

Abstract
The aim of this study is to explore the role of thioredoxin-2 (Trx2) in autophagy and apoptosis during myocardial ischemia-reperfusion (I/R) injury in vivo. In the study, adult male Sprague-Dawley rats were assigned to four groups at random and pretreated with normal saline (sham operation and I/R groups) and either a control lentivirus (Lv-GFP-N) or one expressing Trx2 (Lv-GFP-Trx2). Sevendays after pretreatment, rat MIRI models were produced via occlusion of the left anterior descending coronary artery for 30 min followed by reperfusion for 6 h. Hearts and blood were harvested to assess efficiency of lentivirus transfection via immunofluorescence staining, quantitative RT-PCR and western blotting, oxidative stress via the malondialdehyde level and superoxide dismutase activity, myocardial damage via myocardial enzymelevels and histopathological staining, myocardial apoptosis via TUNEL assays and western blotting, and myocardial autophagy viawestern blotting. Our results showed thatthe delivery of Lv-GFP-Trx2 into the myocardium remarkably increased Trx2 expression. The upregulation of Trx2 contributed to alleviation of oxidative stress, attenuation of myocardial histological damage, reduced leakage of myocardial enzyme and decrease in infarct size. Moreover, the overexpression of Trx2 was significantly associated with thedecreased incidence of apoptosis via ASK1-dependent intrinsic mitochondrial apoptotic pathwayand autophagy via the mammalian target of rapamycin (mTOR) pathway. The study indicates that upregulation of Trx2 protectsthe myocardium from MIRI and isinvolved inthe inhibition of apoptosis and autophagy. Therefore, Trx2 isa promising therapeutic strategy for attenuating MIRI.
Keywords: Trx2, ischemia and reperfusion, apoptosis, autophagy
Introduction
As an emergency manifestation of ischaemic heart disease (IHD), ST-segment elevation myocardial infarction (STEMI) has high mortality and causes disability worldwide [1]. Timely and complete reperfusion is the most effective way to reduce infarct size and limit subsequent ventricular remodeling [2], however, reperfusion per se contributes to additional irreversible injury to the myocardium and affects the final infarct size, and is therefore termed myocardial ischemia/reperfusion (I/R) injury [3].Therefore, the ‘last frontier’ of reperfusion therapy is to prevent and treat lethal reperfusion injury [4].
During I/R injury, oxidative damage plays a crucial role in starting the generation of excess reactive oxygen species (ROS) and eventually progresses, causingcell death due to necrosis, apoptosis and autophagy [5]. Cell necrosis is thought to be an unregulated mode of cell death [6,7]. However, more regulated modes of cell death also occur in infarcted myocardium. Apoptosis is characterized by its typical DNA fragmentation and lack of an inflammatory response [8]. It is energydependent and in-itiated extrinsically by sarcolemmal receptors and intrinsically through cytochrome c released from damaged mitochondria [8]. Autophagy is a dynamic catabolic conserved lysosomal degradation pathway which degrades and recycles old or damaged proteins and organelles [9]. It also plays a critical role through maintaining cellular homoeostasis under physiological and pathological conditions [9]. Its role in myocardial I/Rinjury (MIRI) is less clear [10]. Much evidence illustrates that autophagy is activated during ischemia and further induced during myocardial reperfusion [11]. In addition, necrptosis, which has features of both necrosis and apoptosis, is initiated through the activation of specific receptor-interacting kinases [12]. Although the relative contribution of each cell death mode to infarct size in MIRI remains unclear [6,7], a role for mitochondria is critical to all of them [7]. Intracellular ROS are mainly derived from mitochondrial superoxide from the monoelectronic reduction of oxygen [13]. Superoxide dismutase (SOD) efficiently dismutatessuperoxide to hydrogen peroxide (H2O2). Thus, mitochondria are the major site generating H2O2. Therefore, it is critical to tightly regulate mitochondrial H2O2 and the cell death signaling pathway, which would be specific targets for pharmacological cardioprotection [7].
Besides the glutathione (GSH) system, the thioredoxin (Trx) system is also crucial to protect against oxidative stress in mitochondria [14]. The Trx system in mitochondria is composed of Trx2, Trxreductase 2 (TrxR2) and peroxiredoxin (Prx) [15]. Trx2 has been reported to be ubiquitously expressed with high distribution in metabolically active tissues such ascardiac muscle, skeletal muscle,and brain [16]. Along with Prx3, Trx2 is critical for eliminating H2O2 to maintaina reducing mitochondrial matrix environment [17]. Previous studies have reported that Trx2 is more stable than other thioredoxins in mammals because it lacks structural cysteine residues except for the two located at the active site [18] and plays a more important role in preventing mitochondrial dysfunction than the GSH system [19]. In addition, Trx2 is related to control of the intrinsic mitochondrial apoptotic pathway [17]. It is recently suggested that Trx2 is essential for maintaining cardiac function via suppressing mitochondrial ROS production as well as ASK1-dependent apoptosis during dilated cardiomyopathy and heart failure [16].
In our previous study, we had found that Trx2 protects cardiomyocytes under OGD/R by inhibiting autophagy and apoptosis [20]. Based on the data in vitro, we examined the potential of Trx2 gene therapy and further explored the underlying mechanisms involved in apoptosis and autophagyin a rat MIRI model in vivo.
Materials and methods
Lentiviral constructs
The lentivirus used in the study was commercially constructed by Bio-Link (Shanghai, China). Lentiviral vectors encoding GFP-Trx2 (Lv-GFP-Trx2) or GFP as control (Lv-GFP-N) were constructed using routine procedures [21]. In brief, the plasmid vectors were extracted at high purity with an endotoxin-free solution and then were co-transfected into 293T cells. The medium was replaced with complete medium 8 h after transfection. After 48 h of culture, the supernatant, rich in lentivirus particles, was collected and filtered through a 0.45 μm filter (Millipore). Finally, the viruswas concentrated by ultracentrifugation and the titer was assayed. The final titer was 2 × 109 TU/ml.
Animals and experimental design
Adult male Sprague-Dawley rats (SPF grade, n=40) weighing 240-260 g were provided by Shanghai Songlian Laboratory Animal Farms (production license SCXK2007-0011). All animal experiments wereapproved by the ethics committee of Xinhua Hospital, affiliated with Shanghai Jiaotong University School of Medicine (Shanghai, China; approval no. XHEC-F-2016-012). The rats were housed in a clean and quiet environment at controlled temperature (24-26°C) and humidity (55% ± 5%) and a 12 h light/12 h dark cycle. Water and food were available ad libitum. After adaptive feeding for a week, the rats were assigned randomly to four groups (10 for each group): the sham operation (SO) groupwith normal saline (without left anterior descending occlusion), the myocardial I/R group with normal saline (I/R group), the myocardial I/R group with Lv-GFP-N (Lv-G-N group), and the myocardial I/R group with Lv-GFP-Trx2 (Lv-G-T group) (Figure 1).
Figure 1.
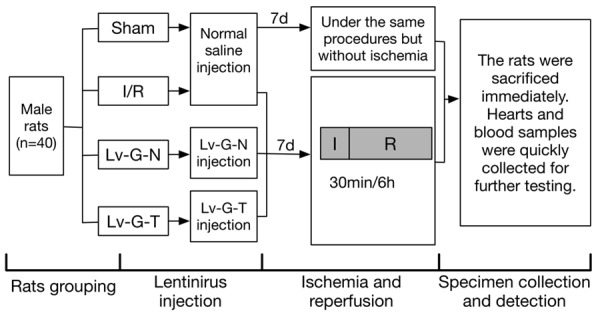
Diagram of experimental protocol. Shown are the grouping of experimental animals and the time pointsof lentiviral injection, ischemia/reperfusionand specimen collection. Lv-G-N=Lv-GFP-N, Lv-G-T=Lv-GFP-Trx2.
Lentivirus transduction in vivo and rat myocardial I/R model
Recombinant lentiviral vectors, Lv-GFP-N (1 × 107 TU/30 µl/rat) or Lv-GFP-Trx2 (1 × 107 TU/30 µl/rat), and an equal volume of normal saline were prepared and kept on ice. After being anaesthetized by intraperitoneal injection of pentobarbital sodium (50 mg/kg), the rats were fixed on the operating board. A tracheal cannula was inserted and connected to a small animal mechanical ventilator (75 breaths/min). A left thoracotomy was performed between the fourth and fifth ribs and the heartwas exposed. After incising the pericardium, intramyocardial injections were performed via a 50 μl microinjector with a 30-gauge needle (Hamilton, Reno, NV, USA) at fiveseparate sites of the left ventricular anterior wall. Lastly, the chest was sutured rapidly. Sevendays afterintramyocardial injection, myocardial I/R models were established as previously reported [22].The proximal left anterior descending (LAD) coronary artery was identified and ligated with a 6-0 silk suture for 30 min followed by 6 h of reperfusion by untying the suture. The success of myocardial I/R models was determined by visible bleaching of the ischemia myocardium and ST segment changes on the ECG. Hearts and blood samples were quickly collected for further testing.
Immunofluorescence staining
We detected the efficiency oflentiviral transfection by immunofluorescence staining as previously described [23]. Briefly, the median third part of the heart sample was fixed in 4% paraformaldehyde followed by 30% sucrose at 4°C, embedded and frozen in OCT solution (Optimal Cutting Temperature, Sakura, Torrance, CA, US) and 5 μm thick sections obtained with a Leica CM3000 cryostat. Finally, the nuclei of the cardiomyocytes were stained with 4’, 6’-diamidino-2-phenylindole (DAPI, Beyotime Biotech, Shanghai, China). Images were obtained using a fluorescence microscope (IX83, Olympus Corporation, Tokyo, Japan).
Quantitative real-time PCR (qRT-PCR)
qRT-PCR was performed using standard methods as described previously [22]. In brief, total RNA was isolated from myocardial samples using TRIzolreagent (TaKaRa, Dalian, China). cDNA was synthesized via reverse transcription with PrimeScript RT Master Mix (Takara). qRT-PCR was performed using SYBR Premix Ex Taq (TaKaRa) according to the manufacturer’s instructions. β-actin was used for normalization. Reactions were conducted using the ABI PRISM 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The primers used in this study for qRT-PCR were forward primer 5’-AGAGCCTCGCCTTTGCCGAT-3’ and reverse primer 5’-TGCCAGATTTTCTCCATGTCGT-3’ for β-actin; forward primer 5’-TGCTGGTGGTCTAACTGGAAC-3’ and reverse primer 5’-TCAATGGCAAGGTCTGTGTG-3’ for Trx2.
Histopathological staining
Hematoxylin and eosin (H&E) staining was conducted as described previously to observe histological changes of heart tissue samples [22]. After myocardial I/R, the heart was isolated, fixed, embedded and cut into 5 μm slices. Next, slices were stained with H&E dyes and examined under light microscopy. The Evans blue/2,3,5-triphenyl tetrazolium chloride (TTC) double staining method was performed to determine the myocardial infarct size. At the end of reperfusion, the LAD wasligated again and Evans blue dye was injected via the aorta to visualize the area at risk, showing no infiltration with the dye. After harvesting and washing with normal saline, the heart was frozen at -20°C for 30 min and then sliced. Then they were incubated with 1% TTC buffer at 37°C for 10 min and fixed in 4% paraformaldehyde overnight. Images were acquired with a scanner and analyzed with Image-Pro Plus 6.0 software (Media Cybernetic, USA).
Detection of CK, LDH, SOD and MDA in serum
At the end of the perfusion, the blood samples were immediately collectedfrom the carotid and the serum samples were isolated by centrifugation (3000 rpm/min) for 15 min at 4°C. Hearts were harvested and frozened immediately in liquid nitrogen and then were ground in a liquid nitrogen-chilled mortar and pestle. Creatine kinase (CK) and lactate dehydrogenase (LDH) activity were assayed to measure the extent of myocardial injury in serum, and the malondialdehyde (MDA) leveland the superoxide dismutase (SOD) activity were assayed to reflect the level of oxidative stress in homogenized cardiac tissue samples using commercially available kits (Nanjing Jiancheng BioInst, Nanjing, China).
TUNEL assay
Terminal deoxy nucleotidyltransferased UTP nick-end labeling (TUNEL) staining was conducted to detect cell apoptosis in myocardial tissues. The median third of the heart was sliced along a cross-section, embedded in paraffin, then cut into 5-µm thick slices. TUNEL staining was performed after antigen retrieval with a commercial kit (Roche Applied Science, Basel, Switzerland) following the manufacturer’s instructions. The cardiomyocytes were stained with c-TnI (Sigma) and the total nuclei were stained with DAPI (Beyotime Biotech, Shanghai, China). Images were acquired under afluorescence microscope, and five random fields of each sample were obtained in the peri-infarct area. The apoptotic index (AI) was determined by counting the percentage of apoptotic to total myocytes.
Western blotting
Proteins were extracted from myocardial tissue of ischemic penumbra by lysis bufferand phosphatase inhibitors (Beyotime Biotechnology, Shanghai, China). The concentration of protein samples were measured with a BCA assay kit (Beyotime Institute of Biotechnology, Shanghai, China). For western blotting, equal amounts of protein (50 μg/lane) were separated by SDS-PAGE. After polyvinylidenedifluoride (PVDF) membranes (Millipore) were blotted with proteins, they were blocked and then incubated with the corresponding primary antibodies overnight at 4°C. Antibodieswere as follows: rabbit anti-Trx2 (1:10,000, Abcam, Cambridge, UK, Cat. No. ab185544), rabbit anti-Bax (1:5000, Abcam, Cat. No. ab32503), rabbit anti-Bcl-2 (1:1000, Abcam, Cat. No. ab59348), rabbit anti-cleaved caspase3 (1:1000, Cell Signaling Technology, Danvers, MA, USA, Cat. No. 9664), rabbit anti-phospho-ASK1 (1:1000, Abcam, Cat. No. ab195821), rabbit anti-ASK1 (1:1000, Abcam, Cat. No. ab131506), rabbit anti-Beclin-1 (1:1000, Cell Signaling Technology, Cat. No. 3495), rabbit anti-LC3A/B (1:1000, Abcam, Cat. No. ab48394), rabbit anti-mTOR (1:1000, Cell Signaling Technology, Cat. No. 2983), rabbit anti-phospho-mTOR (1:1000, Cell Signaling Technology, Cat. No. 2971), rabbit anti-Akt (1:1000, Cell Signaling Technology, Cat. No. 4691), rabbit anti-phospho-Akt (1:1000, Cell Signaling Technology, Cat. No. 4060), rabbit anti-AMPK (1:1000, Cell Signaling Technology, Cat. No. 5832), rabbit anti-phospho-AMPK (1:1000, Cell Signaling Technology, Cat. No. 2535), and mouse anti-β-actin (1:5000, Abcam, Cat. No. ab8226). After the PVDF membranes were incubated with appropriate secondary antibodies, protein bands were visualized by a chemiluminescence imaging analysis system, and then quantitated via ChemiDoc XRS+ software (Bio-Rad Laboratories, Hercules, CA, USA).
Statistical analysis
Statistical analysis was conducted with Prism 6.0 software (GraphPad). All experiments were repeated independently at least three times,and data presented as means ± standard deviation (SD). Statistical comparisons were performed using Student’s t-test. P values < 0.05 were considered statistically significant.
Results
Efficiency of lentivirus transfection
To examine whether Trx2 could alleviate MIRI, we constructed lentivirus vectors, Lv-GFP-Trx2 and Lv-GFP-N, and injected them into the myocardium of rat models. GFP expression was examined to determine the efficiency of Lv-GFP-Trx2 and Lv-GFP-N transfection. Compared with the sham control, the GFP signals were clearly visible, which indicated successful transfection and strong expression of Lv-GFP-Trx2 and Lv-GFP-N (Figure 2).
Figure 2.
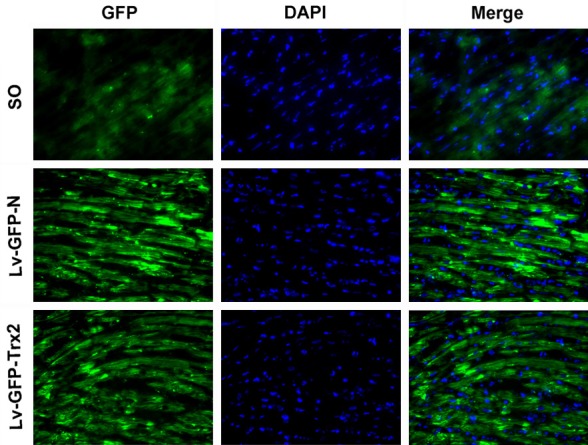
Efficiency of lentiviral transfection into the rat myocardium. Representativeimagesof immunofluorescence staining (original magnification, x400) for GFP (green) and DAPI-labeled cardiomyocyte nuclei (blue). Either Lv-GFP-N or Lv-GFP-Trx2 was injected seven days prior toimmunofluorescence staining.
Lv-GFP-Trx2 transfection in vivo increased Trx2 expression
To further assess transgene expression, the mRNA and protein levels of Trx2 were measured respectively by qRT-PCR and western blotting. After 6 h of reperfusion, the mRNA (Figure 3A) and protein (Figure 3B) levels of myocardial Trx2 were significantly lower in the I/R group than the SO group. Trx2 expression showed no statistical difference, in contrast to the Lv-G-N group; however, Lv-GFP-Trx2 transfection remarkably increased Trx2 expression in I/R myocardium, compared with the I/R and Lv-G-N groups.
Figure 3.
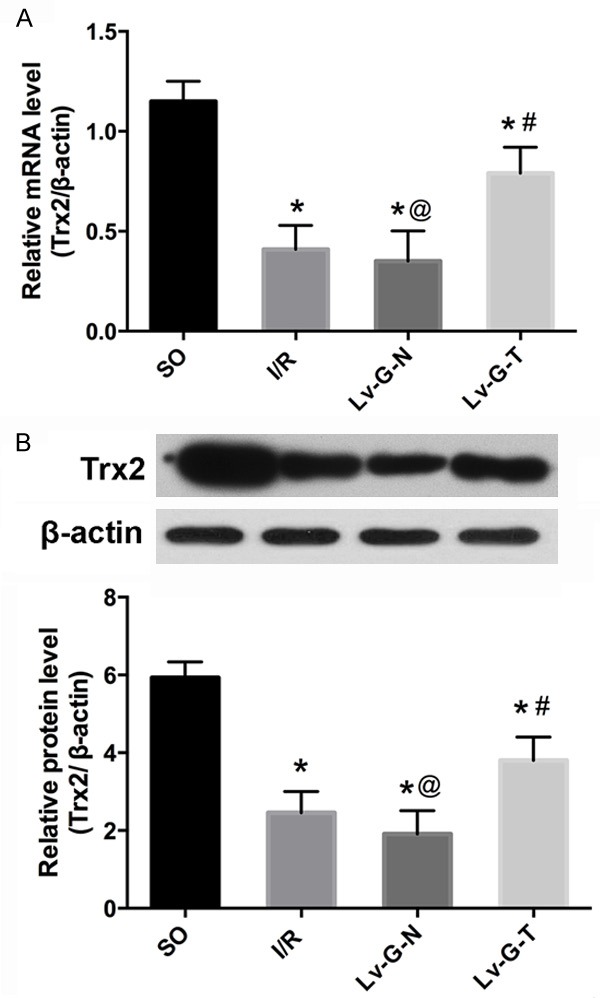
Lv-GFP-Trx2 transfection into the rat myocardium increases the expression of Trx2. A. Quantitative RT-PCR of Trx2. B. Western blots and quantitation of Trx2 in the four experimental groups. Trx2 expression was reduced in the rat model of MIRI, while delivery of Lv-GFP-Trx2 into the myocardium increased the expression of Trx2. *p < 0.05 vs. SO group, #p < 0.05 vs. I/RorLv-GFP-N group, @p>0.05 vs. I/R group.
Up-regulation of Trx2 attenuated oxidative stress and MIRI in rat models
To explore the role of Trx2 in protecting against oxidative stress in the MIRI model, the MDA level (Figure 4A) and SOD activity (Figure 4B) were assayed. As expected, the MDA level was increased while SOD activity was decreased in serum (I/R group vs. SO group, P < 0.05). However, the tendency was reversed notably by Trx2 overexpression. In addition, there was no significant difference on the MDA levelor SOD activitybetween the Lv-G-N group and I/R group.
Figure 4.

Up-regulation of Trx2 attenuates MIRI. A, B. Lv-GFP-Trx2 led to a decrease in MDA level and increase in SOD level in heart tissue after I/R injury. C, D. Lv-GFP-Trx2 led to reductions in CK and LDH levels in serum after myocardial I/R injury. E. Representative images of Evans blue/2, 3, 5-triphenyl tetrazolium chloridedouble staining to determine areas of myocardial infarction. Blue-stained area: non-ischemic tissue; red-stained area: non-infarct tissue; pale white area, infarcted tissue. F. The infarct size is presented as a percentage of the area at risk. G. Representative images of H&E staining (original magnification, x400). *p < 0.05 vs. SO group, #p < 0.05 vs. I/RorLv-GFP-N group, @p>0.05 vs. I/R group.
To further determine whether up-regulation of Trx2 alleviates MIRI-induced myocardial damage, we assayed myocardial enzyme levels, performed myocardial histopathology analysis and measured myocardial infarct size. With regard to myocardial enzymes, low activity of CK (Figure 4C) and LDH (Figure 4D) was detected in the SO group; MIRI increased the leakage of CK and LDH into serum (I/R group vs. SO group, P < 0.05); however, the elevation in serum CK and LDH levels was remarkably suppressed by injection of Lv-GFP-Trx2. In addition, treatment with the control vector (Lv-GFP-N) had no effect on the leakage of CK or LDH induced by I/R injury compared with theI/R group.
Concerning the histopathological changes, H&E staining (Figure 4G) demonstrated that normal myocardial tissue structure and shape were maintained in the SO group, while clusters of infiltrating inflammatory cells, as well as extensive edema, enlarged intercellular spaces and myocyte necrosis were evident in the myocardial tissue of the I/R group. The pathological changes described above were significantly improved in the I/R myocardium transduced with Lv-GFP-Trx2; moreover, no significant histopathological differences were found in myocardial tissue of the Lv-G-N group compared with the I/R group.
The myocardial infarct size is shown in Figure 4E and 4F. No myocardial infarcts were observed in rats of the SO group, while clear, sizable infarctswere observed in rats of the I/R group. Moreover, transduction with Lv-GFP-N did not affect infarct size compared with the I/R group (P>0.05). However, intramyocardial transduction of Lv-GFP-Trx2 led to a significant reduction in myocardial infarct size (vs. I/R or Lv-G-N group, P < 0.05).
Trx2 reduced apoptosis of cardiomyocytes in MIRI
Firstly, we detected the number of TUNEL-positive cardiomyocytes to evaluate the anti-apoptotic potency of Trx2 in the rat MIRI model. As shown in Figure 5A, 5B, the number of TUNEL-positive cardiomyocytes was higher in all three I/R groups than in the SO group, while the overexpression of Trx2 significantly repressed the AI (P < 0.05); Moreover, transduction with Lv-GFP-N did not affect AIcompared with the I/R group (P>0.05). Secondly, we determined the expression of proteins related to the endogenous mitochondrial apoptosis pathway. As Figure 5C, 5D shows, the overexpression of Trx2 partly repressed the activation of apoptosis during MIRI, evidenced by the increased expression of anti-apoptotic Bcl-2 protein, reduced expression of pro-apoptotic Bax protein and activation of p-ASK1 compared with the Lv-G-N group. In line with the TUNEL assay, intramyocardial transduction of Lv-GFP-Trx2 also markedly reduced caspase3 activation (Figure 5C, 5D). In addition, transduction with Lv-GFP-N did not affect expression of those related proteins compared with the I/R group (P>0.05).
Figure 5.
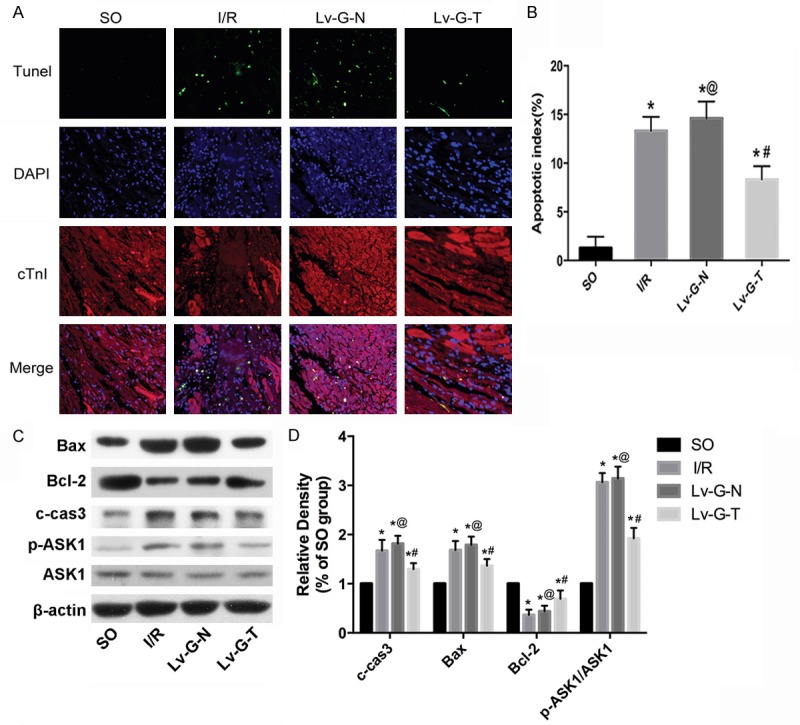
Trx2 reduces the apoptosis ofcardiomyocytes in I/R rat myocardia. (A) Representative TUNEL staining images (original magnification, x400). (B) Quantification of the mean apoptotic index (AI) in each group. (C) Western blotting analysis of cleaved caspase 3, Bcl-2, Bax and ASK1. (D) Quantitation of (C) as fold change by normalizing to control (arbitrarily set as 1.0). β-actin was used as an internal control. *p < 0.05 vs. SO group, #p < 0.05 vs. I/RorLv-GFP-N group, @p>0.05 vs. I/R group.
Trx2 repressed autophagic signaling during MIRI
Western blotting (Figure 6) showed that the expression of autophagy-related proteins LC3II/LC3I and Beclin-1 increased significantly after MIRI compared with the SO group. MIRI following transduction with Lv-GFP-N had no significant effect (Lv-G-N vs. I/R group, P>0.05). However, Trx2 overexpression led to a significant decrease in LC3II/LC3I and Beclin-1 (Lv-G-Trx2 group vs. Lv-G-N group, P < 0.05). Moreover, we demonstrated potential molecular mechanisms for the regulation of Trx2 on autophagy. Figure 6 shows that the phosphorylation of mammalian target of rapamycin (mTOR) was decreased by I/R treatment and that Trx2 overexpression enhanced the phosphorylation of mTOR. The upstream signaling molecules Aktkinase and AMP-activated protein kinase (AMPK) were both involved. Our results indicated that Trx2 induction enhanced the activation of Akt and decreased the activation of AMPK to increase the phosphorylation of mTOR, eventually leading to the inhibition of autophagy (Figure 6).
Figure 6.
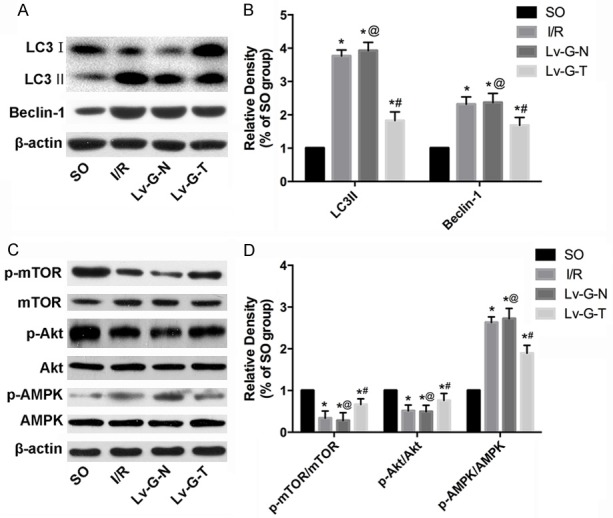
Trx2 suppresses autophagy of cardiomyocytes via mTOR-dependent pathway in I/R rat myocardia. (A) Western blotting analysis of LC3, Beclin1. (B) Quantitation of (A) as fold change by normalizing to control (arbitrarily set as 1.0). (C) Western blotting analysis of mTOR, Akt and AMPK. (D) Quantitation of (C) as fold change by normalizing to control (arbitrarily set as 1.0). β-actin was used as an internal control. *p < 0.05 vs. SO group, #p < 0.05 vs. I/RorLv-GFP-N group, @p>0.05 vs. I/R group.
Discussion
Definitive therapy for IHD always turns to perfusion; however, MIRI has posed a formidable continued barrier to effective treatment, and additional cardioprotective strategies are needed in this context [4,7]. As a novel therapeutic modality, gene therapy has shown promise for cardiovascular diseases [21]. Our data indicates that Trx2 expression decreased in the rat MIRI model and Trx2 overexpression by means of lentivirus transduction protected against MIRI via reducing oxidative stress and alleviating myocardial apoptosis and autophagy.
Initial gene transfer experiments were performed using adenoviral vectors for cardiovascular disease [21,24,25], but adenoviral-based gene delivery has only a limited duration of transgene expression andexerts a strong immune response [26,27]. Therefore, lentivirus was selected for our experiments because it has the capacity to integrate into the rat myocardium for a long time and triggers a minor immune response [21,28]. Our data demonstrated successful transfection oflentivirus into myocardium, and the downregulation of Trx2 expression induced by MIRI could be partly reversed by Trx2 overexpression via lentivirus injection.
Trx2 is essential for cell viability, the control of mitochondrial ROS homeostasis and regulation of apoptosis [29]. Previously it was reported that human Trx2 deficiency causes early-onset neurodegeneration because of the impairment of mitochondrial redox homeostasis [29], and the amount of Trx2 is lower in the auditory cortex of aging rats, which may be related to the pathogenesis of central presbycusis via the Trx2-ASK1 signalling pathway [30]. In heart, it was reported that overexpression of Trx2 decreases superoxide levels and prevents cardiomyocyte hypertrophy in an angiotensin II-induced vascular dysfunction model [31]. Moreover, it was recently observed that Trx2 expression was reduced and ASK1-dependent apoptosis was increased in human hearts with dilated cardiomyopathy (DCM), as well as that mice with a cardiac-specific deletion (Trx2-cKO) developed DCM with an increase in ROS production and ASK1-dependent apoptosis in the myocardium [16]. In our study, we demonstrated that Trx2 overexpression alleviated MIRI-induced myocardial damage, which was confirmed by reduced myocardial enzyme levels, improvement inhistopathological changes and alleviation of myocardial infarct size. Also, SOD is considered as a first line of defense against exogenous or endogenous oxidative damage [30]. As the final product of lipid peroxidation, the accumulation of MDA reflects the acceleration of oxidative stress [30]. Our results showed that Trx2 induction relieved oxidative stress with partial reversion of the increased MDA levels and decreased SOD activity in MIRI rats.
In this work, TUNEL assaysshowed that Trx2 could prevent cardiomyocyte apoptosis, which is consistent with previous research [16,18]. ASK1 is considered as a key regulator of apoptosis induced by oxidative stress in ischemic cardiomyocytes [32], and ASK1-induced apoptosis is executed by mitochondria-dependent caspase activation [33]. ASK1 has been shown to bind with Trx2 to regulate apoptosis [34]. The intrinsic mitochondria apoptotic pathway is regulated by the Bcl-2 family [35]. Previous research has found that Bax, a proapoptotic protein of the Bcl-2 family, is required for all cases of apoptosis mediated by the intrinsic pathway and stimulates the release of apoptogenic mitochondrial proteins into the cytosol [36]. It was observed that the myocardial infarct size was reduced 50% in the I/R model of Bax knockout mice [37]. Antiapoptotic Bcl-2 protein can exert its effect either independently or via direct interactions with Bax and other pro-apoptotic proteins [38]. Consistent with these studies, we found that Trx2 overexpression notably attenuated ASK1-dependent apoptosis, with an increase in Bcl-2 and decrease in p-ASK1, c-caspase3, and Bax. Most lines of evidence suggest that ROS represent important mediators of autophagy [39]. During reperfusion, increased ROS generation leads to uncontrolled excessive induction of autophagy, contributing to autophagiccardiomyocyte death [40]. ROS could oxidize autophagy protein 4 (Atg4) and decrease its activity, contributing to the cleavage of microtubule-associated protein light chain with subsequent LC3Il ipidationto form LC3II and initiate autophagy [41]. Beclin-1 is also strongly induced by ROS, mediating autophagy during reperfusion [40]. Our results showed that the expression of autophagy related proteins LC3II/LC3I and Beclin-1 increased in rats with MIRI, whereas Trx2 could reduce autophagy. As a highly conserved serine/threonine kinase regulating protein synthesis and cell growth, mTOR has been evidenced as a key regulator of autophagy [41]. Many diverse signalling pathways converge on mTOR to regulate autophagy [41,42]. Notably, mTOR is activated to inhibit autophagy by the PI3K/Akt signaling pathway [41]. Also, AMPK is the link between energy status and mTOR activity, and it is activated to inhibit mTOR and thus promote autophagy during metabolic stress [40]. Our findings showed that both the Akt/mTOR and AMPK/mTOR pathways are involved in the regulation of autophagy by Trx2 in rat MIRI. Although autophagy is a double-edged sword inmyocardial I/R injury [40,43], a growing amount of evidence recently has indicated that autophagy is induced and protective during myocardial ischemia, whereas it is further enhanced and detrimental during myocardial reperfusion in I/R models [11,40,44]. Beneficial effects of autophagy could contribute to ATP generation and cellular homeostasis, while autophagy is hyperactivated under accelerated oxidative stress to delete necessary organelles or proteins, leading to autophagic cell death [45]. Crosstalk between autophagy and apoptosis may be another factor in the detrimental effects of autophagy [40]. The link between autophagy and the Bcl-2 family may be a good example; Beclin-1-dependent autophagy is related to Bcl-2 down-regulation because Bcl-2 inhibits Beclin1-mediated activation of autophagy [41]. Notably, Bcl-2 protein is also involved in the regulation of apoptosis, and reduced expression of Bcl-2 may contribute to apoptotic cell death [40]. In this work, we also suggest that the decrease in Beclin-1 and the increase in Bcl-2 induced by Trx2 overexpression are consistent with the alleviation of apoptosis and autophagy.
In conclusion, we demonstrated that inducing the expression of Trx2 via a recombinant lentiviral vector could alleviate MIRI by reducing oxidative stress and inhibiting apoptosis and autophagy in a rat model. Therefore, Trx2 gene delivery is a potential therapeutic modality for the treatment of MIRI.
Acknowledgements
This work was supported by a Nature Science Foundation of China Grant (No.81170124).
Disclosure of conflict of interest
None.
References
- 1.Mendis S, Davis S, Norrving B. Organizational update: the world health organization global status report on noncommunicable diseases 2014; one more landmark step in the combat against stroke and vascular disease. Stroke. 2015;46:e121–122. doi: 10.1161/STROKEAHA.115.008097. [DOI] [PubMed] [Google Scholar]
- 2.Heusch G, Libby P, Gersh B, Yellon D, Bohm M, Lopaschuk G, Opie L. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet. 2014;383:1933–1943. doi: 10.1016/S0140-6736(14)60107-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 4.Bulluck H, Yellon DM, Hausenloy DJ. Reducing myocardial infarct size: challenges and future opportunities. Heart. 2016;102:341–348. doi: 10.1136/heartjnl-2015-307855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kristensen SD, Laut KG, Fajadet J, Kaifoszova Z, Kala P, Di Mario C, Wijns W, Clemmensen P, Agladze V, Antoniades L, Alhabib KF, De Boer MJ, Claeys MJ, Deleanu D, Dudek D, Erglis A, Gilard M, Goktekin O, Guagliumi G, Gudnason T, Hansen KW, Huber K, James S, Janota T, Jennings S, Kajander O, Kanakakis J, Karamfiloff KK, Kedev S, Kornowski R, Ludman PF, Merkely B, Milicic D, Najafov R, Nicolini FA, Noc M, Ostojic M, Pereira H, Radovanovic D, Sabate M, Sobhy M, Sokolov M, Studencan M, Terzic I, Wahler S, Widimsky P. Reperfusion therapy for ST elevation acute myocardial infarction 2010/2011: current status in 37 ESC countries. Eur Heart J. 2014;35:1957–1970. doi: 10.1093/eurheartj/eht529. [DOI] [PubMed] [Google Scholar]
- 6.Orogo AM, Gustafsson AB. Cell death in the myocardium: my heart won’t go on. IUBMB Life. 2013;65:651–656. doi: 10.1002/iub.1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ibanez B, Heusch G, Ovize M, Van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol. 2015;65:1454–1471. doi: 10.1016/j.jacc.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 8.Heusch G, Boengler K, Schulz R. Inhibition of mitochondrial permeability transition pore opening: the Holy Grail of cardioprotection. Basic Res Cardiol. 2010;105:151–154. doi: 10.1007/s00395-009-0080-9. [DOI] [PubMed] [Google Scholar]
- 9.Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell. 2014;159:1263–1276. doi: 10.1016/j.cell.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh KK, Yanagawa B, Quan A, Wang R, Garg A, Khan R, Pan Y, Wheatcroft MD, Lovren F, Teoh H, Verma S. Autophagy gene fingerprint in human ischemia and reperfusion. J Thorac Cardiovasc Surg. 2014;147:1065–1072. e1061. doi: 10.1016/j.jtcvs.2013.04.042. [DOI] [PubMed] [Google Scholar]
- 11.Zeng C, Li H, Fan Z, Zhong L, Guo Z, Guo Y, Xi Y. Crocin-elicited autophagy rescues myocardial ischemia/reperfusion injury via paradoxical mechanisms. Am J Chin Med. 2016;44:515–530. doi: 10.1142/S0192415X16500282. [DOI] [PubMed] [Google Scholar]
- 12.Zhou W, Yuan J. SnapShot: necroptosis. Cell. 2014;158:464–464. e461. doi: 10.1016/j.cell.2014.06.041. [DOI] [PubMed] [Google Scholar]
- 13.Holzerova E, Prokisch H. Mitochondria: much ado about nothing? How dangerous is reactive oxygen species production? Int J Biochem Cell Biol. 2015;63:16–20. doi: 10.1016/j.biocel.2015.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H, Go YM, Jones DP. Mitochondrial thioredoxin-2/peroxiredoxin-3 system functions in parallel with mitochondrial GSH system in protection against oxidative stress. Arch Biochem Biophys. 2007;465:119–126. doi: 10.1016/j.abb.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 15.Mahmood DF, Abderrazak A, El Hadri K, Simmet T, Rouis M. The thioredoxin system as a therapeutic target in human health and disease. Antioxid Redox Signal. 2013;19:1266–1303. doi: 10.1089/ars.2012.4757. [DOI] [PubMed] [Google Scholar]
- 16.Huang Q, Zhou HJ, Zhang H, Huang Y, Hinojosa-Kirschenbaum F, Fan P, Yao L, Belardinelli L, Tellides G, Giordano FJ, Budas GR, Min W. Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation. 2015;131:1082–1097. doi: 10.1161/CIRCULATIONAHA.114.012725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu J, Holmgren A. Thioredoxin system in cell death progression. Antioxid Redox Signal. 2012;17:1738–1747. doi: 10.1089/ars.2012.4650. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka T, Hosoi F, Yamaguchi-Iwai Y, Nakamura H, Masutani H, Ueda S, Nishiyama A, Takeda S, Wada H, Spyrou G, Yodoi J. Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J. 2002;21:1695–1703. doi: 10.1093/emboj/21.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lowes DA, Galley HF. Mitochondrial protection by the thioredoxin-2 and glutathione systems in an in vitro endothelial model of sepsis. Biochem J. 2011;436:123–132. doi: 10.1042/BJ20102135. [DOI] [PubMed] [Google Scholar]
- 20.Li YY, Xiang Y, Zhang S, Wang Y, Yang J, Liu W, Xue FT. Thioredoxin-2 protects against oxygen-glucose deprivation/reperfusion injury by inhibiting autophagy and apoptosis in H9c2 cardiomyocytes. Am J Transl Res. 2017;9:1471–1482. [PMC free article] [PubMed] [Google Scholar]
- 21.Mattila M, Koskenvuo J, Soderstrom M, Eerola K, Savontaus M. Intramyocardial injection of SERCA2a-expressing lentivirus improves myocardial function in doxorubicin-induced heart failure. J Gene Med. 2016;18:124–133. doi: 10.1002/jgm.2885. [DOI] [PubMed] [Google Scholar]
- 22.Wang HB, Yang J, Ding JW, Chen LH, Li S, Liu XW, Yang CJ, Fan ZX, Yang J. RNAi-Mediated Down-Regulation of CD47 Protects against Ischemia/Reperfusion-Induced Myocardial Damage via Activation of eNOS in a Rat Model. Cell Physiol Biochem. 2016;40:1163–1174. doi: 10.1159/000453170. [DOI] [PubMed] [Google Scholar]
- 23.Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, Diwan A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125:3170–3181. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hajjar RJ, Kang JX, Gwathmey JK, Rosenzweig A. Physiological effects of adenoviral gene transfer of sarcoplasmic reticulum calcium ATPase in isolated rat myocytes. Circulation. 1997;95:423–429. doi: 10.1161/01.cir.95.2.423. [DOI] [PubMed] [Google Scholar]
- 25.Yang Y, Duan W, Lin Y, Yi W, Liang Z, Yan J, Wang N, Deng C, Zhang S, Li Y, Chen W, Yu S, Yi D, Jin Z. SIRT1 activation by curcumin pretreatment attenuates mitochondrial oxidative damage induced by myocardial ischemia reperfusion injury. Free Radic Biol Med. 2013;65:667–679. doi: 10.1016/j.freeradbiomed.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Ly H, Kawase Y, Yoneyama R, Hajjar RJ. Gene therapy in the treatment of heart failure. Physiology (Bethesda) 2007;22:81–96. doi: 10.1152/physiol.00037.2006. [DOI] [PubMed] [Google Scholar]
- 27.Toivonen R, Koskenvuo J, Merentie M, Soderstrom M, Yla-Herttuala S, Savontaus M. Intracardiac injection of a capsid-modified Ad5/35 results in decreased heart toxicity when compared to standard Ad5. Virol J. 2012;9:296. doi: 10.1186/1743-422X-9-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niwano K, Arai M, Koitabashi N, Watanabe A, Ikeda Y, Miyoshi H, Kurabayashi M. Lentiviral vector-mediated SERCA2 gene transfer protects against heart failure and left ventricular remodeling after myocardial infarction in rats. Mol Ther. 2008;16:1026–1032. doi: 10.1038/mt.2008.61. [DOI] [PubMed] [Google Scholar]
- 29.Holzerova E, Danhauser K, Haack TB, Kremer LS, Melcher M, Ingold I, Kobayashi S, Terrile C, Wolf P, Schaper J, Mayatepek E, Baertling F, Friedmann Angeli JP, Conrad M, Strom TM, Meitinger T, Prokisch H, Distelmaier F. Human thioredoxin 2 deficiency impairs mitochondrial redox homeostasis and causes early-onset neurodegeneration. Brain. 2016;139:346–354. doi: 10.1093/brain/awv350. [DOI] [PubMed] [Google Scholar]
- 30.Sun HY, Hu YJ, Zhao XY, Zhong Y, Zeng LL, Chen XB, Yuan J, Wu J, Sun Y, Kong W, Kong WJ. Age-related changes in mitochondrial antioxidant enzyme Trx2 and TXNIP-Trx2-ASK1 signal pathways in the auditory cortex of a mimetic aging rat model: changes to Trx2 in the auditory cortex. FEBS J. 2015;282:2758–2774. doi: 10.1111/febs.13324. [DOI] [PubMed] [Google Scholar]
- 31.Widder JD, Fraccarollo D, Galuppo P, Hansen JM, Jones DP, Ertl G, Bauersachs J. Attenuation of angiotensin II-induced vascular dysfunction and hypertension by overexpression of Thioredoxin 2. Hypertension. 2009;54:338–344. doi: 10.1161/HYPERTENSIONAHA.108.127928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerczuk PZ, Breckenridge DG, Liles JT, Budas GR, Shryock JC, Belardinelli L, Kloner RA, Dai W. An apoptosis signal-regulating kinase 1 inhibitor reduces cardiomyocyte apoptosis and infarct size in a rat ischemia-reperfusion model. J Cardiovasc Pharmacol. 2012;60:276–282. doi: 10.1097/FJC.0b013e31825ea0fa. [DOI] [PubMed] [Google Scholar]
- 33.Hatai T, Matsuzawa A, Inoshita S, Mochida Y, Kuroda T, Sakamaki K, Kuida K, Yonehara S, Ichijo H, Takeda K. Execution of apoptosis signal-regulating kinase 1 (ASK1)-induced apoptosis by the mitochondria-dependent caspase activation. J Biol Chem. 2000;275:26576–26581. doi: 10.1074/jbc.M003412200. [DOI] [PubMed] [Google Scholar]
- 34.Zhang R, Al-Lamki R, Bai L, Streb JW, Miano JM, Bradley J, Min W. Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ Res. 2004;94:1483–1491. doi: 10.1161/01.RES.0000130525.37646.a7. [DOI] [PubMed] [Google Scholar]
- 35.Schwarz M, Andrade-Navarro MA, Gross A. Mitochondrial carriers and pores: key regulators of the mitochondrial apoptotic program? Apoptosis. 2007;12:869–876. doi: 10.1007/s10495-007-0748-2. [DOI] [PubMed] [Google Scholar]
- 36.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hochhauser E, Kivity S, Offen D, Maulik N, Otani H, Barhum Y, Pannet H, Shneyvays V, Shainberg A, Goldshtaub V, Tobar A, Vidne BA. Bax ablation protects against myocardial ischemia-reperfusion injury in transgenic mice. Am J Physiol Heart Circ Physiol. 2003;284:H2351–2359. doi: 10.1152/ajpheart.00783.2002. [DOI] [PubMed] [Google Scholar]
- 38.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 39.Shao FY, Du ZY, Ma DL, Chen WB, Fu WY, Ruan BB, Rui W, Zhang JX, Wang S, Wong NS, Xiao H, Li MM, Liu X, Liu QY, Zhou XD, Yan HZ, Wang YF, Chen CY, Liu Z, Chen HY. B5, a thioredoxin reductase inhibitor, induces apoptosis in human cervical cancer cells by suppressing the thioredoxin system, disrupting mitochondrion-dependent pathways and triggering autophagy. Oncotarget. 2015;6:30939–30956. doi: 10.18632/oncotarget.5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sadoshima J. The role of autophagy during ischemia/reperfusion. Autophagy. 2008;4:402–403. doi: 10.4161/auto.5924. [DOI] [PubMed] [Google Scholar]
- 41.Aviv Y, Shaw J, Gang H, Kirshenbaum LA. Regulation of autophagy in the heart: “you only live twice”. Antioxid Redox Signal. 2011;14:2245–2250. doi: 10.1089/ars.2010.3479. [DOI] [PubMed] [Google Scholar]
- 42.Mizushima N. [Autophagy and apoptosis] . Nihon Rinsho Meneki Gakkai Kaishi. 2000;23:527–530. [PubMed] [Google Scholar]
- 43.Cao DJ, Gillette TG, Hill JA. Cardiomyocyte autophagy: remodeling, repairing, and reconstructing the heart. Curr Hypertens Rep. 2009;11:406–411. doi: 10.1007/s11906-009-0070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang B, Zhong S, Zheng F, Zhang Y, Gao F, Chen Y, Lu B, Xu H, Shi G. N-n-butyl haloperidol iodide protects cardiomyocytes against hypoxia/reoxygenation injury by inhibiting autophagy. Oncotarget. 2015;6:24709–24721. doi: 10.18632/oncotarget.5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valentim L, Laurence KM, Townsend PA, Carroll CJ, Soond S, Scarabelli TM, Knight RA, Latchman DS, Stephanou A. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J Mol Cell Cardiol. 2006;40:846–852. doi: 10.1016/j.yjmcc.2006.03.428. [DOI] [PubMed] [Google Scholar]