

Abstract
In this study, the potential clinical effects of thalidomide on bleomycin-induced pulmonary fibrosis were investigated. A Sprague-Dawley rats’ model of pulmonary fibrosis induced by an intratracheal instillation of bleomycin was adopted. The rats in thalidomide treated groups were intraperitoneally injected with thalidomide (10, 20, 50 mg/kg) daily for 28 days, while the rats in control and bleomycin treated groups were injected with a saline solution. The effects of thalidomide on pulmonary injury were evaluated by the lung wet/dry weight ratios, cell counts, and histopathological examination. Inflammation of lung tissues was assessed by measuring the levels of interleukin (IL)-6, IL-8, tumor necrosis factor (TNF)-α, and transforming growth factor (TGF)-β in bronchoalveolar lavage fluid (BALF). Oxidative stress was evaluated by detecting the levels of reactive oxygen species (ROS), superoxide dismutase (SOD), total antioxidant capacity (T-AOC), and malondialdehyde (MDA) in lung tissue. The results indicated that thalidomide treatment remarkably attenuated bleomycin-induced pulmonary fibrosis, oxidative stress and inflammation in rats’ lung. The anti-inflammatory and anti-oxidative effects of thalidomide were also found in human lung fibroblasts. Thalidomide administration significantly stimulated the activity of thioredoxin reductase, while other enzymes or proteins involved in biologic oxidation-reduction equilibrium were not affected. Our findings indicate that thalidomide-mediated suppression of fibro-proliferation may contribute to the anti-fibrotic effect against bleomycin-induced pulmonary fibrosis. The mechanisms are related to the inhibition of oxidative stress and inflammatory response. In summary, these results may provide a rationale to explore clinical application of thalidomide for the prevention of pulmonary fibrosis.
Keywords: Thalidoamide, pulmonary fibrosis, oxidative stress, inflammation, thioredoxin reductase
Introduction
Diffuse interstitial lung disease is characterized by varying degrees of inflammation and fibrosis resulting in derangement of the gas-exchange units of the lung. Around half of interstitial lung diseases are of unknown aetiology and are classified as idiopathic interstitial pneumonias [1]. By far the most common one is idiopathic pulmonary fibrosis, the prognosis of which is worse than that of many cancers [2,3]. Pulmonary fibrosis has an aggressive course and is usually fatal at an average of 3 to 6 years after the onset of symptoms [3-6]. Pulmonary fibrosis refers to a series of lung diseases characterized by inflammation, fibroblast proliferation and excessive collagen deposition. Although the mechanisms underlying pulmonary fibrosis are not clearly understood, current evidence suggests that it is associated with the inflammation and oxidative stress [7-9].
Thalidomide (α-N-phthalimidoglutarimide, as shown in Figure 1) was originally introduced in 1954 as a sedative/hypnotic agent but was removed from the market for its teratogenic effects [10]. In contrast, its extra effects such as anti-inflammatory, immunomodulatory, and antiangiogenic activity have been reported [11-13]. Previous studies have demonstrated the efficacy of thalidomide or its analogs in the treatment of erythema nodosum leprosum, rheumatoid arthritis, Castleman’s disease, Crohn’s disease, renal cell carcinoma, and myelodysplastic disease [14-16]. In recent years, thalidomide has been reported efficacious to treat pulmonary fibrosis [17-20]. However, the mechanisms involved were not fully investigated.
Figure 1.
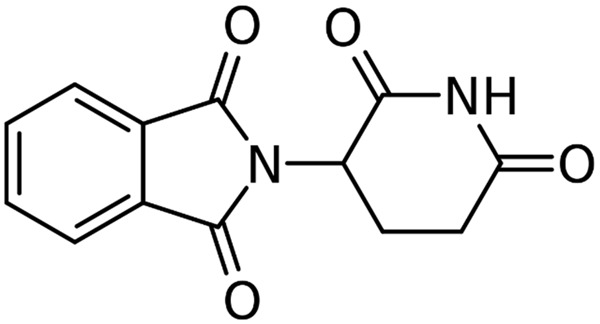
Chemical structure of thalidomide.
A number of key factors were found in the pathogenesis of pulmonary fibrosis including inflammation and oxidative stress. Inflammation is the initial response following lung injury. The inflammatory cells accumulate and release pro-inflammatory cytokines, growth factors, which regulate the proliferation and secretary activity of alveolar fibroblasts [21,22]. Increasing evidence showed that oxidative stress is involved in the pathogenesis of pulmonary fibrosis. Markers of oxidative stress have been identified in patients and aberrant antioxidant activity has been demonstrated to exacerbate pulmonary fibrosis in animal models. Therefore, anti-inflammation and anti-oxidant strategies are introduced to treat pulmonary fibrosis [23,24].
Cytosolic thioredoxin (Trx), thioredoxin reductase (TrxR) and nicotinamide adenine dinucleotide phosphate (NADPH) comprise the mammalian Trx system, which plays powerful roles in defense mechanism against oxidative stress, nitrosative stress and in redox regulation [25,26]. Trx is a small redox-active protein that is ubiquitously present in mammalian and is one of the defense proteins induced in response to various oxidative stress conditions [27,28]. The reduction of oxidized Trx by NADPH is catalyzed by seleno protein TrxR. TrxR may catalyze the NADPH-dependent reduction of H2O2, lipid hydroperoxides and dehydroascorbate as well [29-31]. In addition to its potent anti-oxidative effect, Trx system also has anti-inflammatory properties, mainly because of its ability to inhibit neutrophil chemotaxis to inflammatory sites and to suppress the expression and activation of the macrophage migration-inhibitory factors [32,33]. Because of its desirable anti-oxidative and anti-inflammatory properties, Trx system could be a new and potentially effective therapeutic target for the treatment of pulmonary fibrosis.
In the present study, the effects of thalidomide on pulmonary fibrosis in rats and human lung fibroblasts were investigated. Here we show that thalidomide prevents bleomycin-induced pulmonary fibrosis in rats. The underlying regulatory mechanisms associated with the potential anti-inflammatory and anti-oxidant effects of thalidomide were also investigated.
Materials and methods
Reagents
Thalidomide, bleomycin sulfate, DMSO, and chloral hydrate were purchased from Sigma (St. Louis, MO, USA). Fetal calf serum (FCS) and RPMI 1640 medium were purchased from HyClone (USA). The antibodies to TrxR, TrX, GR, GPx, and β-tubulin were purchased from Cell Signaling Technology (USA).
Animals
Sprague-Dawley rats were obtained from Shanghai SLAC Laboratory Animal Co. Ltd. (Shanghai, China) and maintained in our specific pathogen-free animal facility. Animals used in the studies were housed in wire-mesh-bottomed cages in a 12 h light/dark cycle and kept in a room at a constant temperature of 22-24°C and a relative humidity of 60%. Food and water were provided ad libitum. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All protocols for animal use and euthanasia were reviewed and approved by the General Hospital of Tianjin Medical University Committee on Animal Resources committee (TMU-2015023). All efforts were made to minimize suffering.
Establishment of bleomycin-induced pulmonary fibrosis in rats
The rats were randomly divided into different groups: control group, bleomycin treated group, bleomycin plus thalidomide treated groups. Briefly, all rats were anesthetized with 10% chloral hydrate. Then, the rats were administered a single intratracheal instillation of bleomycin dissolved in saline (5 mg/kg body weight), while an equal volume of saline was instillated into the rats from the control group. The rats of the bleomycin plus thalidomide groups received an intraperitoneal injection of thalidomide at a dose of 10, 20 and 50 mg/kg body weight, respectively, every day for a total of 28 days, while the rats of control and bleomycin treated groups were injected with a saline solution. The rats were sacrificed at the 28th day after the first bleomycin treatment. The bronchoalveolar lavage fluids were collected and the lungs were excised for further study.
Immunohistochemistry
Rats were sacrificed at day 28 after the first instillation of bleomycin, and the lungs were fixed in 4% paraformaldehyde, dehydrated, and embedded in paraffin and sectioned into 5 μm slices. Then the sections were stained with hematoxylin and eosin (H&E), and examined under a light microscope (1:100; BD Pharmingen).
Detection of lung wet/dry weight ratios
The lungs of the rats were removed and weighed immediately, and following dried in an incubator at 60°C for 72 h. The pulmonary edema was estimated as a wet/dry weight ratio.
Cell counting in bronchoalveolar lavage fluid (BALF)
The BALF was centrifuged at 12,000 rpm for 10 min at 4°C, and the supernatant was stored immediately at -80°C until analysis. The sediment cells were resuspended in 50 μl PBS and 10 μl of the cell suspension was made into the smear on a glass slide. The cell smear was air-dried, fixed in methanol for 15 min, and stained with Giemsa solution. Then total cells, neutrophils, macrophages, and lymphocytes were counted with a hemocytometer (Hausser Scientific, Horsham, PA, USA).
Biochemical analysis of BALF
The levels of interleukin (IL)-6, IL-8, tumor necrosis factor (TNF)-α, and transforming growth factor (TGF)-β in BALF were measured by Enzyme-linked Immunosorbent Assay (ELISA) kits in accordance with the manufacture’s instructions.
Measurement of oxidative stress
The lung homogenates were centrifuged for 30 min at 12,000 rpm at 4°C. The serum levels of reactive oxygen species (ROS), superoxide dismutase (SOD), total antioxidant capacity (T-AOC), and malondialdehyde (MDA) in lung tissue were measured according to the instructions of detection kits (Jiancheng Bioengineering Institute, Nanjing, China).
Cell line and cultures
Human embryonal lung fibroblastic cell line WI38VA-13 and human fetal lung fibroblasts cell line IMR-90 were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured in RPMI 1640 medium supplemented with 10% (v/v) heat-inactivated FCS, incubated in 5% CO2 incubator at 37°C. Cells were passaged every 2-3 days and used at about 80-90% of the confluence.
To detect the inflammatory mediators and secreted proteins, human fibroblasts were seeded on 6-well plates (3×105 cells/well), serum starved for 2 h and then exposed to 10 ng/mL IL-1β for 2 h before treatment with thalidomide. For thalidomide treated groups, cells were treated with thalidomide, which was diluted in DMSO and added to the growth medium to yield the final DMSO solvent concentration 0.05% (v/v). For control group, cells were treated with the same concentration of DMSO. In preliminary experiments, this final concentration of DMSO had no gross effect on WI38VA-13 and IMR-90 cells.
RNA extraction and quantitative real time PCR (RT-PCR)
Total mRNA was purified using the RNeasy MiniPrep Kit (Qiagen, USA). The gene expression levels of IL-6, IL-8, TNF-α, and TGF-β were measured by Superscript II First-Strand Synthesis kit (Life Technologies, USA) in accordance with the manufacture’s instructions. Results were presented as gene expression relative to IL-1β pre-stimulation (100%).
Cell culture medium was collected after 24 h and the level of IL-6 protein expression was analyzed by ELISA according to the producer’s protocol (Human IL-6 ELISA set, BD Biosciences, USA). Briefly, 96-well plates were coated by kit capture antibody at 4°C overnight, washed and blocked in kit assay diluent. After washing, samples with controls and standards were incubated on the plates for 2 h at room temperature. Then plates were washed, kit detection antibody, streptavidin horseradish peroxidase (HRP, Cell Signaling, USA) and substrate solution were applied according to the producer’s protocol. Absorbance was measured within 30 min at 450 nm with 570 nm correction after the addition of a kit stop solution. Results were presented as protein expression relative to IL-1β pre-stimulation (100%).
Measurement of TrxR, GR, Trx, and GPx protein expression.
Immunoblot analysis was carried out by western blot as previously described [34,35]. Cells were washed three times with ice-cold PBS and harvested in RIPA lysis buffer with 1 mM PMSF, and then lysed on ice for 5 min. The homogenate was centrifuged at 12,000 rpm for 30 min at 4°C and supernatant was collected for analysis. Equal protein extracts were separated by SDS-PAGE, then electrophoretically transferred to PVDF membranes. Incubation with primary and secondary antibodies was performed in Tris-buffered saline containing 5% non-fat dry milk for 2 h or more. After incubation, membranes were washed in Tris-buffered saline containing 0.1% Tween 20. Blots were reprobed with an antibody against β-tubulin as a loading control of protein loading and transfer.
Determination of TrxR and Trx activity in cell lysates by insulin reduction assay
Freshly collected cell lysates were used to determine cellular Trx and TrxR activities as described previously [36]. Briefly, to measure TrxR activity, in each well of a 96-well plate, 25 μg of cell lysate was incubated in a final volume of 50 μl containing 85 mM Hepes (pH 7.6), 0.3 mM insulin, 10 μM Trx, 2.5 mM EDTA and 660 μM NADPH for 40 min at 37°C. Controls were also set up. 200 μl of 1 mM DTNB in 6 M guanidine-HCl, 200 mM Tris-HCl pH 8.0 solution was added to quench the reaction. The amount of free thiols generated from insulin reduction was determined by DTNB reduction at 412 nm using the VersaMax microplate reader. To measure Trx activity, procedures were carried out similarly to those for determining cellular TrxR activity, except that the cell lysates were incubated with TrxR in place of Trx. Controls containing lysates and all reaction reagents except recombinant rat TrxR for each lysate sample were also set up. For each sample, Trx or TrxR activity was calculated as the absorbance at 412 nm subtracted from that of the corresponding control and expressed as a percentage of the activity measured in DMSO-treated cells.
Determination of GPx activity in cell lysates by GPx activity assays
For determination of cellular GPx activity, thalidomide (1-50 μM) was incubated with lysates of fibroblasts (25 μg protein), 20 nM GR, 1 mM GSH and 200 μM NADPH in a volume of 100 μl phosphate buffer (0.1 M sodium phosphate, 2 mM EDTA pH 7.5) for 1 h at room temperature. H2O2 solution in phosphate buffer was added to initiate the reaction (final concentration 1.5 mM). NADPH consumption was monitored at 340 nm using the VersaMax microplate reader. The results were calculated based on change in absorbance in the initial 3 min and presented as a percentage of GPx activity of drug-treated sample over that of DMSO-treated sample.
Determination of GR activity in cell lysates by glutathione reduction assay
To determine cellular GR activity, 25 μg of cell lysate was mixed with a solution of GSSG and NADPH in phosphate buffer to a final volume of 200 μl (final GSSG and NADPH concentrations 1 mM and 200 μM respectively). The enzyme activity was determined by measuring the decrease in absorbance at 340 nm for 10 min at 37°C and expressed as a percentage of the enzyme activity of that of the DMSO-treated sample.
Statistical analysis
The data were expressed as means ± standard deviation (SD). T-test was used to compare the difference between the two groups. Statistical analyses between three or more groups were analyzed by one-way analysis of variance (ANOVA) by using SPSS software version 16.0 (IBM, Armonk, NY). Values for P less than 0.05 were considered statistically significant.
Results
Thalidomide attenuated bleomycin induced pulmonary fibrosis
The effect of thalidomide on bleomycin-induced lung fibrosis in rats were examined. The histological changes of the rats’ lung tissues at the 28th day were shown in Figure 2. In the lung tissues from the bleomycin-treated rats, pulmonary interalveolar septa became thickened and infiltrated by inflammatory cells, with collagen depositions in the interstitium disclosed by H&E staining. Administration of thalidomide resulted in inhibiting the collagen deposition in the bleomycin-treated rat lung tissues. The pathological changes including damaged lung structure and lung interstitium were attenuated by the administration of thalidomide in a dose-dependent manner.
Figure 2.
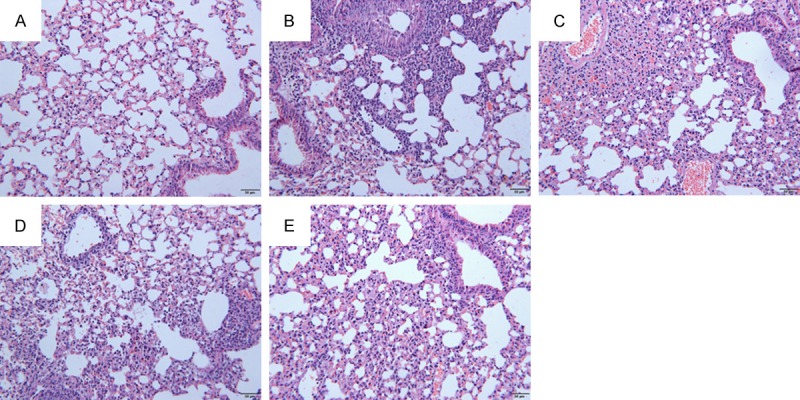
Representative photographs of the effects of thalidomide against bleomycin-induced pathological changes (HE stain). At the 28th day interstitial fibrosis was evident in rat’s lung from the bleomycin treated group (B), compared with control group (A). Fibrotic areas were obviously reduced in lungs of the thalidomide treated group (C, D, E, respectively) relative to those of bleomycin treated groups at day 28. Scale bar = 50 μm.
Thalidomide reduced wet/dry weight ratios of bleomycin-treated rat’s lung
The lung wet/dry ratios were evaluated to indicate the pulmonary edema. As shown in Figure 3, compared to the control group, the lung wet/dry ratio significantly increased in the bleomycin-treated group, and thalidomide administration significantly decreased the lung wet/dry ratios.
Figure 3.
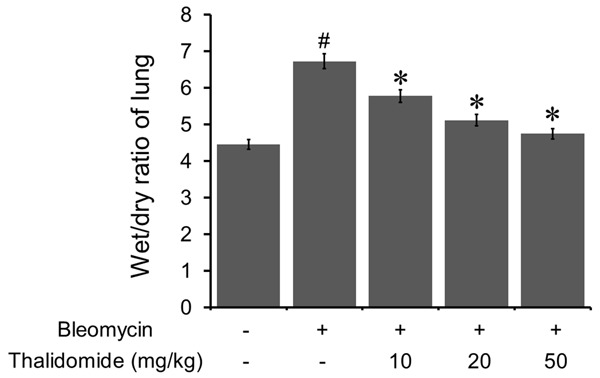
Thalidomide ameliorated bleomycin-induced wet/dry ratio changes in the lung. Bleomycin treatment induced a significant increase of the wet/dry ratio in the lung. However, thalidomide administration reduced the wet/dry ratio in a dose dependent manner. All data were shown as mean ± SD of eight rats. #P < 0.05 vs. control group, *P < 0.05 vs. bleomycin treated group.
Thalidomide attenuated the inflammatory responses in bleomycin-induced pulmonary fibrosis
To determine the effects of thalidomide on bleomycin-induced pulmonary inflammation responses in rats, the inflammatory cell counts in BALF were assayed. As shown in Table 1, compared with untreated control, the number of total cells, neutrophils, macrophages, and lymphocytes in BALF were significantly increased after bleomycin treatment, which, however, were rescued by thalidomide administration.
Table 1.
Effect of thalidomide on bleomycin induced changes in total and differential cell counts in the BALF of rats
| Group | Total cells (×105/ml) | Neutrophils (×104/ml) | Macrophages (×105/ml) | Lymphocytes (×104/ml) |
|---|---|---|---|---|
| Control | 2.21 ± 0.55 | 4.93 ± 0.47 | 1.40 ± 0.53 | 3.24 ± 0.64 |
| Bleomycin | 12.30 ± 1.38# | 22.11 ± 2.15# | 7.92 ± 1.88# | 21.69 ± 2.43# |
| Thalidomide (10 mg/kg) | 9.23 ± 1.24* | 14.24 ± 2.81* | 6.93 ± 1.92 | 17.76 ± 2.13 |
| Thalidomide (20 mg/kg) | 7.82 ± 0.94* | 8.93 ± 1.11* | 5.91 ± 1.69* | 10.17 ± 2.36* |
| Thalidomide (50 mg/kg) | 5.45 ± 0.87* | 6.16 ± 1.18* | 3.97 ± 0.95* | 8.64 ± 2.38* |
Values are mean ± SD.
P < 0.05 vs. Control group;
P < 0.05 vs. bleomycin treated group.
Effects of thalidomide on the expressions of IL-6, IL-8, TNF-α, and TGF-β mRNA in bleomycin-treated rat’s lung tissues
The mRNA levels of IL-6, IL-8, TNF-α, and TGF-β from lung tissues of rats after injection of bleomycin were analyzed by RT-PCR and shown to be significantly elevated compared with control rats, which were obviously suppressed by the administration of thalidomide in a dose-dependent manner Figure 4.
Figure 4.
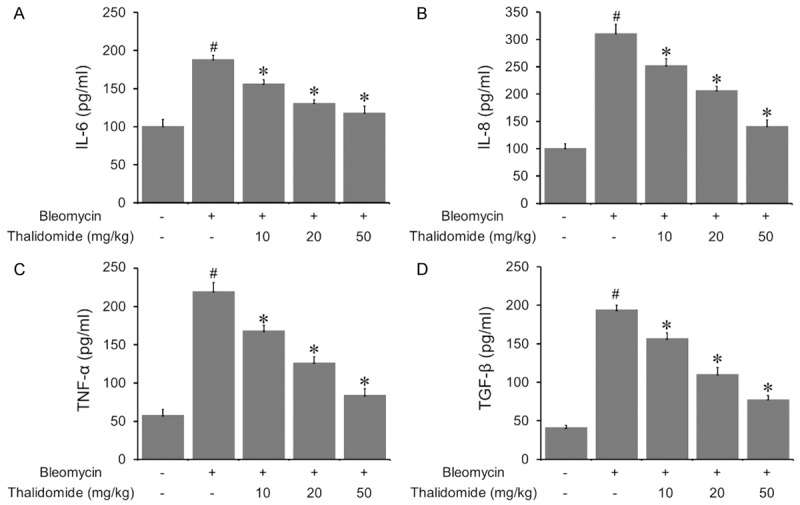
Effects of thalidomide on inflammatory mediators in BALF. Cytokines including IL-6, IL-8, TNF-α, and TGF-β in lung homogenates and serum were determined using ELISA. All these cytokines were significantly increased by bleomycin treatment. However, thalidomide reduced the values of these cytokines significantly in a dose dependent manner. All the data were shown as mean ± SD of eight rats. #P < 0.05 vs. control group, *P < 0.05 vs. bleomycin treated group.
Thalidomide attenuated the oxidative stress in bleomycin-induced pulmonary fibrosis
The levels of ROS, SOD, T-AOC, and MDA in pulmonary tissue were detected to evaluate the oxidative stress of bleomycin-induced pulmonary fibrosis. As shown in Figure 5, MDA and ROS levels were significantly elevated in bleomycin treated group, in which SOD and T-AOC levels were decreased. In contrast, after thalidomide administration, the levels of MDA and ROS were significantly reduced while the levels of SOD and T-AOC were increased.
Figure 5.
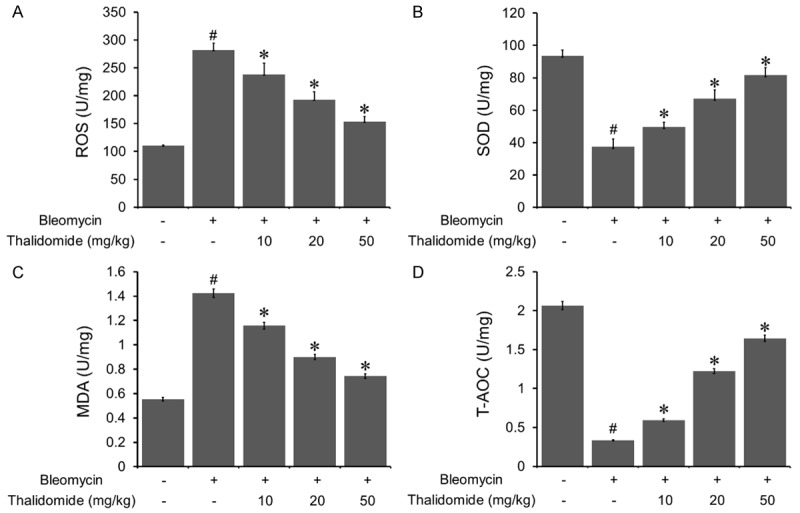
Effects of thalidomide on oxidative stress induced by bleomycin. Bleomycin treatment increased the values of ROS and MDA significantly. While the administration of thalidomide reduced the values of ROS and MDA in a dose dependent manner. Thalidomide could rescue the decrease of the values of SOD and T-AOC induced by bleomycin treatment. Data were shown as mean ± SD. #P < 0.05 vs. control group, *P < 0.05 vs. bleomycin treated group.
Thalidomide exhibited anti-inflammatory effect in human fibroblasts
The effect of thalidomide on the expression of inflammatory mediators in human fibroblasts was examined as while. A significant inhibition of mRNA expressions of IL-6, IL-8, TNF-α, and NGF-β was detected upon thalidomide treatment (Figure 6A). Due to its strong regulation on the mRNA level and pathological relevance, IL-6 was chosen to be measured in cell culture media by ELISA. Thalidomide treatment significantly decreased IL-6 secretion into the medium, compared to the IL-1β pre-stimulated cells (Figure 6B).
Figure 6.
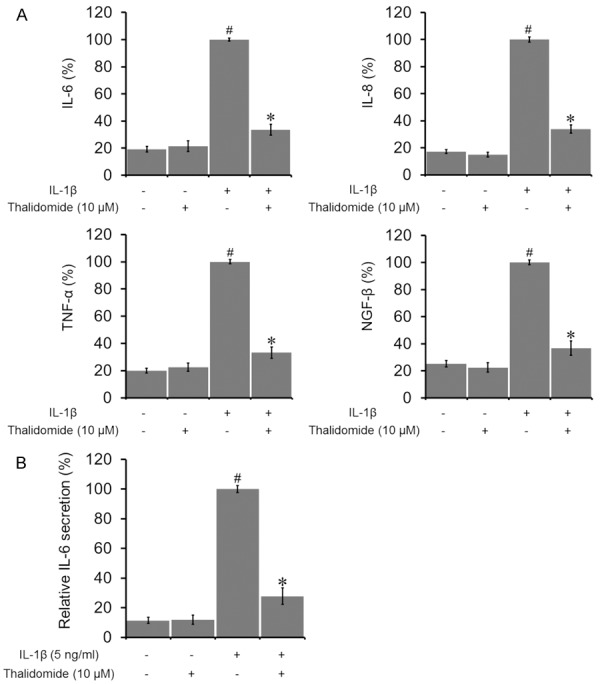
Effects of thalidomide on inflammatory mediators in human lung fibroblasts. (A) Thalidomide significantly reduced mRNA expressions of IL-6, IL-8, TNF-α, and NGF-β in human lung fibroblastic WI38VA-13 cells. Cells were cultured without any treatment, treated with 10 μM thalidomide alone, 5 ng/mL of IL-1β alone, or treated with both thalidomide and IL-1β for 24 h. (B) IL-6 protein secretion in cell culture media was also significantly decreased. Data were presented by the mean ± SD. #P < 0.05 vs control. *P < 0.05 vs IL-1β alone.
Thalidomide up-regulated the expression level of TrxR in human fibroblasts
The effect of thalidomide on oxidation relative proteins were evaluated. Western blotting results showed that thalidomide administration up-regulated the expression level of TrxR. However, the expression levels of Trx, GR, and GPx were not affected, as shown in Figure 7A.
Figure 7.
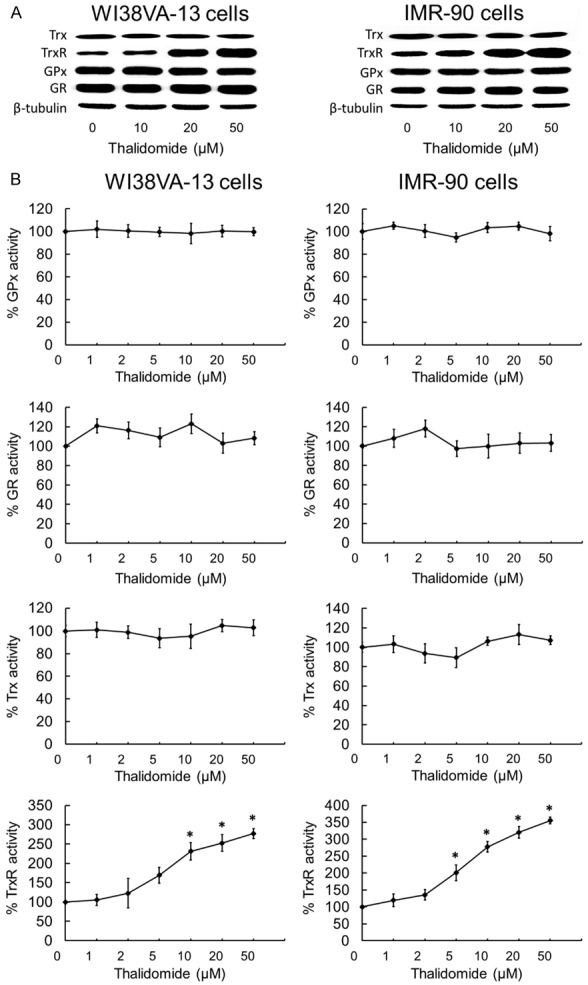
Effects of thalidomide on the expression levels and cellular activities of redox proteins. (A) The lysates of cells treated with thalidomide were analyzed by Western blotting with indicated antibodies for TrxR, GR, Trx and GPx expression. Western blot images are representative of three independent experiments. The results showed that thalidomide administration up-regulated TrxR expression dose-dependently. However, the expression levels of GR, Trx, and GPx were not affected after treating with thalidomide in both WI38VA-13 and IMR-90 cells. (B) Dose-dependent effects of thalidomide on TrxR, GR, Trx and GPx activities in WI38VA-13 and IMR-90 cells. Lysates of WI38VA-13 and IMR-90 cells treated with indicated concentrations of thalidomide for 10 h were assessed for activities of TrxR, GR, Trx and GPx. Enzyme activities were expressed as a percentage of those in DMSO-treated cells. All data points are means ± SD of two to four independent experiments. *Statistically significant difference (P < 0.05) in enzyme activity as compared to DMSO control.
Thalidomide activated the activity of TrxR in human fibroblasts
The effect of thalidomide on TrxR in human fibroblasts was evaluated. After 30 min of incubation in the presence of NADPH, the activation of mammalian TrxR by thalidomide was evaluated in the 5,5’-dithiobis(2-nitrobenzoic acid) acid (DTNB) reduction assay. As shown in Figure 7B, thalidomide stimulated TrxR activity in both cell lines, and IMR-90 cells were generally more susceptible to thalidomide. These results suggested that TrxR activation could potentially serve as an underlying mechanism for at least part of the anti-oxidant effects of thalidomide.
Discussion
Thalidomide experienced an infamous history, since its application as an antiemetic during the first trimester of pregnancy had caused serious birth defects [10]. In recent years, thalidomide has been “rediscovered” as a potent immunomodulatory, antiinflammatory, antiangiogenic, and anti-cancer compound [11-13]. Its immunomodulatory properties indicate its potential in therapy for lung diseases [37,38]. In this study, we investigated the mechanism underlying the effect of thalidomide on pulmonary fibrosis.
Following bleomycin administration, the number of total cells, macrophages, and lymphocytes in BALF were significantly raised, which represented a stimulation of inflammatory responses [39]. Reduction of these cells counts in BALF by thalidomide was also detected in a dose-dependent manner. IL-6, IL-8, TNF-α, and TGF-β play important roles in pulmonary fibrosis, which stimulates the proliferation of fibroblasts, increasing collagen aggregation and synthesis of other cytokines [40,41]. Our results showed that thalidomide prevented bleomycin-induced elevation of IL-6, IL-8, TNF-α and TGF-β in a dose-dependent manner.
Accumulating evidence has indicated that oxidative stress is a potential cause of fibrosis in a variety of organs [42], and there is an imbalance between the generation of ROS and the capacity to detoxify these intermediates [43]. Accordingly, in the present study, oxidative stress was estimated by detecting ROS, SOD, T-AOC, and MDA levels in lung tissue. SOD is an important member of the antioxidant enzymatic defense system and the level of T-AOC reflects the overall cellular endogenous antioxidative capability [44]. And MDA level reflects the degree of organic lipid peroxidation, which indicated the severity of damage to cell membranes [45]. It was shown that in the bleomycin group, MDA and ROS levels were substantially increased while SOD and T-AOC levels were reduced. In contrast, thalidomide remarkably decreased the levels of MDA and ROS, and simultaneously enhanced the levels of SOD and T-AOC. Those results suggested that thalidomide could significantly suppress bleomycin-induced inflammation and oxidative stress, which might explain the protective mechanisms against pulmonary fibrosis induced by bleomycin.
Trx, TR, and NADPH composed Trx system, a very important anti-oxidative system that has been reported many effects, such as regulating cellular reduction/oxidation (redox) status and cell proliferation/cell survival processes [28]. The Trx system has also been reported to regulate the pathologic processes of several kinds of tumors [46], as well involved in cardiovascular disease, heart failure, stroke, inflammation, metabolic syndrome, and other diseases [47]. However, Trx’s function in pulmonary diseases has not been thoroughly investigated. Trx expression increased dramatically in asthma and chronic obstructive pulmonary disease, which represents an attempt to protect the lung from injury [48,49]. Trx has also been proven anti-fibrosis effects in bleomycin-induced fibrosis both in rats and in mice models [50,51], and suppression of Trx system contributed to silica-induced oxidative stress and pulmonary fibrogenesis in rats [52]. It was also discovered that the expression of Trx increased in the lungs of pulmonary fibrosis patients, compared with the controls [53]. Human serum albumin Trx fusion protein inhibited the increase of inflammatory cells in bronchoalveolar lavage fluid, pulmonary inflammatory cytokines, and oxidative stress markers. In addition, post-treatment of human serum albumin Trx had a suppressive effect against bleomycin-induced fibrosis [54]. All of these results proved that Trx might play a key role in the process of pulmonary fibrosis. However, it has not been revealed the approach through Trx and TrxR exert their affect in pulmonary fibrosis.
In the present study, we found that thalidomide up-regulated the expression level of TrxR without affecting the expression levels of GR, Trx, and GPx. The intracellular TrxR activity was significantly activated by thalidomide. Its selectivity towards cellular TrxR activation over related antioxidant enzymes GR, Trx and GPx suggested that thalidomide showed anti-oxidant activity via targeting TrxR. TrxR activation mediated by thalidomide led to cellular Trx reduction, decreased oxidative stress and inhibition of inflammation.
Furthermore, we found that thalidomide exerted direct anti-fibrotic activities in bleomycin-induced pulmonary fibrosis in rats, which was evaluated by the histological changes of the rats’ lung tissues, the wet/dry lung weight ratios and by the oxidant stress of lung tissue. This effect was supposed to be associated with decreased expression of inflammatory mediators, and is likely to include both its immunosuppressive and anti-oxidant effects.
Conclusions
In summary, our study provided direct evidence to prove that in bleomycin-induced pulmonary fibrosis, both inflammatory mediators’ production and oxidative stress could significantly inhibited by thalidomide, through an activation of TrxR. Further investigation should be carried out to identify the accurate mechanism of thalidomide’s impact on Trx system in pulmonary fibrosis.
Acknowledgements
This work was supported by the Chinese National Science and technology support program (Grant No. 2015BAI12B00; 2015BAI12B11).
Disclosure of conflict of interest
None.
References
- 1.Flaherty KR, Travis WD, Colby TV, Toews GB, Kazerooni EA, Gross BH, Jain A, Strawderman RL, Flint A, Lynch JP, Martinez FJ. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am J Respir Crit Care Med. 2001;164:1722–1727. doi: 10.1164/ajrccm.164.9.2103074. [DOI] [PubMed] [Google Scholar]
- 2.Coultas DB, Zumwalt RE, Black WC, Sobonya RE. The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med. 1994;150:967–972. doi: 10.1164/ajrccm.150.4.7921471. [DOI] [PubMed] [Google Scholar]
- 3.Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med. 2001;345:517–525. doi: 10.1056/NEJMra003200. [DOI] [PubMed] [Google Scholar]
- 4.Mason RJ, Schwarz MI, Hunninghake GW, Musson RA NHLBI Workshop Summary. Pharmacological therapy for idiopathic pulmonary fibrosis. Past, present, and future. Am J Respir Crit Care Med. 1999;160:1771–1777. doi: 10.1164/ajrccm.160.5.9903009. [DOI] [PubMed] [Google Scholar]
- 5.Fernandez IE, Eickelberg O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet. 2012;380:680–688. doi: 10.1016/S0140-6736(12)61144-1. [DOI] [PubMed] [Google Scholar]
- 6.Puglisi S, Torrisi SE, Giuliano R, Vindigni V, Vancheri C. What we know about the pathogenesis of idiopathic pulmonary fibrosis. Semin Respir Crit Care Med. 2016;37:358–367. doi: 10.1055/s-0036-1580693. [DOI] [PubMed] [Google Scholar]
- 7.Sisson TH, Mendez M, Choi K, Subbotina N, Courey A, Cunningham A, Dave A, Engelhardt JF, Liu X, White ES, Thannickal VJ, Moore BB, Christensen PJ, Simon RH. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181:254–263. doi: 10.1164/rccm.200810-1615OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koegelenberg CF, Ainslie GM, Dheda K, Allwood BW, Wong ML, Lalloo UG, Abdool-Gaffar MS, Khalfey H, Irusen EM. Recommendations for the management of idiopathic pulmonary fibrosis in south africa: a position statement of the south african thoracic society. J Thorac Dis. 2016;8:3711–3719. doi: 10.21037/jtd.2016.12.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest. 2012;122:2756–2762. doi: 10.1172/JCI60323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma NL, Sharma VC, Mahajan VK, Shanker V, Ranjan N, Gupta M. Thalidomide: an experience in therapeutic outcome and adverse reactions. J Dermatol Treat. 2007;18:335–340. doi: 10.1080/09546630701386993. [DOI] [PubMed] [Google Scholar]
- 11.Franks ME, Macpherson GR, Figg WD. Thalidomide. Lancet. 2004;363:1802–1811. doi: 10.1016/S0140-6736(04)16308-3. [DOI] [PubMed] [Google Scholar]
- 12.D’Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A. 1994;91:4082–4085. doi: 10.1073/pnas.91.9.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Desanctis JB, Mijares M, Suarez A, Compagnone R, Garmendia J, Moreno D, Salazar-Bookaman M. Pharmacological properties of thalidomide and its analogues. Recent Pat Inflamm Allergy Drug Discov. 2010;4:144–148. doi: 10.2174/187221310791163026. [DOI] [PubMed] [Google Scholar]
- 14.Zhou S, Wang F, Tze-Chen H, Wu JM, Wu E. Thalidomide-a notorious sedative to a wonder anticancer drug. Curr Med Chem. 2013;20:4102–4108. doi: 10.2174/09298673113209990198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang XY, Chen Y, Du Q. Successful treatment of thalidomide for recurrent bleeding due to gastric angiodysplasia in hereditary hemorrhagic telangiectasia. Eur Rev Med Pharmacol Sci. 2013;17:1114–1116. [PubMed] [Google Scholar]
- 16.Garrido Serrano A, Leon R, Sayago M, Márquez JL. Thalidomide treatment in cirrhotic patients with severe anemia secondary to vascular malformations. Dig Dis Sci. 2012;57:1112–1113. doi: 10.1007/s10620-011-1971-9. [DOI] [PubMed] [Google Scholar]
- 17.Amirshahrokhi K. Anti-inflammatory effect of thalidomide in paraquat-induced pulmonary injury in mice. Int Immunopharmacol. 2013;17:210–215. doi: 10.1016/j.intimp.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 18.Horton MR, Hallowell RW. Revisiting thalidomide: fighting with caution against idiopathic pulmonary fibrosis. Drugs Today (Barc) 2012;48:661–671. doi: 10.1358/dot.2012.48.10.1855760. [DOI] [PubMed] [Google Scholar]
- 19.Horton MR, Santopietro V, Mathew L, Horton KM, Polito AJ, Liu MC, Danoff SK, Lechtzin N. Thalidomide for the treatment of cough in idiopathic pulmonary fibrosis: a randomized trial. Ann Intern Med. 2012;157:398–406. doi: 10.7326/0003-4819-157-6-201209180-00003. [DOI] [PubMed] [Google Scholar]
- 20.Tabata C, Tabata R, Kadokawa Y, Hisamori S, Takahashi M, Mishima M, Nakano T, Kubo H. Thalidomide prevents bleomycin-induced pulmonary fibrosis in mice. J Immunol. 2007;179:708–714. doi: 10.4049/jimmunol.179.1.708. [DOI] [PubMed] [Google Scholar]
- 21.Tabata C, Kubo H, Tabata R, Wada M, Sakuma K, Ichikawa M, Fujita S, Mio T, Mishima M. All-trans retinoic acid modulates radiation-induced proliferation of lung fibroblasts via IL-6/IL-6R system. Am J Physiol. 2006;290:597–606. doi: 10.1152/ajplung.00282.2005. [DOI] [PubMed] [Google Scholar]
- 22.Tabata C, Kadokawa Y, Tabata R, Takahashi M, Okoshi K, Sakai Y, Mishima M, Kubo H. Alltrans-retinoic acid prevents radiation- or bleomycin-induced pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174:1352–1360. doi: 10.1164/rccm.200606-862OC. [DOI] [PubMed] [Google Scholar]
- 23.Kliment CR, Oury TD. Oxidative stress, extracellular matrix targets, and idiopathic pulmonary fibrosis. Free Radic Biol Med. 2010;49:707–717. doi: 10.1016/j.freeradbiomed.2010.04.036. [DOI] [PubMed] [Google Scholar]
- 24.Cheresh P, Kim SJ, Tulasiram S, Kamp DW. Oxidative stress and pulmonary fibrosis. Biochim Biophys Acta. 2013;1832:1028–1040. doi: 10.1016/j.bbadis.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim YC, Yamaguchi Y, Kondo N, Masutani H, Yodoi J. Thioredoxin dependent redox regulation of the antioxidant responsive element (ARE) in electrophile response. Oncogene. 2003;22:1860–1865. doi: 10.1038/sj.onc.1206369. [DOI] [PubMed] [Google Scholar]
- 26.Holmgren A, Johansson C, Berndt C, Lönn ME, Hudemann C, Lillig CH. Thiol redox control via thioredoxin and glutaredoxin systems. Biochem Soc Trans. 2005;33:1375–1377. doi: 10.1042/BST0331375. [DOI] [PubMed] [Google Scholar]
- 27.Holmgren A. Thioredoxin. Annu Biochem Rev. 1985;54:237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 28.Lu J, Holmgren A. Thioredoxin system in cell death progression. Antioxid Redox Signal. 2012;17:1738–1747. doi: 10.1089/ars.2012.4650. [DOI] [PubMed] [Google Scholar]
- 29.Zhong L, Holmgren A. Mammalian thioredoxin reductases as hydroperoxide reductases. Methods Enzymol. 2002;347:236–243. doi: 10.1016/s0076-6879(02)47023-1. [DOI] [PubMed] [Google Scholar]
- 30.Bjornstedt M, Hamberg M, Kumar S, Xue J, Holmgren A. Human thioredoxin reductase directly reduces lipid hydroperoxides by NADPH and selenocystine strongly stimulates the reaction via catalytically generated selenols. J Biol Chem. 1995;270:11761–11764. doi: 10.1074/jbc.270.20.11761. [DOI] [PubMed] [Google Scholar]
- 31.May JM, Mendiratta S, Hill KE, Burk RF. Reduction of dehydroascorbate to ascorbate by the selenoenzyme thioredoxin reductase. J Biol Chem. 1997;272:22607–22610. doi: 10.1074/jbc.272.36.22607. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura H, Herzenberg LA, Bai J, Araya S, Kondo N, Nishinaka Y, Herzenberg LA, Yodoi J. Circulating thioredoxin suppresses lipopolysaccharideinduced neutrophil chemotaxis. Proc Natl Acad Sci U S A. 2001;98:15143–15148. doi: 10.1073/pnas.191498798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamaki H, Nakamura H, Nishio A, Nakase H, Ueno S, Uza N, Kido M, Inoue S, Mikami S, Asada M, Kiriya K, Kitamura H, Ohashi S, Fukui T, Kawasaki K, Matsuura M, Ishii Y, Okazaki K, Yodoi J, Chiba T. Human thioredoxin-1 ameliorates experimental murine colitis in association with suppressed macrophage inhibitory factor production. Gastroenterology. 2006;131:1110–1121. doi: 10.1053/j.gastro.2006.08.023. [DOI] [PubMed] [Google Scholar]
- 34.Fan H, Liang Y, Jiang B, Li X, Xun H, Sun J, He W, Lau HT, Ma X. Curcumin inhibits intracellular fatty acid synthase and induces apoptosis in human breast cancer MDA-MB-231 cells. Oncol Rep. 2016;35:2651–2656. doi: 10.3892/or.2016.4682. [DOI] [PubMed] [Google Scholar]
- 35.Nie F, Liang Y, Jiang B, Li X, Xun H, Sun J, He W, Lau HT, Ma X. Apoptotic effect of tannic acid on fatty acid synthase over-expressed human breast cancer cells. Tumor Biol. 2016;37:2137–2143. doi: 10.1007/s13277-015-4020-z. [DOI] [PubMed] [Google Scholar]
- 36.Kaminska KK, Bertrand HC, Tajima H, Stafford WC, Cheng Q, Chen W, Wells G, Arner ES, Chew EH. Indolin-2-one compounds targeting thioredoxin reductase as potential anticancer drug leads. Oncotarget. 2016;7:4023351. doi: 10.18632/oncotarget.9579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X, Qian L, Nan H, Cui M, Hao X, Du Y. Function of the transforming growth factor-β1/c-Jun N-terminal kinase signaling pathway in the action of thalidomide on a rat model of pulmonary fibrosis. Exp Ther Med. 2014;7:669–674. doi: 10.3892/etm.2013.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tabata C, Tabata R, Takahashi Y, Nakamura K, Nakano T. Thalidomide prevents cigarette smoke extract-induced lung damage in mice. Int Immunopharmacol. 2015;25:511–517. doi: 10.1016/j.intimp.2015.02.036. [DOI] [PubMed] [Google Scholar]
- 39.Mouratis MA, Aidinis V. Modeling pulmonary fibrosis with bleomycin. Curr Opin Pulm Med. 2011;17:355–361. doi: 10.1097/MCP.0b013e328349ac2b. [DOI] [PubMed] [Google Scholar]
- 40.Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208:1339–1350. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Denis M. Interleukin-6 in mouse hypersensitivity pneumonitis: changes in lung free cells following depletion of endogenous IL-6 or direct administration of IL-6. J Leukocyte Biol. 1992;52:197–201. doi: 10.1002/jlb.52.2.197. [DOI] [PubMed] [Google Scholar]
- 42.Caspary WJ, Lanzo DA, Niziak C. Effect of deoxyribonucleic acid on the production of reduced oxygen by bleomycin and iron. Biochemistry. 1982;21:334–338. doi: 10.1021/bi00531a021. [DOI] [PubMed] [Google Scholar]
- 43.Todd NW, Luzina IG, Atamas SP. Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair. 2012;5:11. doi: 10.1186/1755-1536-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Del Rio D, Stewart AJ, Pellegrini N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr Metab Cardiovas. 2005;15:316–328. doi: 10.1016/j.numecd.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 45.Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop Dj, Horwitz E. Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 46.Powis G, Kirkpatrick DL. Thioredoxin signaling as a target for cancer therapy. Curr Opin Pharmacol. 2007;7:392–397. doi: 10.1016/j.coph.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 47.Mahmood DF, Abderrazak A, El Hadri K, Simmet T, Rouis M. The thioredoxin system as a therapeutic target in human health and disease. Antioxid Redox Signal. 2013;19:1266–1303. doi: 10.1089/ars.2012.4757. [DOI] [PubMed] [Google Scholar]
- 48.Zhang S, Xu N, Nie J, Dong L, Li J, Tong J. Proteomic alteration in lung tissue of rats exposed to cigarette smoke. Toxicol Lett. 2008;178:191–196. doi: 10.1016/j.toxlet.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 49.Ito W, Kobayashi N, Takeda M, Ueki S, Kayaba H, Nakamura H, Yodoi J, Chihara J. Thioredoxin in allergic inflammation. Int Arch Allergy Immunol. 2011;155:142–146. doi: 10.1159/000327501. [DOI] [PubMed] [Google Scholar]
- 50.Gon Y, Sasada T, Matsui M, Hashimoto S, Takagi Y, Iwata S, Wada H, Horie T, Yodoi J. Expression of thioredoxin in bleomycin-injured airway epithelium: possible role of protection against bleomycin induced epithelial injury. Life Sci. 2001;68:1877–1888. doi: 10.1016/s0024-3205(01)00980-8. [DOI] [PubMed] [Google Scholar]
- 51.Hoshino T, Nakamura H, Okamoto M, Kato S, Araya S, Nomiyama K, Oizumi K, Young HA, Aizawa H, Yodoi J. Redox-active protein thioredoxin prevents proinflammatory cytokineor bleomycin-induced lung injury. Am J Respir Crit Care Med. 2003;168:1075–1083. doi: 10.1164/rccm.200209-982OC. [DOI] [PubMed] [Google Scholar]
- 52.Zhu Z, Yang G, Wang Y, Yang J, Gao A, Niu P, Tian L. Suppression of thioredoxin system contributes to silica-induced oxidative stress and pulmonary fibrogenesis in rats. Toxicol Lett. 2013;222:289–294. doi: 10.1016/j.toxlet.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 53.Tiitto L, Kaarteenaho-Wiik R, Sormunen R, Holmgren A, Pääkkö P, Soini Y, Kinnula VL. Expression of the thioredoxin system in interstitial lung disease. J Pathol. 2003;201:363–370. doi: 10.1002/path.1435. [DOI] [PubMed] [Google Scholar]
- 54.Kodama A, Watanabe H, Tanaka R, Tanaka H, Chuang VT, Miyamoto Y, Wu Q, Endo M, Hamasaki K, Ishima Y, Fukagawa M, Otagiri M, Maruyama T. A human serum albuminthioredoxin fusion protein prevents experimental contrast-induced nephropathy. Kidney Int. 2013;83:446–454. doi: 10.1038/ki.2012.429. [DOI] [PubMed] [Google Scholar]