we provide preclinical data indicating that a newer generation of anti-AR compound, enzalutamide, strongly prevents AR-mediated urothelial carcinogenesis.
The AR, a member of the nuclear receptor superfamily, regulates gene expression in response to ligands in target cells [24,25]. Unliganded AR is mainly located in the cytoplasm and associates with heat-shock proteins. Upon binding of androgens, the AR undergoes a conformational change within the ligand-binding domain and dissociates from the heat-shock proteins. Activated receptors then form homodimers and translocate to the nucleus. In the nucleus, the dimerized AR-ligand complex initiates gene transcription of androgen-regulated genes, which is usually mediated by interaction with transcriptional co-activators and/or co-repressors [26,27]. Non-androgenic molecules, such as growth factors and cytokines, have also been shown to activate the AR in the absence of androgens principally in prostate cancer cells [28]. Our previous studies in bladder cancer cells have demonstrated that androgens [9,19,29] and epidermal growth factor [30] are able to induce AR transcriptional activity as well as their growth. In addition, it has been suggested that androgens up- or down-regulate a variety of molecules/pathways, including those known to involve urothelial tumorigenesis (e.g. Akt [31], β-catenin [32,33], CD24 [34], c-myc [13,32], cyclins D1/D3/E [13,35], ELK1 [36], epidermal growth factor receptor/ERBB2 [31], ERK1/2 [31], FGFR3 [13], GATA3 [13], PTEN [13], p21 [10,13], p53 [10,13], and UGT1A [12]), in non-neoplastic or neoplastic urothelial cells. In some of these studies, flutamide and/or bicalutamide were shown to antagonize the effects of androgens on the expression/activity of AR or its upstream pathways/downstream targets [9,12,13,19,29-32,34,36]. We here confirmed some of these previous observations and further demonstrated that enzalutamide inhibited the expression and transcriptional activity of AR in MCA-exposed SVHUC cells. We also found that enzalutamide, hydroxyflutamide, and bicalutamide similarly up- and down-regulated the expression of tumor suppressor genes (i.e. p53, p27, PTEN) and oncogenes (i.e. c-myc, cyclin D1, cyclin E), respectively, in SVHUC-AR cells undergoing neoplastic transformation. Meanwhile, AR overexpression in SVHUC cells resulted in induction of tumorigenic activity in vitro, as we previously described [13], as well as in vivo shown for the first time.
As aforementioned, there are obvious advantages of enzalutamide treatment, over that with flutamide or bicalutamide, in men with prostate cancer, presumably derived from not only its higher binding affinity for the AR but also unique actions on the AR including the prevention of AR nuclear translocation and AR binding to DNA or co-activators [16-18]. As a result, enzalutamide is widely prescribed for the treatment of metastatic castration-resistant prostate cancer, especially in those who are resistant to flutamide and/or bicalutamide therapy [18]. We confirmed that hydroxyflutamide and bicalutamide strongly prevented neoplastic transformation of AR-positive urothelial cells. We further demonstrated that enzalutamide also inhibited urothelial tumorigenesis and that its effects were similar to those of hydroxyflutamide and bicalutamide. Therefore, although androgen-induced AR translocation into the nucleus of urothelial cells has been documented [13,32], these additional mechanisms of enzalutamide actions for inhibiting urothelial tumorigenesis may be of little significance. Indeed, enzalutamide was found to effectively prevent AR nuclear translocation in SVHUC-AR cells undergoing malignant transformation, while hydroxyflutamide and bicalutamide did not significantly alter it. Blockade of androgen-AR binding by classic anti-androgens may thus sufficiently prevent AR-induced urothelial tumorigenesis. More importantly, our current data, together with previous observations, indicate that androgen deprivation therapy with anti-AR drugs may be a promising approach for chemoprevention of bladder tumor recurrence. Meanwhile, the effects of enzalutamide on the functions of AR as well as androgen-mediated expression/activity of AR targets in non-neoplastic urothelial cells need to be further investigated. It should also be noteworthy that the majority of non-neoplastic urothelial cells versus less than half of urothelial cancers were shown to express the AR [6,7,37], suggesting that AR could be lost during tumorigenesis and/or tumor growth.
In conclusion, AR signals were confirmed to induce urothelial carcinogenesis. In addition, 3 AR antagonists, enzalutamide, flutamide, and bicalutamide, were found to similarly inhibit neoplastic transformation of AR-positive urothelial cells, while only enzalutamide could prevent AR nuclear translocation. These findings enhance the feasibility of androgen deprivation therapy, using, for instance, enzalutamide, as a potential chemopreventive approach for urothelial tumors.
Acknowledgements
This study was supported by Astellas Pharma Global Development and Medivation, Inc. (ENZA-13J19 to H.M.).
Disclosure of conflict of interest
None.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 3.Hartge P, Harvey EB, Linehan WM, Silverman DT, Sullivan JW, Hoover RN, Fraumeni JF Jr. Unexplained excess risk of bladder cancer in men. J Natl Cancer Inst. 1990;82:1636–1640. doi: 10.1093/jnci/82.20.1636. [DOI] [PubMed] [Google Scholar]
- 4.Miyamoto H, Zheng Y, Izumi K. Nuclear hormone receptor signals as new therapeutic targets for urothelial carcinoma. Curr Cancer Drug Targets. 2012;12:14–22. doi: 10.2174/156800912798888965. [DOI] [PubMed] [Google Scholar]
- 5.Hsu I, Vitkus S, Da J, Yeh S. Role of oestrogen receptors in bladder cancer development. Nat Rev Urol. 2013;10:317–326. doi: 10.1038/nrurol.2013.53. [DOI] [PubMed] [Google Scholar]
- 6.Ide H, Miyamoto H. Steroid hormone receptor signals as prognosticators for urothelial tumor. Dis Markers. 2015;2015:840640. doi: 10.1155/2015/840640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mizushima T, Tirador KA, Miyamoto H. Androgen receptor activation: a prospective therapeutic target for bladder cancer. Expert Opin Ther Targets. 2017;21:249–257. doi: 10.1080/14728222.2017.1280468. [DOI] [PubMed] [Google Scholar]
- 8.Imada S, Akaza H, Ami Y, Koiso K, Ideyama Y, Takenaka T. Promoting effects and mechanisms of action of androgen in bladder carcinogenesis in male rats. Eur Urol. 1997;31:360–364. doi: 10.1159/000474484. [DOI] [PubMed] [Google Scholar]
- 9.Miyamoto H, Yang Z, Chen YT, Ishiguro H, Uemura H, Kubota Y, Nagashima Y, Chang YJ, Hu YC, Tsai MY, Yeh S, Messing EM, Chang C. Promotion of bladder cancer development and progression by androgen receptor signals. J Natl Cancer Inst. 2007;99:558–568. doi: 10.1093/jnci/djk113. [DOI] [PubMed] [Google Scholar]
- 10.Hsu JW, Hsu I, Xu D, Miyamoto H, Liang L, Wu XR, Shyr CR, Chang C. Decreased tumorigenesis and mortality from bladder cancer in mice lacking urothelial androgen receptor. Am J Pathol. 2013;182:1811–1820. doi: 10.1016/j.ajpath.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imaoka S, Yoneda Y, Sugimoto T, Ikemoto S, Hiroi T, Yamamoto K, Nakatani T, Funae Y. Androgen regulation of CYP4B1 responsible for mutagenic activation of bladder carcinogens in the rat bladder: detection of CYP4B1 mRNA by competitive reverse transcription-polymerase chain reaction. Cancer Lett. 2001;166:119–123. doi: 10.1016/s0304-3835(00)00572-3. [DOI] [PubMed] [Google Scholar]
- 12.Izumi K, Zheng Y, Hsu JW, Chang C, Miyamoto H. Androgen receptor signals regulate UDPglucuronosyltransferases in the urinary bladder: a potential mechanism of androgen-induced bladder carcinogenesis. Mol Carcinog. 2013;52:94–102. doi: 10.1002/mc.21833. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Ishiguro H, Kawahara T, Miyamoto Y, Izumi K, Miyamoto H. GATA3 in the urinary bladder: suppression of neoplastic transformation and down-regulation by androgens. Am J Cancer Res. 2014;4:461–473. [PMC free article] [PubMed] [Google Scholar]
- 14.Shiota M, Yokomizo A, Takeuchi A, Imada K, Kiyoshima K, Inokuchi J, Tatsugami K, Ohga S, Nakamura K, Honda H, Naito S. Secondary bladder cancer after anticancer therapy for prostate cancer: reduced comorbidity after androgen-deprivation therapy. Oncotarget. 2015;6:14710–14719. doi: 10.18632/oncotarget.3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Izumi K, Taguri M, Miyamoto H, Hara Y, Kishida T, Chiba K, Murai T, Hirai K, Suzuki K, Fujinami K, Ueki T, Udagawa K, Kitami K, Moriyama M, Miyoshi Y, Tsuchiya F, Ikeda I, Kobayashi K, Sato M, Morita M, Noguchi K, Uemura H. Androgen deprivation therapy prevents bladder cancer recurrence. Oncotarget. 2014;5:12665–12674. doi: 10.18632/oncotarget.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL. Development of a secondgeneration antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawahara T, Miyamoto H. Androgen receptor antagonists in the treatment of prostate cancer. Clin Immunol Endocr Metab Drugs. 2014;1:11–19. [Google Scholar]
- 18.Bambury RM, Scher HI. Enzalutamide: development from bench to bedside. Urol Oncol. 2015;33:280–288. doi: 10.1016/j.urolonc.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 19.Kawahara T, Ide H, Kashiwagi E, El-Shishtawy KA, Li Y, Reis LO, Zheng Y, Miyamoto H. Enzalutamide inhibits androgen receptor-positive bladder cancer cell growth. Urol Oncol. 2016;34:432, e15–e23. doi: 10.1016/j.urolonc.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 20.Izumi K, Li Y, Ishiguro H, Zheng Y, Yao JL, Netto GJ, Miyamoto H. Expression of UDP-glucuronosyltransferase 1A in bladder cancer: association with prognosis and regulation by estrogen. Mol Carcinog. 2014;53:314–324. doi: 10.1002/mc.21978. [DOI] [PubMed] [Google Scholar]
- 21.Kawahara T, Kashiwagi E, Li Y, Zheng Y, Miyamoto Y, Netto GJ, Ishiguro H, Miyamoto H. Cyclosporine A and tacrolimus inhibit urothelial tumorigenesis. Mol Carcinog. 2016;55:161–169. doi: 10.1002/mc.22265. [DOI] [PubMed] [Google Scholar]
- 22.Ishiguro H, Izumi K, Kashiwagi E, Zheng Y, Li Y, Kawahara T, Miyamoto H. Semenogelin I promotes prostate cancer cell growth via functioning as an androgen receptor coactivator and protecting against zinc cytotoxicity. Am J Cancer Res. 2015;5:738–747. [PMC free article] [PubMed] [Google Scholar]
- 23.Reznikoff CA, Loretz LJ, Christian BJ, Wu SQ, Meisner LF. Neoplastic transformation of SV40-immortalized human urinary tract epithelial cells by in vitro exposure to 3-methylcholanthrene. Carcinogenesis. 1988;9:1427–36. doi: 10.1093/carcin/9.8.1427. [DOI] [PubMed] [Google Scholar]
- 24.Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;10:2208–2219. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- 25.Culig Z. Targeting the androgen receptor in prostate cancer. Expert Opin Pharmacother. 2014;15:1427–1437. doi: 10.1517/14656566.2014.915313. [DOI] [PubMed] [Google Scholar]
- 26.Rahman M, Miyamoto H, Chang C. Androgen receptor coregulators in prostate cancer: mechanisms and clinical implications. Clin Cancer Res. 2004;10:2208–2219. doi: 10.1158/1078-0432.ccr-0746-3. [DOI] [PubMed] [Google Scholar]
- 27.Shiota M, Yokomizo A, Fujimoto N, Naito S. Androgen receptor cofactors in prostate cancer: potential therapeutic targets of castration-resistant prostate cancer. Curr Cancer Drug Targets. 2011;11:870–881. doi: 10.2174/156800911796798904. [DOI] [PubMed] [Google Scholar]
- 28.Lamont KR, Tindall DJ. Minireview: alternative activation pathways for the androgen receptor in prostate cancer. Mol Endocrinol. 2011;25:897–907. doi: 10.1210/me.2010-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng Y, Ishiguro H, Ide H, Inoue S, Kashiwagi E, Kawahara T, Jalalizadeh M, Reis LO, Miyamoto H. Compound A inhibits bladder cancer growth predominantly via glucocorticoid receptor transrepression. Mol Endocrinol. 2015;29:1486–1497. doi: 10.1210/me.2015-1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Izumi K, Zheng Y, Li Y, Zaengle J, Miyamoto H. Epidermal growth factor induces bladder cancer cell proliferation through activation of androgen receptor. Int J Oncol. 2012;41:1587–1592. doi: 10.3892/ijo.2012.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng Y, Izumi K, Yao JL, Miyamoto H. Dihydrotestosterone upregulates the expression of epidermal growth factor receptor and ERBB2 in androgen receptor-positive bladder cancer cells. Endocr-Relat Cancer. 2011;18:451–464. doi: 10.1530/ERC-11-0010. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Zheng Y, Izumi K, Ishiguro H, Ye B, Li F, Miyamoto H. Androgen activates β-catenin signaling in bladder cancer cells. Endocr-Relat Cancer. 2013;20:293–304. doi: 10.1530/ERC-12-0328. [DOI] [PubMed] [Google Scholar]
- 33.Lin C, Yin Y, Stemler K, Humphrey P, Kibel AS, Mysorekar IU, Ma L. Constitutive β-catenin activation induces male-specific tumorigenesis in the bladder urothelium. Cancer Res. 2013;73:5914–5925. doi: 10.1158/0008-5472.CAN-12-4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Overdevest JB, Knubel KH, Duex JE, Thomas S, Nitz MD, Harding MA, Smith SC, Frierson HF, Conaway M, Theodorescu D. CD24 expression is important in male urothelial tumorigenesis and metastasis in mice and is androgen regulated. Proc Natl Acad Sci U S A. 2012;109:E3588–E3596. doi: 10.1073/pnas.1113960109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu JT, Han BM, Yu SQ, Wang HP, Xia SJ. Androgen receptor is a potential therapeutic target for bladder cancer. Urology. 2010;75:820–827. doi: 10.1016/j.urology.2009.10.041. [DOI] [PubMed] [Google Scholar]
- 36.Kawahara T, Shareef HK, Aljarah AK, Ide H, Li Y, Kashiwagi E, Netto GJ, Zheng Y, Miyamoto H. ELK1 is up-regulated by androgen in bladder cancer cells and promotes tumor progression. Oncotarget. 2015;6:29860–29876. doi: 10.18632/oncotarget.5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kashiwagi E, Fujita K, Yamaguchi S, Fushimi H, Ide H, Inoue S, Mizushima T, Reis LO, Sharma R, Netto GJ, Nonomura N, Miyamoto H. Expression of steroid hormone receptors and its prognostic significance in urothelial carcinoma of the upper urinary tract. Cancer Biol Ther. 2016;17:1188–1196. doi: 10.1080/15384047.2016.1235667. [DOI] [PMC free article] [PubMed] [Google Scholar]