

Abstract
Abnormal proliferation of vascular smooth muscle cells (VSMCs) contributes to the development of cardiovascular diseases. Studies have showed the great impact of microRNAs (miRNAs) on the cell proliferation in VSMCs. This study examined the in vitro functional roles of miR-665 in the VSMCs and explored the underlying molecular mechanisms. The mRNA and protein expression levels were determined by qRT-PCR and western blot assays, respectively. CCK-8, transwell invasion and wound healing assays were performed to measure VSMCs proliferation, invasion and migration, respectively. The miR-665 targeted-3’UTR of fibroblast growth factor 9 (FGF9) and myocyte enhancer factor 2D (MEF2D) was confirmed by luciferase reporter assay. Platelet-derived growth factor-bb (PDGF-bb) and 20% serum promoted cell proliferation and suppressed the expression of miR-665 in VSMCs. In vitro functional assays demonstrated that miR-665 inhibited VSMCs proliferation, invasion and migration. Bioinformatics analysis showed that FGF9 and MEF2D were found to be downstream targets of miR-665. Luciferase report assay confirmed that FGF9 and MEF2D 3’UTRs are direct targets of miR-665, and miR-665 overexpression suppressed both the mRNA and protein expression levels of FGF9 and MEF2D. Furthermore, rescue experiments showed that enforced expression of FGF9 or MEF2D attenuated the inhibitory effects of miR-665 on VSMCs proliferation. More importantly, overexpression of miR-665 also suppressed the mRNA and protein expression levels of β-catenin, c-myc and cyclin D1. In summary, miR-665 suppressed the VSMCs proliferation, invasion and migration via targeting FGF9 and MEF2D, and the in vitro effects of miR-665 on VSMCs may be associated with modulation of Wnt/β-catenin signaling activities.
Keywords: Vascular smooth muscle cells, miR-665, cell proliferation, FGF9, MEF2D, Wnt/β-catenin
Introduction
Cardiovascular diseases, such as hypertension, coronary artery disease, atherosclerosis and congestive heart failure, are a major cause of death worldwide [1]. Vascular smooth muscle cells (VSMCs) are important components of the blood vessels, and VSMCs are responsible for the contraction and relaxation of blood vessels under different stimuli, including mechanical injury, growth factors, hemodynamic alteration, and ligand-receptor signaling [2,3]. Studies have shown that abnormal cell proliferation, invasion and migration, and disruption of inflammatory signaling in VSMCs largely contribute to various cardiovascular diseases including atherosclerosis, hypertension and restenosis [4,5]. In this regard, to better understand the molecular mechanisms underlying the inappropriate proliferation of VSMCs may be useful for us to identify new therapeutic targets for the treatment of cardiovascular diseases.
Recently, microRNAs (miRNAs) were found to play diverse cellular functions including cell proliferation, differentiation and cell apoptosis. MiRNAs are a class of short noncoding RNAs, and they can negatively regulate expression of targeted genes by inducing translation repression or mRNA degradation via complementing with the 3’ untranslated region (3’UTR) of mRNAs [6]. The functional roles of miRNAs in VSMCs have been elucidated in a variety of studies. MiRNAs such as miR-541, miR-146b-5p, miR-181b were found to promote VSMCs proliferation [7-9]; while miRNAs such as miR-124, miR-503, and miR-140 had inhibitory effects on the VSMCs proliferation [10-12]. Our previous studies also showed that miR-379 inhibited cell proliferation, invasion and migration of VSMCs via targeting the 3’UTR of insulin-like factor-1 [13]. Previous studies have showed that miR-665 was down-regulated in the cardiomyocytes from the chronic heart failure [14], and miR-665 was demonstrated to be involved in the regulation of the expression of cardioprotective cannabinoid receptor CB2 in patients with severe heart failure [14], suggesting that miR-665 may play important roles in the regulation of cardiovascular functions. MiR-665 was also found to suppress the invasion and metastasis of osteosarcoma [15], and in the prostate cancer, miR-665 was shown to be associated with the tumor metastasis [16]. In addition, up-regulation of miR-665 promotes apoptosis and colitis in inflammatory bowel disease by repressing the endoplasmic reticulum stress components [17]. Though miR-665 was implicated in the involvement of cardiovascular disease, the exact mechanisms of miR-665 in the regulation of cardiovascular functions, particularly VSMCs functions were largely unknown [18].
Fibroblast growth factor 9 (FGF9) is a potent mitogen secreted from bone marrow cells, and it belongs to the FGF family. Studies have demonstrated that FGF9 was associated with lots of vessel biological processes. FGF9 was found to promote smooth muscle cells wrapping microvessels in implants through platelet-derived growth factor Rβ and sonic hedgehog, which are important in the pathophysiology of blood vessels [19]. In addition, studies found that FGF9 markedly stimulated both the proliferation of neointimal smooth muscle cells and the activation of extracellular signal-related kinases 1/2 [20]. Recently, FGF9 was found to be downstream target of miR-182, which prevents VSMCs dedifferentiation [21]. Taken together, these studies implicated the important roles of FGF9 in the pathophysiology of blood vessels.
Myocyte enhancer factor 2D (MEF2D), a member of the MEF2 family, was originally identified as a muscle-specific factor that binds an A/T-rich consensus sequence associated with a large number of genes expressed in the skeletal and cardiovascular systems [22]. In the cardiovascular research, MEF2D was found to be required for the survival of cardiomyocytes [23], and overexpression of MEF2D prolonged the survival of cardiomyocytes [24]. Studies by using transgenic mice that have enforced overexpression of MEF2D implicated the important function for MEF2D in stress-dependent cardiac growth and reprogramming of gene expression in the adult heart [25]. In the cancer studies, MEF2D promoted cell proliferation in various types of cancer cells, including liver cancer, lung cancer and gastric cancer [26-28], and MEF2D also promoted tumor angiogenesis in vitro and in vivo in the colorectal cancer [29]. In the VSMCs, the activation of MEF2D by big mitogen-activated protein kinase 1 (BMK1) involved in the up-regulation of c-jun and co-activation of p38, which resulted in the enhanced proliferation VSMCs [30], suggesting that MEF2D may promote VSMCs proliferation.
The Wnt signaling pathway plays important roles in the regulation of cell proliferation and embryonic development, and β-catenin is one of the key mediators in the pathway. Studies demonstrated that VSMCs proliferation was promoted through the transfection of a degradation-resistant β-catenin transgene into rats [31], and inhibition of β-catenin suppressed cell proliferation of VSMCs and also down-regulated the expression of cyclin D1 [32]. In addition, activation of Wnt/β-catenin signaling pathway also induces cell proliferation and survival in human endothelial cells [33]. These studies established the importance of Wnt/β-catenin signaling in regulation of VSMCs functions.
In this study, the effects of miR-665 on VSMCs proliferation, invasion and migration were examined, and the potential molecular mechanisms were also explored. PDGF-bb or 20% serum treatment promotes VSMCs proliferation and down-regulated the expression of miR-665. Gain-of-function and loss-of-function assays were performed to reveal the effects of miR-665 on VSMCs proliferation, invasion and migration. The bioinformatics analysis was employed to predict the potential downstream targets of miR-665, and FGF9 and MEF2D were selected for further experimentation, as their potential roles in the regulation of VSMCs proliferation. FGF9 and MEF2D have been suggested to be interacted with Wnt/β-catenin signaling [27,34], and Wnt/β-catenin signaling also was suggested to be involved in the VSMCs proliferation, therefore, the effects of miR-665 on the Wnt/β-catenin was also examined by using quantitative real-time PCR (qRT-PCR) and western blot assays.
Materials and methods
Cell culture
VSMCs cell lines (human aortic smooth muscle cells) were obtained from the Cell bank of Chinese Academy of Science (Shanghai, China), and the cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone, GE Health Care, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific, Waltham, USA). Cells were maintained in a humidified atmosphere with 5% CO2 at 37°C before experimentation.
MiRNAs and plasmids
MiR-665 mimics, miR-665 inhibitors and their respective controls were synthesized by Ribobio (Guangzhou, China). The empty vector pcDNA3.1, pcDNA3.1-fibroblast growth factor 9 (FGF9), and pcDNA3.1-myocyte-specific enhancer factor 2D (MEF2D) were purchased from Shanghai BlueGene Biotech (Shanghai, China).
Transfection, platelet-derived growth factor-bb (PDGF-bb), 20% serum treatment, LiCl treatment
Cells were transfected with miR-665 mimics, miR-665 inhibitor, or their controls using Lipofectamine 2000 (Invitrogen, Carlsbad, USA) according to the manufacturer’s instructions. Co-transfection of miR-665 mimics and pcDNA-3.1-FGF9 or pcDNA3.1-MEF2D was also performed by using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. PDGF-bb (Sigma, St.Louis, USA) treatment was performed at a concentration of 30 ng/ml for 24 h, or 20% serum treated VSMCs for 24 h, and then CCK-8 assay was performed at 0, 24, 48 and 72 h after PDGF-bb or 20% serum treatment. LiCl treatment was performed by treating VSMCs at a concentration of 25 mM.
RNA preparation and quantitative real-time PCR analysis
Total RNAs were isolated from VSMCs by using Trizol reagent (Invitrogen). The PrimeScript RT Master Mix kit (Takara, Dalian, China) was used to obtain cDNA for mRNA detection, whereas PrimerScript II 1st Strand cDNA synthesis kit (Takara) was applied to reverse transcribe RNA for miRNA detection. Real time PCR was performed using SYBR Green PCR Kit (Takara) according to the manufacturer’s instructions. U6 was used as an internal control for miR-665, and GAPDH was used as an internal control for other genes. The relative expressions of genes were calculated based on Comparative Ct method.
Cell proliferation assay
For the CCK-8 assay, cells were seeded in 96-well plates (5 × 103 cells/well) and incubated for 24 h, and then treated with PDGF-bb or 20% serum; or transfected with miR-665 mimics, miR-665 inhibitor or their respective controls; or co-transfected with miR-665 mimics and pcDNA3.1-FGF9 or pcDNA3.1-MEF2D. At 0 h, 24 h, 48 h, and 72 h after PDGF-bb or 20% serum treatment, or at 48 h after transfection, CCK-8 kit (Beyotime) was used to detect cell proliferation index according to manufacturer’s instructions.
Cell invasion assay
For the invasion assay, cells were seeded in 24-well transwell plates (5 × 104 cells/well) and incubated for 24 h, and then transfected with miR-665 mimics, miR-665 inhibitor or their respective controls. Cells were re-suspended in serum-free DMEM and grown in the upper chambers with Matrigel-coated membrane (BD Bioscience, San Jose, USA), and 500 μl of DMEM containing 10% FBS was added into the bottom of the chambers. Cells were allowed to migrated through the 8 μm polyethylene terephthalate membrane for 24 h, cells passed through the membrane were fixed in 4% formaldehyde and stained with 0.1% crystal violet.
Wound healing assay
For the wound healing assay, cells were seeded in 6-well plates (5 × 105 cells/well), 48 h after transfection, a pipette tip was used to create a wound. The cells were then cultured in serum-free medium. Cells migrated into wound surface and the average distance of migrating cells was determined under an inverted microscopy at 0 h and 48 h.
Luciferase reporter assay
The pmirGLO vectors containing wild type or mutant miR-665 binding site in FGF9 3’UTR and MEF2D 3’UTR were synthesized by Ribobio (Guangzhou, China). VSMCs were seeded into 24-well plates 24 h before transfection and then co-transfected with 50 ng of wild type or mutant luciferase vector containing FGF9 3’UTR or MEF2D 3’UTR and 20 μM miR-665 mimics, miR-665 inhibitor or their respective controls. After 48 h, luciferase activity was assayed by using the Dual-luciferase Reporter Assay System (Promega, Madison, USA).
Western blot
Proteins were extracted from whole cell lysates and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, then transferred to a polyvinylidene fluoride membrane. The following primary antibodies were used: rabbit anti-FGF9 (1:1000; Abcam, Cambridge, UK), rabbit anti-MEF2D (1:1000, Abcam), rabbit anti-β-catenin (1:4000, Cell Signaling Technology, Danvers, USA), rabbit anti-c-myc (1:3000, Cell Signaling Technology), rabbit anti-cyclin D1 (1:2500, Cell Signaling Technology) and mouse anti-β-actin (1:5000; Abcam). Membranes were then incubated with the respective horseradish peroxidase-conjugated secondary antibodies (1:4000; Abcam). The bands were visualized by exposing the membranes under a ChemoDoc XRS detection system (Bio-Rad, Hercules, USA). The whole gel blot for the detected proteins were shown in Figures S1, S2 and S3.
Statistical analysis
All statistical analysis was carried out using GraphPad Prism version 6 (GraphPad Prism version 6.0, Inc. La Jolla, USA). The significant differences between two groups were analyzed by t-test, and the significant differences among more than two groups were analyzed by one-way ANOVA followed by Bonferroni’s multiple comparison tests. All data are expressed as the mean values of at least three independent replicates ± SEM, and P<0.05 was considered to be statistically significant.
Results
The effects of PDGF-bb or 20% serum treatment on the cell proliferation and expression levels of miR-665
In the present study, the VSMCs were treated with PDGF-bb (30 ng/ml) or 20% serum, and at 0, 24, 48 and 72 h after treatments, the cell proliferative ability was measured by CCK-8 assay. In the PDGF-bb-treated VSMCs, the cell proliferative ability was significantly enhanced at 24, 48, and 72 h post-treatment (Figure 1A); in the 20% serum-treated VSMCs, the cell proliferative ability was also significantly enhanced at 48 and 72 h post-treatment (Figure 1B). In order to examine the potential effects of miR-665 on the function of VSMCs, the expression levels of miR-665 were measured by qRT-PCR at 72 h after PDGF-bb (30 ng/ml) or 20% serum treatments, and the results showed that PDGF-bb treatment significantly suppressed the expression levels of miR-665 in the VSMCs (Figure 1C), and the expression level of miR-665 was also decreased in the VSMCs received 20% serum treatment (Figure 1D).
Figure 1.
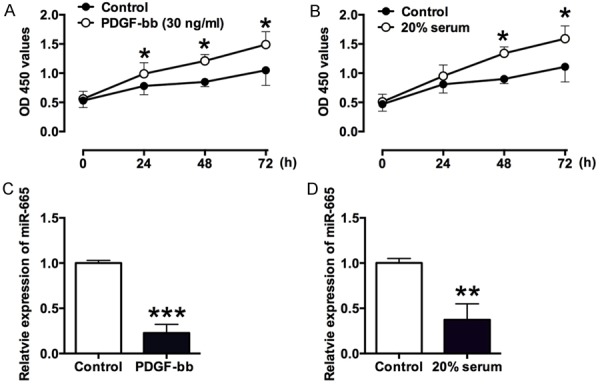
The effects of PDGF-bb and 20% serum on cell proliferation and miR-665 expression in VSMCs. A. PDGF-bb (30 ng/ml) significantly promotes VSMCs proliferation at 24, 48 and 72 h after treatment, as measured by CCK-8 assay. B. 20% serum significantly promotes VSMCs proliferation at 48 and 72 h after treatment, as measured by CCK-8 assay. C. The expression of miR-665 as measured by qRT-PCR was down-regulated at 72 h post PDGF-bb (30 ng/ml) treatment. D. The expression of miR-665 as measured by qRT-PCR was down-regulated at 72 h post 20% serum treatment. Data represents the mean values of three independent replicates ± SEM; significant differences relative to control group were shown as *P<0.05, **P<0.01, ***P<0.001.
The effects of miR-665 on the cell proliferation, invasion and migration in VSMCs
In order to further explore the in vitro functional roles of miR-665 in VSMCs, the gain-of-function and loss-of-function studies were performed. Overexpression of miR-665 in the VSMCs was achieved by transfecting VSMCs with miR-665 mimics, and down-regulation of miR-665 in the VSMCs was achieved by transfecting VSMCs with miR-665 inhibitor. As shown in Figure 2A, the expression level of miR-665 in VSMCs was increased by more than 10 fold after miR-665 mimics transfection (Figure 2A); while miR-665 inhibitor transfection significantly suppressed the expression level of miR-665 in VSMCs (Figure 2B). Furthermore, the CCK-8 assay was performed to determine the cell proliferative ability in VSMCs transfected with miR-665 mimics or miR-665 inhibitor, or their respective controls. MiR-665 mimics transfection significantly decreased the OD 450 values in VSMCs when compared to control group (Figure 2C); miR-665 inhibitor transfection increased the OD 450 values in VSMCs when compared to control group (Figure 2D), suggesting the miR-665 suppresses the cell proliferative ability of VSMCs. The cell invasion and cell migration in VSMCs were assessed by cell invasion and wound healing assays, respectively. The number of invaded cells in VSMCs transfected with miR-665 mimics was significantly decreased when compared to control group (Figure 2E), and miR-665 inhibitor transfection significantly increased the number of invaded cells (Figure 2F), suggesting that miR-665 suppresses the VSMCs invasion. For the wound healing assay, miR-665 mimics transfection significantly decreased the percentage of repaired wound (Figure 2G), while miR-665 inhibitor transfection enhanced the wound repair in VSMCs (Figure 2H), suggesting that miR-665 inhibits the VSMCs migration.
Figure 2.
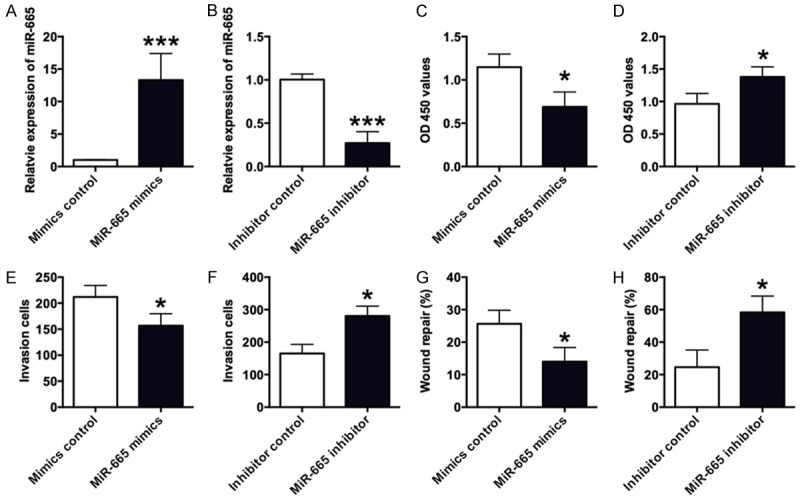
The effects of miR-665 on VSMCs proliferation, invasion and migration. A and B. The expression level of miR-665 in VSMCs was measured by qRT-PCR assay 48 h after cells transfected with miR-665 mimics, miR-665 inhibitor, or their respective controls. MiR-665 mimics transfection increased the miR-665 expression, and miR-665 inhibitor transfection suppressed the miR-665 expression level. C and D. The VSMCs proliferation was measured by CCK-8 assay 48 h after cells transfected with miR-665 mimics, miR-665 inhibitor, or their respective controls. MiR-665 mimics transfection increased VSMCs proliferation, and miR-665 inhibitor transfection suppressed the VSMCs proliferation. E and F. The VSMCs invasion was measured by transwell assay 48 h after cells transfected with miR-665 mimics, miR-665 inhibitor or their respective controls. MiR-665 mimics transfection increased VSMCs invasion, and miR-665 inhibitor transfection suppressed VSMCs invasion. G and H. The VSMCs migration was measured by wound healing assay 48 h after cells transfected with miR-665 mimics, miR-665 inhibitor, or their respective controls. MiR-665mimics transfection enhanced VSMCs migration, and miR-665 inhibitor transfection suppressed VSMCs migration. Data represents the mean values of three independent replicates ± SEM; significant differences relative to control group were shown as *P<0.05, ***P<0.001.
FGF9 and MEF2D were potential downstream targets of miR-665
As miRNAs regulate the gene expression via targeting the 3’UTR of the genes, the downstream targets of miR-665 were predicted by using TargetScan (www.targetscan.org). In the predicted results, a list of potential genes was found, and in the present study, FGF9 and MEF2D were chosen for further investigation because of their potential functions roles in VSMCs. To validate whether FGF9 is a target of miR-665, the FGF9 3’UTR fragment containing wild-type or mutant miR-665-binding sequence was sub-cloned to the Firefly luciferase reporter gene (Figure 3A). When miR-665 mimics were co-transfected with the wild type reporter plasmid, the relative luciferase activity of the reporter containing wild type FGF9 3’UTR was significantly suppressed; and co-transfection of miR-665 inhibitor and the report plasmid significantly increased the luciferase activity of the reporter containing wild type FGF9 3’UTR (Figure 3B); while the luciferase activity of the reporter containing mutant FGF9 3’UTR was unaffected (Figure 3C). In addition, qRT-PCR and western blot were performed to examine the effect of miR-665 overexpression on the mRNA and protein levels of FGF9 in VSMCs, miR-665 mimics transfection significantly suppressed the mRNA and protein expression level of FGF9 (Figure 3D). To further investigate the effect of FGF9 on VSMCs proliferation, pcDNA3.1-FGF9 and miR-665 mimics were co-transfected into VSMCs, and enforced expression of FGF9 sufficiently attenuated the inhibitory action of miR-665 on VSMCs proliferation (Figure 3E).
Figure 3.
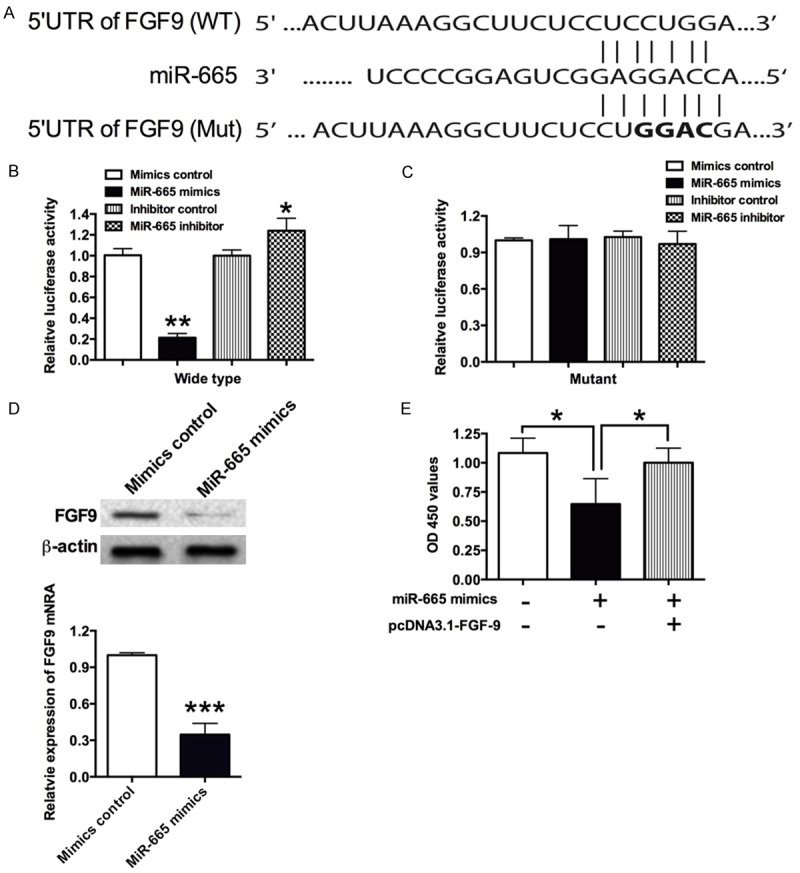
FGF9 is a direct target of miR-665. A. The seed sequence of wild type (WT) 3’UTR or mutant (Mut) 3’UTR of FGF9 that is complementary to miR-665; B and C. VSMCs were co-transfected with miR-665 mimics or its control with wild type or mutant 3’UTR of FGF9, and luciferase activity was detected at 48 h post transfection; D. The mRNA and protein expression level of FGF9 in VSMCs was determined by qRT-PCR and western blot, respectively, at 48 h after cells transfected with miR-665 mimics or its control. MiR-665 mimics transfection suppressed the mRNA and protein levels of FGF9. E. The cell proliferation of VSMCs was measured by CKK-8 assay at 48 h after cells co-transfected with miR-665 mimics and pcDNA3.1-FGF9 or their respective controls. Data represents the mean values of three independent replicates ± SEM; significant differences relative to control group were shown as *P<0.05, **P<0.01, ***P<0.001.
To validate whether MEF2D is a target of miR-665, similar experiments as shown above were performed. When miR-665 mimics were co-transfected with the wild type reporter plasmid, the relative luciferase activity of the reporter containing wild type MEF2D 3’UTR was significantly suppressed (Figure 4A and 4B); and co-transfection of miR-665 inhibitor and the report plasmid significantly increased the luciferase activity of the reporter containing wild type MEF2D 3’UTR (Figure 4A and 4B); while the luciferase activity of the reporter containing mutant MEF2D 3’UTR was unaffected (Figure 4A and 4C). In addition, qRT-PCR and western blot results showed that miR-665 mimics transfection significantly suppressed the mRNA and protein expression levels of MEF2D (Figure 4D). The rescue experiments further reveal that forced overexpression of MEF2D also significantly restored the inhibitory effects of miR-665 on the cell proliferation in the VSMCs (Figure 4E).
Figure 4.
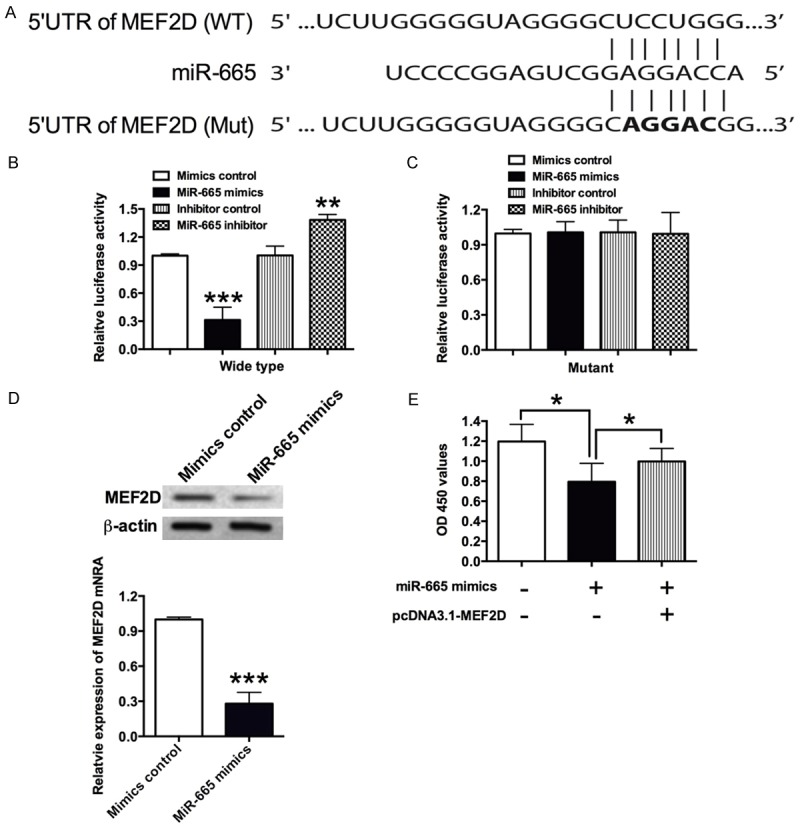
MEF2D is a direct target of miR-665. A. The seed sequence of wild type (WT) 3’UTR or mutant (Mut) 3’UTR of MEF2D that is complementary to miR-665. B and C. VSMCs were co-transfected with miR-665 mimics or its control with wild type or mutant 3’UTR of MEF2D, and luciferase activity was detected at 48 h post transfection. D. The mRNA and protein expression levels of MEF2D in VSMCs was determined by qRT-PCR and western blot, respectively, at 48 h after cells transfected with miR-665 mimics or its control. MiR-665 mimics transfection suppressed the mRNA and protein levels of MEF2D. E. The VSMCs proliferation was measured by CKK-8 assay at 48 h after cells co-transfected with miR-665 mimics and pcDNA3.1-MEF2D or their respective controls. Data represents the mean values of three independent replicates ± SEM; significant differences relative to control group were shown as *P<0.05, **P<0.01, ***P<0.001.
The effects of miR-665 on the Wnt/β-catenin signaling
As Wnt signaling was found to play important roles in VSMCs proliferation. In this study, the effects of miR-655 on the Wnt/β-catenin signaling was examined by qRT-PCR and western blot, and miR-665 mimics transfection significantly suppressed the mRNA expression level of β-catenin, c-myc and cyclin D1 in VSMCs, and pretreatment with the Wnt agonist, LiCl, significantly attenuated the inhibitory effects of miR-665 on the mRNA expression level of β-catenin, c-myc and cyclin D1 in VSMCs (Figure 5A-C). Western blot results also showed that the protein levels of β-catenin, c-myc and cyclin D1 were decreased in VSMCs transfected with miR-665 mimics, and pretreatment with LiCl increased the protein levels of β-catenin, c-myc and cyclin D1 in VSMCs transfected with miR-665 mimics when compared to cells only treated with miR-665 mimics (Figure 5D).
Figure 5.
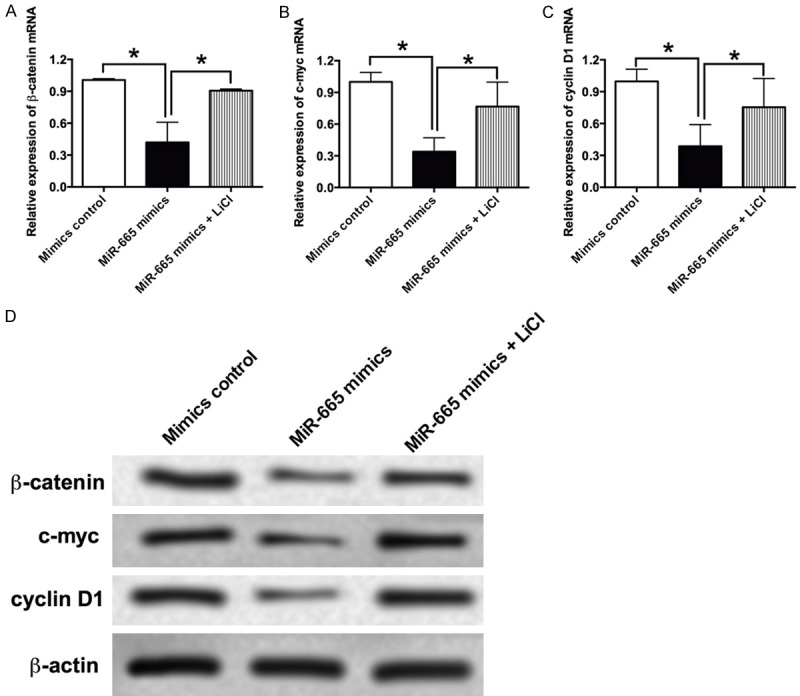
The effects of miR-665 on the Wnt/β-catenin signaling in VSMCs. The mRNA expression levels of (A) β-catenin, (B) c-myc and (C) cyclin D1 in VSMCs after treatment with miR-665 mimics or miR-665 mimics + LiCl were determined by qRT-PCR assay. (D) The protein levels of β-catenin, c-myc and cyclin D1 in VSMCs after treatment with miR-665 mimics or miR-665 mimics + LiCl were determined by western blot assay. Data represents the mean values of three independent replicates ± SEM; significant differences relative to control group were shown as *P<0.05.
Discussion
Recent studies have showed that aberrant expression of miRNAs was found in stenotic and atherosclerotic arteries, where the neointimal lesion was formed [1]. However, the molecular mechanisms of the aberrant expression of miRNAs in cardiovascular diseases were largely unexplored. In this study, our results showed that PDGF-bb or 20% serum treatment enhanced cell proliferation and also suppressed the expression levels of miR-665 in the VSMCs. In vitro functional assays showed that overexpression of miR-665 suppressed VSMCs proliferation, invasion and migration, and down-regulation of miR-665 enhanced the VSMCs proliferation, invasion and migration. In addition, the downstream targets (FGF9 and MEF2D) of miR-665 were further confirmed by luciferase reporter assays. Overexpression of miR-665 suppressed the mRNA and protein expression levels of FGF9 and MEF2D in VSMCs. The enforced expression of FGF9 and MEF2D also attenuated the inhibitory effects of miR-665 on VMSCs proliferation. In vitro qRT-PCR and western blot results also showed that overexpression of miR-665 suppressed the activity of Wnt/β-catenin signaling pathway.
The aberrant expression of miRNAs was found to be important in the regulation of VSMCs proliferation, invasion and migration, which has been confirmed in numerous studies. MiR-136 promotes cell proliferation of VSMCs via targeting PPP2R2A in atherosclerosis [35]. MiR-181b was found to activate the PI3K and MAPK pathways, which results in enhancing the cell proliferation of VSMCs [36]. On the other hand, miR-145 was found to inhibit VSMCs proliferation by targeting CD40 [37]; and miR-129-5p was found to inhibit cell proliferation of VSMCs via targeting Wnt5a [38]. Although there is limited evidence to indicate the functional role of miR-665 in cardiovascular diseases, miR-665 was found to suppress the cell proliferation, cell invasion and cell migration in osteosarcoma via targeting RAB23 [15], in addition, in the chronic heart failure study, miR-665 expression is significantly decreased in the cardiomyocytes, and downregulation of miR-665 may contribute to a compensatory response of the diseased myocardium [14]. Our results showed that overexpression of miR-665 suppressed VSMCs proliferation, cell invasion and migration, and down-regulation of miR-665 enhanced VSMCs proliferation, cell invasion and migration. Taken together, these results suggested that miR-665 had inhibitory effects on the VSMCs proliferation, cell invasion and migration.
Like other miRNAs, miR-665 also exerts its functions in VSMCs via targeting the 3’UTR of specific genes. The bioinformatics analysis and luciferase reporter assays showed that FGF9 and MEF2D are downstream targets of miR-665 in VSMCs. FGF9, a member of the FGF family, is a potent mitogen secreted from bone marrow cells [39]. Studies have shown that proliferation of VSMCs after arterial injury depends on the interaction between FGF2 and FGF9 [20]. Studies showed that FGF9 delivery during angiogenesis produces durable, vaso-responsive microvessels wrapped by VSMCs [40]. FGF9 was also found to activate progenitor cells and enhance angiogenesis in the infarcted diabetic heart [41]. More importantly, miR-182 was found to prevent VSMCs cell dedifferentiation via FGF9/PDGFR-beta signaling [21]. In this study, we further demonstrated that miR-665 suppressed VSMCs proliferation via targeting FGF9. MEF2D, a member of the MEF2 family, is a major transcription activator in the muscle development [42]. Studies have found that MEF2D overexpression was involved in the progression of several types of cancers including colorectal cancer, liver cancer, lung cancer, osteosarcoma and leukemia [28,43-46], and overexpression of MEF2D prolonged the survival of cardiomyocytes [24]. There is little evidence about the role of MEF2D in the VSMCs proliferation, however, studies have suggested that the activation of MEF2D by BMK1 is required for the up-regulation of c-jun, which is an important transcriptional factor in the serum-induced cell proliferation in VSMCs [30]. Our results showed that overexpression of miR-665 suppressed the mRNA and protein expression levels of MEF2D in VSMCs, and further rescue experiments showed that enforced expression of MEF2D prevented the inhibitory effects of miR-665 on VSMCs proliferation, suggesting the miR-665 suppressed VSMCs proliferation via targeting MEF2D.
Evidence from previous studies implicated that both FGF9 and MEF2D had potential interactions with the Wnt/β-catenin signaling pathway. In terms of FGF9, FGF9 regulates β-catenin mediated Wnt signaling in lung mesenchyme [47], and FGF9 and FGF/Wnt/β-catenin signaling pathway was also found to play important roles in ablative laser-induced wound healing processes [34]. In terms of MEF2D, suppression of MEF2D was found to significantly inactivated Wnt/β-catenin signaling pathway in gastric cancer cells [27], and loss of MEF2D expression in animal cap cells from Xenopus caused a significant reduction in the membrane β-catenin complexes [48]. Therefore, in the present study, we are tempted to further explore the effects of miR-665 on the activity of Wnt/β-catenin signaling based on the facts that miR-665 affected the expression of both FGF9 and MEF2D in VSMCs. The Wnt family consists of 19 conserved genes, and these genes were found to encode for the secreted glycoprotein ligands for Frizzled receptors [49]. The canonical Wnt signaling pathway is largely regulated via the balance of β-catenin regulation [49]. Studies have showed that Wnt/β-catenin signaling interacts with TGF-β/Smad3 signaling to promote cell proliferation in VSMCs [18], and Wnt/β-catenin signaling activation also promotes VSMCs to osteogenictrans differentiation and calcification via regulating Runx2 gene expression [49]. In addition, studies demonstrated that VSMCs proliferation was promoted through the transfection of a degradation-resistant β-catenin transgene into rats [31], and inhibition of β-catenin suppressed cell proliferation of VSMCs and also down-regulated the expression of cyclin D1 [32]. Our results showed that overexpression of miR-665 suppressed the Wnt/β-catenin signaling, and this effect could be prevented with LiCl treatment. Our data suggested that miR-665 may regulate VSMCs proliferation via modulating the Wnt/β-catenin signaling.
Conclusions
In summary, the present study demonstrated that miR-665 suppressed the VSMCs proliferation, invasion and migration via targeting FGF9 and MEF2D, and the in vitro effects of miR-665 on VSMCs may be associated with modulation of Wnt/β-catenin signaling activities as summarized in Figure 6. The present study may provide novel therapeutic target for combating cardiovascular diseases such as atherosclerosis.
Figure 6.
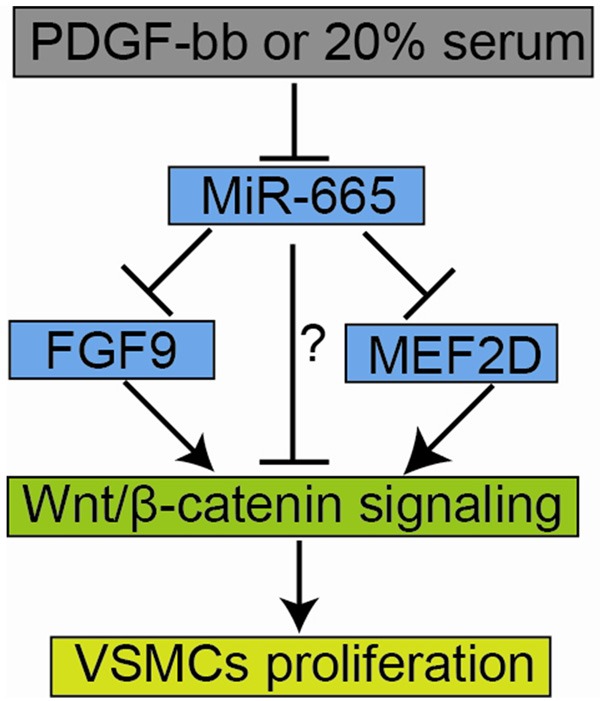
Schematic illustration demonstrating the role of miR-665 in VSMCs. PDGF-bb or 20% serum treatment suppressed miR-665 expression. MiR-665 inhibited Wnt/β-catenin signaling activities possibly via suppressing FGF9 or MEF2D or by other unidentified mechanism, and the inhibition of FGF9, MEF2D and the activities of Wnt/β-catenin signaling may contribute to the suppression of VSMCs proliferation.
Acknowledgements
This project was supported by the social development and technology research project of Shaanxi Province (2015SF080).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Gurha P. MicroRNAs in cardiovascular disease. Curr Opin Cardiol. 2016;31:249–254. doi: 10.1097/HCO.0000000000000280. [DOI] [PubMed] [Google Scholar]
- 2.Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118:692–702. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porter KE, Riches K. Vascular smooth muscle as a target for novel therapeutics. Curr Diab Rep. 2015;15:72. doi: 10.1007/s11892-015-0647-9. [DOI] [PubMed] [Google Scholar]
- 4.Huang CH, Ciou JS, Chen ST, Kok VC, Chung Y, Tsai JJ, Kurubanjerdjit N, Huang CF, Ng KL. Identify potential drugs for cardiovascular diseases caused by stress-induced genes in vascular smooth muscle cells. Peer J. 2016;4:e2478. doi: 10.7717/peerj.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tykocki NR, Boerman EM, Jackson WF. Smooth muscle ion channels and regulation of vascular tone in resistance arteries and arterioles. Compr Physiol. 2017;7:485–581. doi: 10.1002/cphy.c160011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robinson HC, Baker AH. How do microRNAs affect vascular smooth muscle cell biology? Curr Opin Lipidol. 2012;23:405–411. doi: 10.1097/MOL.0b013e32835719a1. [DOI] [PubMed] [Google Scholar]
- 7.Yang F, Xu Z, Duan S, Luo M. MicroRNA-541 promotes the proliferation of vascular smooth muscle cells by targeting IRF7. Am J Transl Res. 2016;8:506–515. [PMC free article] [PubMed] [Google Scholar]
- 8.Wang H, Jiang M, Xu Z, Huang H, Gong P, Zhu H, Ruan C. miR-146b-5p promotes VSMC proliferation and migration. Int J Clin Exp Pathol. 2015;8:12901–12907. [PMC free article] [PubMed] [Google Scholar]
- 9.Hori D, Dunkerly-Eyring B, Nomura Y, Biswas D, Steppan J, Henao-Mejia J, Adachi H, Santhanam L, Berkowitz DE, Steenbergen C, Flavell RA, Das S. miR-181b regulates vascular stiffness age dependently in part by regulating TGF-beta signaling. PLoS One. 2017;12:e0174108. doi: 10.1371/journal.pone.0174108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choe N, Kwon DH, Shin S, Kim YS, Kim YK, Kim J, Ahn Y, Eom GH, Kook H. The microRNA miR-124 inhibits vascular smooth muscle cell proliferation by targeting S100 calcium-binding protein A4 (S100A4) FEBS Lett. 2017;591:1041–1052. doi: 10.1002/1873-3468.12606. [DOI] [PubMed] [Google Scholar]
- 11.Bi R, Ding F, He Y, Jiang L, Jiang Z, Mei J, Liu H. miR-503 inhibits platelet-derived growth factor-induced human aortic vascular smooth muscle cell proliferation and migration through targeting the insulin receptor. Biomed Pharmacother. 2016;84:1711–1716. doi: 10.1016/j.biopha.2016.10.081. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Xu J. MiR-140-5p regulates hypoxia-mediated human pulmonary artery smooth muscle cell proliferation, apoptosis and differentiation by targeting Dnmt1 and promoting SOD2 expression. Biochem Biophys Res Commun. 2016;473:342–348. doi: 10.1016/j.bbrc.2016.03.116. [DOI] [PubMed] [Google Scholar]
- 13.Li K, Wang Y, Zhang A, Liu B, Jia L. miR-379 inhibits cell proliferation, invasion, and migration of vascular smooth muscle cells by targeting Insulin-Like Factor-1. Yonsei Med J. 2017;58:234–240. doi: 10.3349/ymj.2017.58.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohnle P, Schutz SV, Schmidt M, Hinske C, Hubner M, Heyn J, Beiras-Fernandez A, Kreth S. MicroRNA-665 is involved in the regulation of the expression of the cardioprotective cannabinoid receptor CB2 in patients with severe heart failure. Biochem Biophys Res Commun. 2014;451:516–521. doi: 10.1016/j.bbrc.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 15.Dong C, Du Q, Wang Z, Wang Y, Wu S, Wang A. MicroRNA-665 suppressed the invasion and metastasis of osteosarcoma by directly inhibiting RAB23. Am J Transl Res. 2016;8:4975–4981. [PMC free article] [PubMed] [Google Scholar]
- 16.Sadeghi M, Ranjbar B, Ganjalikhany MR, F MK, Schmitz U, Wolkenhauer O, Gupta SK. MicroRNA and transcription factor gene regulatory network analysis reveals key regulatory elements associated with prostate cancer progression. PLoS One. 2016;11:e0168760. doi: 10.1371/journal.pone.0168760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li M, Zhang S, Qiu Y, He Y, Chen B, Mao R, Cui Y, Zeng Z, Chen M. Upregulation of miR-665 promotes apoptosis and colitis in inflammatory bowel disease by repressing the endoplasmic reticulum stress components XBP1 and ORMDL3. Cell Death Dis. 2017;8:e2699. doi: 10.1038/cddis.2017.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DiRenzo DM, Chaudhary MA, Shi X, Franco SR, Zent J, Wang K, Guo LW, Kent KC. A crosstalk between TGF-beta/Smad3 and Wnt/betacatenin pathways promotes vascular smooth muscle cell proliferation. Cell Signal. 2016;28:498–505. doi: 10.1016/j.cellsig.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–1312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agrotis A, Kanellakis P, Kostolias G, Di Vitto G, Wei C, Hannan R, Jennings G, Bobik A. Proliferation of neointimal smooth muscle cells after arterial injury. Dependence on interactions between fibroblast growth factor receptor-2 and fibroblast growth factor-9. J Biol Chem. 2004;279:42221–42229. doi: 10.1074/jbc.M408121200. [DOI] [PubMed] [Google Scholar]
- 21.Dong N, Wang W, Tian J, Xie Z, Lv B, Dai J, Jiang R, Huang D, Fang S, Tian J, Li H, Yu B. MicroRNA-182 prevents vascular smooth muscle cell dedifferentiation via FGF9/PDGFRbeta signaling. Int J Mol Med. 2017;39:791–798. doi: 10.3892/ijmm.2017.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Firulli AB, Miano JM, Bi W, Johnson AD, Casscells W, Olson EN, Schwarz JJ. Myocyte enhancer binding factor-2 expression and activity in vascular smooth muscle cells. Association with the activated phenotype. Circ Res. 1996;78:196–204. doi: 10.1161/01.res.78.2.196. [DOI] [PubMed] [Google Scholar]
- 23.Desjardins CA, Naya FJ. Antagonistic regulation of cell-cycle and differentiation gene programs in neonatal cardiomyocytes by homologous MEF2 transcription factors. J Biol Chem. 2017;292:10613–10629. doi: 10.1074/jbc.M117.776153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Estrella NL, Clark AL, Desjardins CA, Nocco SE, Naya FJ. MEF2D deficiency in neonatal cardiomyocytes triggers cell cycle re-entry and programmed cell death in vitro. J Biol Chem. 2015;290:24367–24380. doi: 10.1074/jbc.M115.666461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim Y, Phan D, van Rooij E, Wang DZ, McAnally J, Qi X, Richardson JA, Hill JA, Bassel-Duby R, Olson EN. The MEF2D transcription factor mediates stress-dependent cardiac remodeling in mice. J Clin Invest. 2008;118:124–132. doi: 10.1172/JCI33255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li GJ, Zhao GQ, Yang JP, Zhou YC, Yang KY, Lei YJ, Huang YC. Effect of miR-1244 on cisplatin-treated non-small cell lung cancer via MEF2D expression. Oncol Rep. 2017;37:3475–3483. doi: 10.3892/or.2017.5624. [DOI] [PubMed] [Google Scholar]
- 27.Xu K, Zhao YC. MEF2D/Wnt/beta-catenin pathway regulates the proliferation of gastric cancer cells and is regulated by microRNA-19. Tumour Biol. 2016;37:9059–9069. doi: 10.1007/s13277-015-4766-3. [DOI] [PubMed] [Google Scholar]
- 28.Kong J, Liu X, Li X, Wu J, Wu N, Chen J, Fang F. Pokemon promotes the invasiveness of hepatocellular carcinoma by enhancing MEF2D transcription. Hepatol Int. 2016;10:493–500. doi: 10.1007/s12072-015-9697-y. [DOI] [PubMed] [Google Scholar]
- 29.Xiang J, Sun H, Su L, Liu L, Shan J, Shen J, Yang Z, Chen J, Zhong X, Avila MA, Yan X, Liu C, Qian C. Myocyte enhancer factor 2D promotes colorectal cancer angiogenesis downstream of hypoxia-inducible factor 1alpha. Cancer Lett. 2017;400:117–126. doi: 10.1016/j.canlet.2017.04.037. [DOI] [PubMed] [Google Scholar]
- 30.Zhao M, Liu Y, Bao M, Kato Y, Han J, Eaton JW. Vascular smooth muscle cell proliferation requires both p38 and BMK1 MAP kinases. Arch Biochem Biophys. 2002;400:199–207. doi: 10.1016/S0003-9861(02)00028-0. [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Xiao Y, Mou Y, Zhao Y, Blankesteijn WM, Hall JL. A role for the beta-catenin/T-cell factor signaling cascade in vascular remodeling. Circ Res. 2002;90:340–347. doi: 10.1161/hh0302.104466. [DOI] [PubMed] [Google Scholar]
- 32.Quasnichka H, Slater SC, Beeching CA, Boehm M, Sala-Newby GB, George SJ. Regulation of smooth muscle cell proliferation by betacatenin/T-cell factor signaling involves modulation of cyclin D1 and p21 expression. Circ Res. 2006;99:1329–1337. doi: 10.1161/01.RES.0000253533.65446.33. [DOI] [PubMed] [Google Scholar]
- 33.Masckauchan TN, Shawber CJ, Funahashi Y, Li CM, Kitajewski J. Wnt/beta-catenin signaling induces proliferation, survival and interleukin-8 in human endothelial cells. Angiogenesis. 2005;8:43–51. doi: 10.1007/s10456-005-5612-9. [DOI] [PubMed] [Google Scholar]
- 34.Zheng Z, Kang HY, Lee S, Kang SW, Goo B, Cho SB. Up-regulation of fibroblast growth factor (FGF) 9 expression and FGF-WNT/betacatenin signaling in laser-induced wound healing. Wound Repair Regen. 2014;22:660–665. doi: 10.1111/wrr.12212. [DOI] [PubMed] [Google Scholar]
- 35.Zhang CF, Kang K, Li XM, Xie BD. MicroRNA-136 promotes vascular muscle cell proliferation through the ERK1/2 pathway by targeting PPP2R2A in atherosclerosis. Curr Vasc Pharmacol. 2015;13:405–412. doi: 10.2174/1570161112666141118094612. [DOI] [PubMed] [Google Scholar]
- 36.Li TJ, Chen YL, Gua CJ, Xue SJ, Ma SM, Li XD. MicroRNA 181b promotes vascular smooth muscle cells proliferation through activation of PI3K and MAPK pathways. Int J Clin Exp Pathol. 2015;8:10375–10384. [PMC free article] [PubMed] [Google Scholar]
- 37.Guo X, Li D, Chen M, Chen L, Zhang B, Wu T, Guo R. miRNA-145 inhibits VSMC proliferation by targeting CD40. Sci Rep. 2016;6:35302. doi: 10.1038/srep35302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Liu Z, Zhou M, Liu C. MicroRNA-129-5p inhibits vascular smooth muscle cell proliferation by targeting Wnt5a. Exp Ther Med. 2016;12:2651–2656. doi: 10.3892/etm.2016.3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naruo K, Seko C, Kuroshima K, Matsutani E, Sasada R, Kondo T, Kurokawa T. Novel secretory heparin-binding factors from human glioma cells (glia-activating factors) involved in glial cell growth. Purification and biological properties. J Biol Chem. 1993;268:2857–2864. [PubMed] [Google Scholar]
- 40.Frontini MJ, Nong Z, Gros R, Drangova M, O’Neil C, Rahman MN, Akawi O, Yin H, Ellis CG, Pickering JG. Fibroblast growth factor 9 delivery during angiogenesis produces durable, vasoresponsive microvessels wrapped by smooth muscle cells. Nat Biotechnol. 2011;29:421–427. doi: 10.1038/nbt.1845. [DOI] [PubMed] [Google Scholar]
- 41.Singla D, Wang J. Fibroblast growth factor-9 activates c-Kit progenitor cells and enhances angiogenesis in the infarcted diabetic heart. Oxid Med Cell Longev. 2016;2016:5810908. doi: 10.1155/2016/5810908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Black BL, Olson EN. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol. 1998;14:167–196. doi: 10.1146/annurev.cellbio.14.1.167. [DOI] [PubMed] [Google Scholar]
- 43.Gu Z, Churchman M, Roberts K, Li Y, Liu Y, Harvey RC, McCastlain K, Reshmi SC, Payne-Turner D, Iacobucci I, Shao Y, Chen IM, Valentine M, Pei D, Mungall KL, Mungall AJ, Ma Y, Moore R, Marra M, Stonerock E, Gastier-Foster JM, Devidas M, Dai Y, Wood B, Borowitz M, Larsen EE, Maloney K, Mattano LA Jr, Angiolillo A, Salzer WL, Burke MJ, Gianni F, Spinelli O, Radich JP, Minden MD, Moorman AV, Patel B, Fielding AK, Rowe JM, Luger SM, Bhatia R, Aldoss I, Forman SJ, Kohlschmidt J, Mrozek K, Marcucci G, Bloomfield CD, Stock W, Kornblau S, Kantarjian HM, Konopleva M, Paietta E, Willman CL, Loh ML, Hunger SP, Mullighan CG. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun. 2016;7:13331. doi: 10.1038/ncomms13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su L, Luo Y, Yang Z, Yang J, Yao C, Cheng F, Shan J, Chen J, Li F, Liu L, Liu C, Xu Y, Jiang L, Guo D, Prieto J, Avila MA, Shen J, Qian C. MEF2D transduces microenvironment stimuli to ZEB1 to promote epithelial-mesenchymal transition and metastasis in colorectal cancer. Cancer Res. 2016;76:5054–5067. doi: 10.1158/0008-5472.CAN-16-0246. [DOI] [PubMed] [Google Scholar]
- 45.Yu H, Sun H, Bai Y, Han J, Liu G, Liu Y, Zhang N. MEF2D overexpression contributes to the progression of osteosarcoma. Gene. 2015;563:130–135. doi: 10.1016/j.gene.2015.03.046. [DOI] [PubMed] [Google Scholar]
- 46.Zhao X, Liu M, Li D. Oleanolic acid suppresses the proliferation of lung carcinoma cells by miR-122/Cyclin G1/MEF2D axis. Mol Cell Biochem. 2015;400:1–7. doi: 10.1007/s11010-014-2228-7. [DOI] [PubMed] [Google Scholar]
- 47.Yin Y, White AC, Huh SH, Hilton MJ, Kanazawa H, Long F, Ornitz DM. An FGF-WNT gene regulatory network controls lung mesenchyme development. Dev Biol. 2008;319:426–436. doi: 10.1016/j.ydbio.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Katz Imberman S, Kolpakova A, Keren A, Bengal E. Myocyte enhancer factor 2D regulates ectoderm specification and adhesion properties of animal cap cells in the early Xenopus embryo. FEBS J. 2015;282:2930–2947. doi: 10.1111/febs.13331. [DOI] [PubMed] [Google Scholar]
- 49.Cai T, Sun D, Duan Y, Wen P, Dai C, Yang J, He W. WNT/beta-catenin signaling promotes VSMCs to osteogenic transdifferentiation and calcification through directly modulating Runx2 gene expression. Exp Cell Res. 2016;345:206–217. doi: 10.1016/j.yexcr.2016.06.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.