

Abstract
In cancer cells the small compounds erastin and RSL3 promote a novel type of cell death called ferroptosis, which requires iron‐dependent accumulation of lipid reactive oxygen species. Here we assessed the contribution of lipid peroxidation activity of lipoxygenases (LOX) to ferroptosis in oncogenic Ras‐expressing cancer cells. Several 12/15‐LOX inhibitors prevented cell death induced by erastin and RSL3. Furthermore, siRNA‐mediated silencing of ALOX15 significantly decreased both erastin‐induced and RSL3‐induced ferroptotic cell death, whereas exogenous overexpression of ALOX15 enhanced the effect of these compounds. Immunofluorescence analyses revealed that the ALOX15 protein consistently localizes to cell membrane during the course of ferroptosis. Importantly, treatments of cells with ALOX15‐activating compounds accelerated cell death at low, but not high doses of erastin and RSL3. These observations suggest that tumor ferroptosis is promoted by LOX‐catalyzed lipid hydroperoxide generation in cellular membranes.
Keywords: Cell death, glutathione peroxidase, iron, Ras, reactive oxygen species
Ferroptosis is a cell death program that is distinct from apoptosis and necroptosis.1, 2 During ferroptosis, glutathione (GSH) depletion resulting from inhibition of cystine uptake or inactivation of the GSH‐dependent lipid peroxidase GPX4 causes iron‐dependent accumulation of reactive oxygen species (ROS) and lipid ROS that result in cell death.3, 4 Ferroptosis can be triggered by a number of small molecules, including erastin and (1S, 3R)‐RSL3, which directly inhibit the cell surface cystine/glutamate antiporter (system xc −) and GPX4, respectively.3, 4 Recent studies reported that the anti‐neoplastic drugs sorafenib and altretamine also inhibit system xc − and GPX4, respectively.5, 6, 7 Although oncogenic RAS expression sensitizes cells to ferroptosis, in some cases RAS‐driven NRF2 activation induces increased anti‐oxidant gene expression that results in increased resistance to cell death.4, 8 Meanwhile, wild type p53 expression can promote ferroptosis by suppressing the system xc − expression, which suggests that ferroptosis may have a tumor‐suppressive role.9
Specific inhibitors have been developed to investigate cell death signaling pathways involved in ferroptosis and its role in several diseases.1 Ferrostatins and liproxstatins, compounds that are related to lipophilic antioxidants, selectively blocked ferroptosis induced by GPX4 inhibition.3, 10 Thus, GPX4‐mediated reduction of toxic lipid peroxides is critical for suppressing ferroptosis in normal and cancer cells. GPX4 essentially regulates membrane peroxide levels that, in turn, affect lipoxygenase (LOX) activity.11 In mouse embryonic fibroblasts (MEF), Gpx4 depletion leads to 12/15‐lox‐dependent cell death, whereas 12/15‐lox deficient cells were resistant to GSH depletion.12 In contrast, Alox15 gene deletion did not rescue mice that expressed inactive GPX4 from embryonic lethality13 or protect Gpx4‐deficient mice from acute renal damage.10 The finding that several LOX inhibitors are effective in preventing erastin‐induced ferroptosis in MEF and pancreatic cancer cells indicates a possible role for LOX in chemical‐induced ferroptosis.10, 14
Lipoxygenase are nonheme iron‐containing dioxygenases that catalyze the stereospecific insertion of oxygen into polyunsaturated fatty acids (PUFA) such as arachidonic acid and linoleic acid.15 Although most LOX prefer free fatty acids as a substrate, some isoforms, including rabbit 15‐LOX, can oxygenate esterified polyenoic fatty acids in cellular membranes. Human LOX are expressed in a variety of tissues and several tumors, and have been implicated in cancer,16 although the role of LOX in cancer is unclear. Cancer cells accumulate iron, and iron provisioned from extracellular pathways mediated by transferrin receptors and from intracellular pathways involved in ferritinophagy is essential for ferroptotic cell death.17, 18, 19 Iron may directly catalyze the formation of lipid radicals and the propagation of chain lipid peroxidation, but LOX‐mediated generation of lipid hydroperoxides is also involved in cell death progression. In this study, we examined the expression and dynamics of several LOX isoforms during ferroptosis in a human fibrosarcoma cell line.
Materials and Methods
Chemicals
Erastin was purchased from Calbiochem (Darmstadt, Germany). Baicalein and PD146176 were purchased from Cayman Chemical and Santa Cruz Biotech, respectively. 6‐hydroxy‐2,5,7,8‐tetramethylchroman‐2‐carboxylic acid (Trolox) was purchased from Tokyo Chemical Industries (Tokyo). (1S, 3R)‐RSL3 was synthesized as described previously.19
Synthesis of an ALOX15 activator
The ALOX15 activator (E)‐1‐(7‐benzylidene‐3‐phenyl‐3,3a,4,5,6,7‐hexahydroindazol‐2‐yl)‐2‐(4‐methylpiperazin‐1‐yl)ethanone (Fig. 1, compound 4) was synthesized according to a previously described method.20 The key intermediate, (E)‐1‐(7‐benzylidene‐3‐phenyl‐3,3a,4,5,6,7‐hexahydro‐indazole‐2‐yl)‐2‐chloroethanone (compound 3), was synthesized from 2,6‐bis(benzylidene)‐1‐cyclohexanone (compound 1) according to a previous report.21 Briefly, K2CO3 (83.2 mg, 0.602 mmol, 2.0 eq.), KI (15.0 mg, 0.090 mmol, 0.3 eq.) and 1‐methylpiperazine (50.3 mL, 0.451 mmol, 1.5 eq) were added to a stirred solution of compound 3 (111 mg, 0.301 mmol) in CH3CN (10 mL). The reaction mixture was refluxed for 3 h. After the reaction mixture was cooled to room temperature, the insoluble solid was removed by filtration and the filtrate was concentrated in vacuo. The residue was extracted by ethyl acetate and the organic layer was washed with brine before drying over anhydrous Na2SO4. After filtering to remove the Na2SO4, the filtrate was concentrated in vacuo to yield the crude product. The remaining starting material (compound 3) was removed as a crystalline solid from hexane‐ethyl acetate. The mother liquor was concentrated in vacuo and the resulting residue was lyophilized to yield ALOX15 activator (compound 4) as a white solid with 69% yield. The compound structure was confirmed by recording electrospray‐ionization mass spectra (ESI‐MS) using an Applied Biosystems API‐2000 mass spectrometer. ESI‐MS(m/z): detected, 429.4 [MH]+ C27H33N4O calculated, 429.3.
Figure 1.
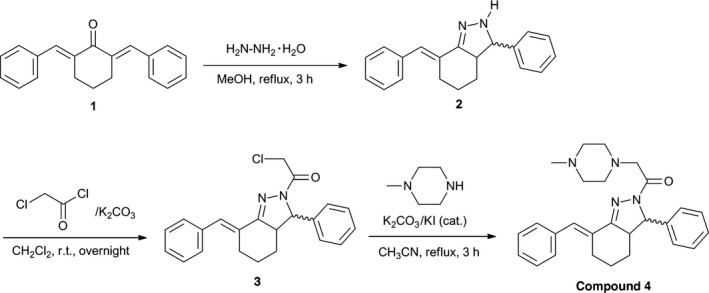
Synthesis of an ALOX15 activator. The ALOX15 activator (Compound 4) was synthesized as described in the Materials and Methods.
Cell culture and ferroptosis induction
HT1080 (human fibrosarcoma) and Panc‐1 (human pancreatic carcinoma) cells were cultured in DMEM containing 10% FBS. Calu‐1 (human non‐small cell lung cancer) cells were cultured in Eagle's minimum essential medium with Earle's salts. For ferroptosis induction, cells were plated at 1.0 × 105 cells per well on six‐well plates and treated with erastin or RSL‐3, as described previously.19 The cell death rate was determined by a trypan blue dye‐exclusion assay.
Plasmid construction
Full‐length ALOX12 and ALOX15 genes were amplified by PCR using the HT1080 and the 293 cDNA library, respectively, as a template. Truncated fragments of ALOX15 (110‐662, lacking the N‐terminal domain) were constructed by PCR using the primer 5′‐CGGAATTCATGGAAGGCACCGGCCGCACTGT‐3′. All fragments were subcloned into pcDNA3 (Invitrogen, CA, USA) with monomeric red fluorescent protein (mRFP) (a kind gift from Dr Tsien, University of California, San Diego).
siRNA and transfection
siRNA against human ALOX15 was designed and synthesized by Integrated DNA Technologies (hs.Ri.ALOX15.13). Transfections were performed using HilyMax reagent (Dojindo Mol Tech, Kumamoto, Japan) and Lipofectamin 3000 reagent (Invitrogen), as described previously.19
Western blot analysis
Immunoblotting was performed as described previously.19 Blotted membranes were blocked with 5% skim milk for 30 min, and incubated with primary antibodies (1:1000 mouse anti‐15‐lipoxygenase‐1 3G8, Abcam [Tokyo, Japan], 1:1500 mouse anti‐5‐lipoxygenase, BD Bioscience, or 1:4000 rabbit anti‐ALOX12 C‐terminal, Abgent [CA, USA]).
Fluorescence microscopy
HT1080 cells were cultured on Lab‐Tek chamber glass slides at 37°C for 48 h. Indirect immunofluorescence staining was performed as described previously.22 Cells were fixed with 4% paraformaldehyde and permeabilized with 0.05% Triton‐X‐100 and then incubated with primary antibodies followed by incubation with Alexa 488‐conjugated, species‐specific anti‐IgG secondary antibodies (Molecular Probes, OR, USA). Anti‐4‐hydroxy‐2‐nonenal mouse antibody (clone HNEJ‐2, JalCA, Shizuoka, Japan) was diluted before use (1:25). Intrinsic mRFP and antibody staining signals were observed with an FV10i confocal microscope (Olympus, Tokyo, Japan) or with an epifluorescence microscope (Olympus BX‐50) equipped with a SenSys charge‐coupled device camera (Photometrics). Fluorescence images were collected by a single rapid scan with identical parameters (e.g. contrast and brightness) for all samples.
Cellular lipoxygenase assay
HT1080 cells were treated with 10 μM ALOX15 activator or dimethylsulfoxide (DMSO) at 37°C for 2 h. Collected cells were washed and suspended in PBS containing protease inhibitors (0.5 mM phenylmethylsulfonyl fluoride, 5 μg/mL aprotinin, 5 μg/mL leupeptin and 5 μg/mL pepstatin). Cells were sonicated three times by the sonic dismembrator for a membrane‐associated protein preparation. The cell preparations were submitted to the lipoxygenase activity assay as described previously.23 Cell extracts (10 μg each) were incubated with 1 mM linoleic acid as substrate in 0.1 M phosphate buffer at room temperature. The resultant 13‐HpODE was quantified by the reversed phase HPLC. The chromatographic conditions were as follows: column, TSKgel ODS‐80Ts QA (2.0 mm internal diameter × 150 mm) (Tosoh, Japan); eluent, 0.05% formic acid containing 60% acetonitrile; flow rate, 0.2 mL/min; column temperature, 35°C; detection, 234 nm.
Statistical analysis
Data are presented as mean ± SE. Nonparametric data were compared using the Mann–Whitney U‐test or the Kruskal–Wallis test followed by the Steel test.
Results
We previously showed that treatment of HT1080 fibrosarcoma cells with the ferroptosis inducers erastin and RSL3 for 5–6 h and 2–3 h, respectively, resulted in the emergence of ROS around the Golgi/endosome compartments before ROS levels began to increase markedly.19 Furthermore, Dixon et al. found that erastin‐induced ferroptosis can be prevented by the addition of the specific ferroptosis inhibitor ferrostatin‐1 added 6 h later.3 Here we first examined whether late addition of inhibitors can block RSL3‐induced ferroptosis in HT1080 cells. The lipophilic anti‐oxidant Trolox protected cells from death when it was added 3 h after RSL3 addition, but not at 4 h (Fig. 2a). We next confirmed lipid oxidant generation by immunofluorescence using an antibody to 4‐hydroxy‐2‐nonenal (4‐HNE), which is a major aldehyde product of ω‐6 PUFA peroxidation.24 Very low immunoreactive signals for 4‐HNE were detected in HT1080 cells under basal conditions (Fig. 2b). Consistent with results obtained using the fluorescent lipid peroxidation sensor C11‐BODIPY 581/591,19 4‐HNE levels gradually increased within 3 h after RSL3 addition and were prominent around 4 h later (Fig. 2b). These results suggest that oxidative generation of 4‐HNE from PUFA is associated with the execution of ferroptotic cell death.
Figure 2.
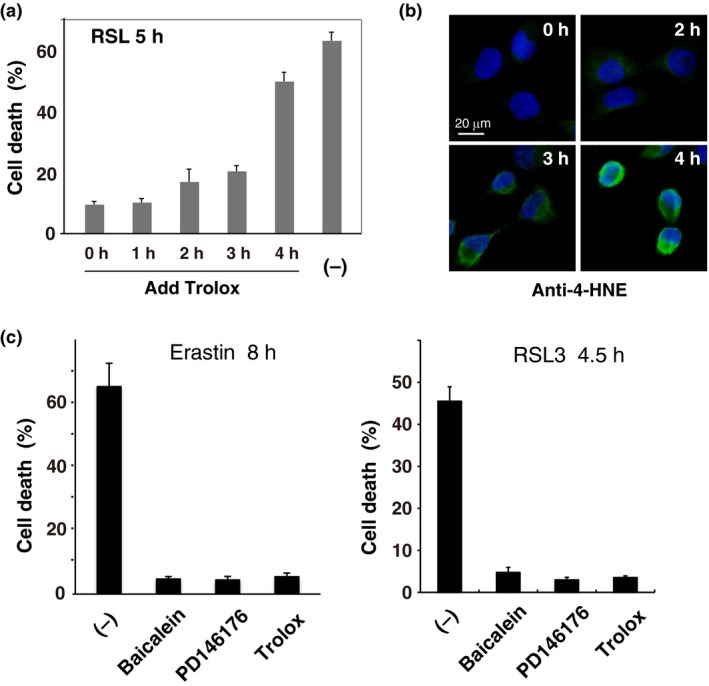
Ferroptotic cell death involves the accumulation of oxidized polyunsaturated fatty acids (PUFA) fragments, and can be blocked by lipoxygenase (LOX) inhibitors. (a) HT1080 cells were treated with 1 μM RSL3 in the presence or absence of Trolox (0.2 mM) for 5 h. Trolox was added either at the same time as RSL3 (0 h) or 1–4 h later. The cell death rate was determined by trypan blue dye‐exclusion assay. Results are the mean ± SE of three independent experiments. (b) Cells were treated with RSL3 for the indicated time. The cells were fixed and immunostained by anti‐4‐hydroxy‐2‐nonenal (4‐HNE) antibody. Fluorescence signals were visualized by microscopy with constant fluorescence parameters. Experiments were repeated three times with reproducible results. Bar, 20 μm. (c) HT1080 cells were treated with 10 μM erastin or 1 μM RSL3 in the presence or absence of each inhibitor for the indicated time. Baicalein (10 μM); PD146176 (5 μM); Trolox (0.1 mM). The cell death rate was determined as in (a). Results are the mean ± SE of four independent experiments.
To address the involvement of PUFA oxygenation by LOX in ferroptosis, we used several LOX inhibitors. We confirmed the previous observation14 that the 12/15‐LOX inhibitor baicalein prevented erastin‐induced cell death, and found that it protected cells from RSL3 toxicity (Fig. 2c and Fig. S1). Similarly, the selective ALOX15 inhibitor PD146176 prevented both erastin‐induced and RSL3‐induced ferroptosis in HT1080 (Fig. 2c), Calu‐1 (Fig. S1a) and Panc‐1 cells (Fig. S1b). In contrast, the ALOX5 inhibitors caffeic acid and MK‐886 had no effect on cell death (data not shown). These results indicate that the ALOX12 or ALOX15 may both be involved in ferroptotic cell death in oncogenic Ras‐expressing cells, although the specificity of these inhibitors is unclear.16
To assess the levels of LOX proteins during ferroptosis, we prepared HT1080 cell lysates and subjected them to immunoblot analysis. ALOX12 protein expression was induced by either erastin or RSL3 treatment, and the protein levels appeared to be stable even during the late phase of ferroptosis (Fig. 3a). In contrast, ALOX15, ALOX5 and even β‐actin levels were decreased in dying cells, although the drug treatment with Trolox did not induce decreases in these proteins (Fig. S2). Therefore, decreased proteins may have resulted from unspecific degradation following cell membrane disruption. We confirmed these observations by immunofluorescent analysis, which showed very faint signals for ALOX12 in untreated HT1080 cells but its membrane‐associated signals accumulation in cells treated with erastin or RSL3 (Fig. 3b). ALOX15 was continuously localized on the plasma membrane in control cells as well as erastin‐treated and RSL3‐treated cells, suggesting that ALOX15 rather than ALOX12 generates lipid hydroperoxides in ferroptotic HT1080 cells.
Figure 3.
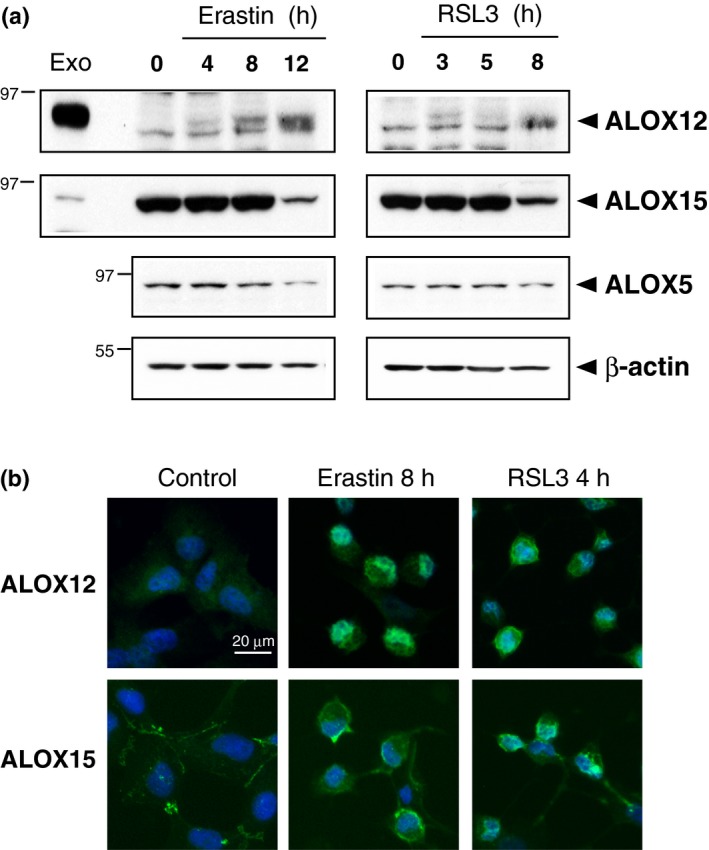
Expression and intracellular localization of lipoxygenases (LOX) enzymes during ferroptosis. (a) HT1080 cells were treated with 10 μM erastin or 1 μM RSL3 for the indicated time. After washing the cell surface, cell extracts (20 μg) were prepared and subjected to immunoblot analysis with antibodies to ALOX12, ALOX15, ALOX5 and β‐actin. Lysates (2 μg) from cells that were transiently transfected with plasmids carrying ALOX12 or ALOX15 were loaded as a control (Exo). (b) Cells were treated with erastin (8 h) or RSL3 (4 h). The cells were fixed and immunostained by anti‐ALOX12 or ALOX15 antibody. Fluorescence signals were visualized by microscopy with constant fluorescence parameters. Experiments were repeated three times with reproducible results. Bar, 20 μm.
Next, the silencing effect of three siRNA, si‐15LO‐1, si‐15LO‐2 and si‐15LO‐3, was assessed by co‐expression with mRFP‐tagged ALOX15. si‐15LO‐2 had the most effective knockdown activity (Fig. 4a). Specific knockdown of endogenous ALOX15 was also verified by immunoblotting (Fig. 4b). ALOX15 knockdown cells treated with either erastin or RSL3 had a significant decrease in the cell death rate (Fig. 4c), suggesting that ALOX15 is required for full sensitivity of HT1080 cells to ferroptosis‐inducing compounds (FIN). Suppression of ferroptosis following ALOX15 silencing was similarly detected in both Calu‐1 lung cancer cells (Fig. 4d) and the non‐tumor fibroblast cell line OUMS that was stably transfected with HRASV12 mutant (data not shown).
Figure 4.
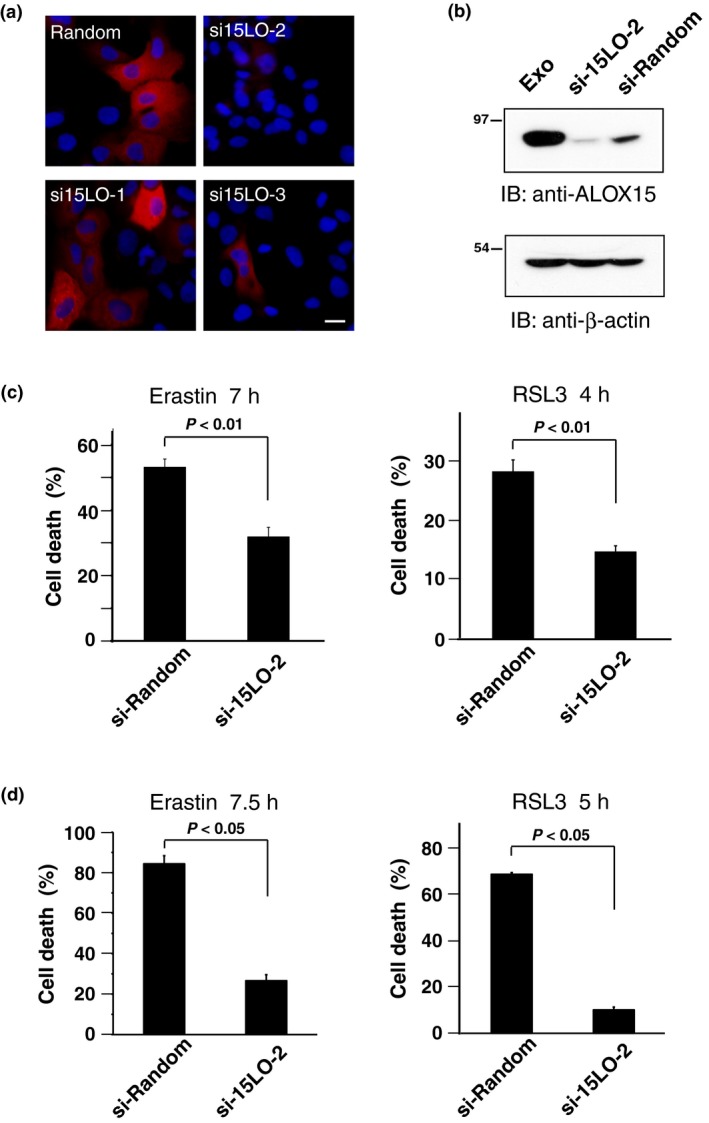
Specific knockdown of ALOX15‐1 expression attenuates ferroptosis in HT1080 cells. (a) HT1080 cells were transiently transfected with mRFP‐tagged ALOX15 plasmid plus either control siRNA (Random) or human ALOX15 siRNA (si15LO‐1, si15LO‐2, si15LO‐3). After 48 h, cells were fixed and red fluorescence signals were visualized by microscopy with constant fluorescence parameters. Bar, 10 μm. (b) HT1080 cells transfected with either siRandom or si15LO‐2, or cells expressing ALOX15 (Exo) were lysed. Each cell lysate was analyzed by immunoblotting with anti‐ALOX15 antibody. The expression of β‐actin was examined as a control. (c) HT1080 cells transfected with either siRandom or si15LO‐2 were incubated with 10 μM erastin or 1 μM RSL3 for the indicated time. The cell death rate was determined by trypan blue dye‐exclusion assay. Results are the mean ± SE of four independent experiments. (d) Calu‐1 cells transfected with either siRandom or si15LO‐2 were incubated with 10 μM erastin or 1 μM RSL3 for the indicated time. The cell death rate was determined as in (c). Results are the mean ± SE of three independent experiments.
We next analyzed the effect of LOX overexpression on ferroptotic cell death. Fluorescence microscopy of cells expressing mRFP‐tagged ALOX12 or ALOX15 at extremely high levels showed cell death without FIN treatments (Fig. 5a). Notably, cells with exogenous expression of ALOX15 had an increased cell death rate following treatment with low concentrations of erastin and RSL3 (Fig. 5b). Similar results were obtained with Calu‐1 cells (Fig. S3). LOX have a two‐domain structure consisting of a small N‐terminus and a catalytic C‐terminal domain.15 An ALOX15 N‐terminal domain‐deletion mutant (ΔN) that is defective in membrane targeting showed weak enhancing activity (Fig. 5b), indicating that ALOX15 localization and interaction with lipid membranes is essential for ferroptosis.
Figure 5.
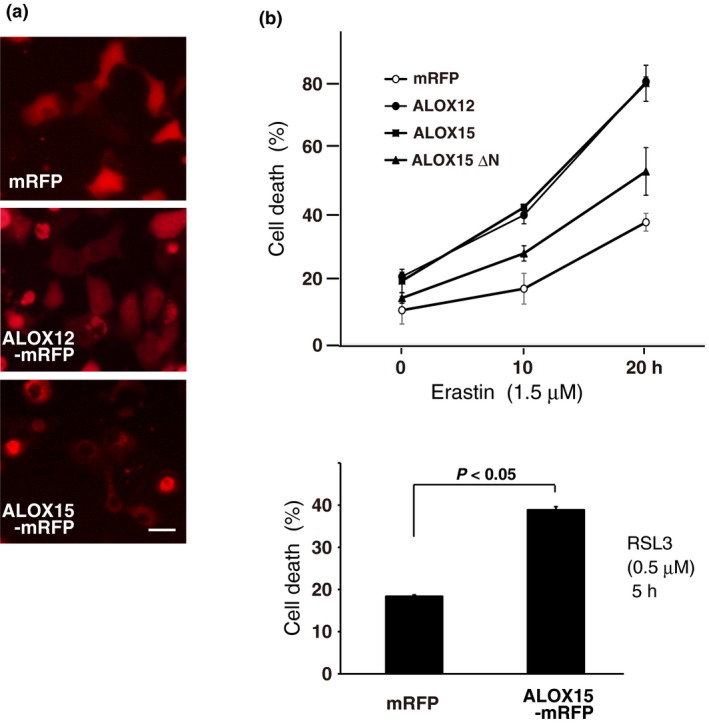
Exogenous expression of lipoxygenases (LOX) enhances drug‐induced ferroptosis. (a) HT1080 cells were transiently transfected with expression plasmids encoding mRFP, or mRFP‐tagged ALOX12 or ALOX15. After 24 h, red fluorescence signals were visualized by microscopy with constant fluorescence parameters. Bar, 20 μm. (b) Cells transfected with mRFP, ALOX12‐mRFP, ALOX15‐mRFP or ALOX15 with the N‐terminus deleted (ΔN)‐mRFP were treated with 1.5 μM erastin for the indicated time (upper graph). Cells transfected with mRFP or ALOX15‐mRFP were treated with 0.5 μM RSL3 for 5 h (lower graph). The cell death rate was determined by trypan blue dye‐exclusion assay. Results are the mean ± SE of three independent experiments.
ALOX15 expression is likely to be associated with the cytotoxicity of FIN compounds. To examine this possibility, we used an activator of ALOX1520 to define the role of its enzymatic activity in ferrpotosis. Activating cellular lipoxygenase by the compound (E)‐1‐(7‐benzylidene‐3‐phenyl‐3,3a,4,5,6,7‐hexahydroindazol‐2‐yl)‐2‐(4‐methylpiperazin‐1‐yl)ethanone was first confirmed (Fig. 6a). Next, the toxicity of the compound towards HT1080 cells was examined. The LOX activator alone did not cause ferroptosis, although at a higher concentration (>20 μM) the compound induced cell death (presumably apoptosis) at the 24‐h time point (Fig. 6b). Meanwhile, the compound clearly enhanced ferroptotic cell death when cells were treated with low concentrations of RSL3 (<1.0 μM) (Fig. 6c). Enhancement of ferroptosis by the activator was similarly detected in other tumor cell lines, Calu‐1 and Panc‐1 (Fig. 6d,e). These observations suggest that ALOX15 regulates FIN‐induced ferroptosis in conjunction with GPX4.
Figure 6.
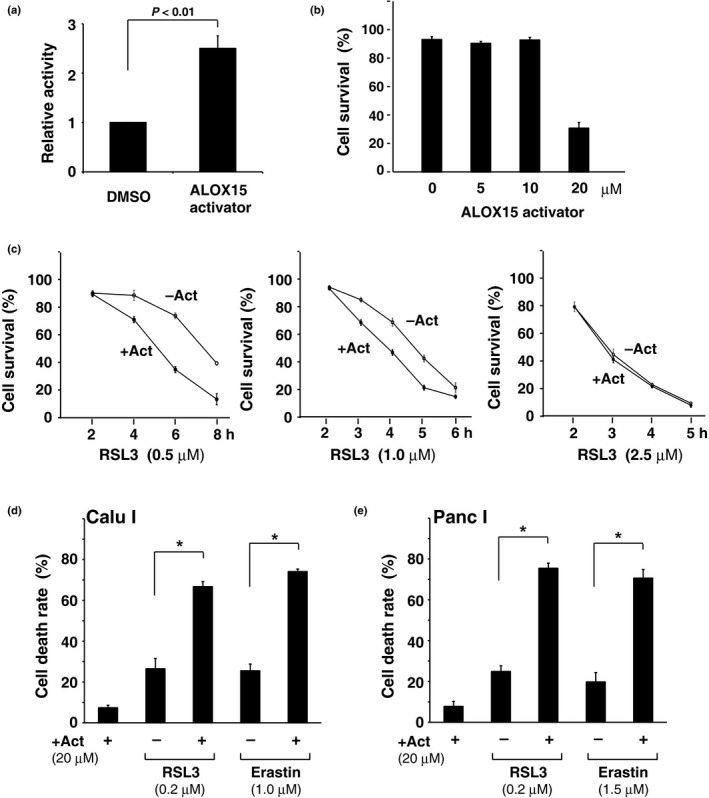
ALOX15 activator enhances ferroptosis of cells treated with low‐dose drugs. (a) HT1080 cells were treated with an ALOX15 activating compound (10 μM) for 2 h. Intracellular lipoxygenase (LOX) activity was determined for each cell extract, as described in the Materials and Methods. Results are the mean ± SE of five independent experiments. (b) HT1080 cells were treated with an ALOX15 activator at the indicated concentrations. After 24 h, the cell survival rate was determined by trypan blue dye‐exclusion assay. Results are the mean ± SE of three independent experiments. (c) HT1080 cells were treated with 0.5, 1 or 2.5 μM RSL3 in the presence or absence of an ALOX15 activator (Act; 10 μM) for the indicated time. The cell survival rate was determined as in (b). Results are the mean ± SE of three independent experiments. (d) Calu‐1 cells were treated with 0.2 μM RSL3 or 1 μM erastin in the presence or absence of the ALOX15 activator for 6.5 h. The cell death rate was determined by trypan blue dye‐exclusion assay. Results are the mean ± SE of three independent experiments. (e) Panc‐1 cells were treated with 0.2 μM RSL3 or 1.5 μM erastin with or without the activator for 7 h. The cell death rate was determined as in (d). *P < 0.05; The Mann–Whitney U‐test.
Discussion
Cell death by ferroptosis is characterized by intracellular iron‐dependency and accumulation of lipid ROS.1, 2, 3, 4 In earlier studies, sensitivity to ferroptosis‐inducing drugs (e.g. erastin and RSL3) could be explained by the finding that oncogenic Ras‐expressing tumor cells accumulate iron by regulating autophagy and functional lysosome pathways.17, 19 In fact, treatment with autophagy inhibitors or compounds that alter lysosomal function prevented ferroptotic cell death in several cancer cell lines. Moreover, oxidized membranes detected by fluorescent probes were observed at perinuclear compartments during the early phase of ferroptosis.19 Therefore, free iron from endosomes/lysosomes is presumably involved in lipid ROS generation via certain pathways.
Oxidized PUFA produced by two distinct pathways, LOX‐catalyzed and free radical‐mediated lipid oxidation, can lead to execution of cell death.25 LOX show lipid peroxidation activity with iron in the ferric state (Fe3+). Here we showed that at least one LOX enzyme, ALOX15, contributes to drug‐induced ferroptosis in HT1080 cells through the generation of hydroperoxides in cellular membranes. Both specific inhibitors and siRNA‐mediated knockdown of ALOX15 significantly decreased erastin‐induced and RSL3‐induced cell death (Figs. 2c and 4c). Moreover, overexpression of wild type ALOX15 but not an N‐terminal truncated form enhanced erastin‐induced ferroptosis (Fig. 5), and specific activator treatments also accelerated cell death induced by low‐dose FIN drugs (Fig. 6). Immunofluorescence analyses revealed that the ALOX15 protein consistently localizes to cellular membranes during the drug treatments (Fig. 3b). Thus, most ALOX15 protein is presumably active and may contribute to ferroptotic cell death via production of lipid hydroperoxides in cell membranes. Of note, the novel p53 target gene SAT1 was recently shown to induce ferroptosis in conjunction with ALOX15 expression.26
Our data do not exclude the contributions to ferroptosis by other LOX. 12/15‐LOX inhibitors prevented cell death, whereas ALOX12 overexpression significantly enhanced cell death (Figs 2c and 5b). Interestingly, ALOX12 expression was gradually elevated during the erastin or RSL3 treatments, and was stable in the late stage of ferroptosis, although ALOX12 expression in the steady state was nearly undetectable by our antibody (Fig. 3). Meanwhile, the ALOX5 inhibitors caffeic acid and MK‐866 did not suppress the drug‐induced ferroptosis (data not shown). Therefore, in HT1080 cells the activities of ALOX12 and ALOX5 appear to be associated with drug‐induced ferroptosis to a lesser degree than ALOX15. A recent report demonstrated that siRNA pools specific for ALOX15B and ALOXE3 inhibited erastin‐induced cell death in HT1080 cells and that knockdown of all ALOX genes in G401 cells prevented cell death induced by imidazole ketoerastin but not by RSL3. 27 In our study, both specific inhibitors and silencing of ALOX15 were effective in RSL3‐treated HT1080 cells (Figs 2c and 4c). Furthermore, the inhibitors prevented RSL3‐induced cell death in Calu‐1 and Panc‐1 (Fig. S1), and ALOX15 knockdown caused a significant decrease in Calu‐1 ferroptosis (Fig. 4d). These results suggest that at least the ALOX15 protein is involved in FIN‐induced ferroptosis in cancer cells, although other LOX may contribute to it in each cell line.
ALOX15‐catalyzed oxidation of linoleic acid and arachidonic acid produces 13‐hydroperoxyoctadecadienoic acid (HPODE) and 15‐hydroperoxyeicosatetraenoic acid (HPETE), respectively, both of which are considered to be intermediates for formation of 4‐HNE.28 A recent report demonstrated that increased release of 5‐hydroxyeicosatetraenoic acid (5‐HETE), 11‐HETE and 15‐HETE was detected in cells in which Gpx4 was deleted10 and, furthermore, cells treated with deuterated PUFA had suppressed ferroptosis.27 These observations suggested that PUFA oxidation is essential for ferroptotic cell death. We showed that the ALOX15 activator accelerated low‐dose FIN‐induced cell death but did not affect that induced by higher doses of drugs (Fig. 6). In intracellular membranes, lipid peroxidizing and lipid peroxide‐reducing activities are in balance, which may be regulated by ALOX15 and GPX4. Our data suggested that increased production of lipid hydroperoxides in cellular membranes by LOX could promote susceptibility to ferroptosis. Our immunofluorescence assays showed that 4‐HNE clearly accumulates on cellular membranes in ferroptotic HT1080 cells (Fig. 2b). Profound accumulation of 4‐HNE thus appears to be a point of no return in the ferroptotic pathway (Fig. 2a,b). Because 4‐HNE is a highly reactive aldehyde and can alter many cellular constituents,24 unregulated accumulation of 4‐HNE from ω‐6 PUFA oxidation may directly contribute to ferroptotic cell death.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting information
Fig. S1. Lipoxygenase (LOX) inhibitors prevent cell death induced by erastin or RSL3 in Calu‐1 and Panc‐1 cells. Calu‐1 (a) or Panc‐1 (b) were treated with 10 μM erastin or 1 μM RSL3 in the presence or absence of each inhibitor for the indicated time.
{kind=link}
Fig. S2. Expression of lipoxygenase (LOX) enzymes in cells treated by erastin or RSL3 with a ferroptosis inhibitor. HT1080 cells were treated with erastin or RSL3 in the presence of Trolox. Cell extracts were subjected to immunoblot analysis.
{kind=link}
Fig. S3. Exogenous expression of ALOX15 enhances drug‐induced ferroptosis in Calu‐1 cells. Calu‐1 cells transfected with mRFP or mRFP‐tagged ALOX15 were treated with 1.5 μM erastin or 0.2 μM RSL3 for 9 h.
{kind=link}
Acknowledgments
We thank Dr Oda (Gunma University), Ms M. Hosoi, M. Takeda, A. Oaku, M. Shimizu, N. Sakaya, K. Hasebe and R. Torii for their generous support. This work was supported by the joint research program of the Institute for Molecular and Cellular Regulation, Gunma University (grant numbers 16034 and 16035) and a grant from the Gunma University Medical Innovation project.
Cancer Sci 108 (2017) 2187–2194
Funding Information
Institute for Molecular and Cellular Regulation, Gunma University (16034 and 16035), grant from the Gunma University Medical Innovation project.
References
- 1. Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 2016; 15: 348–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci 2016; 73: 2195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dixon SJ, Lemberg KM, Lamprecht MR et al Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell 2012; 149: 1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang WS, SriRamaratnam R, Welsch ME et al Regulation of ferroptotic cancer cell death by GPX4. Cell 2014; 156: 317–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lachaier E, Louandre C, Godin C et al Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res 2014; 34: 6417–22. [PubMed] [Google Scholar]
- 6. Dixon SJ, Patel DN, Welsch M et al Pharmacological inhibition of cystine‐glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014; 3: e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Woo JH, Shimoni Y, Yang WS et al Elucidating compound mechanism of action by network perturbation analysis. Cell 2015; 162: 441–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun X, Ou Z, Chen R et al Activation of the p62‐Keap1‐NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016; 63: 173–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jiang L, Kon N, Li T et al Ferroptosis as a p53‐mediated activity during tumour suppression. Nature 2015; 520: 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Friedmann Angeli JP, Schneider M, Proneth B et al Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 2014; 16: 1180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med 2003; 34: 145–69. [DOI] [PubMed] [Google Scholar]
- 12. Seiler A, Schneider M, Forster H et al Glutathione peroxidase 4 senses and translates oxidative stress into 12/15‐lipoxygenase dependent‐ and AIF‐mediated cell death. Cell Metab 2008; 8: 237–48. [DOI] [PubMed] [Google Scholar]
- 13. Brutsch SH, Wang CC, Li L et al Expression of inactive glutathione peroxidase 4 leads to embryonic lethality, and inactivation of the Alox15 gene does not rescue such knock‐in mice. Antioxid Redox Signal 2015; 22: 281–93. [DOI] [PubMed] [Google Scholar]
- 14. Xie Y, Song X, Sun X et al Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem Biophys Res Commun 2016; 473: 775–80. [DOI] [PubMed] [Google Scholar]
- 15. Kuhn H, Saam J, Eibach S, Holzhutter HG, Ivanov I, Walther M. Structural biology of mammalian lipoxygenases: enzymatic consequences of targeted alterations of the protein structure. Biochem Biophys Res Commun 2005; 338: 93–101. [DOI] [PubMed] [Google Scholar]
- 16. Kuhn H, Banthiya S, van Leyen K. Mammalian lipoxygenases and their biological relevance. Biochim Biophys Acta 2015; 1851: 308–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron‐dependent, nonapoptotic cell death in oncogenic‐RAS‐harboring cancer cells. Chem Biol 2008; 15: 234–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell 2015; 59: 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Torii S, Shintoku R, Kubota C et al An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J 2016; 473: 769–77. [DOI] [PubMed] [Google Scholar]
- 20. Meng H, McClendon CL, Dai Z et al Discovery of novel 15‐lipoxygenase activators to shift the human arachidonic acid metabolic network toward inflammation resolution. J Med Chem 2016; 59: 4202–9. [DOI] [PubMed] [Google Scholar]
- 21. Khalaf AA, El‐Shafei AK, El‐Sayed AM. Synthesis of some new bicyclic pyrazoline derivatives. J Heterocyclic Chem 1982; 19: 609–12. [Google Scholar]
- 22. Hou N, Mogami H, Kubota‐Murata C, Sun M, Takeuchi T, Torii S. Preferential release of newly synthesized insulin assessed by a multi‐label reporter system using pancreatic beta‐cell line MIN6. PLoS ONE 2012; 7: e47921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koshiishi I, Tsuchida K, Takajo T, Komatsu M. Radical scavenger can scavenge lipid allyl radicals complexed with lipoxygenase at lower oxygen content. Biochem J 2006; 395: 303–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Uchida K. 4‐Hydroxy‐2‐nonenal: a product and mediator of oxidative stress. Prog Lipid Res 2003; 42: 318–43. [DOI] [PubMed] [Google Scholar]
- 25. Magtanong L, Ko PJ, Dixon SJ. Emerging roles for lipids in non‐apoptotic cell death. Cell Death Differ 2016; 23: 1099–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ou Y, Wang SJ, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53‐mediated ferroptotic responses. Proc Natl Acad Sci USA 2016; 113: E6806–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA 2016; 113: E4966–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luczaj W, Gegotek A, Skrzydlewska E. Antioxidants and HNE in redox homeostasis. Free Radic Biol Med 2016; 111: 87–101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Lipoxygenase (LOX) inhibitors prevent cell death induced by erastin or RSL3 in Calu‐1 and Panc‐1 cells. Calu‐1 (a) or Panc‐1 (b) were treated with 10 μM erastin or 1 μM RSL3 in the presence or absence of each inhibitor for the indicated time.
Fig. S2. Expression of lipoxygenase (LOX) enzymes in cells treated by erastin or RSL3 with a ferroptosis inhibitor. HT1080 cells were treated with erastin or RSL3 in the presence of Trolox. Cell extracts were subjected to immunoblot analysis.
Fig. S3. Exogenous expression of ALOX15 enhances drug‐induced ferroptosis in Calu‐1 cells. Calu‐1 cells transfected with mRFP or mRFP‐tagged ALOX15 were treated with 1.5 μM erastin or 0.2 μM RSL3 for 9 h.