

Abstract
Albuminuria is an independent risk factor for renal interstitial fibrosis (RIF). Glomerular-filtered albumin in endocytic and non-endocytic pathways may injure proximal tubular epithelial cells (PTECs) via megalin and TGFβRII, respectively. Since megalin and TGFβRII are both modified by post-translational core fucosylation, which plays a critical role in RIF. Thus, we sought to identify whether core fucosylation is a potential target for reducing albumin-induced injury to PTECs. We constructed a human PTEC-derived cell line (HK-2 cells) and established an in vitro model of bovine serum albumin (BSA) injury. RNAi was used to inhibit the expression of megalin, TGFβRII, and Fut8. Western blotting, immunostaining, ELISA, lectin blotting, and fluorescence-activated cell sorting were used to identify BSA-induced endocytic and non-endocytic damage in HK-2 cells. Fut8 is a core fucosylation-related gene, which is significantly increased in HK-2 cells following an incubation with BSA. Fut8 siRNA significantly reduced the core fucosylation of megalin and TGFβRII and also inhibited the activation of the TGFβ/TGFβRII/Smad2/3 signaling pathway. Furthermore, Fut8 siRNA could reduce monocyte chemotactic protein-1, reactive oxygen species, and apoptosis, as well as significantly decrease the fibronectin and collagen I levels in BSA-overloaded HK-2 cells. Core fucosylation inhibition was more effective than inhibiting either megalin or TGFβRII for the prevention of albumin-induced injury to PTECs. Our findings indicate that post-translational core fucosylation is essential for the albumin-induced injury to PTECs. Thus, the inhibition of core fucosylation could effectively alleviate albumin-induced endocytic and non-endocytic injury to PTECs. Our study provides a potential therapeutic target for albuminuria-induced injury.
Keywords: Albuminuria, core fucosylation, megalin, TGFβRII, proximal tubular epithelial cell, FUT8
Introduction
Albuminuria is strongly associated with progressive kidney tubulo-interstitial damage and chronic kidney disease [1,2]. An effective intervention strategy to inhibit albumuria-induced renal tubular interstitial damage is lacking [3].
It has been reported that albuminuria may cause tubular injury by overstressing the endocytic system [4,5]. The role of megalin, a major albumin endocytic receptor, includes the following functions: 1) mediation of excessive endocytosis of albumin which induces apoptosis; 2) increasing levels of inflammatory cytokines (e.g., monocyte chemotactic protein-1 [MCP-1]); 3) regulation upon activation of normal T cell expressionand secreting factors (RANTES) and reactive oxygen species (ROS) in the kidneys, which contribute to tubulo-interstitial damage [6,7]. In addition to the endocytic pathway, glomerular-filtered albumin could damage proximal tubular epithelial cells (PTECs) via a non-endocytic pathway. It has been reported that PTECs were induced to secrete transforming growth factor-1 (TGF-β1) following exposure to bovine serum albumin (BSA), even when albumin endocytosis was inhibited [8]. It has been well-established that TGF-β1 mediates the activation of the TGFβ1/TGFβRII/Smad2/3 signaling pathways, which contribute to renal interstitial fibrosis (RIF) [9-11]. Thus, albumin could cause injury to PTECs through both endocytic, as well as non-endocytic pathways. Therefore, we investigated the manner in which this plasma protein could regulate both the endocytic and the non-endocytic pathways to establish a more effective strategy for the prevention and treatment of albuminuria injury in patients with kidney disease.
Glycosylation is a critical type of post-translational modification which exhibits profound effects on physiological and pathological processes (e.g., cell proliferation, migration, and apoptosis) [12,13]. Core fucosylation is uniquely catalyzed by α-1,6-fucosyltransferase (FUT8) in mammals, which is formed of alpha1,6-fucose linked to the core N-acetylglucosamine of N-linked glycans. In addition, core fucosylation is a unique protein glycosylation pattern associated with several biological and pathological functions (e.g., cellular signaling transduction, cellular migration, and inflammatory responses) [14,15]. Moreover, it has been reported that core fucosylated proteins are associated with many types of cancers, including pancreatic, lung, ovarian, and prostate cancers [16-20]. Our group previously found that the expression of core fucosylation was significantly upregulated in rats with unilateral ureter obstruction (UUO) [21,22]. Moreover, inhibiting core fucosylation could suppress RIF in UUO rats, suggesting that core fucosylation plays an important role in RIF [21,22]. Although it has been established that albuminuria contributes to RIF in patients with kidney disease and TGFβRII and megalin are modified by core fucosylation [23,24], there remains uncertainty as to whether core fucosylation plays a key role in albumin-induced damage to PTECs.
To explore the role of core fucosylation in albumin-induced injury to PTECs, we incubated HK-2 cells with BSA and investigated the role of core fucosylation by silencing its specific transfer enzyme FUT8 at different time points. To our knowledge, there has been no previous report describing the role of core fucosylation in albumin overload injury to PTECs.
Materials and methods
Cell culture
Immortalized human renal proximal tubular epithelial cells (HK-2) were purchased from the American Type Culture Collection (Manassas, VA, USA) and cultured with DMEM/F12 medium supplemented with 10% FBS at a plating density of 2 × 105 cells/well in six-well plates (Costar, Corning Incorporated; Corning, NY, USA) at 37°C and 5% CO2 in a humidified atmosphere. For all experiments, the cells were cultured without FBS for 12 h, after which the cells were randomly divided into the following six groups: 1) Con, cells cultured in DMEM/F12 medium; 2) Mock, cells transiently transfected with 30 nM scrambled siRNA for 24 h; 3) BSA, cells treated with 10 mg/mL BSA for 4 h or 24 h; 4) BSAF, cells transfected with 30 nM Fut8 siRNA for 24 h, and then incubated with 10 mg/mL BSA for 4 h or 24 h; 5) BSAM, cells transfected with 30 nM megalin siRNA for 24 h, and then incubated with 10 mg/mL BSA for 4 h or 24 h; and 6) BSAT, cells transfected with 30 nM TGFβRII siRNA for 24 h, and then incubated with 10 mg/mL BSA for 24 h.
Transient transfection of Fut8 siRNA, megalin siRNA, and TGFβRII siRNA
RNAi was performed using three predesigned siRNAs, anti-Fut8 siRNA, anti-TGFβRII siRNA, and anti-megalin siRNA (Takara, Co. Dalian, China) denoted as Fut8 siRNA (5’-GGUGCAUGUUGAAGAACAUTT-3’), TGFβRII siRNA (5’-GGUCGCUUUGCUGAGGUCUTT-3’), and megalin siRNA (5’-GCUAUUGUAUUAGAUCCUU-3’). For the transfection, the cells were seeded into six-well culture plates and incubated for 24 h to allow cell division to occur in an antibiotic-free medium. Three chemically synthesized siRNAs targeting Fut8, megalin, and TGFβRII were combined with the transfection reagent and added to the cell culture wells in accordance with the manufacturer’s instructions.
Western blotting
The treated cells were harvested in RIPA buffer. Lysates were clarified by centrifugation at 15,000 × g for 10 min at 4°C, and the protein concentrations were determined using a BCA protein assay kit. Protein samples were denatured at 100°C for 3 min, separated on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and electroblotted onto polyvinylidene difluoride membranes (Bio-Rad; Hercules, CA, USA). The blots were probed with the appropriate primary antibodies at 4°C overnight to detect the expression of FUT8 (1:400), megalin (1:400), MCP-1 (1:200), TGF-β1 (1:200), and TGFβRII (1:500). The blots were washed, incubated with an HRP-labeled secondary antibody for 1 h at room temperature, and imaged via ECL detection (Amersham Biosciences). Band intensity was quantified using Labworks Image Acquisition and Analysis software (Basingstoke, UK).
Fluorescence-activated cell sorting and confocal fluorescence microscopy
For all experiments, 1 × 105 HK-2 cells were assessed for the uptake of fluorescent-labeled BSA using a FACSCaliburTM instrument (Becton Dickinson; Hubbardston, MA, USA). Briefly, the cells were incubated with 100 µg/mL TRITC-BSA for 60 min at 37°C, whereas the control cells were incubated without TRITC-BSA. TRITC-BSA endocytosis was arrested by washing the cells five times with ice-cold phosphate buffered saline (PBS). The results are presented as the mean fluorescence intensity (MFI). For confocal fluorescence microscopy, HK-2 cells were incubated with 100 µg/mL TRITC-BSA at 37°C for 60 min, and then washed five times with ice-cold PBS and fixed with 4% formaldehyde. A confocal fluorescent microscope was used to observe the endocytosis of TRITC-BSA.
Lectin immunoprecipitation assays
The cells were washed twice with ice-cold PBS and lysed in cold RIPA lysis buffer. The lysates were clarified by centrifugation (12,000 × g for 20 min at 4°C), and 500 µg of the total protein was incubated with 2 µg of anti-megalin or anti-TGFβRII antibody at 4°C for 2 h. A negative control without the antibody was also included. Sepharose-protein G beads (20 μL) were added and incubated at 4°C overnight. The beads were collected by centrifugation (12,000 × g for 30 s) and washed three times with ice-cold RIPA buffer. Equal amounts of protein-bound beads were then subjected to 12% SDS-PAGE for lectin blotting.
Lectin blotting
Whole cell lysates or immunoprecipitated megalin were subjected to 12% SDS-PAGE and transferred to PVDF membranes. The membranes were then blocked with 5% BSA in Tris-buffered saline containing 0.05% Tween-20 (TBST) overnight at 4°C, and then incubated with LCA (1:1000; specifically recognizes Fuc-1,6 GlcNAc) in TBST for 1 h at room temperature. After washing three times with TBST, the membranes were incubated in TBST containing LCA-Biotin (1:200) for 1 h at room temperature. After washing four times with TBST, lectin-reactive proteins were detected using an ECL kit.
Apoptotic changes detected by Annexin V-FITC
HK-2 cell monolayers were detached by a brief incubation with Trypsin-EDTA. The cells were then resuspended in binding buffer (BD Pharmingen; San Diego, CA, USA) and incubated with Annexin V-FITC for 15 min at room temperature in the dark, followed by staining with propidium iodide. The cells were then analyzed within 1 h using a FACSCalibur flow cytometer. A contour diagram of FITC-Annexin V/PI double staining was derived using flow cytometry. The total apoptotic proportion includes the percentage of cells that were Annexin V+/PI- and Annexin V+/PI+; CellQuest software (Becton Dickinson) was used for the data analysis.
MCP-1 and TGF-β1 enzyme-linked immunosorbent assay (ELISA)
MCP-1 and TGF-β1 ELISA kits (Santa Cruz, CA, USA) was used to detect TGF-β1 and MCP-1 levels in HK-2 cells. The absorbance measurements for all samples and standards were performed in triplicate, and were read at a wavelength of 450 nm using an ELISA plate reader (Bio-Rad Model 680, Hercules, CA, USA). The concentration of MCP-1 and TGF-β1 for each sample was determined by extrapolation from a standard curve derived from the standards provided by the manufacturer. The level of secreted MCP-1 and TGF-β1 was corrected based on the total cellular protein. All samples and standards were measured in duplicate.
ROS production detected by DCFDA fluorescence dye
ROS production was measured using 2,7-dichlorofluorescein diacetate (DCFDA) fluorescence dye. The cells were incubated with 1 µM DCFDA at 37°C for 30 min. The level of fluorescence intensity was measured using a FACSCaliburTM instrument with an excitation wavelength of 485 nm and an emission wavelength of 535 nm.
Analysis of p-Smad2/3 immunofluorescence
HK-2 cells were incubated at 4°C overnight with a goat anti-p-Smad2/3 antibody (1:150) followed by an incubation with an FITC-rabbit anti-goat antibody (1:100) for 1 h at room temperature. After washing with 1 × PBS, the cells were observed using a fluorescent microscope.
Statistical analysis
All values are expressed as the mean ± standard deviation. The differences between the experimental samples were assessed using an ANOVA with a post-hoc analysis using a Tukey’s t-test. P values < 0.05 were considered to be statistically significant.
The SPSS 20.0 software (IBM, Armonk, NY, USA) was applied for statistical analysis.
Results
BSA activates injury pathways and increases FUT8 expression
To determine the damage induced by BSA exposure, we performed Western blotting to measure the expression of megalin, MCP-1, TGFβRII, p-Smad2/3, and FUT8 following incubation with BSA at different time points. The expression of megalin remained unchanged for up to 6 h. Following a 24 h incubation, the expression of megalin decreased to 55% ± 2% of the control (Figure 1A). The expression of MCP-1 was significantly increased following a 4 h BSA incubation (P < 0.01), although this level was lower than an incubation for 6 h (P < 0.05) (Figure 1B). The expression of TGFβRII, p-Smad2/3, and FUT8 gradually increased in a time-dependent manner following a BSA incubation for 24 h (P < 0.01) (Figure 1C-E).
Figure 1.
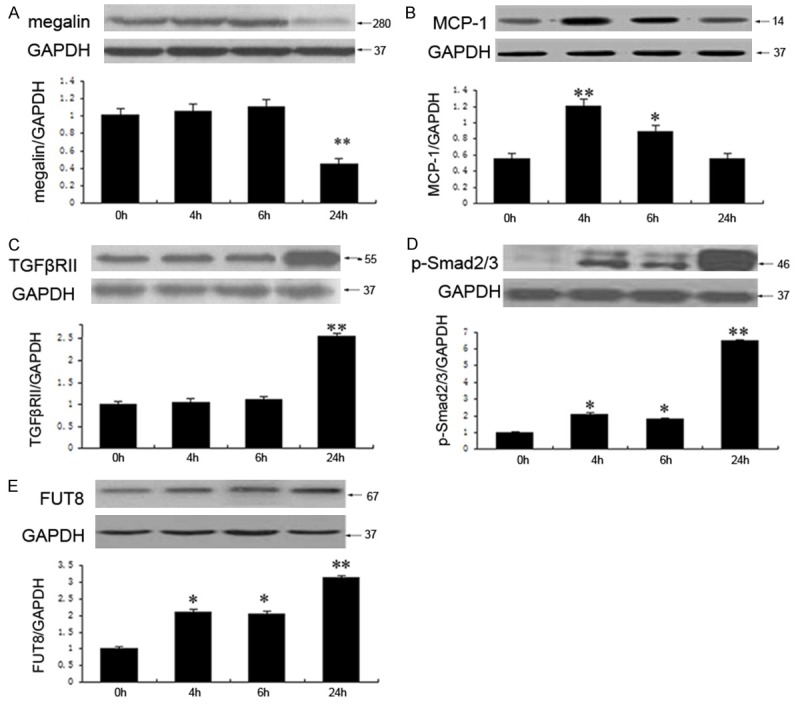
BSA activates injury-induced pathways and increases the level of FUT8 expression. The expression of megalin remained unaltered for up to 6 h, whereas it declined to 55% ± 2% after an incubation with BSA for 24 h (A). MCP-1 expression reached a peak value after BSA incubation for 4 h and decreased after a 6 h incubation (B). TGFβRII expression remained unaltered following a BSA incubation for 6 h, whereas it increased 1.5-fold over the original levels after a 24 h incubation (C). P-Smad2/3 increased in a time-dependent manner from 0 h to 24 h (D). The expression of FUT8 increased in a time-dependent manner from 0 h to 24 h (E). The values are expressed as the mean ± SD. *P < 0.05, **P < 0.01, each group (except for the Control group) vs. normal group. All experiments were conducted in triplicate.
Inhibition of core fucosylation suppression of BSA endocytosis
To further study the effect of core fucosylation on BSA endocytosis, we performed Western blotting, immunoprecipitation, fluorescence-activated cell sorting (FACS), and confocal microscopy to observe the effect of siRNAs on BSA endocytosis. As shown in Figure 2A, 2B, incubation with BSA significantly upregulated BSA endocytosis in HK-2 cells in the BSA-treatment group (P < 0.05). Treatment with Fut8 siRNA significantly inhibited core fucosylation and suppressed BSA endocytosis in HK-2 cells in the BSAF group (BSA together with Fut8 siRNA) (P < 0.05). In addition, we observed that Fut8 siRNA, but not TGFβ siRNA, exhibited the same inhibitory effect on BSA endocytosis as megalin siRNA. As shown in Figure 2C, megalin was modified by core fucose, and its core fucosylation was upregulated by BSA in the BSA-treatment group (P < 0.05).
Figure 2.

Fut8 siRNA suppressed BSA endocytosis. Flow cytometry showed that Fut8 siRNA markedly inhibited intensity of fluorescence from BSA-FITC in the BSAF group compared with the BSA group (A). The confocal analysis revealed that the intensity of red fluorescence of TRITC-BSA was significantly increased in the BSA group, while it was downregulated in response to Fut8 siRNA in the BSAF group (B). The immunoprecipitation and lectin blot analysis results showed that Fut8 siRNA significantly reduced the level of megalin core fucosylation following a 4 h BSA incubation (C). BSAF: BSA with Fut8 siRNA; BSAM: BSA with megalin siRNA; BSAT BSA with TGFβRII siRNA. Scale marks: 25 μm. Green: ZO-1 antibody. Red: TRITC-BSA. The values are expressed as the means ± SD. Original magnification, 400 ×; *P < 0.05, each group (except for the Control group) vs. normal group. ##P < 0.01, each group (except for the Control group) vs. BSA group.
Inhibiting core fucosylation suppresses the activation of the TGFβ/TGFβRII/Smad2/3 signaling pathway
We next investigated whether inhibiting core fucosylation could suppress the activation of the TGFβ/TGFβRII/Smad2/3 signaling pathway after incubating with BSA for 24 h. Western blotting and immunoprecipitation was performed to determine the level of core fucosylation in HK-2 cells. Immunofluorescence was performed to detect the level of p-Smad2/3 and its translocation in HK-2 cells. As shown in Figure 3, BSA significantly upregulated the level of TGFβRII core fucosylation. Moreover, BSA also increases the level of p-Smad2/3, as well as p-Smad2/3 nuclear translocation in the BSA-treatment group (P < 0.05). Fut8 siRNA markedly decreased core fucosylation and markedly reduced the expression of p-Smad2/3, as well as its nuclear translocation in the BSAF group (Figure 3B and 3C; P < 0.05). Fut8 siRNA exhibited the same effect on p-Smad2/3 and its nuclear translocation as TGFβRII siRNA (Figure 3C); however, megalin siRNA did not exhibit such effects (Figure 3B).
Figure 3.

Fut8 siRNA suppressed the activation of the TGFβ/TGFβRII/Smad2/3 signaling pathway. Immunoprecipitation and lectin blotting proved that Fut8 siRNA could markedly decrease the core fucosylation of TGFβRII without altering its protein expression. In contrast, megalin siRNA had no effect on either TGFβRII expression or core fucosylation following a 24 h incubation (A). Western blot assays showed that BSA-upregulated p-Smad2/3 was significantly downregulated in the BSAF group compared to the BSA group (B). Immunofluorescence staining indicated that p-Smad2/3 was weakly expressed in the cytoplasm of normal HK-2 cells, while it was increased and translocated into the nuclei following BSA stimulation. Fut8 siRNA inhibited the expression and nuclear translocation of p-Smad2/3 in BSAF group (C). BSAF: BSA with Fut8 siRNA; BSAM: BSA with megalin siRNA; BSAT BSA with TGFβRII siRNA. Scale bars: 100 μm. Green: FITC. Blue: DAPI. Protein expression in each sample was normalized to GAPDH expression. The values are expressed as the means ± SD. *P < 0.05, **P < 0.01, each group (except for the Control group) vs. normal group. #P < 0.05, ##P < 0.01, each group (except for the Control group) vs. BSA group.
Inhibition of core fucosylation inhibits albumin-induced inflammation and oxidative stress
To investigate the effect of inhibiting core fucosylation on inflammation and oxidative stress, we performed Western blotting and an ELISA to determine the expression of MCP-1, TGFβ1, NF-κB, and the levels of ROS, respectively. The results revealed that BSA significantly increased the expression of MCP-1 and NF-κB following a 4 h or 24 h incubation (Figure 4A-C, 4G-I; P < 0.01). In addition, the level of ROS also increased following an incubation for 4 h or 24 h (Figure 4D and 4J; P < 0.01). Fut8 siRNA significantly inhibited the upregulation of MCP-1, NF-κB, and ROS in the BSAF group (Figure 4A-D, 4G-J; P < 0.01) . Megalin siRNA could also significantly reduce the expression of MCP-1, NF-κB, and the level of ROS in the BSAM (BSA together with megalin siRNA) group (Figure 4A-D, 4G-J; P < 0.05), whereas TGFβRII siRNA had little effect on them in the BSAT (BSA together with TGFβRII siRNA) group. Of note, megalin and Fut8 siRNA could markedly upregulate the expression of TGFβ1 in the BSAF and BSAM groups (Figure 4E, 4F, 4K, 4L) (P < 0.01).
Figure 4.

Fut8 siRNA inhibits albumin-induced inflammation and oxidative stress. MCP-1, NF-κB, and TGF-β1 were determined by Western blot and ELISA. ROS generation (%) was measured using a ROS sensitive fluorometric probe 2,7-dichlorofluorescein diacetate (DCFDA) by flow cytometric analysis. MCP-1 (A, B, G, and H), ROS (D and J), and NF-κB (C and I) was significantly increased in HK-2 cells from the BSA group in the 4 h and 24 h incubation, all of which were reversed by Fut8 siRNA. BSA increased the expression of TGF-β1, whereas megalin siRNA and Fut8 siRNA further increased the expression of TGF-β1 after the 4 h (E and F) and 24 h (K and L) incubation periods. BSAF: BSA with Fut8 siRNA; BSAM: BSA with megalin siRNA; BSAT BSA with TGFβRII siRNA. Protein expression in each sample was normalized to GAPDH expression. The values are expressed as the means ± SD. *P < 0.05, **P < 0.01, for each group (except for the Control group) vs. normal group. #P < 0.05, ##P < 0.01, for each group (except for the Control group) vs. BSA group.
Inhibition of core fucosylation suppresses fibronectin and collagen I
To confirm the effect of core fucosylation on the accumulation of the HK-2 extracellular matrix, we performed Western blotting to determine the expression of fibronectin (FN) and collagen I (Col I) I after BSA incubation for 4 h or 24 h. The results revealed that the expression of FN (Figure 5A) and Col I (Figure 5B) showed no significant intergroup differences following a 4 h incubation. In contrast, after 24 h, stimulation with BSA increased the expression of FN (Figure 5C) and Col I (Figure 5D). Treatment with Fut8 siRNA significantly downregulated the expression of FN and Col I in the BSAF group (Figure 5; P < 0.01), whereas megalin siRNA exhibited virtually no effect on the expression of these molecules.
Figure 5.

Fut8 siRNA inhibited the upregulation of fibronectin and collagen I. The Western blot showed that the expression of fibronectin (FN) (A), and collagen I (Col I) (B) showed no significant intergroup differences following an incubation for 4 h. BSA increased the expression of FN (C) and Col I (D) following BSA incubation for 24 h. Fut8 siRNA significantly downregulated the expression of FN and Col I in the BSAF group (C and D). BSAF: BSA with Fut8 siRNA; BSAM: BSA with megalin siRNA; BSAT BSA with TGFβRII siRNA. Scale bars: 100 μm. The values are expressed as the means ± SD. *P < 0.05, **P < 0.01, for each group (except for the Control group) vs. normal group. #P < 0.05, ##P < 0.01, for each group (except for the Control group) vs. BSA group.
Inhibition of core fucosylation suppresses albumin-induced cellular apoptosis
To investigate the effect of an inhibition of core fucosylation on cellular apoptosis, we performed FACS analysis to determine cellular apoptosis following an incubation with BSA for 4 h or 24 h. We observed that cellular apoptosis was significantly upregulated following BSA incubation for 4 h or 24 h in the BSAF group (Figure 6A and 6B; P < 0.01). In addition, Fut8 siRNA could significantly downregulate the level of cellular apoptosis in the BSAF group (Figure 6A and 6B; P < 0.01). Moreover, it was observed that Fut8 siRNA is more efficient at reducing cellular apoptosis compared to TGFβRII and megalin siRNA.
Figure 6.

Fut8 siRNA decreased cellular apoptosis. A contour diagram of FITC-Annexin V/PI dual staining by flow cytometry. The three quadrants represent the different cell conditions. The upper right quadrant, nonviable, late apoptotic, and necrotic cells (FITC+/PI+); lower left quadrant, viable cells (FITC-/PI-); and lower right quadrant, early apoptotic cells (FITC+/PI-). FACS analysis also revealed that the BSA incubation increased apoptosis in the BSA group following the incubation of HK-2 cells for 4 h (A) and 24 h (B). Fut8 siRNA significantly downregulated cellular apoptosis in the BSAF group after BSA incubation for 4 h or 24 h. BSAF: BSA with Fut8 siRNA; BSAM: BSA with megalin siRNA; BSAT BSA with TGFβRII siRNA. Scale bars: 100 μm. Protein expression in each sample was normalized to GAPDH expression. The values are expressed as the mean ± SD. *P < 0.05, **P < 0.01, for each group (except for the Control group) vs. normal group. #P < 0.05, ##P < 0.01, for each group (except for the Control group) vs. BSA group.
Discussion
Recent research on glycosylation has garnered immense interest in drug development, post-translational glycosylation is becoming an attractive biomarker and treatment target for some diseases [25,26]. Core fucosylation is a pivotal post-translational modification that has a profound effect on the regulation of various physiological processes [18]; however, there are only a limited number of studies on core glycosylation with respect to various kidney diseases. Moreover, there is a lack of research on glycosylation in albuminuria. Our study reveals the critical role of core fucosylation in PTEC albumin injury and confirms that it is a potential therapeutic target in albuminuria-induced injury in vitro.
First, we found that BSA endocytosis increases in HK-2 cells after BSA incubation. This finding indicates that BSA activates the albumin-induced endocytic injury pathway. Moreover, an incubation with BSA increased the expression of TGFβRII and p-Smad2/3, and induced the nuclear translocation of p-Smad2/3. This finding suggests that BSA activates the albumin-induced non-endocytic injury pathway. Of note, FUT8 expression and the core fucosylation of TGFβRII and megalin in PTECs was upregulated. Thus, this indicates that core fucosylation may play an important role in albumin-induced injury to PTECs.
We next investigated the role of core fucosylation in albumin-induced endocytic injury, and found that Fut8 siRNA could significantly inhibit the core fucosylation of megalin and BSA endocytosis. In addition, Fut8 siRNA has the same effect on HK-2 cells with megalin siRNA. Since albumin endocytosis is considered to be attributed to megalin [13-15], we hypothesize that core fucosylation may affect albumin-induced endocytic injury via regulating the function of megalin. Moreover, we also studied the effect of core fucosylation on the non-endocytic injury pathway. We observed that suppressing core fucosylation could inhibit the TGFβ/TGFβRII/Smad2/3 signaling pathway activated by BSA incubation. This suggests that core fucosylation is essential for activation of the TGFβ/TGFβRII/Smad2/3 signaling pathway following albumin-induced injury. In accordance with the findings described above, we hypothesize that core fucosylation may affect albumin-induced non-endocytic injury via regulating the function of TGFβRII. It is important to note that core fucosylation is ubiquitous. In addition to megalin and TGFβRII, core fucosylation may affect other proteins involved in the process of albumin-induced injury. In addition, our experimental conditions were not fit for evaluating the effect of core fucosylation on TGFβRII, megalin, and corresponding ligand binding occurs via both direct and indirect mechanisms. This study provides a basis for further explorations to elucidate the mechanisms of this process.
In accordance with previous research [8], our study demonstrated that TGF-β1 levels were upregulated even after inhibiting BSA endocytosis. Since it is known that TGF-β1 can activate the TGFβ/TGFβRII/Smad2/3 signaling pathway; inhibiting only one injury-induced pathway does not completely block albumin-induced injury. In accordance with the above results, we regard core fucosylation as a potential therapeutic target for preventing endocytic and non-endocytic injuries. This viewpoint is further confirmed by our results which showed that inhibiting core fucosylation could be more efficient than either megalin siRNA or TGFβRII siRNA at suppressing inflammation, oxidative stress, cellular apoptosis, and the upregulation of FN and Col I. Our findings suggest that targeting core glycosylation may be a more effective method for treating typical renal diseases derived from albuminuria (e.g., diabetic nephropathy).
In summary, this study provides novel insight into the mechanistic role of glycosylation in albumin-induced injury to PTECs. Moreover, these findings suggest that the regulation of post-translational core fucosylation may be a potential therapeutic target for the treatment of albumin-induced injury.
Acknowledgements
This work was supported by grants from the Natural Science Foundation of Liaoning Province (No. 2013028048 and No. 20170540267 to Dapeng Wang), the National Natural Science Foundation of China (NSFC) (No. 81530021 to Hongli Lin), the Scientific Research Project of Liaoning Provincial Education Department (No. L2015139 to Ming Fang and No. L2014349 to Nan Shen).
Disclosure of conflict of interest
None.
References
- 1.Lambers Heerspink HJ, Gansevoort RT. Albuminuria is an appropriate therapeutic target in patients with CKD: the pro view. Clin J Am Soc Nephrol. 2015;10:1079–88. doi: 10.2215/CJN.11511114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson DW, Jones GR, Mathew TH, Ludlow MJ, Chadban SJ, Usherwood T, Polkinghorne K, Colagiuri S, Jerums G, Macisaac R, Martin H Australasian Proteinuria Consensus Working Group. Chronic kidney disease and measurement of albuminuria or proteinuria: a position statement. Med J Aust. 2012;197:224–225. doi: 10.5694/mja11.11468. [DOI] [PubMed] [Google Scholar]
- 3.Smink PA, Lambers Heerspink HJ, Gansevoort RT, de Jong PE, Hillege HL, Bakker SJ, de Zeeuw D. Albuminuria, estimated GFR, traditional risk factors, and incident cardiovascular disease: the PREVEND (Prevention of Renal and Vascular Endstage Disease) study. Am J Kidney Dis. 2012;60:804–811. doi: 10.1053/j.ajkd.2012.06.017. [DOI] [PubMed] [Google Scholar]
- 4.Saito A, Sato H, Iino N, Takeda T. Molecular mechanisms of receptor-mediated endocytosis in the renal proximal tubular epithelium. J Biomed Biotechnol. 2010;2010:403272. doi: 10.1155/2010/403272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okamura K, Dummer P, Kopp J, Qiu L, Levi M, Faubel S, Blaine J. Endocytosis of albumin by podocytes elicits an inflammatory response and induces apoptotic cell death. PLoS One. 2013;8:e54817. doi: 10.1371/journal.pone.0054817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akour AA, Kennedy MJ, Gerk P. Receptor-mediated endocytosis across human placenta: emphasis on megalin. Mol Pharm. 2013;10:1269–78. doi: 10.1021/mp300609c. [DOI] [PubMed] [Google Scholar]
- 7.Imig JD, Ryan MJ. Immune and inflammatory role in renal disease. Compr Physiol. 2013;3:957–76. doi: 10.1002/cphy.c120028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diwakar R, Pearson AL, Colville-Nash P, Brunskill NJ, Dockrell ME. The role played by endocytosis in albumin-induced secretion of TGF-beta1 by proximal tubular epithelial cells. Am J Physiol Renal Physiol. 2007;292:F1464–1470. doi: 10.1152/ajprenal.00069.2006. [DOI] [PubMed] [Google Scholar]
- 9.Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1009–22. doi: 10.1152/ajpregu.00809.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M, Chen S, McGowan TA, Sharma K. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci U S A. 2000;97:8015–8020. doi: 10.1073/pnas.120055097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Russo LM, Comper WD, Osicka TM. Mechanism of albuminuria associated with cardiovascular disease and kidney disease. Kidney Int Suppl. 2004:S67–8. doi: 10.1111/j.1523-1755.2004.09218.x. [DOI] [PubMed] [Google Scholar]
- 12.Takahashi M, Hasegawa Y, Gao C, Kuroki Y, Taniguchi N. N-glycans of growth factor receptors: their role in receptor function and disease implications. Clin Sci (Lond) 2016;130:1781–92. doi: 10.1042/CS20160273. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi M, Kuroki Y, Ohtsubo K, Taniguchi N. Core fucose and bisecting GlcNAc, the direct modifiers of the N-glycan core: their functions and target proteins. Carbohydr Res. 2009;344:1387–1390. doi: 10.1016/j.carres.2009.04.031. [DOI] [PubMed] [Google Scholar]
- 14.Zhao YY, Takahashi M, Gu JG, Miyoshi E, Matsumoto A, Kitazume S, Taniguchi N. Functional roles of N-glycans in cell signaling and cell adhesion in cancer. Cancer Sci. 2008;99:1304–1310. doi: 10.1111/j.1349-7006.2008.00839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noda K, Miyoshi E, Uozumi N, Yanagidani S, Ikeda Y, Gao C, Suzuki K, Yoshihara H, Yoshikawa K, Kawano K, Hayashi N, Hori M, Taniguchi N. Gene expression of alpha1-6 fucosyltransferase in human hepatoma tissues: a possible implication for increased fucosylation of alphafetoprotein. Hepatology. 1998;28:944–952. doi: 10.1002/hep.510280408. [DOI] [PubMed] [Google Scholar]
- 16.Okuyama N, Ide Y, Nakano M, Nakagawa T, Yamanaka K, Moriwaki K, Murata K, Ohigashi H, Yokoyama S, Eguchi H, Ishikawa O, Ito T, Kato M, Kasahara A, Kawano S, Gu J, Taniguchi N, Miyoshi E. Fucosylated haptoglobin is a novel marker for pancreatic cancer: a detailed analysis of the oligosaccharide structure and a possible mechanism for fucosylation. Int J Cancer. 2006;118:2803–2808. doi: 10.1002/ijc.21728. [DOI] [PubMed] [Google Scholar]
- 17.Chen CY, Jan YH, Juan YH, Yang CJ, Huang MS, Yu CJ, Yang PC, Hsiao M, Hsu TL, Wong CH. Fucosyltransferase 8 as a functional regulator of nonsmall cell lung cancer. Proc Natl Acad Sci U S A. 2013;110:630–635. doi: 10.1073/pnas.1220425110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geng F, Shi BZ, Yuan YF, Wu XZ. The expression of core fucosylated E-cadherin in cancer cells and lung cancer patients: prognostic implications. Cell Res. 2004;14:423–33. doi: 10.1038/sj.cr.7290243. [DOI] [PubMed] [Google Scholar]
- 19.Saldova R, Royle L, Radcliffe CM, Abd Hamid UM, Evans R, Arnold JN, Banks RE, Hutson R, Harvey DJ, Antrobus R, Petrescu SM, Dwek RA, Rudd PM. Ovarian cancer is associated with changes in glycosylation in both acute-phase proteins and IgG. Glycobiology. 2007;17:1344–1356. doi: 10.1093/glycob/cwm100. [DOI] [PubMed] [Google Scholar]
- 20.Tabarés G, Radcliffe CM, Barrabés S, Ramírez M, Aleixandre RN, Hoesel W, Dwek RA, Rudd PM, Peracaula R, de Llorens R. Different glycan structures in prostate-specific antigen from prostate cancer sera in relation to seminal plasma PSA. Glycobiology. 2006;16:132–145. doi: 10.1093/glycob/cwj042. [DOI] [PubMed] [Google Scholar]
- 21.Lin H, Wang D, Wu T, Dong C, Shen N, Sun Y, Sun Y, Xie H, Wang N, Shan L. Blocking core fucosylation of TGF-beta1 receptors downregulates their functions and attenuates the epithelial-mesenchymal transition of renal tubular cells. Am J Physiol Renal Physiol. 2011;300:F1017–25. doi: 10.1152/ajprenal.00426.2010. [DOI] [PubMed] [Google Scholar]
- 22.Shen N, Lin H, Wu T, Wang D, Wang W, Xie H, Zhang J, Feng Z. Inhibition of TGF-beta1-receptor posttranslational core fucosylation attenuates rat renal interstitial fibrosis. Kidney Int. 2013;84:64–77. doi: 10.1038/ki.2013.82. [DOI] [PubMed] [Google Scholar]
- 23.Morelle W, Haslam SM, Ziak M, Roth J, Morris HR, Dell A. Characterization of the N-linked oligosaccharides of megalin (gp330) from rat kidney. Glycobiology. 2000;10:295–304. doi: 10.1093/glycob/10.3.295. [DOI] [PubMed] [Google Scholar]
- 24.Wang X, Inoue S, Gu J, Miyoshi E, Noda K, Li W, Mizuno-Horikawa Y, Nakano M, Asahi M, Takahashi M, Uozumi N, Ihara S, Lee SH, Ikeda Y, Yamaguchi Y, Aze Y, Tomiyama Y, Fujii J, Suzuki K, Kondo A, Shapiro SD, Lopez-Otin C, Kuwaki T, Okabe M, Honke K, Taniguchi N. Dysregulation of TGF-beta1 receptor activation leads to abnormal lung development and emphysemalike phenotype in core fucose-deficient mice. Proc Natl Acad Sci U S A. 2005;102:15791–15796. doi: 10.1073/pnas.0507375102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim YS, Yoo HS, Ko JH. Implication of aberrant glycosylation in cancer and use of lectin for cancer biomarker discovery. Protein Pept Lett. 2009;16:499–507. doi: 10.2174/092986609788167798. [DOI] [PubMed] [Google Scholar]
- 26.Contessa JN, Bhojani MS, Freeze HH, Ross BD, Rehemtulla A, Lawrence TS. Molecular imaging of N-linked glycosylation suggests glycan biosynthesis is a novel target for cancer therapy. Clin Cancer Res. 2010;16:3205–3214. doi: 10.1158/1078-0432.CCR-09-3331. [DOI] [PMC free article] [PubMed] [Google Scholar]