

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a life‐threatening disease and there is an urgent need to develop improved therapeutic approaches. The role of c‐Jun N‐terminal kinase (JNK) in PDAC stroma is not well defined even though dense desmoplastic reactions are characteristic of PDAC histology. We aimed to explore the role of JNK in PDAC stroma in mice. We crossed Ptf1a Cre/+;Kras G12D/+ mice with JNK1 −/− mice to generate Ptf1a Cre/+ ;Kras G12D/+ ;JNK1 −/− (Kras;JNK1−/−) mice. Tumor weight was significantly lower in Kras;JNK1−/− mice than in Kras;JNK1+/− mice, whereas histopathological features were similar. We also transplanted a murine PDAC cell line (mPC) with intact JNK1 s.c. into WT and JNK1 −/− mice. Tumor diameters were significantly smaller in JNK1 −/− mice. Phosphorylated JNK (p‐JNK) was activated in α‐smooth muscle actin (SMA)‐positive cells in tumor stroma, and mPC‐conditioned medium activated p‐JNK in tumor‐associated fibroblasts (TAF) in vitro. Relative expression of Ccl20 was downregulated in stimulated TAF. Ccl20 is an important chemokine that promotes CD8+ T‐cell infiltration by recruitment of dendritic cells, and the number of CD8+ T cells was decreased in Kras;JNK1+/− mice compared with Kras;JNK1−/− mice. These results suggest that the cancer secretome decreases Ccl20 secretion from TAF by activation of JNK, and downregulation of Ccl20 secretion might be correlated with reduction of infiltrating CD8+ T cells. Therefore, we concluded that inhibition of activated JNK in pancreatic tumor stroma could be a potential therapeutic target to increase Ccl20 secretion from TAF and induce accumulation of CD8+ T cells, which would be expected to enhance antitumor immunity.
Keywords: Ccl20, JNK, Kras, molecular targeted therapy, pancreatic cancer
Pancreatic ductal adenocarcinoma (PDAC) is a deadly disease with a 5‐year relative survival rate of 7%. This is largely as a result of the difficulty of early detection of PDAC, and patients are usually diagnosed at an advanced stage.1 Although there has been some improvement in survival with novel chemotherapy regimens,2, 3 PDAC is still a life‐threatening disease and there is an urgent need to develop improved therapeutic approaches.
Besides the well‐known mutations such as Kras, TP53, CDKN2A, and SMAD4, genomic DNA sequence analysis in the last decade has revealed novel mutated genes associated with Hedgehog, Wnt/Notch, and c‐Jun N‐terminal kinase (JNK) signaling pathways in PDAC.4 JNK was initially identified as a member of the MAPK pathway, which regulates phosphorylation of c‐Jun.5 It has been reported that JNK signaling pathways play various roles in inflammation, apoptosis, cell survival, and cell proliferation. In PDAC and other malignant tumors, such as prostate cancer, hepatocellular carcinoma, and gastric cancer, activated JNK has been reported to be involved in cancer development.6, 7, 8, 9 The role of JNK in PDAC cells has been well examined in multiple papers demonstrating that upregulated JNK in PDAC cells is important for accelerating an invasive, migratory, and metastatic phenotype, as well as for enhancing survival of circulating tumor cells and acquisition of chemoresistant ability in cancer stem cells.10, 11, 12 Furthermore, JNK inhibitor SP600125 suppresses PDAC cell proliferation and shows therapeutic effects in murine PDAC.9 Therefore, JNK is considered a potential therapeutic target.
Dense desmoplastic reactions and fibrosis are characteristic of PDAC histology,13 and the role of JNK in PDAC stroma is not well defined. Desmoplasia and fibrosis are considered to be derived from pancreatic stellate cells.14, 15 This type of tumor microenvironment enhances stiffness and increases hydrostatic pressure, resulting in impairment of drug delivery to the tumor. In addition, the microenvironment is thought to mediate a hypoxic microenvironment and infiltration of inflammatory cells, which may support PDAC progression.16, 17 In spite of this evidence, which suggests that the tumor stroma may also be a potential therapeutic target, treatment targeting tumor stroma by depletion of myofibroblasts failed in a murine model of PDAC; the treatment resulted in the development of poorly differentiated and invasive tumors and resulted in shorter survival.18, 19 Taken together, it is still unclear whether PDAC stroma is friend or foe,20 but total depletion of PDAC stroma leads to unfavorable results. The relationship between tumor cells and stroma is likely different depending on the context; thus, further examination is needed to better understand the complex interaction.
In the present study, we deleted JNK1 activation in tumor stroma using a s.c. xenograft tumor model transplanted into JNK1 knockout mice and noted that JNK1 depletion in PDAC stroma suppressed tumor progression. Moreover, we examined the effect of JNK inhibition in tumor‐associated fibroblasts (TAF) and secretion of related cytokines and chemokines to explore possible underlying mechanisms.
Materials and Methods
Mice
Ptf1a Cre/+;LSL‐Kras G12D/+ mice21 and JNK1 knockout (JNK1 −/−) mice22, 23 were previously described. All mice were maintained in filter‐topped cages and fed on autoclaved food and water at the Institute for Adult Diseases, Asahi Life Foundation, and Yokohama City University, Graduate School of Medicine, according to National Institutes of Health (NIH) guidelines. All experiments were approved by the Ethics Committee for Animal Experimentation of the Yokohama City University and the Institute for Adult Diseases, Asahi Life Foundation.
Reagents
We purchased JNK inhibitor SP600125 from Cayman Chemical (Ann Arbor, MI, USA), and transforming growth factor alpha (TGF‐α) from R&D Systems (Minneapolis, MN, USA). Primary antibodies used in immunohistochemical and immunofluorescence assays included rabbit anti‐phospho‐JNK antibody (1:50; Cell Signaling Technology, Danvers, MA, USA), rat anti‐mouse CD45 antibody (1:50; BD Biosciences, San Jose, CA, USA), rat anti‐mouse F4/80 antibody (1:100; eBioscience, San Diego, CA, USA), rabbit anti‐Ki‐67 antibody (1:100; Abcam, Cambridge, UK), mouse anti‐α‐smooth muscle actin (α‐SMA) antibody (1:50; Santa Cruz, Santa Cruz, CA, USA), biotin hamster anti‐mouse CD11c antibody (1:100; BD Biosciences), rabbit anti‐cleaved caspase3 antibody (1:1600; Cell Signaling Technology) and rabbit anti‐CD8 antibody (clone EP1150Y, 1:250; Epitomics, Burlingame, CA, USA). Primary antibodies used in immunoblotting analysis included rabbit anti‐phospho‐JNK antibody (1:1000; Cell Signaling Technology), rabbit anti‐JNK antibody (1:1000; Cell Signaling Technology), rabbit anti‐phospho‐Stat3 antibody (1:2000; Cell Signaling Technology) and mouse anti‐Stat3 antibody (1:1000; Cell Signaling Technology).
Cell culture
A murine pancreatic cancer cell line (K399), established from Ptf1a Cre/+;LSL‐Kras G12D/+;Tgfbr2flox/flox mice, was kindly provided by Dr Ijichi, University of Tokyo.24 TAF were established from Ptf1a Cre/+;LSL‐Kras G12D/+ mice.14 K399 cells were cultured in RPMI‐1640 medium (Wako, Osaka, Japan) and TAF were cultured in DMEM medium (Gibco BRL, Grand Island, NY, USA); 10% FBS (Biosera, Inc., Miami, FL, USA) and 2% penicillin‐streptomycin (Thermo Fisher Scientific, Waltham, MA, USA) were added to both. K399 conditioned medium (supernatant) was prepared as follows: K399 cells were cultured to confluence in a 10‐cm dish, washed twice with PBS (Gibco BRL), and cultured for 24 h in fresh RPMI‐1640 medium. The medium was collected and sterilized by filtering through a 0.22‐μm filter (Merck Millipore, Billerica, MA, USA). Control RPMI‐1640 medium was poured into a 10‐cm dish at the same time that the K399 medium was changed, placed in the incubator for 24 h, and sterilized by the same procedures as the supernatant.
TAF stimulation by supernatant
TAF were plated in six‐well or 12‐well plates and cultured to confluence. Each well was washed twice with PBS and fresh DMEM medium, mixed with control medium or supernatant at a ratio of 1:1, was added. For JNK signaling inhibition, TAF were preincubated with SP600125 (20 μM) for 1 h before supernatant stimulation. For immunoblotting analysis of JNK, TAF were incubated for 15, 30, 60, 90, 120 min and harvested using lysis buffer. For immunoblotting analysis of Stat3, TAF were incubated for 30 and 60 min and harvested using lysis buffer. For quantitative RT‐PCR, TAF were incubated for 8 h and harvested using ISOGEN2 (Wako).
Subcutaneous tumor xenograft model
K399 cells (2 × 106) were injected s.c. into the interscapular and interiliac regions of wild‐type (WT) or JNK1 −/− mice (four separate regions per mouse; n = 2 per group). When tumors became palpable, diameter of tumors was measured periodically (approximately every week) between day 11 and day 68.
Histopathological examination
Pancreatic tissues were fixed in buffered 10% formalin. Embedding in paraffin blocks, preparation of paraffin sections, H&E, and Picro‐Sirius Red staining (PSR) were done by Sept Sapie, KK (Tokyo, Japan). Area of each normal acinar and graded pancreatic intraepithelial neoplasia (PanIN) lesion (1, 2, and 3) was measured and the percentage respectively calculated.21 PSR was scored using the following scale: 0 = absent; 1 = mild; 2 = moderate; 3 = severe.( 25 ).
Immunohistochemical examination
Standard procedures were applied for immunohistochemical examination. In brief, after deparaffinization and rehydration, endogenous peroxidase was blocked with 3% hydrogen peroxide for 20 min at room temperature. For heat‐mediated antigen retrieval, with the exception of anti‐CD45 antibody, slides were processed for 15 min at 121°C in an autoclave or twice for 10 min each at 500 W in a microwave oven in 10 mM citrate buffer (pH 6.0). Retrievagen A (BD Biosciences) was used for the slides with anti‐CD45 antibody at 89°C for 10 min. Slides were then incubated with the primary antibodies at the appropriate concentration (described earlier in Reagents) overnight at 4°C. Secondary antibodies, including biotinylated anti‐rabbit, anti‐mouse, and anti‐rat antibodies, were applied for 30 min at room temperature; all secondary antibodies were purchased from Vector Laboratories (Burlingame, CA, USA) and were diluted 1:200. The VECTASTAIN ABC kit (Vector Laboratories) was used according to the manufacturer's directions, and slides were allowed to react with diaminobenzidine (Muto Pure Chemicals, Tokyo, Japan) solution. Hematoxylin was used as a counterstain. Number of cells staining positive for CD45, F4/80, CD11c, CD8, and cleaved caspase3 were counted and averaged. Ki‐67‐positive nuclei in PanIN lesions were counted and divided by the total number of nuclei in the PanIN lesion. α‐SMA‐positive areas were measured in pixels by ImageJ26 and averaged.
Immunofluorescence examination
Slides were deparaffinized and rehydrated. For heat‐mediated antigen retrieval, slides were processed for 15 min in an autoclave at 121°C. Slides were incubated with the primary antibodies at the appropriate concentrations (see Reagents) overnight at 4°C. Alexa Fluor® 488‐ or 594‐conjugated secondary antibodies (1:500; Life Technologies, Carlsbad, CA, USA) were applied for 1 h at room temperature, protected from light, and covered with mounting medium containing DAPI (Vector Laboratories). Images were taken using a fluorescence microscope (BZ‐9000; Keyence, Osaka, Japan).
Immunoblotting analysis
Cell lysates were prepared using a tissue protein extraction reagent (Thermo Fisher Scientific). Immunoblotting analysis was carried out on the lysates, which were separated by SDS‐PAGE and transferred to Immobilon‐P membranes (Merck Millipore). The membranes were incubated in PVDF blocking reagent for Can Get Signal immunostain (Toyobo, Osaka, Japan) at room temperature for 1 h to block non‐specific reactions, overnight at 4°C with primary antibodies at the appropriate concentrations (see Reagents). Then, HRP‐conjugated secondary antibodies were applied to the membrane for 1 h and photographs of the image were taken on LAS‐3000 (Fujifilm, Tokyo, Japan) using ECL prime Western blotting detection reagent (GE Healthcare, Chicago, IL, USA).
Quantitative RT‐PCR (qRT‐PCR)
RNA was extracted from TAF and pancreatic tissues using ISOGEN2 (Wako). Extracted RNA was reverse transcribed to generate cDNA using a high‐capacity RNA‐to‐cDNA kit (Thermo Fisher Scientific). qRT‐PCR, using the cDNA, was carried out with fast SYBR green master mix (Thermo Fisher Scientific) according to the manufacturer's directions in 96‐well plates on a 7900HT Fast Real‐Time PCR System (Applied Biosystems, Waltham, MA, USA). Relative expression levels of each gene were calculated using the delta Ct method normalizing the data based on the endogenous reference (GAPDH). Primer sequences are shown in Table S1.
Pancreatic acinar isolation and collagen culture
The pancreas was removed, washed twice with ice‐cold PBS, minced, and digested with collagenase I (37°C with shaker). Digested pancreatic pieces were washed and pipetted through a mesh. Acinar cells were pelleted (200 g for 2 min at 4°C) and resuspended in medium (1% FBS and 1 μg/mL dexamethasone). Acinar cells were cultured with or without TGF‐α (50 ng/mL) or SP600125 (10 μM).
Statistical analysis
Significant differences were detected using Student's t‐test. P‐values < 0.05 were considered statistically significant.
Results
Effect of JNK1 deletion in Kras‐mediated pancreatic tumors
To analyze the effect of JNK1 in Kras‐mediated pancreatic tumors, we crossed Ptf1a Cre/+ mice with LSL‐Kras G12D/+ and JNK1 −/− mice to generate Ptf1a Cre/+ ;LSL‐Kras G12D/+ ;JNK1 −/− (Kras;JNK1−/−) mice. Littermates, Ptf1a Cre/+ ;LSL‐Kras G12D/+ ;JNK1 +/− (Kras;JNK1+/−), were used as controls (Fig. 1a). There were no phenotypical differences between Kras;JNK1+/− mice and Ptf1a Cre/+ ;LSL‐Kras G12D/+ mice (data not shown). Mice were killed at 6 months of age and ratio of pancreas weight to bodyweight (pancreas/B.W., %) was measured. In Kras;JNK1−/− mice, the pancreas/B.W. ratio was significantly smaller than in Kras;JNK1+/− mice (P = 0.001) (Fig. 1b). Interestingly, despite this result, we noted no histopathological differences between Kras;JNK1+/− and Kras;JNK1−/− mice. There were no significant differences in the rate of PanIN formation (Fig. 2a); therefore, we hypothesized that JNK1 deletion did not have any effects on PanIN formation. To confirm this hypothesis, we harvested acinar cells from WT mice and cultured them in collagen with or without TGF‐α and/or JNK inhibitor SP600125. Stimulation of the acinar cells by TGF‐α resulted in induction of acinar‐to‐ductal metaplasia (ADM), both with or without SP600125 (Fig. 3a). Furthermore, acinar cells harvested from JNK1−/− mice could not avoid ADM (Fig. 3b). These results suggest that JNK1 is non‐essential for ADM and PanIN formation.
Figure 1.
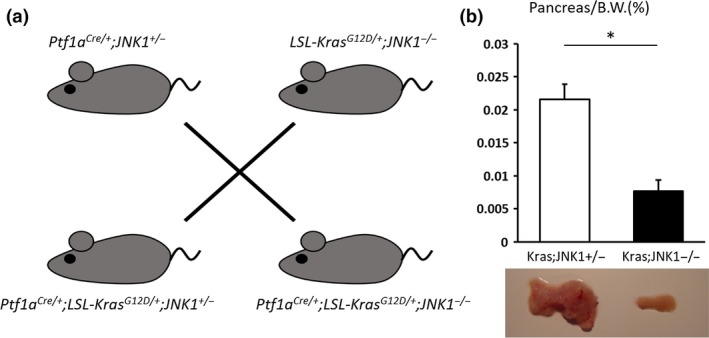
Effect of c‐Jun N‐terminal kinase1 (JNK1) deletion in a Kras‐mediated murine pancreatic tumor model. (a) Schematic depiction of mouse mating. We crossed Ptf1a Cre/+ with LSL‐Kras G12D/+ and JNK1 −/− mice to generate Ptf1a Cre/+ ;LSL‐Kras G12D/+ ;JNK1 +/− mice (Kras;JNK1+/−) and Ptf1a Cre/+ ;LSL‐Kras G12D/+ ;JNK1 −/− mice (Kras;JNK1−/−). (b) Ratio of pancreas weight to bodyweight (Pancreas/B.W.) was measured (upper panel). Typical examples of pancreas from Kras;JNK1−/− and Kras;JNK1+/− mice (lower panel). Data shown represent mean ± SEM (*P = 0.001, Student's t‐test).
Figure 2.
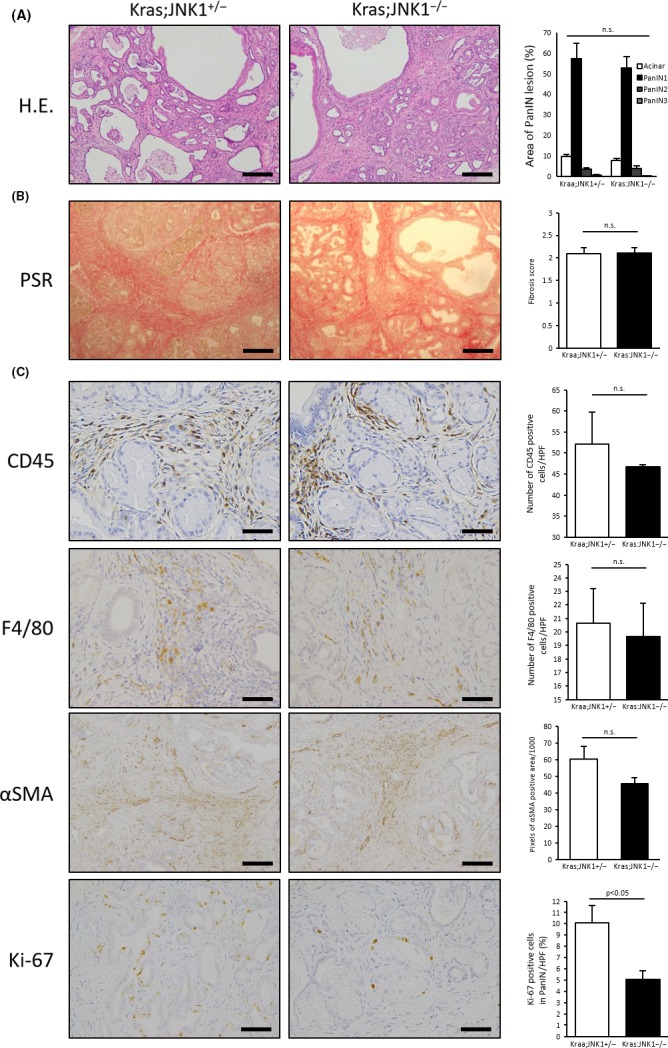
Histopathological analysis of c‐Jun N‐terminal kinase1 (JNK1) deletion in Kras‐mediated murine pancreatic tumors. (a) (Left) H&E in the pancreas of Kras;JNK1+/− and Kras;JNK1−/− mice is shown (magnification, ×200; scale bar, 100 μm). (Right) Each area of normal acinar and graded pancreatic intraepithelial neoplasia (PanIN) lesions (1, 2, and 3) was measured and the percentage calculated, respectively. Results are mean ± SEM of five random views for each sample (Kras;JNK1+/−, n = 4; Kras;JNK1−/−, n = 6). (b) (Left) Picro‐Sirius Red staining (PSR) was evaluated and scored as follows: 0 = absent; 1 = mild; 2 = moderate; 3 = severe (magnification, ×200; scale bar, 100 μm). (Right) Results are mean ± SEM of five random views (n = 6 in each group). (c) (Left) Immunohistochemical staining for CD45, F4/80, alpha smooth muscle actin (α‐SMA) and Ki‐67 are shown. Number of stained cells was counted and averaged in CD45‐ and F4/80‐positive cells (magnification, ×400; scale bar, 50 μm). (Right) Results are mean ± SEM of five random views (CD45: Kras;JNK1+/−, n = 3; Kras;JNK1−/−, n = 4. F4/80: n = 4 in each group). Ki‐67‐stained nuclei in PanIN lesions were counted and divided by the total number of nuclei in PanIN lesions (magnification, ×400; scale bar, 50 μm). Averaged results are mean ± SEM of five random views (n = 4 in each group). α‐SMA‐positive areas were measured in pixels by ImageJ and averaged (magnification, ×200; scale bar, 100 μm). Results are mean ± SEM of six random views (n = 3 in each group) (n.s., not significant).
Figure 3.
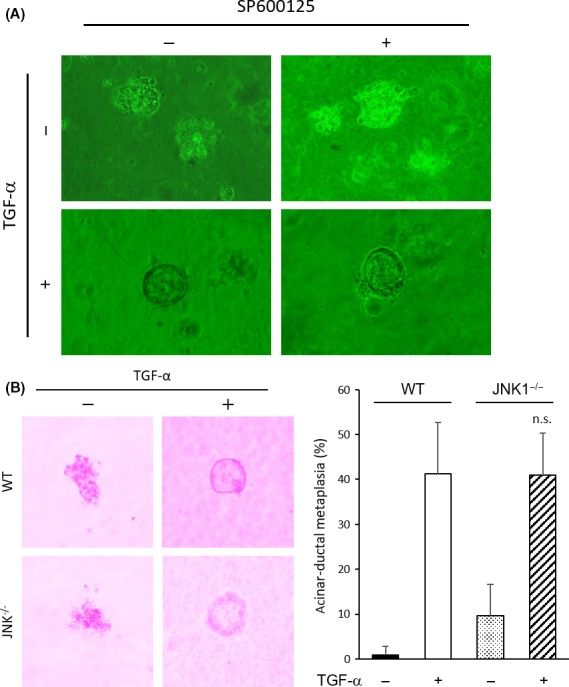
Impact of c‐Jun N‐terminal kinase1 (JNK1) deletion on induction of acinar‐to‐ductal metaplasia (ADM). (a) Acinar cells were isolated from WT mouse pancreas and cultured with or without transforming growth factor (TGF)‐β (50 ng/mL) or SP600125 (10 μM). (b) (Left) Acinar cells were isolated from the pancreas of WT or JNK1−/− mice and cultured with or without TGF‐α (50 ng/mL). (Right) Percentage of ADM was measured. Results are mean ± SD (WT, n = 10; JNK1−/−, n = 4) (P = 0.97, Student's t‐test) (n.s., not significant).
Histopathological analysis of tumor stroma showed no significant differences in the amount of PSR‐stained collagen fibers (Fig. 2b), the number of infiltrated CD45‐ and F4/80‐stained cells, and the area of α‐SMA‐stained lesions (Fig. 2c). Therefore, JNK1 does not play an important role in the development of tumor stroma. However, the number of Ki‐67‐stained cells in PanIN lesions was significantly greater in Kras;JNK1+/− mice than in Kras;JNK1−/− mice (P < 0.05) (Fig. 2c). These results suggest that JNK inhibition reduces tumor growth without changing tumor structure. These results are in accordance with previous reports showing that the JNK inhibitor, SP600125, inhibits pancreatic cancer growth in vivo 27 and has a therapeutic effect on murine pancreatic ductal adenocarcinoma.9
JNK activation in Kras‐mediated pancreatic tumor stroma
As the effect of JNK on pancreatic cancer cells has already been reported,9, 27 we focused on pancreatic tumor stroma to determine whether it has an effect on tumor progression. We noted that not only PanIN cells but also stromal cells were partially stained with phosphorylated‐JNK (p‐JNK) antibodies (Fig. 4a). This finding indicates that JNK is activated in the pancreatic tumor microenvironment. For further investigation, we transplanted K399 cells s.c. onto the backs of WT or JNK1−/− mice. In this model, transplanted K399 cells had intact JNK1 activation whereas other components, such as fibroblasts, immune cells, and endothelial cells, lacked JNK1. We measured the diameter of each s.c. tumor for 68 days, and the sizes of xenografts transplanted into JNK1−/− mice were significantly smaller than those in WT mice after 38 days (Fig. 4b). As the majority of p‐JNK‐stained cells in stroma were spindle‐shaped, we considered them fibroblasts (Fig. 4a, arrowhead). To elucidate the p‐JNK‐stained cell types, we carried out co‐immunofluorescent staining for p‐JNK with α‐SMA or F4/80, and both stains colocalized with p‐JNK staining (Fig. 4c,d). Although both fibroblasts and macrophages were stained with p‐JNK, the tumor stroma was rich in α‐SMA ‐stained cells compared with F4/80‐stained cells. Therefore, we focused on fibroblasts for further investigation.
Figure 4.
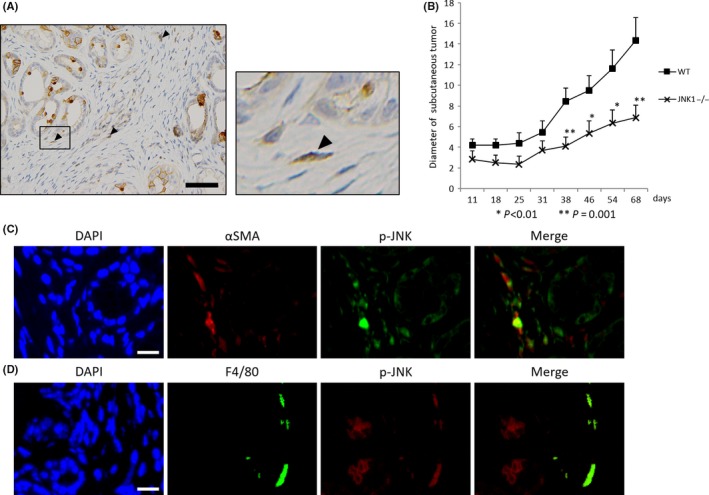
Importance of phosphorylated c‐Jun N‐terminal kinase (p‐JNK) in the tumor microenvironment. (a) Immunohistochemical staining of p‐JNK is shown. Pancreatic intraepithelial neoplasia (PanIN) cells and stromal cells are stained with anti p‐JNK antibody (arrowhead) (scale bar, 50 μm). (b) K399: Kras‐mediated murine pancreatic cancer cell line was transplanted s.c. onto the backs of WT or JNK1−/− mice (four separate regions in each mouse; n = 2 in each group). Diameters of s.c. tumors were measured and averaged at the indicated days. Results are mean ± SEM (*P < 0.01, **P = 0.001, Student's t‐test). (c) Co‐immunofluorescence staining for alpha smooth muscle actin (α‐SMA) and p‐JNK shows colocalization of α‐SMA and p‐JNK (scale bar, 15 μm). (d) Co‐immunofluorescence staining for F4/80 and p‐JNK shows colocalization of F4/80 and p‐JNK (scale bar, 15 μm).
p‐JNK activation in tumor‐associated fibroblasts and their expression of related cytokines and chemokines
We established TAF derived from Kras‐mediated murine pancreatic tumors and stimulated them with K399‐conditioned medium. p‐JNK activation in TAF lysate was evaluated by immunoblotting analysis, and we found that p‐JNK was activated by K399‐conditioned medium after 15 and 30 min (Fig. 5a). Prolonged JNK activation was also observed at 24 h after stimulation (Fig. S1a). This JNK activation is not K399‐conditioned medium specific; the conditioned medium produced by another murine pancreatic cancer cell line (EPPK1, established from Ptf1a Cre/+;LSL‐Kras G12D/+ mice, materials and methods are shown in Doc. S1) could also activate JNK in TAF (Fig. S1b). These results suggest that the cancer secretome activated JNK in TAF directly. To investigate the role of JNK activation in TAF, we next examined the expression of cytokines and chemokines that have been reported to be related to pancreatic ductal adenocarcinoma progression, including chemokine ligand (Ccl)2, Ccl20, chemokine (C‐X‐C motif) ligand (Cxcl) 1, Cxcl2, Cxcl10, Cxcl12, interleukin (IL)‐1β, IL‐6, TGF‐β1, tumor necrosis factor‐α (TNF‐α), bone morphogenetic protein (BMP), CD40 ligand (CD40L), granulocyte/macrophage–colony‐stimulating factor (GM‐CSF; CSF2), granulocyte colony‐stimulating factor (CSF3), epidermal growth factor (EGF), IL‐2, IL‐4, IL‐12b, IL‐13, IL‐17A, Nodal, TNF‐related apoptosis‐inducing ligand (Trail), and vascular endothelial growth factor C (VEGFC) (Figs 5b; S2). Expression of Ccl20 and Cxcl1 were downregulated by K399 condition medium, and SP600125 had a rescue effect. Furthermore, Ccl20 expression was significantly upregulated in Kras;JNK1−/− pancreatic tumors compared with Kras;JNK1+/− tumors. In contrast, Cxcl1 expression was not significantly upregulated in Kras;JNK1−/− tumors (Fig. 6a). Ccl20 expression is reported to be regulated through activated signal transducers and activator of transcription 3 (Stat3),28 and JNK signaling is considered to be a negative regulator of Stat3 activation.29 Therefore, we investigated the involvement of the JNK‐Stat3 pathway by immunoblotting analysis, and we found that phosphor‐Stat3 (p‐Stat3) was certainly downregulated in TAF stimulated by K339 conditioned medium (Fig. 5c). Ccl20 is one of the chemokines that recruits dendritic cells (DC). In addition, it has been reported that DC are responsible for cross‐priming CD8+ T cells, which are cytotoxic effector cells.30, 31 Therefore, we focused on these immune cells for further investigation.
Figure 5.
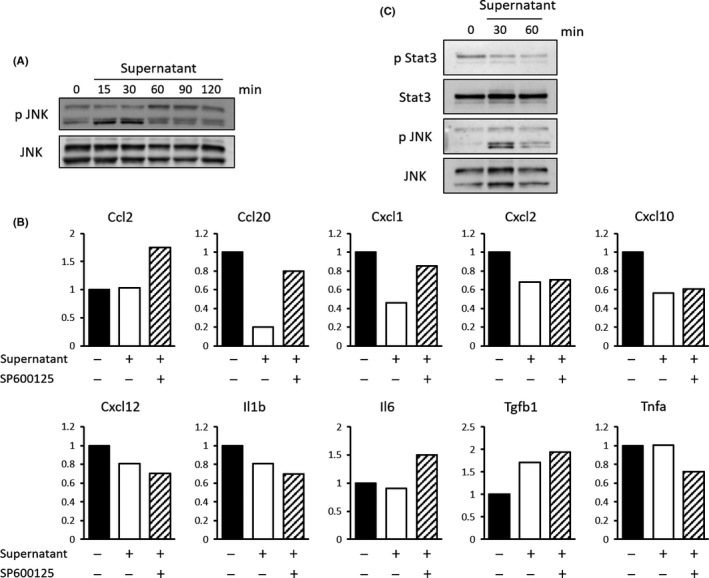
Effect of activated phosphorylated c‐Jun N‐terminal kinase (p‐JNK) in tumor‐associated fibroblasts (TAF). (a) TAF, fibroblasts derived from Kras‐mediated murine pancreatic cancers, were stimulated by K399‐conditioned medium and harvested at the indicated times (top). Immunoblotting analyses of stimulated TAF lysate with anti‐p‐JNK and anti‐JNK antibodies are shown. (b) TAF were stimulated by control medium or K399‐conditioned medium with or without SP600125 (20 μM) and harvested after 8 h. Relative mRNA expression of indicated cytokines and chemokines is shown. (c) TAF were stimulated by K399‐conditioned medium and harvested at the indicated times (top). Immunoblotting analyses of stimulated TAF lysate with anti‐p‐Stat3, anti‐Stat3, anti‐p‐JNK and anti‐JNK antibodies are shown.
Figure 6.
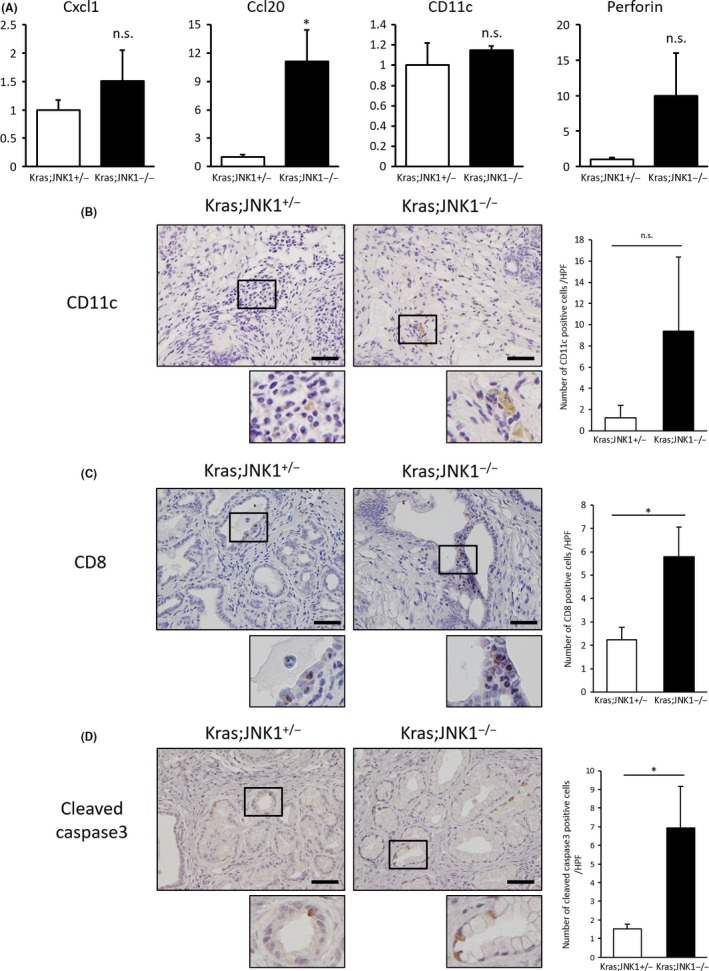
Chemokine ligand (Ccl)20 expression, CD11c positive cells and CD8 + T cell recruitment in pancreatic tumor tissue. (a) mRNA was isolated from pancreatic tumors of Kras;JNK1+/− and Kras;JNK1−/− mice. Relative mRNA expression of chemokine (C‐X‐C motif) ligand (Cxcl)1, Ccl20, Cd11c, and perforin are shown (n = 4 in Kras;JNK1+/− group and n = 3 in Kras;JNK1−/− group) (Cxcl1, P = 0.21; Ccl20, *P < 0.05; Cd11c, P = 0.60; perforin, P = 0.16; Student's t‐test) (n.s., not significant). (b) (Left) Immunohistochemical staining using anti‐CD11c is shown (magnification, ×400; scale bar, 50 μm). (Right) Number of antibody‐stained cells was counted and averaged. Results are mean ± SEM of five random views (n = 2 in each group) (P < 0.37, Student's t‐test). (c) (Left) Immunohistochemical staining using anti‐CD8 antibody is shown (magnification, ×400; scale bar, 50 μm). (Right) Number of antibody‐stained cells was counted and averaged. Results are mean ± SEM of eight random views (n = 5 in each group) (*P < 0.05, Student's t‐test). (d) (Left) Immunohistochemical staining using anti‐cleaved caspase‐3 antibody is shown (magnification, ×400; scale bar, 50 μm). (Right) Number of antibody‐stained cells was counted and averaged. Results are mean ± SEM of five random views (n = 3 in Kras;JNK1+/− group and n = 5 in Kras;JNK1−/− group) (*P < 0.05, Student's t‐test).
Ccl20 upregulation and recruitment of CD8+ T cells in Kras;JNK1−/− mice
To determine the role of Ccl20 in Kras;JNK1−/− pancreatic tumors, we carried out qRT‐PCR analysis for CD11c and perforin. CD11c is a marker for DC,31 and perforin is a pore‐forming protein that induces apoptosis and destroys cells and plays important roles in the cytotoxic activity of CD8+ T cells and natural killer cells.32 Although CD11c and perforin expression were not statistically different in Kras;JNK1−/− mice compared with Kras;JNK1+/−, CD11c expression showed a tendency of slight upregulation and perforin expression showed a strong tendency of upregulation in Kras;JNK1−/− mice (Fig. 6a). Immunohistochemical analysis showed CD11c‐stained cell infiltration in the tumor stroma, and there was a tendency for a larger number of CD11c‐stained cells infiltrating in Kras;JNK1−/− pancreatic tumors (Fig. 6b). To elucidate whether cytotoxic T‐cell recruitment was accelerated in Kras;JNK1−/− pancreatic tumors, we carried out immunohistochemical staining using an anti‐CD8 antibody. Numbers of CD8‐stained cells and cleaved caspase‐3‐positive PanIN cells were significantly greater in Kras;JNK1−/− mice than in Kras;JNK1+/− mice (P < 0.05) (Fig. 6c,d), which should be another reason, in addition to the numerous Ki‐67‐stained cells in PanIN lesions, that the pancreas/B.W. ratio of Kras;JNK1−/− mice was smaller than in Kras;JNK1+/− mice. To summarize our results, in pancreatic tumor stroma, p‐JNK in TAF is activated by the cancer secretome, which downregulates Ccl20 expression. This leads to a decrease in the recruitment of DC and CD8+ T cells, thereby resulting in accelerated tumor growth (Fig. 7).
Figure 7.
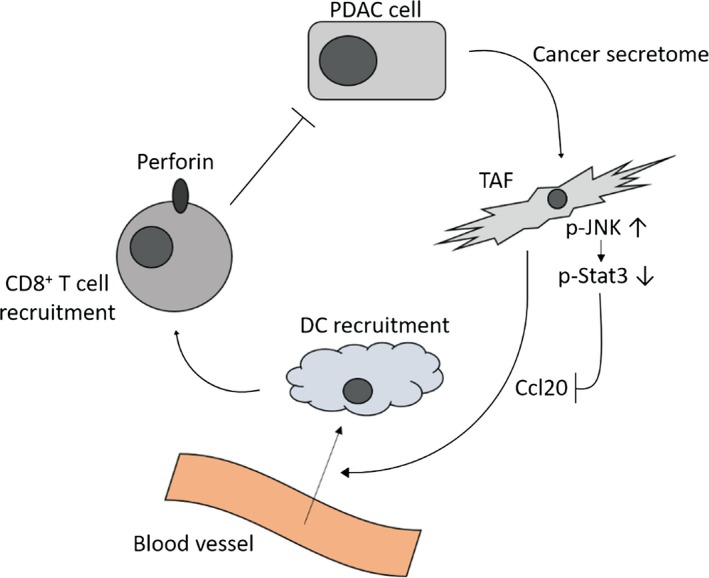
Schematic depiction of the relationship between phosphorylated c‐Jun N‐terminal kinase (p‐JNK) and chemokine ligand (Ccl)20. The cancer secretome upregulates p‐JNK activation and downregulates p‐Stat3 activation in tumor‐associated fibroblasts (TAF), leading to downregulation of Ccl20 secretion and reduction of dendritic cells (DC) and CD8 + T cell recruitment in pancreatic tumors.
Discussion
We demonstrated here that JNK activation is important to promote pancreatic tumor progression, not only in tumor cells but also in tumor stroma, especially in fibroblasts; however, the initiation and formation of PanIN was not affected by inhibition of JNK in vitro or knockout of JNK1 in vivo. Toste et al. have reported that gemcitabine treatment activates JNK and p38 signaling pathways in pancreatic cancer‐associated fibroblasts (CAF).33 These gemcitabine‐treated CAF secrete multiple inflammatory mediators, resembling a senescence‐associated secretory phenotype that is considered to promote tumor progression. Additionally, inhibition of stress‐associated MAPK signaling suppresses induction of the senescence‐associated secretory phenotype in CAF. Therefore, inhibition of JNK has therapeutic potential for PDAC patients by targeting both the tumor cells and the stromal component, and it could be a promising choice for both monotherapy and combination therapy with current chemotherapeutic regimens.
There are currently no commercially available JNK inhibitors, but a few oral JNK inhibitors, including CC‐930 (tanzisertib)34 and AS602801 (bentamapimod),35 recently entered clinical trials. CC‐930 is being tested in idiopathic pulmonary fibrosis and high‐dose CC‐930 caused elevation of hepatic transaminases when used for long‐term treatment.36 AS602801 is being tested in endometriosis and demonstrated favorable safety and tolerability after 5 months of administration.37, 38 Both diseases are benign, chronic inflammatory diseases, and the efficacy of the inhibitors has not been examined in malignant tumor patients; our results suggest that it is worth examining the safety and tolerability in those patients.
The effect of JNK activation on Ccl20 expression is controversial: Kao et al. reported that JNK inhibition upregulates Ccl20 expression and secretion induced by IL‐17 in human airway epithelium;39 Hosokawa et al. reported that JNK inhibition does not affect Ccl20 production induced by IL‐1β in human gingival fibroblasts;40 Rhee et al. reported that JNK inhibition does not alter Toll‐like receptor 5‐induced Ccl20 expression;41 and Kanda et al. reported that prolactin‐mediated JNK activation induces Ccl20 production by human keratinocytes.42 Therefore, the effect of JNK activation on Ccl20 expression may be context‐dependent. In our model, K399‐conditioned medium suppressed Ccl20 expression by TAF, and addition of a JNK inhibitor rescued the expression levels; thus, JNK activation in TAF, stimulated by the pancreatic cancer secretome, downregulated Ccl20 expression in our model.
Ccl20 expression is considered to be regulated through nuclear factor kappa B (NF‐κB), CCAAT/enhancer‐binding proteins, activator protein 1 (AP‐1), specificity protein 1 (Sp‐1), and activated Stat3.28 In this study, it is interesting that whereas AP‐1 is one of the downstream elements of the JNK signaling pathway, Ccl20 expression was downregulated in JNK‐activated TAF, and downregulated Stat3 activation should be the mechanism responsible for Ccl20 downregulation. It is still not clear why the Stat3 pathway but not AP‐1 is the dominant regulator of Ccl20 expression in TAF.
Despite our findings showing that Ccl20 expression correlated with the upregulation of DC markers and cytotoxic T‐cell infiltration, which would be expected to induce tumor apoptosis and to increase antitumor effects, there are several reports showing that Ccl20 has supportive rather than suppressive effects on PDAC cells. Campbell et al. reported that Ccl20 promotes PDAC cell invasion,43 and Liu et al. reported that Ccl20, derived from tumor‐associated macrophages, enhances PDAC cell growth and metastatic ability.44 Moreover, Rubie et al. showed that Ccl20 was significantly upregulated in human PDAC tissue and associated with advanced T stage based on the UICC TNM classification.45 However, in other carcinomas, such as esophageal, gastric, ovarian, and colorectal cancers, upregulation of Ccl20 expression and infiltration of DC and CD8+ T cells represent a positive prognostic factor for survival.46, 47, 48, 49 The present study shows, for the first time, that Ccl20 may exhibit antitumor effects by recruiting DC and CD8+ T cells to pancreatic tumor stroma. Tumor immunity has recently been highlighted as a therapeutic target, and nivolumab, (an antibody against programmed death 1 receptor) targeting an immune checkpoint, has recently become widely available for the treatment of melanoma and other carcinomas.50, 51, 52 Accumulation of antitumor immune cells in the tumor stroma is expected to increase the efficacy of immune checkpoint therapy; thus, JNK inhibition could be a favorable choice in combination with immune checkpoint therapy.
In summary, results from the present study suggest that activated JNK suppresses Ccl20 expression in TAF and inhibits infiltration of antitumor immune cells (DC and CD8+ T cells). JNK inhibition rescues Ccl20 expression in TAF and increases the number of antitumor immune cells. Thus, JNK is an attractive target for PDAC treatment.
Disclosure Statement
Authors declare no conflicts of interest for this article.
Supporting information
Doc. S1. Supplementary materials and methods: Cell culture, tumor‐associated fibroblast (TAF) stimulation by K399 conditioned medium to examine prolonged JNK activation, TAF stimulation by EPPK1 conditioned medium.
Table S1. List of the primers used for qRT‐PCR.
Fig. S1. (a) Tumor‐associated fibroblasts (TAF) were stimulated by K399‐conditioned medium and harvested at the indicated times (top), to evaluate the long‐term effect. (b) TAF were stimulated by EPPK1‐conditioned medium and harvested at the indicated times (top) (EPPK1: murine pancreatic cancer cell line established from Ptf1a Cre/+ ;LSL‐Kras G12D/+ mice). Immunoblotting analyses of stimulated TAF lysate with anti‐p‐JNK and anti‐JNK antibodies are shown.
Fig. S2. Tumor‐associated fibroblasts (TAF) were stimulated by control medium or K399‐conditioned medium with or without SP600125 (20 μM) and harvested after 8 h. Relative mRNA expression of indicated cytokines and chemokines is shown.
Acknowledgment
We thank Y. Yamashita for her technical assistance.
Cancer Sci 108 (2017) 2156–2165
Funding Information
None.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016; 66: 7–30. [DOI] [PubMed] [Google Scholar]
- 2. Rajeshkumar NV, Yabuuchi S, Pai SG et al Superior therapeutic efficacy of nab‐paclitaxel over cremophor‐based paclitaxel in locally advanced and metastatic models of human pancreatic cancer. Br J Cancer 2016; 115: 442–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Conroy T, Desseigne F, Ychou M et al FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011; 364: 1817–25. [DOI] [PubMed] [Google Scholar]
- 4. Jones S, Zhang X, Parsons DW et al Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008; 321: 1801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Derijard B, Hibi M, Wu IH et al JNK1: a protein kinase stimulated by UV light and Ha‐Ras that binds and phosphorylates the c‐Jun activation domain. Cell 1994; 76: 1025–37. [DOI] [PubMed] [Google Scholar]
- 6. Ouyang X, Jessen WJ, Al‐Ahmadie H et al Activator protein‐1 transcription factors are associated with progression and recurrence of prostate cancer. Cancer Res 2008; 68: 2132–44. [DOI] [PubMed] [Google Scholar]
- 7. Wang J, Tai G. Role of C‐Jun N‐terminal kinase in hepatocellular carcinoma development. Target Oncol 2016; 11: 723–38. [DOI] [PubMed] [Google Scholar]
- 8. Shibata W, Maeda S, Hikiba Y et al c‐Jun NH2‐terminal kinase 1 is a critical regulator for the development of gastric cancer in mice. Cancer Res 2008; 68: 5031–9. [DOI] [PubMed] [Google Scholar]
- 9. Takahashi R, Hirata Y, Sakitani K et al Therapeutic effect of c‐Jun N‐terminal kinase inhibition on pancreatic cancer. Cancer Sci 2013; 104: 337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yuan XP, Dong M, Li X, Zhou JP. GRP78 promotes the invasion of pancreatic cancer cells by FAK and JNK. Mol Cell Biochem 2015; 398: 55–62. [DOI] [PubMed] [Google Scholar]
- 11. Suzuki S, Okada M, Shibuya K et al JNK suppression of chemotherapeutic agents‐induced ROS confers chemoresistance on pancreatic cancer stem cells. Oncotarget 2015; 6: 458–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ebelt ND, Cantrell MA, Van Den Berg CL. c‐Jun N‐terminal kinases mediate a wide range of targets in the metastatic cascade. Genes Cancer 2013; 4: 378–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kleeff J, Korc M, Apte M et al Pancreatic cancer. Nat Rev Dis Primers 2016; 2: 16022. [DOI] [PubMed] [Google Scholar]
- 14. Apte MV, Haber PS, Applegate TL et al Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut 1998; 43: 128–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bachem MG, Schunemann M, Ramadani M et al Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005; 128: 907–21. [DOI] [PubMed] [Google Scholar]
- 16. Liu H, Ma Q, Xu Q et al Therapeutic potential of perineural invasion, hypoxia and desmoplasia in pancreatic cancer. Curr Pharm Des 2012; 18: 2395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tang D, Wang D, Yuan Z et al Persistent activation of pancreatic stellate cells creates a microenvironment favorable for the malignant behavior of pancreatic ductal adenocarcinoma. Int J Cancer 2013; 132: 993–1003. [DOI] [PubMed] [Google Scholar]
- 18. Rhim AD, Oberstein PE, Thomas DH et al Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014; 25: 735–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ozdemir BC, Pentcheva‐Hoang T, Carstens JL et al Depletion of carcinoma‐associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014; 25: 719–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gore J, Korc M. Pancreatic cancer stroma: friend or foe? Cancer Cell 2014; 25: 711–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hingorani SR, Petricoin EF, Maitra A et al Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003; 4: 437–50. [DOI] [PubMed] [Google Scholar]
- 22. Sabapathy K, Kallunki T, David JP, Graef I, Karin M, Wagner EF. c‐Jun NH2‐terminal kinase (JNK)1 and JNK2 have similar and stage‐dependent roles in regulating T cell apoptosis and proliferation. J Exp Med 2001; 193: 317–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine‐driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005; 121: 977–90. [DOI] [PubMed] [Google Scholar]
- 24. Ijichi H, Chytil A, Gorska AE et al Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas‐specific blockade of transforming growth factor‐beta signaling in cooperation with active Kras expression. Genes Dev 2006; 20: 3147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takano S, Kimura T, Yamaguchi H, Kinjo M, Nawata H. Effects of stress on the development of chronic pancreatitis. Pancreas 1992; 7: 548–55. [DOI] [PubMed] [Google Scholar]
- 26. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012; 9: 671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ennis BW, Fultz KE, Smith KA et al Inhibition of tumor growth, angiogenesis, and tumor cell proliferation by a small molecule inhibitor of c‐Jun N‐terminal kinase. J Pharmacol Exp Ther 2005; 313: 325–32. [DOI] [PubMed] [Google Scholar]
- 28. Zhao L, Xia J, Wang X, Xu F. Transcriptional regulation of CCL20 expression. Microbes Infect 2014; 16: 864–70. [DOI] [PubMed] [Google Scholar]
- 29. Lim CP, Cao X. Serine phosphorylation and negative regulation of Stat3 by JNK. J Biol Chem 1999; 274: 31055–61. [DOI] [PubMed] [Google Scholar]
- 30. Schutyser E, Struyf S, Van Damme J. The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev 2003; 14: 409–26. [DOI] [PubMed] [Google Scholar]
- 31. Le Borgne M, Etchart N, Goubier A et al Dendritic cells rapidly recruited into epithelial tissues via CCR6/CCL20 are responsible for CD8 + T cell crosspriming in vivo. Immunity 2006; 24: 191–201. [DOI] [PubMed] [Google Scholar]
- 32. Osinska I, Popko K, Demkow U. Perforin: an important player in immune response. Cent Eur J Immunol 2014; 39: 109–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Toste PA, Nguyen AH, Kadera BE et al Chemotherapy‐induced inflammatory gene signature and protumorigenic phenotype in pancreatic CAFs via stress‐associated MAPK. Mol Cancer Res 2016; 14: 437–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Plantevin Krenitsky V, Nadolny L, Delgado M et al Discovery of CC‐930, an orally active anti‐fibrotic JNK inhibitor. Bioorg Med Chem Lett 2012; 22: 1433–8. [DOI] [PubMed] [Google Scholar]
- 35. Ferrandi C, Richard F, Tavano P et al Characterization of immune cell subsets during the active phase of multiple sclerosis reveals disease and c‐Jun N‐terminal kinase pathway biomarkers. Mult Scler 2011; 17: 43–56. [DOI] [PubMed] [Google Scholar]
- 36. van der Velden JL, Ye Y, Nolin JD et al JNK inhibition reduces lung remodeling and pulmonary fibrotic systemic markers. Clin Transl Med 2016; 5: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Okada M, Kuramoto K, Takeda H et al The novel JNK inhibitor AS602801 inhibits cancer stem cells in vitro and in vivo. Oncotarget 2016; 7: 27021–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hussein M, Chai DC, Kyama CM et al c‐Jun NH2‐terminal kinase inhibitor bentamapimod reduces induced endometriosis in baboons: an assessor‐blind placebo‐controlled randomized study. Fertil Steril 2016; 105: 815–24. e5. [DOI] [PubMed] [Google Scholar]
- 39. Kao CY, Huang F, Chen Y et al Up‐regulation of CC chemokine ligand 20 expression in human airway epithelium by IL‐17 through a JAK‐independent but MEK/NF‐kappaB‐dependent signaling pathway. J Immunol 2005; 175: 6676–85. [DOI] [PubMed] [Google Scholar]
- 40. Hosokawa Y, Hosokawa I, Shindo S, Ozaki K, Matsuo T. IL‐22 enhances CCL20 production in IL‐1beta‐stimulated human gingival fibroblasts. Inflammation 2014; 37: 2062–6. [DOI] [PubMed] [Google Scholar]
- 41. Rhee SH, Keates AC, Moyer MP, Pothoulakis C. MEK is a key modulator for TLR5‐induced interleukin‐8 and MIP3alpha gene expression in non‐transformed human colonic epithelial cells. J Biol Chem 2004; 279: 25179–88. [DOI] [PubMed] [Google Scholar]
- 42. Kanda N, Shibata S, Tada Y, Nashiro K, Tamaki K, Watanabe S. Prolactin enhances basal and IL‐17‐induced CCL20 production by human keratinocytes. Eur J Immunol 2009; 39: 996–1006. [DOI] [PubMed] [Google Scholar]
- 43. Campbell AS, Albo D, Kimsey TF, White SL, Wang TN. Macrophage inflammatory protein‐3alpha promotes pancreatic cancer cell invasion. J Surg Res 2005; 123: 96–101. [DOI] [PubMed] [Google Scholar]
- 44. Liu B, Jia Y, Ma J et al Tumor‐associated macrophage‐derived CCL20 enhances the growth and metastasis of pancreatic cancer. Acta Biochim Biophys Sin (Shanghai) 2016; 48: 1067–74. [DOI] [PubMed] [Google Scholar]
- 45. Rubie C, Frick VO, Ghadjar P et al CCL20/CCR6 expression profile in pancreatic cancer. J Transl Med 2010; 8: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu L, Pan K, Zheng HX et al IL‐17A promotes immune cell recruitment in human esophageal cancers and the infiltrating dendritic cells represent a positive prognostic marker for patient survival. J Immunother 2013; 36: 451–8. [DOI] [PubMed] [Google Scholar]
- 47. Ohtani H, Nakayama T, Yoshie O. In situ expression of the CCL20‐CCR6 axis in lymphocyte‐rich gastric cancer and its potential role in the formation of lymphoid stroma. Pathol Int 2011; 61: 645–51. [DOI] [PubMed] [Google Scholar]
- 48. Sato E, Olson SH, Ahn J et al Intraepithelial CD8 + tumor‐infiltrating lymphocytes and a high CD8 + /regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA 2005; 102: 18538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Naito Y, Saito K, Shiiba K et al CD8 + T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res 1998; 58: 3491–4. [PubMed] [Google Scholar]
- 50. Wolchok JD, Kluger H, Callahan MK et al Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013; 369: 122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brahmer J, Reckamp KL, Baas P et al Nivolumab versus docetaxel in advanced squamous‐cell non‐small‐cell lung cancer. N Engl J Med 2015; 373: 123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Motzer RJ, Escudier B, McDermott DF et al Nivolumab versus everolimus in advanced renal‐cell carcinoma. N Engl J Med 2015; 373: 1803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Doc. S1. Supplementary materials and methods: Cell culture, tumor‐associated fibroblast (TAF) stimulation by K399 conditioned medium to examine prolonged JNK activation, TAF stimulation by EPPK1 conditioned medium.
Table S1. List of the primers used for qRT‐PCR.
Fig. S1. (a) Tumor‐associated fibroblasts (TAF) were stimulated by K399‐conditioned medium and harvested at the indicated times (top), to evaluate the long‐term effect. (b) TAF were stimulated by EPPK1‐conditioned medium and harvested at the indicated times (top) (EPPK1: murine pancreatic cancer cell line established from Ptf1a Cre/+ ;LSL‐Kras G12D/+ mice). Immunoblotting analyses of stimulated TAF lysate with anti‐p‐JNK and anti‐JNK antibodies are shown.
Fig. S2. Tumor‐associated fibroblasts (TAF) were stimulated by control medium or K399‐conditioned medium with or without SP600125 (20 μM) and harvested after 8 h. Relative mRNA expression of indicated cytokines and chemokines is shown.