

Abstract
The highly selective multi‐targeted agent sorafenib is an inhibitor of a number of intracellular signaling kinases with anti‐proliferative, anti‐angiogenic and pro‐apoptotic effects in various types of tumors, including human non‐small cell lung cancer (NSCLC). Betulin displays a broad spectrum of biological and pharmacological properties, including anticancer and chemopreventive activity. Combination of drugs with different targets is a logical approach to overcome multilevel cross‐stimulation among key signaling pathways in NSCLC progression. NSCLC cell lines, A549, H358 and A427, with different KRAS mutations, and normal human peripheral blood lymphocyte cells, were treated with sorafenib and betulinic acid alone and in combination. We examined the effect of different combined treatments on viability (MTS test), proliferation and apoptotic susceptibility based on flow cytometry, alterations in signaling pathways by western blotting and colony‐forming ability. The combination of sorafenib with betulinic acid had a strong effect on the induction of apoptosis of different NSCLC cell lines. In addition, this combination was not toxic for human peripheral blood lymphocytes. Combination treatment changed the expression of proteins involved in the mitochondrial apoptosis pathway and induced apoptotic death by caspase activation. Importantly, combination treatment with low drug concentrations tremendously reduced the colony‐forming ability of A549, H358 and A427 cells, as compared to both compounds alone. In this study, we showed that combination therapy with low concentrations of sorafenib and betulinic acid had the capacity to induce high levels of cell death and abolish clonogenic activity in some NSCLC cell lines regardless of KRAS mutations.
Keywords: Betulinic acid, clonogenic activity, combination therapy, non‐small cell lung cancer, sorafenib
Lung cancer is the second leading cause of cancer‐related mortality worldwide, and more than 1.6 million cases are diagnosed every year.1, 2 Tobacco smoking and exposure to environmental carcinogens have been found to be the major risk factors in the development of this disease.3 Lung cancer can be classically subdivided into small cell lung cancer (SCLC) and three types of non‐small cell lung cancer (NSCLC) that include squamous cell carcinoma, adenocarcinoma and large cell carcinoma.1 Most lung cancer patients are diagnosed at an advanced stage, with a short survival rate.4 Despite considerable advances in our knowledge and experience in the treatment of lung cancer patients, our capacity to effectively fight and treat this disease is still limited. One way to improve these results is to personalize treatment based on tumor molecular characteristics.4, 5
Molecular targeted therapies are currently applied in the treatment regimen of NSCLC, because they have been shown to extend progression‐free survival and improve overall survival.6 Molecular biomarkers, such as epidermal growth factor receptor (EGFR) mutations or amplification,7 echinoderm microtubule‐associated protein‐like 4‐naplastic lymphoma kinase (EML4‐ALK) translocation8 or KRAS9 and PI3KCA mutations,10 can serve as targets for these therapies, and also as indicators of therapeutic outcomes.
Sorafenib is a multi‐targeted kinase inhibitor that has shown efficacy against a wide variety of tumors in preclinical models.11 The anti‐tumor effect of sorafenib is thought to be mediated through its inhibition of the RAS/RAF/ERK pathway involved in cell proliferation as well as its inhibition of VEGFR2‐related angiogenesis.12 It has also been reported that sorafenib induces apoptosis in human leukemia cells and other human tumor cell lines through translation inhibition and downregulation of myeloid cell leukemia‐1 (Mcl‐1) and a Bcl‐2 family member.13 Sorafenib has demonstrated anti‐tumor activity in phase I and II trials as monotherapy14, 15 and combined treatment16, 17 in patients with advanced NSCLC. However, sorafenib in combination with cytotoxic agents for the treatment of NSCLC patients failed to demonstrate significant survival benefit in phase III studies.18 Until now, the precise molecular mechanisms that account for this phenomenon have remained unclear.19
Novel medicinal lead compounds are sought by assaying large compound collections that can be retrieved from natural sources or produced by synthetic chemistry or biotransformation.20 Natural substances derived mostly from plants provide an excellent platform for new drug discovery. Plant pentacyclic lupane‐type triterpenes, such as betulin and betulinic acid, are recognized as chemopreventive agents and are believed to be pharmacologically harmless. Betulinic acid, discovered in 1995 in the stem bark of the plant Zizyphus mauritiana, has been experimentally evaluated for its multiple biological activities.21, 22 Previously reported in vitro and in vivo studies have demonstrated that these compounds have anti‐tumor and antiproliferative properties, and induce apoptosis in various tumor cells.23, 24, 25 Apoptosis is accompanied by caspase activation, mitochondrial membrane alterations and DNA fragmentation.26 Thus far, betulinic acid and other betulin derivatives have been poorly explored against NSCLC,23, 27, 28 but many new studies have shown that they have a potential role in anti‐cancer therapy.29, 30, 31, 32
Recently, studies have shown that a combination of different drugs in tumor patient therapies may increase the efficiency of antitumor response. Combining drugs with different targets is a logical approach to overcome multilevel cross‐stimulation among key pathways in NSCLC progression. In the present study, we hypothesized that combined treatment of sorafenib and betulinic acid could enhance the inhibitory effect on NSCLC cells.
Materials and Methods
Cell culture and reagents
The NSCLC lines, with different KRAS mutations A549 (p.G12S), H358 (p.G12C) and A427 (p.G12D) were purchased from the American Type Culture Collection (Manassas, VA, USA) and cultured in recommended growth media with 10% FBS (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) and Antibiotic Antimycotic Solution (Sigma‐Aldrich, St. Louis, MO, USA). All cell lines were cultured at 37°C in a humidified atmosphere of 5% CO2. The cells were seeded at densities of 1 × 104 cells/0.1 mL (0.32 cm2) (cell viability assay), 6 × 104 cells/0.5 mL (1.9 cm2) (flow cytometry), 1 × 105 cells/3 mL (9.5 cm2) (long‐term colony formation assay, serial replating assay) and 1 × 106 cells/4 mL (21 cm2) (western blotting). The cells were treated with sorafenib (1.3 μg/mL; LC Laboratories), betulinic acid (3 μg/mL; Sigma‐Aldrich Chemistry), and both sorafenib and betulinic acid at 1 day post‐seeding. Three days later, the cells were collected for an appropriate assay.
Cell viability assay
Cell viability was assessed by CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA) according to the manufacturer's protocol. Each treatment within a single experiment was performed in triplicate. Absorbance at 490 nm was recorded by a Wallac 1420 VICTOR2 plate reader (PerkinElmer, Waltham, MA, USA). Data were normalized to untreated control.
Proliferation assay
Cell labeling with CellTrace Far Red was performed according to the protocols provided by the manufacturer (CellTrace Far Red Cell Proliferation Kit, Invitrogen, Molecular Probes, USA). The compound was dissolved in dry DMSO to make a 5‐mM stock solution stored at −20°C until use. Seeded cells were suspended in 1 mL PBS and 1 μL of CellTrace Far Red stock solution was added to a final concentration of 5 μM. Cells were incubated at 37°C and protected from light for 20 min. The cells were washed with warm PBS (with Ca2 + and Mg2 + ) and after 1 h in new clean medium, cells were treated with drugs. CellTrace Far Red produces a stable and well retained fluorescent signal with very little variance between cells within generations, allowing visualization of proliferating cells for up to eight generations. When cells were dividing, CellTrace Far Red distributed equally into daughter cells. Data was acquired on a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) and analyzed using Flowing Software 2.5.1 (Perttu Terho, Turku, Finland).
Annexin V staining
Apoptosis was assessed using an Annexin V Apoptosis Detection Kit (Santa Cruz Biotechnology, Santa Cruz, CA, USA) according to the manufacturer's protocol. Briefly, the cells were stained with Annexin V‐FITC (8 μg/mL) and PI (5 μg/mL) for 15 min at room temperature (RT) in the dark. In between steps, the cells were washed with cold PBS (with Ca2 + and Mg2 + ) containing 2.5% FBS. Data was acquired on a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) and analyzed using Flowing Software 2.5.1 (Perttu Terho, Turku, Finland). Apoptosis was quantified as a percentage of both Annexin V‐positive and Annexin V/PI‐double‐positive cells.
Western blotting
Whole cell lysates were prepared using cold RIPA buffer (150 mM NaCl, 50 mM Tris‐HCl pH 8.0, 1% NP‐40, 0.5% sodium deoxycholate, 1% SDS) supplemented with SigmaFAST Protease Inhibitor Cocktail (Sigma‐Aldrich) and Halt Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). The cell lysates were then sonicated for 10 s at 100% power using a Sonopuls HD 2070 ultrasonic homogenizer (Bandelin, Berlin, Germany) and centrifuged at 10 000 g for 10 min at 4°C to pellet cellular debris. Protein concentration was determined using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) according to the manufacturer's protocol. Absorbance at 570 nm was recorded by a Wallac 1420 VICTOR2 plate reader. Cell lysates with Laemmli sample buffer (50 mM Tris‐HCl pH 6.8, 10% glycerol, 5% 2‐mercaptoethanol, 2% SDS, 0.05% bromophenol blue) were heated for 5 min at 95°C, the proteins were separated by SDS‐PAGE using 8%–12% resolving gels (SDS‐PAGE running buffer: 25 mM Tris, 192 mM glycine, 0.1% SDS) and transferred (semi‐dry transfer) to PVDF membrane (0.45 μm pore size; Merck Millipore, Billerica, MA, USA). In between steps, membranes were washed with TBST (20 mM Tris, 150 mM NaCl, 0.1% Tween 20), blocked either with 1% casein in 0.1 M Tris‐HCl pH 8.0, 214 mM NaCl or 5% BSA/TBST (Sigma‐Aldrich) for 1 at RT or overnight at 4°C and then incubated with primary antibody overnight at 4°C. After probing with HRP‐conjugated secondary antibody for 1 h at RT, proteins of interest were detected using SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific). The following antibodies were used in this study: anti‐caspase‐8 p18 (1:1000, #sc‐7890; Santa Cruz Biotechnology), anti‐caspase‐9 p35 (1:1000, #sc‐8355; Santa Cruz Biotechnology), anti‐PARP (1:1000, #sc‐7150; Santa Cruz Biotechnology), anti‐CHOP (1:2000, #2895; Cell Signaling Technology), anti‐ Bcl‐2 (1:1000, #sc‐7382; Santa Cruz Biotechnology), anti‐ Bcl‐xs/l (1:1000, #sc‐634; Santa Cruz Biotechnology), anti‐ Bax (1:1000, #sc‐6236; Santa Cruz Biotechnology), anti‐ Bak (1:1000, #sc‐832; Santa Cruz Biotechnology), anti‐Akt (1:1000, #4691; Cell Signaling Technology), anti‐phospho‐Akt (1:1000, #4060; Cell Signaling Technology), anti‐mTOR (1:1000, #2983; Cell Signaling Technology), anti‐phospho‐mTOR (1:1000, #2974; Cell Signaling Technology), anti‐ERK1/2 (1:1000, #9102; Cell Signaling Technology), anti‐phospho‐ERK1/2 (1:1000, #9101; Cell Signaling Technology), anti‐actin/HRP (1:2000, #sc‐1615; Santa Cruz Biotechnology), anti‐mouse/HRP (1:2000, #P0447; Dako, Glostrup, Denmark) and anti‐rabbit/HRP (1:2000–3000, #P0048; Dako).
Long‐term colony formation assay
The viable cells were counted using a hemacytometer (trypan blue exclusion method) and seeded in duplicate at a density of 5 × 102 cells/6 mL (21 cm2). The dishes had been pre‐coated with poly‐L‐lysine/PBS (0.001%; Sigma‐Aldrich) and washed twice with PBS (with Ca2 + and Mg2 + ). After 2 weeks, the colonies were fixed and stained with 1% crystal violet/ethanol (Sigma‐Aldrich), documented with an Olympus Stylus SH‐50 camera (Olympus America, USA), and counted manually using ImageJ 1.47 software (National Institutes of Health, Bethesda, MD, USA). The term plating efficiency (PE) indicates the percentage of seeded cells that grow to form colonies. The surviving fraction (SF) is calculated as the ratio between PE of treated and control cells multiplied by 100.
Analysis of drug interaction
The nature of the interactions between drugs studied was analyzed using the combination index method, derived from the median‐effect principle of Chou and Talalay.33 CI values indicate the following: <0.1, very strong synergism; 0.1–0.3, strong synergism; 0.3–0.7, synergism; 0.7–0.85, moderate synergism; 0.85–0.9, slight synergism; 0.9–1.1, nearly additive; 1.1–1.2, slight antagonism; 1.2–1.45, moderate antagonism; 1.45–3.3, antagonism; 3.3–10, strong antagonism; >10, very strong antagonism. The CI value was calculated using CompuSyn software (ComboSyn, Paramus NJ, USA). The CI was defined as follows: CI = (D)1/(Dx)1 + (D)2/(Dx)2 for mutually exclusive drugs. In the denominator, (Dx) is for D1 “alone” that inhibits a system x%, and (Dx)2 is for D2 “alone” that inhibits a system x%. In the numerators, (D)1 + (D)2 “in combination” also inhibit x%.
Statistical analysis
Data are presented as means ± SD of results from at least three independent experiments. Comparisons between two groups: sorafenib treatment group versus combinatorial treatment group and betulinic acid treatment group versus combinatorial treatment group were analyzed by two‐tailed Student's t‐test. Significance was assumed at *P < 0.05 and **P <0.01.
Results
Combination of sorafenib and betulinic acid inhibits growth of non‐small cell lung cancer cell lines
To evaluate the effect of sorafenib, betulinic acid and their combination on the viability of NSCLC cells in vitro, we first examined the effects of this drug on the growth of NSCLC cells using the MTS assay. NSCLC cells were treated either with 1.3 μg/mL sorafenib or 3 μg/mL betulinic acid alone or in combination for 72 h. Sorafenib and betulinic acid alone decreased cell viability by an average of 88.3 ± 4.9 and 62.2 ± 8.8% in A549 cells, 89.2 ± 9.1 and 70.3 ± 7.5% in H358 cells, and 94.2 ± 1.4 and 77.7 ± 5.1% in A427 cells, respectively, but the combination treatment reduced cell viability more effectively to 52.2 ± 3.9% in A549 cells (CI = 0.749), 54.4 ± 5.3% in H358 cells (CI = 0.802) and 34.5 2.6% in A427 cells (CI = 0.497) (Fig. 1a). We next investigated whether the combination of relatively low concentrations of sorafenib and betulinic acid could inhibit NSCLC cell proliferation in vitro using a CellTrace Far Red Cell Proliferation Kit. As shown in Figure 1b, combination treatment significantly reduced the percentage of proliferating cells to 4.15 ± 2.45% in A549 cells (CI = 0.224), 13.59 ± 6.63% in H358 cells (CI = 0.169) and 9.21 ± 1.44% in A427 cells (CI = 0.249), as compared to either compound alone (P < 0.05).
Figure 1.
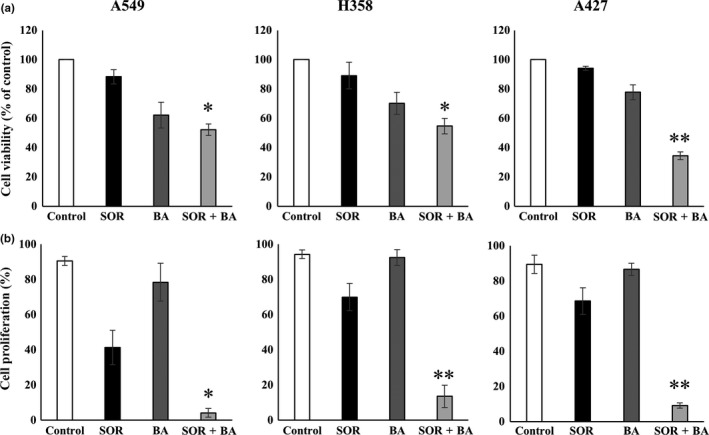
Effects of combination treatment with sorafenib (SOR) and betulinic acid (BA) on the growth of non‐small cell lung cancer (NSCLC) cell lines. (a) Cell viability of A549, H358 and A427 cells after exposure to 1.3 μg/mL sorafenib and 3 μg/mL betulinic acid alone and in combination. (b) Cell proliferation of A549, H358 and A427 cells after treatment with 1.3 μg/mL sorafenib and 3 μg/mL betulinic acid alone and in combination (n = 4). Data are presented as means ± SD normalized to untreated control. *P < 0.05, **P < 0.01 compared with sorafenib treatment group and betulinic acid treatment group.
Combined treatment of sorafenib and betulinic acid induce cell death in non‐small cell lung cancer cell lines
We measured apoptosis‐associated cell death using Annexin V‐FITC/PI double staining and we detected apoptosis‐related genes by western blotting to examine whether cell number reduction could be attributed to increased cell death. Combined treatment with sorafenib and betulinic acid induced apoptosis in 85.1 ± 5.4% of A549 cells, 54.5 ± 8.1% of H358 cells and 88.1 ± 11.9% of A427 cells (Fig. 2a). Treatment with a combination of these drugs led to a marked increase of apoptotic cells when compared to the extent of apoptosis following single drug treatments (P < 0.01) (Fig. 2a).
Figure 2.
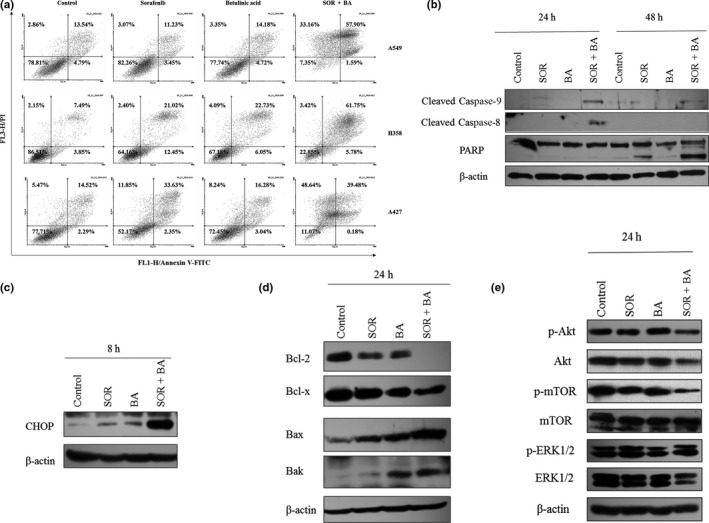
Cytotoxicity effect of combination treatment with sorafenib (SOR) and betulinic acid (BA) on non‐small cell lung cancer (NSCLC) cells. (a) Representative Annexin V‐FITC/PI‐double staining histograms of A549, H358 and A427 cells after treatments with 1.3 μg/mL sorafenib and 3 μg/mL betulinic acid alone and in combination (n = 4). (b) Representative immunoblot of cleaved caspase‐9 and caspase‐8 activation and PARP fragmentation from A549 cells treated with 1.3 μg/mL sorafenib and 3 μg/mL betulinic acid alone and in combination (n = 3). Actin served as a loading control. (c) Representative immunoblot of CHOP expression from A549 cells treated with 1.3 μg/mL sorafenib and 3 μg/mL betulinic acid alone and in combination (n = 3). Actin served as a loading control. (d) Representative immunoblot of bcl‐2 family expression from A549 cells treated with 1.3 μg/mL sorafenib and 3 μg/mL betulinic acid alone and in combination (n = 3). Actin served as a loading control. (e) Representative immunoblots of phospho‐Akt (Ser473), Akt, phospho‐mTOR (Ser2481), mTOR, phospho‐ERK1/2, ERK1/2 expression from A549 cells after 24 h treatment with 1.3 μg/mL sorafenib and 3 μg/mL betulinic acid alone and in combination (n = 3). Actin served as a loading control.
Apoptosis‐related caspase‐9 and caspase‐8 gene activation and PARP cleavage was also measured in A549 cells (Fig. 2b). Combination of sorafenib and betulinic acid resulted in procaspase‐9 and procaspase‐8 cleavage, which led to the appearance of caspase‐9 and caspase‐8 after 24 h and cleaved PARP after 48 h.
The expression patterns of CHOP, Bcl‐2, Bcl‐x, Bax and Bak were determined by western blot analysis to explore the possible mechanism of the proapoptotic effect of combination treatment with sorafenib and betulinic acid. The results (Fig. 2c,d) showed that combined treatment with sorafenib and betulinic acid significantly increased the expression of CHOP and Bax proteins, and decreased the expression of anti‐apoptotic genes, Bcl‐2 and Bcl‐x in live A549 cells when compared to the expression levels in cells treated with sorafenib and betulinic acid alone. In contrast, the expression of Bak was increased by betulinic acid alone.
The PI3K/AKT and MAPK signaling pathways are among the most frequently deregulated pathways in cancer, thereby suggesting a key role in carcinogenesis. Therefore, using western blotting, we evaluated the effect of sorafenib and betulinic acid alone and in combination on the phosphorylation of these proteins. The combination treatment inhibited expression or phosphorylation of Akt and mTOR, whereas MAPK was not affected (Fig. 2e).
Combined but not single sorafenib and betulinic acid treatment markedly reduces colony‐forming ability of non‐small cell lung cancer cells
Subsequently, we investigated whether combinatorial treatments with sorafenib and betulinic acid could impair NSCLC cell colony‐forming ability more effectively than individual compounds. For this purpose, we performed a long‐term colony formation assay. As shown in Figure 3, the clonogenic activity of A549, H358 and A427 cells after combinatorial but not single sorafenib and betulinic acid treatment significantly decreased (P < 0.05). This effect was much stronger in H358 cells (Fig. 3).
Figure 3.

Combinational treatment with sorafenib (SOR) and betulinic acid (BA) significantly reduces the colony‐forming ability of A549, H358 and A427 cells as compared to either compound alone. Long‐term colony formation assay. On the left: representative images of colonies formed by A549, H358 and A427 cells after treatment with 1.3 μg/mL sorafenib and 3 μg/mL betulinic acid alone and in combination. On the right: the surviving fraction (SF) of A549, H358 and A427 cells after treatment with 1.3 μg/mL sorafenib and 3 μg/mL betulinic acid alone and in combination (n = 3). *P < 0.05, **P < 0.01 compared with sorafenib treatment group and betulinic acid treatment group.
Combination treatment with sorafenib and betulinic acid is not toxic to human peripheral blood lymphocytes
Peripheral blood lymphocyte (PBL) cells were treated with 1.3 μg/mL sorafenib and 3 μg/mL betulinic acid alone and in combination. Cell viability was measured after 72 h by the MTS assay. As shown in Figure 4a, combination treatment inhibited cell growth by an average of 67 ± 3.1%, but the percentage of apoptotic cells measured by Annexin V‐FITC/PI‐double staining showed that combinational treatment with sorafenib and betulinic acid did not increase apoptosis in PBL cells.
Figure 4.
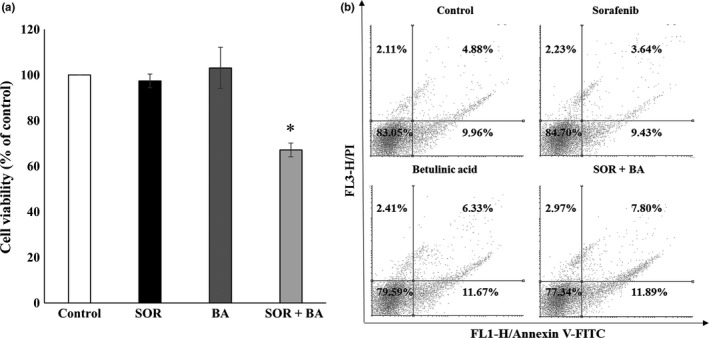
Combination treatment with sorafenib (SOR) and betulinic acid (BA) is not toxic to human peripheral blood lymphocytes (PBL). (a) Cell viability of PBL cells after treatment with sorafenib 1.3 μg/mL and betulinic acid 3 μg/mL alone and in combination (n = 4). Data are presented as means ± SD normalized to untreated control. (b) Representative Annexin V‐FITC/PI‐double staining histograms of PBL cells after treatments with 1.3 μg/mL sorafenib and 3 μg/mL betulinic acid alone and in combination (n = 3). *P < 0.05, **P < 0.01 compared with sorafenib treatment group and betulinic acid treatment group.
Discussion
One of the goals of cancer chemotherapy is to explore and develop therapies that can selectively induce apoptosis in cancer cells. Recent studies have shown that different drug combinations used in the treatment of tumor patients can increase the efficiency of antitumor response. In the present study, we hypothesized that the combination of sorafenib and betulinic acid could enhance the inhibitory effect in NSCLC cells.
Previous studies have shown that sorafenib alone inhibited the growth of NSCLC cells, and its combination with other drugs, such as gemcitabine or erlotinib, could increase the inhibitory effect in NSCLC cells or mouse models.34, 35 The effectiveness of betulinic acid and other betulin derivatives in combination with chemotherapy or other target drugs is poorly explored. Only the synergistic effect of betulin with acyclovir and rimantadine was measured against herpes simplex and influenza viruses.36, 37 To the best of our knowledge, the results of the present study have demonstrated for the first time that the combination of sorafenib and betulinic acid provides synergistic anti‐proliferative and pro‐apoptotic effects in NSCLC cells.
In this study, we found that the exposure of NSCLC cells to sorafenib and betulinic acid more effectively reduced cell viability and clonogenicity, and increased programmed cell death more than individual compounds at the same or even higher concentrations. We evaluated the effects of sorafenib and betulinic acid on the growth of NSCLC cells using two different assay systems, the MTS assay and the CellTrace Far Red Cell Proliferation Kit assay. We found that the potency of combination treatment in cell growth inhibition was similar for NSCLC cells, regardless of the assay system used. The inhibitory effect of sorafenib on tumor cell growth in vitro is mediated by the inhibition of RAF signaling pathways.11 Takezawa et al.12 showed that sorafenib targeted B‐RAF in NSCLC cells with wild‐type KRAS and C‐RAF in cells with mutant KRAS. The literature suggests that betulinic acid has the potential to specifically inhibit cancer cell growth through targeting the phosphoinositide 3‐kinase (PI3K)/AKT signaling pathway.26 In the present study, we also examined this intracellular signaling pathway in NSCLC cell lines following treatment with the combination of sorafenib and betulinic acid. Our results showed that sorafenib combined with betulinic acid inhibited the Akt signaling pathway in A549 cells. One of the consequences of Akt inhibition is the inhibition of mTOR. In turn, no changes were detected in MAPK activation. Thus, the combination of sorafenib with betulinic acid may target the PI3K/AKT pathway and this targeted approach may underlie the synergistic effects revealed here.
Apoptosis is associated with the inhibition of the Bcl‐2 protein family and the activation of the caspase cascade.38 The function of Bcl‐2 protein, also known as pro‐survival Bcl‐2 protein, is to inhibit apoptosis and prolong cell survival. Overexpression of Bcl‐2 protein is associated with a poor response to lung cancer treatment.39 Bax, a pro‐apoptotic protein of the Bcl‐2 family, plays a key role in mediating the apoptotic response.40 The ratio of Bcl‐2 to Bax is commonly considered a determinant in the initiation of apoptosis.41 We observed that combination treatment with sorafenib and betulinic acid could significantly inhibit the downregulation of Bcl‐2 and upregulation of Bax. Kennedy et al.42 demonstrated that Akt inhibited apoptosis and cytochrome c release induced by several pro‐apoptotic Bcl‐2 family members. Suppression of Akt can directly activate Bad. Several papers have reported that suppression of PI3K/AKT signaling induces endoplasmic reticulum (ER) stress and that apoptosis involves Bax/Bak oligomerization. 43 In this study, we have shown that betulinic acid alone increased the expression of Bak. In addition, the Bcl‐2 protein family is essential in mediating the crosstalk between ER and mitochondria to trigger apoptosis under chronic or irreversible ER stress.44 We used CCAAT/enhancer‐binding protein (C/EBP) homologous protein (CHOP) as a marker of ER stress‐mediated apoptosis, the expression of which is induced after combination treatment. CHOP is a transcription factor involved in the ER stress response. The overexpression of CHOP induces apoptosis through downregulation of Bcl‐2 and upregulation of the pro‐apoptotic proteins like Bim, Bax and Puma.45, 46 In our investigation, CHOP was rapidly induced by combination of sorafenib and betulinic acid. We found that combined treatment with sorafenib and betulinic acid induced both caspase 9 and caspase 8 in A549 cells, suggesting that both mitochondrial‐dependent and mitochondrial‐independent factors were responsible for the apoptotic potential. Subsequently, elevated levels of caspases led to the formation of cleaved product (85 kDa) of poly ADP‐ribose polymerase (PARP). The function of PARP is a routine repair of DNA damage and this protein is commonly considered as a biochemical hallmark of apoptosis.47 The annexin V/PE experiment was used to determine the apoptosis in A549, H358 and A427 NSCLC cells. The results showed that the apoptosis rate of the experimental group was significantly increased, as compared to the control group. The obtained results suggest that the possible mechanism of action of both compounds involves inhibition of Akt, which, in turn, induces ER stress, CHOP overexpression, decreasing Bcl2 expression and enhancing Bak expression. This is followed by the activation of caspases and cleavage of PARP in the nucleus.
We also performed a longer‐term clonogenic survival assay and, once again, we found that the combination with sorafenib and betulinic acid inhibited the survival of NSCLC cells. It should be noted that combinatorial treatments with low concentration of sorafenib and betulinic acid have therapeutic potential for NSCLC treatment, because targeting a clonogenic/tumor initiating/stem cell‐like subset of cancer cells is thought to be essential for a successful cancer therapy.48
Clinically, phase III trials of sorafenib in previously treated advanced NSCLC have failed, because they did not significantly increase overall survival and generated unacceptable toxicities.19 We used a low concentration of this drug to avoid this toxicity effect. In this study, we have shown that combination treatment with sorafenib and betulinic acid is not toxic for human PBL.
Recently, several publications have shown a synergistic effect of sorafenib in combination with different compounds against different types of tumors. Sorafenib in combination with histone deacetylase (HDAC) inhibitors exerted antiproliferative effect in vitro and in vivo on anaplastic thyroid carcinoma cell lines.49 Sorafenib combined with gemcitabine or pemetrexed generated a synergistic effect against the A549 cell line.50 The antitumor effect of sorafenib and pemetrexed combination was additionally enhanced by PDE5 inhibitors in NSCLC cells.51 Varinostat, when combined with gefitinib, strongly inhibited the growth of mutant KRAS lung cancer cells and hepatocarcinoma, but the combination of sorafenib and varinostat did not show such an effect.52
In conclusion, the present study showed that sorafenib in combination with betulinic acid enhanced the inhibitory effect on NSCLC cells. Betulinic acid strengthened the anti‐proliferative action of sorafenib, promoting NSCLC cell apoptosis and allowing for the use of lower doses of sorafenib than currently applied. More importantly, the combination of sorafenib and betulinic acid was effective regardless of KRAS mutations. Therefore, it is worthwhile considering this combination treatment for NSCLC, and further evaluation in clinical trials is warranted.
Disclosure Statement
The authors have no conflicts of interest to declare.
Acknowledgments
This work was supported by a grant from the Polish Academy of Sciences (no. 03/2016).
Cancer Sci 108 (2017) 2265–2272
Funding Information
Polish Academy of Sciences, (Grant/Award Number: ‘ no 03/2016‘)
References
- 1. Ettinger DS, Wood DE, Akerley W et al Non‐small cell lung cancer, Version 6.2015. J Natl Compr Canc Netw 2015; 13: 515–24. [DOI] [PubMed] [Google Scholar]
- 2. Kutkowska J, Porebska I, Rapak A. Non‐small cell lung cancer – Mutations, targeted and combination therapy. Postepy Hig Med Dosw (Online) 2017; 71: 431–45. [DOI] [PubMed] [Google Scholar]
- 3. Vineis P, Alavanja M, Buffler P et al Tobacco and cancer: recent epidemiological evidence. J Natl Cancer Inst 2004; 96: 99–106. [DOI] [PubMed] [Google Scholar]
- 4. Politi K, Herbst RS. Lung cancer in the era of precision medicine. Clin Cancer Res 2015; 21: 2213–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cheng L, Alexander RE, Maclennan GT et al Molecular pathology of lung cancer: key to personalized medicine. Mod Pathol 2012; 25: 347–69. [DOI] [PubMed] [Google Scholar]
- 6. Sherwood J, Dearden S, Ratcliffe M, Walker J. Mutation status concordance between primary lesions and metastatic sites of advanced non‐small‐cell lung cancer and the impact of mutation testing methodologies: a literature review. J Exp Clin Cancer Res 2015; 34: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krawczyk P, Ramlau R, Chorostowska‐Wynimko J et al The efficacy of EGFR gene mutation testing in various samples from non‐small cell lung cancer patients: a multicenter retrospective study. J Cancer Res Clin Oncol 2015; 141: 61–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bravaccini S, Tumedei MM, Ulivi P et al ALK translocation detection in non‐small cell lung cancer cytological samples obtained by TBNA or EBUS‐TBNA. Cytopathology 2015; 27: 103–7. [DOI] [PubMed] [Google Scholar]
- 9. Karachaliou N, Mayo C, Costa C et al KRAS mutations in lung cancer. Clin Lung Cancer 2013; 14: 205–14. [DOI] [PubMed] [Google Scholar]
- 10. Miyake N, Chikumi H, Takata M, Nakamoto M, Igishi T, Shimizu E. Rapamycin induces p53‐independent apoptosis through the mitochondrial pathway in non‐small cell lung cancer cells. Oncol Rep 2012; 28: 848–54. [DOI] [PubMed] [Google Scholar]
- 11. Zhang J, Gold KA, Kim E. Sorafenib in non‐small cell lung cancer. Expert Opin Investig Drugs 2012; 21: 1417–26. [DOI] [PubMed] [Google Scholar]
- 12. Takezawa K, Okamoto I, Yonesaka K et al Sorafenib inhibits non‐small cell lung cancer cell growth by targeting B‐RAF in KRAS wild‐type cells and C‐RAF in KRAS mutant cells. Cancer Res 2009; 69: 6515–21. [DOI] [PubMed] [Google Scholar]
- 13. Hong S, Tan M, Wang S, Luo S, Chen Y, Zhang L. Efficacy and safety of angiogenesis inhibitors in advanced non‐small cell lung cancer: a systematic review and meta‐analysis. J Cancer Res Clin Oncol 2015; 141: 909–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dingemans AM, Mellema WW, Groen HJ et al A phase II study of sorafenib in patients with platinum‐pretreated, advanced (Stage IIIb or IV) non‐small cell lung cancer with a KRAS mutation. Clin Cancer Res 2013; 19: 743–51. [DOI] [PubMed] [Google Scholar]
- 15. Wakelee HA, Lee JW, Hanna NH, Traynor AM, Carbone DP, Schiller JH. A double‐blind randomized discontinuation phase‐II study of sorafenib (BAY 43‐9006) in previously treated non‐small‐cell lung cancer patients: eastern cooperative oncology group study E2501. J Thorac Oncol 2012; 7: 1574–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Adjei AA, Molina JR, Mandrekar SJ et al Phase I trial of sorafenib in combination with gefitinib in patients with refractory or recurrent non‐small cell lung cancer. Clin Cancer Res 2007; 13: 2684–91. [DOI] [PubMed] [Google Scholar]
- 17. Lim SM, Cho BC, Kim SW et al A multicenter phase II study of sorafenib in combination with erlotinib in patients with advanced non‐small cell lung cancer (KCSG‐0806). Lung Cancer 2016; 93: 1–8. [DOI] [PubMed] [Google Scholar]
- 18. Scagliotti G, Novello S, von Pawel J et al Phase III study of carboplatin and paclitaxel alone or with sorafenib in advanced non‐small‐cell lung cancer. J Clin Oncol 2010; 28: 1835–42. [DOI] [PubMed] [Google Scholar]
- 19. Paz‐Ares L, Hirsh V, Zhang L et al MISSION Trial – A phase III, multi‐center, placebo‐controlled trial of sorafenib in patients with relapsed or refractory predominantly non‐squamous NSCLC after 2 or 3 previous treatment regimens. J Thorac Oncol 2015; 10: 1745–53. [DOI] [PubMed] [Google Scholar]
- 20. Newman D, Cragg G. Natural products in medicinal chemistry. Bioorg Med Chem 2009; 17: 2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Drag‐Zalesinska M, Wysocka T, Borska S et al The new esters derivatives of betulin and betulinic acid in epidermoid squamous carcinoma treatment – In vitro studies. Biomed Pharmacother 2015; 72: 91–7. [DOI] [PubMed] [Google Scholar]
- 22. Cichewicz RH, Kouzi SA. Chemistry, biological activity, and chemotherapeutic potential of betulinic acid for the prevention and treatment of cancer and HIV infection. Med Res Rev 2004; 24: 90–114. [DOI] [PubMed] [Google Scholar]
- 23. Li XD, Zhang YJ, Han JC. Betulin inhibits lung carcinoma proliferation through activation of AMPK signaling. Tumour Biol 2014; 35: 11153–8. [DOI] [PubMed] [Google Scholar]
- 24. Orchel A, Kulczycka A, Chodurek E et al Influence of betulin and 28‐O‐propynoylbetulin on proliferation and apoptosis of human melanoma cells (G‐361). Postepy Hig Med Dosw (Online) 2014; 68: 191–7. [DOI] [PubMed] [Google Scholar]
- 25. Yi J, Zhu R, Wu J et al In vivo protective effect of betulinic acid on dexamethasone induced thymocyte apoptosis by reducing oxidative stress. Pharmacol Rep 2016; 68: 95–100. [DOI] [PubMed] [Google Scholar]
- 26. Majeed R, Hussain A, Sangwan PL et al PI3K target based novel cyano derivative of betulinic acid induces its signalling inhibition by down‐regulation of pGSK3beta and cyclin D1 and potentially checks cancer cell proliferation. Mol Carcinog 2016; 55: 964–76. [DOI] [PubMed] [Google Scholar]
- 27. Das J, Das S, Paul A, Samadder A, Khuda‐Bukhsh AR. Strong anticancer potential of nano‐triterpenoid from Phytolacca decandra against A549 adenocarcinoma via a Ca(2 + )‐dependent mitochondrial apoptotic pathway. J Acupunct Meridian Stud 2014; 7: 140–50. [DOI] [PubMed] [Google Scholar]
- 28. Liao CR, Kuo YH, Ho YL et al Studies on cytotoxic constituents from the leaves of Elaeagnus oldhamii Maxim. in non‐small cell lung cancer A549 cells. Molecules 2014; 19: 9515–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Krol SK, Kielbus M, Rivero‐Muller A, Stepulak A. Comprehensive review on betulin as a potent anticancer agent. Biomed Res Int 2015; 2015: 584189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang SJ, Liu MC, Xiang HM, Zhao Q, Xue W, Yang S. Synthesis and in vitro antitumor evaluation of betulin acid ester derivatives as novel apoptosis inducers. Eur J Med Chem 2015; 102: 249–55. [DOI] [PubMed] [Google Scholar]
- 31. Potze L, Di Franco S, Grandela C et al Betulinic acid induces a novel cell death pathway that depends on cardiolipin modification. Oncogene 2016; 35: 427–37. [DOI] [PubMed] [Google Scholar]
- 32. Ali‐Seyed M, Jantan I, Vijayaraghavan K, Bukhari SN. Betulinic acid: recent advances in chemical modifications, effective delivery, and molecular mechanisms of a promising anticancer therapy. Chem Biol Drug Des 2016; 87: 517–36. [DOI] [PubMed] [Google Scholar]
- 33. Chou TC, Talalay P. Quantitative analysis of dose‐effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22: 27–55. [DOI] [PubMed] [Google Scholar]
- 34. Pasqualetti G, Ricciardi S, Mey V, Del Tacca M, Danesi R. Synergistic cytotoxicity, inhibition of signal transduction pathways and pharmacogenetics of sorafenib and gemcitabine in human NSCLC cell lines. Lung Cancer 2011; 74: 197–205. [DOI] [PubMed] [Google Scholar]
- 35. Giovannetti E, Labots M, Dekker H et al Molecular mechanisms and modulation of key pathways underlying the synergistic interaction of sorafenib with erlotinib in non‐small‐cell‐lung cancer (NSCLC) cells. Curr Pharm Des 2013; 19: 927–39. [PubMed] [Google Scholar]
- 36. Gong Y, Raj KM, Luscombe CA et al The synergistic effects of betulin with acyclovir against herpes simplex viruses. Antiviral Res 2004; 64: 127–30. [DOI] [PubMed] [Google Scholar]
- 37. Savinova OV, Pavlova NI, Boreko EI. New betulin derivatives in combination with rimantadine for inhibition of influenza virus reproduction. Antibiot Khimioter 2009; 54: 16–20. [PubMed] [Google Scholar]
- 38. Scorrano L, Korsmeyer SJ. Mechanisms of cytochrome c release by proapoptotic BCL‐2 family members. Biochem Biophys Res Commun 2003; 304: 437–44. [DOI] [PubMed] [Google Scholar]
- 39. Groeger AM, Esposito V, De Luca A et al Prognostic value of immunohistochemical expression of p53, bax, Bcl‐2 and Bcl‐xL in resected non‐small‐cell lung cancers. Histopathology 2004; 44: 54–63. [DOI] [PubMed] [Google Scholar]
- 40. Walensky LD. BCL‐2 in the crosshairs: tipping the balance of life and death. Cell Death Differ 2006; 13: 1339–50. [DOI] [PubMed] [Google Scholar]
- 41. Cory S, Adams JM. Killing cancer cells by flipping the Bcl‐2/Bax switch. Cancer Cell 2005; 8: 5–6. [DOI] [PubMed] [Google Scholar]
- 42. Kennedy SG, Kandel ES, Cross TK, Hay N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol 1999; 19: 5800–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lin ML, Chen SS, Huang RY et al Suppression of PI3K/AKT signaling by synthetic bichalcone analog TSWU‐CD4 induces ER stress‐ and Bax/Bak‐mediated apoptosis of cancer cells. Apoptosis 2014; 19: 1637–53. [DOI] [PubMed] [Google Scholar]
- 44. Rodriguez D, Rojas‐Rivera D, Hetz C. Integrating stress signals at the endoplasmic reticulum: the BCL‐2 protein family rheostat. Biochim Biophys Acta 2011; 1813: 564–74. [DOI] [PubMed] [Google Scholar]
- 45. Nishitoh H. CHOP is a multifunctional transcription factor in the ER stress response. J Biochem 2012; 151: 217–9. [DOI] [PubMed] [Google Scholar]
- 46. Lu HY, Chen XQ, Tang W, Wang QX, Zhang J. GRP78 silencing enhances hyperoxia‐induced alveolar epithelial cell apoptosis via CHOP pathway. Mol Med Rep 2017; 16: 1493–501. [DOI] [PubMed] [Google Scholar]
- 47. Mazumder S, Plesca D, Almasan A. Caspase‐3 activation is a critical determinant of genotoxic stress‐induced apoptosis. Methods Mol Biol 2008; 414: 13–21. [DOI] [PubMed] [Google Scholar]
- 48. Morrison R, Schleicher SM, Sun Y et al Targeting the mechanisms of resistance to chemotherapy and radiotherapy with the cancer stem cell hypothesis. J Oncol 2011; 2011: 941876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Park KC, Kim SM, Jeon JY et al Synergistic activity of N‐hydroxy‐7‐(2‐naphthylthio) Heptanomide and sorafenib against cancer stem cells, anaplastic thyroid cancer. Neoplasia 2017; 19: 145–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li J, Wang S, Su ZF, Yuan Y. Synergistic effects of sorafenib in combination with gemcitabine or pemetrexed in lung cancer cell lines with K‐ras mutations. Contemp Oncol (Pozn) 2016; 20: 33–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Booth L, Roberts JL, Poklepovic A, Dent P. PDE5 inhibitors enhance the lethality of [pemetrexed + sorafenib]. Oncotarget 2017; 8: 13464–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jeannot V, Busser B, Vanwonterghem L et al Synergistic activity of vorinostat combined with gefitinib but not with sorafenib in mutant KRAS human non‐small cell lung cancers and hepatocarcinoma. Onco Targets Ther 2016; 9: 6843–55. [DOI] [PMC free article] [PubMed] [Google Scholar]