

Abstract
In recent decades, immunotherapy has undergone extensive developments for oncologic therapy applications. Dendritic cells (DCs) plays a fundamental role in cell-based vaccination immunotherapy against various types of cancer. It involves stimulating innate and adaptive immunity, in particular cytotoxic T-cell mediated antitumor effects, against targeted tumors and has been studied in both preclinical and clinical settings. Nevertheless, clinical outcomes have been unsatisfying. The antitumor response requires sufficient migration of viable DCs from primary administration site to targeted tumors through related lymphatics. The dynamics and mechanisms of the DCs migration still need further investigation. Here, we briefly introduce the current clinically applicable methods for manufacturing DC-based cancer vaccines and their most commonly used non-invasive, real-time tracking approaches. Furthermore, we propose a hypothesis that intraperitoneal injection may improve the efficiency of DC-based cancer vaccine.
Keywords: Dendritic cells, cancer vaccine, image techniques, animal models
Introduction
Dendritic cells (DCs) are specialized antigen-presenting cells (APCs) that have been extensively studied over the past decades as the initiator and modulator of immune response [1]. Since first characterized in 1973 by Ralph Steinman [2,3], the critical role of DCs in immunity, as well as their therapeutic potential in cancer, has been intensively elucidated [4,5]. Antitumoral response of DC-based vaccines leads to the rejection of tumors and strongly depends on the activation of cytotoxic T lymphocytes following capture of antigen, antigen processing, and presentation to effector T cells [6]. The ideal cancer-immunity cycle requires orchestrating the multistep processes as illustrated below (Figure 1 reproduced with permission from Immunity) [7].
Figure 1.
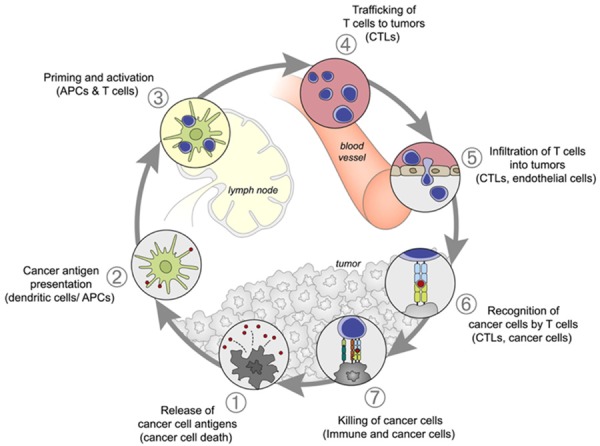
The cancer-immunity cycle. The induction of immune response against cancer can be illustrated as a self-propagating cyclic process, leading to an accumulation of various types of immune-stimulatory factors that in principle should promote and amplify T-cell responses. Inhibitory factors usually produced by cancer result in immune regulatory feedback mechanisms, which may halt the process or limit the immunity. The cycle can be divided into seven main steps, which have been described in the text. The involved primary cell types and anatomic location of each step are also listed. Abbreviations: APCs, antigen presenting cells; CTLs, cytotoxic T lymphocytes.
Cancer vaccines are one of the immunotherapy strategies designed to serve as an inducer of the cancer-immunity cycle. Since first reported by Nature Medicine in 1996 [8], a large amount of clinical trials on DC-based cancer vaccines have been carried out. A search for “dendritic cells and cancer vaccine” on www.clinicaltrials.gov showed 362 registered studies, of which 332 were phase 1 or phase 2 clinical trials. Although long-term benefits have been described in a small number of reports, objective clinical immune response remains quite dismal. The reported maximum rates of conventional objective tumor response are at most 15% [9]. Both immune tolerance secondary to the lack of costimulatory molecules for DCs maturation in peripheral tissues [10,11] and the immune-suppressive environment of the tumor may affect DCs maturation and migration, leading to decreased efficacy cancer vaccine immunotherapy [12].
Numerous basic and clinical studies have already described diverse approaches to specific tumor antigen loading and subsequent migration to draining lymph nodes (LNs) and systemic lymphoid organs [13-16]. Previous studies have also proven the feasibility of DCs maturation in vitro [17,18]. Monitoring DCs migration in vivo will offer important insights into the biomechanism of DCs in antitumor immunity. In this review, we will discuss the sources of DCs subsets, routes of administration, clinically applicable means of DCs labeling, and the most commonly used techniques for non-invasive monitoring in murine models: magnetic resonance imaging (MRI) and positron emission tomography (PET). We will also describe the application of these principles to future clinical cancer immunotherapy.
Source of DCs and DCs classification
DCs occupy a relatively minor part of cell populations in circulating blood, tissues, and organs; cell isolation procedures are also time consuming with low yields [19]. These factors make in vitro generation and differentiation of DCs from precursors a necessary tool for biological research and DC-based immunotherapy. Bone marrow is currently the preferred source of precursor cells for DCs generation and differentiation [20]. First reported in 1986 in rats, protocols for mice were subsequently developed, and extensive efforts have been made to establish a standard for harvesting murine bone marrow derived DCs (Figure 2 reproduced from JoVE) [21].
Figure 2.
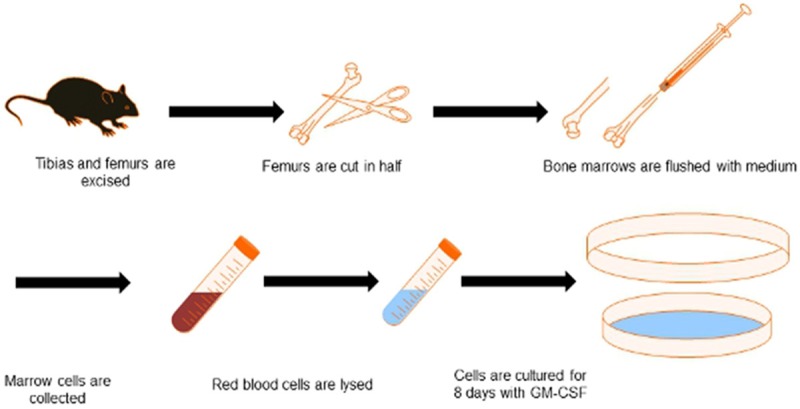
Schematic representation of DCs isolation and generation from mice bone marrow. First, both tibias and femurs are dissected, surrounding tissues are removed, and the long bones are sterilized in ethanol. The bones are cut in half and flushed with medium. After the red blood cells are lysed, bone marrow cells are recovered and cultured for 8 days in the presence of GM-CSF and IL-4 to differentiate them into DCs.
The taxonomy of DCs is quite complicated. Herein, it will just be discussed briefly. DCs are classified by localization, cell-surface phenotype, and specific function [22]. Via different stages of development, DCs can be simply classified as either mature or immature DCs, which differ from each other by localization and function [23]. Immature DCs generally remain in peripheral tissues, recognizing and taking up foreign antigens using phagocytic and endocytic receptors. They are capable of inducing T-cell deletion/anergy and promoting regulatory T cells (Treg cells), resulting in T-cell tolerance [24]. Compared with mature DCs, immature ones are limited in secreting cytokines and activating T cells. In the presence of inflammatory cytokines or adverse signals (such as dying cancer cells or cancer vaccines with tumor antigen), immature DCs will mature, followed by a concomitant decrease in antigen uptake ability and an increase in proinflammatory cytokine secretion, T cells stimulation, and DCs migration to draining LNs [25]. Another primary classification is conventional DCs (cDCs, also termed myeloid DCs) and plasmacytoid DCs (pDCs), which can be discriminated by differential phenotypic expression [23]. cDCs more commonly reside in lymphoid organs, such as spleen, thymus, and LNs, with a subpopulation named migratory DCs found in blood. With a high expression of MHC II and the ability to cross-present exogenous antigens on MHC I, cDCs play an important role in eliciting robust protective immune responses [26]. In contrast, pDCs have a diminished MHC II expression and a relatively poor ability to activate T cells, but elicit a stronger viral infection response through increased Toll-like receptor-7/-9 expression and IFN-α/β production [27].
Nomenclature of DCs is remarkably complex. Even though human and mouse DCs subsets express totally different phenotype, the foundation of classification is similar. Organization of DCs by function in human and mouse parallel each other, and the functional alignment of DCs subsets in these two species will definitely facilitate the translation of immunity understanding from mouse models into human [28].
Methods of tumor antigen loading in DCs
As illustrated in Figure 1, the first step of the cancer-immunity cycle is antigen release from tumor cells and subsequent capture by APCs. Hence, it is necessary to load DCs with targeted tumor antigens or proteins that are distinctively overexpressed in tumor cells as cancer vaccines. DCs can be exploited for cancer vaccines at both the immature and the mature stage [29], and a diverse set of strategies have been utilized to load DCs with tumor antigens, including: (1) DCs loaded with short or long peptides, (2) DCs loaded with proteins, (3) DCs loaded with tumor cell lysates, (4) DCs fused with whole tumor cells, (5) DCs transferred with RNA or DNA, and (6) DCs loaded with neoantigens targeting specific tumor mutation [4,30]. Each of these strategies has advantages and disadvantages, and a scientific debate regarding the ideal method of tumor antigen loading is still ongoing. The goal is to induce maximum innate and adoptive immunity and avoid immune tolerance by the antigen loading method. With emergence of novel techniques for targeting antigens, it is reasonable to expect a more optimal DC-based vaccine loading strategy in the near future [9].
Routes of injection and effectiveness
Various routes of DC-based vaccines administration are reported including subcutaneous (SC), intradermal (ID), intravenous (IV), intraperitoneal (IP), intranodal (IN), intralymphatic (IL), and intratumoral (IT) [31], each of which leads to variable outcomes [32,33]. Several attempts have been made to determine the optimal route of vaccine administration and induction of strong protective immune responses [34-36], yet the milestone has still to be achieved. DCs are region-specific immune cells that target specific sites of the body [37]. The IV and IT routes have been demonstrated to be inefficient by some studies as the DC-based vaccines administered through these methods are not primarily distributed to the LNs [37,38]; this conclusion is still currently under investigation [39]. Several studies have proven that ID administration is more efficient but leads to very low DCs migration rate-maximally up to 5% of injected DCs, while SC administration has a better migration efficiency but poorer outcome [32,40-42]. These two routes are also the most common methods for vaccination in human clinical trials (data from www.clinicaltrials.gov), where low migration rate and low efficacy are major obstacles [43]. Some studies have suggested that the most effective routes of administration may be IN or IL, as DCs migration to draining lymph nodes is crucial for induction of T cells and NK cells. However, the advantage of IN or IL over ID has not been precisely verified due to technical difficulties. IN and IL injections are complex procedures that need ultrasound guidance even when operated by highly experienced clinicians, and only a few of the DCs are correctly injected into the proper position [42,44].
Although IP injection of DC-based cancer vaccines is rarely reported, it may be a potential regimen for gastrointestinal tumors since huge populations of LNs are present throughout the gastrointestinal track, including cisterna chyli, mediastinal LNs, gastric and gastro-omental LNs, hepatic LNs, pancreaticoduodenal LNs, mesenteric LNs, ileocolic LNs, intestinal lymphatic trunk, and thoracic duct. Furthermore, the abdominal cavity also contains spleen and gut-associated lymphoid tissues, which are two other types of secondary lymphoid organs [37]. Together with LNs, they are vital to elicit cellular and humoral immunity [45-47]. Most importantly, these lymphatic tissues are all readily accessible to IP-injected DC-based vaccines. Subsequently, active absorption of the injected DCs by the LNs and lymphatic vessels may be easier for the DCs to migrate to targeted immune tissues. All these factors may accelerate delivery and migration of DC-based cancer vaccines and improve outcomes.
Strategy of labeling and image monitoring of labeled DCs
As mentioned above, imaging of DCs is crucial for visualizing the route of DCs migration and distribution, determining kinetics of the interactions between DCs and other immune cells in the LNs and organs, and ultimately, achieving a comprehensive view of the biological mechanism of DC-based vaccines to help improve clinical outcomes of cancer immunotherapy. We will only discuss clinically feasible non-invasive and real-time measurements of DC-based vaccines imaging and their corresponding cell labeling.
Positron emission tomography (PET) was first established in the clinical setting to monitor and measure enzyme reactions, ligand-receptor interactions, as well as cellular and tissue metabolism [48]. Micro PET has been developed for preclinical research owing to its high sensitivity, precise quantification, and unique assessment of cell viability and function to track immune cells in vivo. Labeling DCs for PET can be divided into two main categories: direct and indirect labeling methods. Though 18F-FDG is feasible for PET imaging despite a short half-life, direct labeling generally requires agents with a relatively long half-life radioisotope, such as 111In-oxiquinolon or 64Cu-PTSM, which may in turn limit their translation to clinic due to continuous radioactivity. Another limitation of direct labeling for PET is the efflux of the labeling agent, which may cause result bias [49]. In contrast, indirect labeling methods such as transfection of reporter genes can overcome the disadvantages of direct labeling, such as loss of labeling with cell proliferation [50]. However, indirect labeling is mostly used in preclinical studies because of the need for genetic editing. In addition to technical difficulties in cell labeling, PET has high costs, low spatial resolution, and poor tissue contrast, which all together restrict PET as the preference of clinical imaging technique.
MRI represents a sophisticated imaging tool with the highest spatial resolution of all non-invasive and real-time imaging modalities and has been widely implemented in preclinical and clinical studies for cell trafficking and migration [51]. Detection of target cells often requires MR contrast agent labeling, which has already been explored by several studies. For direct labeling in vitro and in vivo, the most commonly used contrast agents include biodegradable dextrans, siloxane, citrate, and polymer-coated magnetic iron oxide nanoparticles such as superparamagnetic iron oxide (SPIO, particle size: 50-100 nm) (Figure 3, reproduced from Radiology), ultra-small superparamagnetic iron oxide (USPIO, particle size: 10-50 nm), micrometer-sized iron oxide (MPIOs, particle size: >1 μm), or cross-linked iron oxide (CLIO), all of which lead to hypointensity (negative contrast) in T2-weighted MR images (Figure 4, reproduced from Radiology). These particles have an inherently greater effect on relaxivity in contrast to paramagnetic labeling agents that produce the positive contrast using T1-weighted sequences. Adjunct conjugated monoclonal antibodies or transfection agents have been utilized for more efficient uptake of superparamagnetic agents or paramagnetic contrast agents [52]. Feridex I.V. and Feraheme are commercially available iron-oxide nanoparticles that are approved by Food and Drug Administration (FDA). These labeling methods require phagocytosis for agent uptake and accumulation, which requires relatively immature DCs with greater endocytosis capability. However, it was reported that magnetodendrimers can be universally taken up by different type of cells to avoid insufficient cell internalization [53]. Paramagnetic chemical exchange saturation transfer (PARACEST) agents have been reported as a novel class of MR contrast agents for MR imaging and use a different mechanism from paramagnetic and superparamagnetic iron-oxide agents. PARACEST agents are capable of tracking two particular cell types with two distinct PARACEST markers and specific radiofrequencies in the same experiment [54]. HIV Tat peptide and 19F have also been explored as MR labeling agents, but the improvements of these emerging methods still need to be validated by further studies [55,56]. Indirect labeling requires the insertion of specific exogenous reporter gene for MR detection, which limits its application in clinical settings. As the labeling agents have evolved, innovation in MRI sequences is required. Together with the development of MRI scanners, it could be possible to not only visualize the biodistribution of DC-based vaccines but also quantify migration and even investigate single cell dynamics. A summary of recent researches of PET and MRI-monitored DC-based vaccines in animal models is reported in Table 1. Currently, MRI is more frequently used to visualize homing and engraftment immediately after DCs inoculation in a longitudinal manner.
Figure 3.
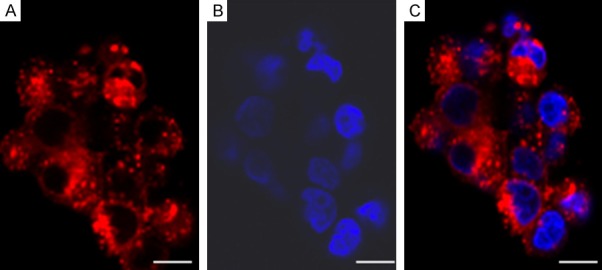
DCs labeled with SPIO in vitro. Images are acquired using fluorescence microscopy. (A) Texas Red particles accumulated in cytoplasm while (B) nuclei are stained blue with DAPI, with coregistration in (C), a merged image. Scale bars: 10 μm.
Figure 4.
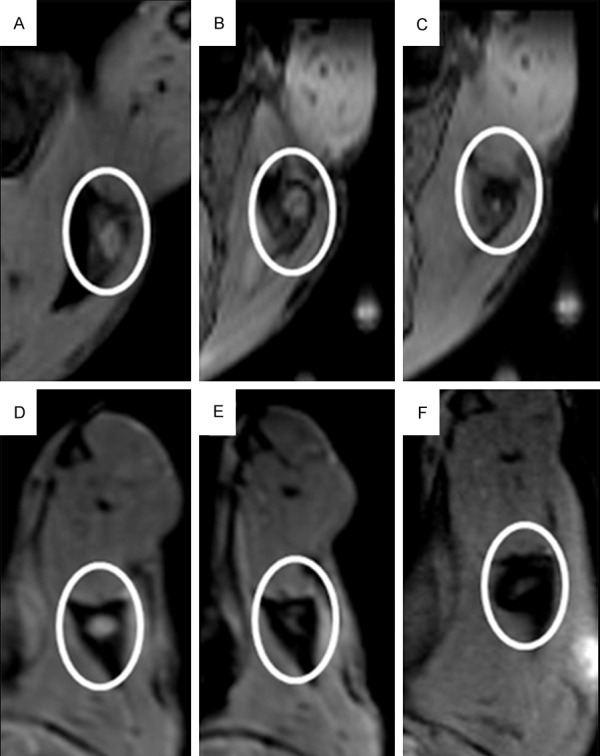
T2-weighted MR images of SPIO-labelled DCs in vivo. Different amounts of DC-based vaccine were injected into left popliteal LN at different time points. (A-C) 1-million DCs were injected at each of 3 consecutive time points: (A) before DCs injection; (B) 6 hours after injection; and (C) 24 hours after injection. In (D-F) 2-million DCs were injected at the same time points as (A-C) respectively.
Table 1.
Recent research in PET and MRI-monitored DC-based vaccines in animal models
| Technique | Labeling | Route of administration | Disease | Reference |
|---|---|---|---|---|
| PET | 18F-FIAU | i.s. | Coccidioidomy cosis | [57] |
| 68Ga-IONP | s.c. | Melanoma | [58] | |
| MRI | Hsp70-SPIONs | s.c. | Glioma | [59] |
| FTH-GFP | s.c. | --- | [60] | |
| N-alkyl-PEI2k-GLY/SPIONs | s.c. | --- | [61] | |
| SPIO | s.c. | --- | [62-66] | |
| SPIO | s.c. | Pancreatic | [67] | |
| Fe NP/IONP | s.c. | Lymphoma | [68] | |
| MNPs/111In-oxine | s.c. | Breast cancer | [69] | |
| HINP | s.c. | --- | [70] | |
| SPIO-EGFP | s.c. | --- | [71] | |
| SPIO-labeled tumor cells | i.d. | Melanoma | [72] |
Abbreviations: i.s.: intranasal; IONP: iron oxide nanoparticles; s.c.: subcutaneous; i.d.: intradermal. Hsp70: heat shock protein 70; SPIONs: superparamagnetic iron oxide nanoparticles; FTH: human ferritin heavy chain; GFP: green fluorescence protein; NP: nanoparticles; IONP: iron oxide nanoparticles; MNPs: paramagnetic nanoparticles; HINP: hybrid imaging nanoprobe (comprised of visible and near-infrared light emitting quantum dots tethered to SPIO).
Conclusion
Immunity plays a central role in the defense against diseases, and DC-based vaccines are postulated as a potential and powerful immunotherapy for cancer. Despite the theoretical possibility of being a complete cure, the clinical outcomes of DC-based cancer vaccine therapy have been rather disappointing. One major obstacle is the low migration rate of injected vaccine cells via different routes. Therefore, assessing the fates of therapeutically administered DCs may be extremely critical to answer important questions and improve this promising approach for cancer therapy.
To date, most of the DC-based cancer vaccines in the clinical settings are administrated subcutaneously, intradermally, intranodally, or intralymphatically, which have been proven to be suboptimal routes. Intraperitoneal injection of DC-based cancer vaccines may be relatively effective for abdominal tumors. The two most frequently used preclinical and clinical imaging methods for non-invasive and real-time monitoring of the distribution and migration of injected DCs are PET and MRI.
With the development of cell labeling and image technologies, in vivo cell tracking performed alone or in a combination should be able to elucidate the dynamics of viable vaccine cells administrated through specific routes and further facilitate the translation from preclinical research to clinical applications with the purpose of boosting immunotherapy outcomes. These advances will likely promote novel DC-based cancer vaccination strategies.
Acknowledgements
This work is supported by the USA National Cancer Institute R01CA209886-01 and R01CA196967.
Disclosure of conflict of interest
None.
References
- 1.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 2.Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. 1973;137:1142–1162. doi: 10.1084/jem.137.5.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. II. Functional properties in vitro. J Exp Med. 1974;139:380–397. doi: 10.1084/jem.139.2.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palucka K, Banchereau J. Dendritic-cellbased therapeutic cancer vaccines. Immunity. 2013;39:38–48. doi: 10.1016/j.immuni.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol. 2016;37:855–865. doi: 10.1016/j.it.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- 7.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 8.Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, Engleman EG, Levy R. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med. 1996;2:52–58. doi: 10.1038/nm0196-52. [DOI] [PubMed] [Google Scholar]
- 9.Bol KF, Schreibelt G, Gerritsen WR, de Vries IJ, Figdor CG. Dendritic cell-based immunotherapy: state of the art and beyond. Clin Cancer Res. 2016;22:1897–1906. doi: 10.1158/1078-0432.CCR-15-1399. [DOI] [PubMed] [Google Scholar]
- 10.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 11.Tarbell KV, Petit L, Zuo X, Toy P, Luo X, Mqadmi A, Yang H, Suthanthiran M, Mojsov S, Steinman RM. Dendritic cell-expanded, islet-specific CD4+ CD25+ CD62L+ regulatory T cells restore normoglycemia in diabetic NOD mice. J Exp Med. 2007;204:191–201. doi: 10.1084/jem.20061631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. 2008;222:162–179. doi: 10.1111/j.1600-065X.2008.00602.x. [DOI] [PubMed] [Google Scholar]
- 13.Adema GJ, de Vries IJ, Punt CJ, Figdor CG. Migration of dendritic cell based cancer vaccines: in vivo veritas? Curr Opin Immunol. 2005;17:170–174. doi: 10.1016/j.coi.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 14.Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5:296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- 15.de Vries IJ, Lesterhuis WJ, Scharenborg NM, Engelen LP, Ruiter DJ, Gerritsen MJ, Croockewit S, Britten CM, Torensma R, Adema GJ, Figdor CG, Punt CJ. Maturation of dendritic cells is a prerequisite for inducing immune responses in advanced melanoma patients. Clin Cancer Res. 2003;9:5091–5100. [PubMed] [Google Scholar]
- 16.McIlroy D, Gregoire M. Optimizing dendritic cell-based anticancer immunotherapy: maturation state does have clinical impact. Cancer Immunol Immunother. 2003;52:583–591. doi: 10.1007/s00262-003-0414-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morse MA, Mosca PJ, Clay TM, Lyerly HM. Dendritic cell maturation in active immunotherapy strategies. Expert Opin Biol Ther. 2002;2:35–43. doi: 10.1517/14712598.2.1.35. [DOI] [PubMed] [Google Scholar]
- 18.Zou GM, Tam YK. Cytokines in the generation and maturation of dendritic cells: recent advances. Eur Cytokine Netw. 2002;13:186–199. [PubMed] [Google Scholar]
- 19.Williams LA, Egner W, Hart DN. Isolation and function of human dendritic cells. Int Rev Cytol. 1994;153:41–103. doi: 10.1016/s0074-7696(08)62188-9. [DOI] [PubMed] [Google Scholar]
- 20.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colonystimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bowers WE, Berkowitz MR. Differentiation of dendritic cells in cultures of rat bone-marrow cells. J Exp Med. 1986;163:872–883. doi: 10.1084/jem.163.4.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collin M, McGovern N, Haniffa M. Human dendritic cell subsets. Immunology. 2013;140:22–30. doi: 10.1111/imm.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Datta J, Terhune JH, Lowenfeld L, Cintolo JA, Xu S, Roses RE, Czerniecki BJ. Optimizing dendritic cell-based approaches for cancer immunotherapy. Yale J Biol Med. 2014;87:491–518. [PMC free article] [PubMed] [Google Scholar]
- 24.Mahnke K, Schmitt E, Bonifaz L, Enk AH, Jonuleit H. Immature, but not inactive: the tolerogenic function of immature dendritic cell. Immunol Cell Biol. 2002;80:477–483. doi: 10.1046/j.1440-1711.2002.01115.x. [DOI] [PubMed] [Google Scholar]
- 25.Wimmers F, Schreibelt G, Skold AE, Figdor CG, De Vries IJ. Paradigm shift in dendritic cellbased immunotherapy: from in vitro generated monocyte-derived DCs to naturally circulating DC subsets. Front Immunol. 2014;5:165. doi: 10.3389/fimmu.2014.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre E, Vu Manh TP, Baranek T, Storset AK, Marvel J, Boudinot P, Hosmalin A, Schwartz-Cornil I, Dalod M. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J Exp Med. 2010;207:1283–1292. doi: 10.1084/jem.20100223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Keeffe M, Hochrein H, Vremec D, Caminschi I, Miller JL, Anders EM, Wu L, Lahoud H, Henri S, Scott B, Hertzog P, Tatarczuch L, Shortman K. Mouse plasmacytoid cells: long-lived cells, heterogeneous in surface phenotype and function, that differentiate into CD8(+) dendritic cells only after microbial stimulus. J Exp Med. 2002;196:1307–1319. doi: 10.1084/jem.20021031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlitzer A, Ginhoux F. Organization of the mouse and human DC network. Curr Opin Immunol. 2014;26:90–99. doi: 10.1016/j.coi.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7:790–802. doi: 10.1038/nri2173. [DOI] [PubMed] [Google Scholar]
- 30.Tureci O, Vormehr M, Diken M, Kreiter S, Huber C, Sahin U. Targeting the heterogeneity of cancer with individualized neoepitope vaccines. Clin Cancer Res. 2016;22:1885–1896. doi: 10.1158/1078-0432.CCR-15-1509. [DOI] [PubMed] [Google Scholar]
- 31.Strioga MM, Felzmann T, Powell DJ Jr, Ostapenko V, Dobrovolskiene NT, Matuskova M, Michalek J, Schijns VE. Therapeutic dendritic cellbased cancer vaccines: the state of the art. Crit Rev Immunol. 2013;33:489–547. doi: 10.1615/critrevimmunol.2013008033. [DOI] [PubMed] [Google Scholar]
- 32.Fong L, Brockstedt D, Benike C, Wu L, Engleman EG. Dendritic cells injected via different routes induce immunity in cancer patients. J Immunol. 2001;166:4254–4259. doi: 10.4049/jimmunol.166.6.4254. [DOI] [PubMed] [Google Scholar]
- 33.Quillien V, Moisan A, Carsin A, Lesimple T, Lefeuvre C, Adamski H, Bertho N, Devillers A, Leberre C, Toujas L. Biodistribution of radiolabelled human dendritic cells injected by various routes. Eur J Nucl Med Mol Imaging. 2005;32:731–741. doi: 10.1007/s00259-005-1825-9. [DOI] [PubMed] [Google Scholar]
- 34.Budimir N, de Haan A, Meijerhof T, Gostick E, Price DA, Huckriede A, Wilschut J. Heterosubtypic cross-protection induced by whole inactivated influenza virus vaccine in mice: influence of the route of vaccine administration. Influenza Other Respir Viruses. 2013;7:1202–1209. doi: 10.1111/irv.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujkuyama Y, Tokuhara D, Kataoka K, Gilbert RS, McGhee JR, Yuki Y, Kiyono H, Fujihashi K. Novel vaccine development strategies for inducing mucosal immunity. Expert Rev Vaccines. 2012;11:367–379. doi: 10.1586/erv.11.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu Q, Talton J, Zhang G, Cunningham T, Wang Z, Waters RC, Kirk J, Eppler B, Klinman DM, Sui Y, Gagnon S, Belyakov IM, Mumper RJ, Berzofsky JA. Large intestine-targeted, nanoparticle-releasing oral vaccine to control genitorectal viral infection. Nat Med. 2012;18:1291–1296. doi: 10.1038/nm.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malik B, Rath G, Goyal AK. Are the anatomical sites for vaccine administration selected judiciously? Int Immunopharmacol. 2014;19:17–26. doi: 10.1016/j.intimp.2013.12.023. [DOI] [PubMed] [Google Scholar]
- 38.Mackensen A, Krause T, Blum U, Uhrmeister P, Mertelsmann R, Lindemann A. Homing of intravenously and intralymphatically injected human dendritic cells generated in vitro from CD34+ hematopoietic progenitor cells. Cancer Immunol Immunother. 1999;48:118–122. doi: 10.1007/s002620050555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilgenhof S, Van Nuffel AM, Benteyn D, Corthals J, Aerts C, Heirman C, Van Riet I, Bonehill A, Thielemans K, Neyns B. A phase IB study on intravenous synthetic mRNA electroporated dendritic cell immunotherapy in pretreated advanced melanoma patients. Ann Oncol. 2013;24:2686–2693. doi: 10.1093/annonc/mdt245. [DOI] [PubMed] [Google Scholar]
- 40.Hao SG, Ye ZM, Yang JC, Bai O, Xiang J. Intradermal vaccination of dendritic cell-derived exosomes is superior to a subcutaneous one in the induction of antitumor immunity. Cancer Biother Radiopharm. 2006;21:146–154. doi: 10.1089/cbr.2006.21.146. [DOI] [PubMed] [Google Scholar]
- 41.Morse MA, Coleman RE, Akabani G, Niehaus N, Coleman D, Lyerly HK. Migration of human dendritic cells after injection in patients with metastatic malignancies. Cancer Res. 1999;59:56–58. [PubMed] [Google Scholar]
- 42.Verdijk P, Aarntzen EH, Lesterhuis WJ, Boullart AC, Kok E, van Rossum MM, Strijk S, Eijckeler F, Bonenkamp JJ, Jacobs JF, Blokx W, Vankrieken JH, Joosten I, Boerman OC, Oyen WJ, Adema G, Punt CJ, Figdor CG, de Vries IJ. Limited amounts of dendritic cells migrate into the TCell area of lymph nodes but have high immune activating potential in melanoma patients. Clin Cancer Res. 2009;15:2531–2540. doi: 10.1158/1078-0432.CCR-08-2729. [DOI] [PubMed] [Google Scholar]
- 43.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 44.Lesterhuis WJ, de Vries IJ, Schreibelt G, Lambeck AJ, Aarntzen EH, Jacobs JF, Scharenborg NM, van de Rakt MW, de Boer AJ, Croockewit S, van Rossum MM, Mus R, Oyen WJ, Boerman OC, Lucas S, Adema GJ, Punt CJ, Figdor CG. Route of administration modulates the induction of dendritic cell vaccine-induced antigenspecific T cells in advanced melanoma patients. Clin Cancer Res. 2011;17:5725–5735. doi: 10.1158/1078-0432.CCR-11-1261. [DOI] [PubMed] [Google Scholar]
- 45.Boyden AW, Legge KL, Waldschmidt TJ. Pulmonary infection with influenza a virus induces site-specific germinal center and T follicular helper cell responses. PLoS One. 2012;7:e40733. doi: 10.1371/journal.pone.0040733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dyer CM, Lew AM. Antigen targeted to secondary lymphoid organs via vascular cell adhesion molecule (VCAM) enhances an immune response. Vaccine. 2003;21:2115–2121. doi: 10.1016/s0264-410x(02)00742-9. [DOI] [PubMed] [Google Scholar]
- 47.Okamoto N, Chihara R, Shimizu C, Nishimoto S, Watanabe T. Artificial lymph nodes induce potent secondary immune responses in naive and immunodeficient mice. J Clin Invest. 2007;117:997–1007. doi: 10.1172/JCI30379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Green MV, Seidel J, Vaquero JJ, Jagoda E, Lee I, Eckelman WC. High resolution PET, SPECT and projection imaging in small animals. Comput Med Imaging Graph. 2001;25:79–86. doi: 10.1016/s0895-6111(00)00057-4. [DOI] [PubMed] [Google Scholar]
- 49.Adonai N, Nguyen KN, Walsh J, Iyer M, Toyokuni T, Phelps ME, McCarthy T, McCarthy DW, Gambhir SS. Ex vivo cell labeling with 64Cupyruvaldehydebis(N4-methylthiosemicarbazone) for imaging cell trafficking in mice with positron-emission tomography. Proc Natl Acad Sci U S A. 2002;99:3030–3035. doi: 10.1073/pnas.052709599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Acton PD, Zhou R. Imaging reporter genes for cell tracking with PET and SPECT. Q J Nucl Med Mol Imaging. 2005;49:349–360. [PubMed] [Google Scholar]
- 51.Modo M, Hoehn M, Bulte JW. Cellular MR imaging. Mol Imaging. 2005;4:143–164. doi: 10.1162/15353500200505145. [DOI] [PubMed] [Google Scholar]
- 52.Kircher MF, Gambhir SS, Grimm J. Noninvasive cell-tracking methods. Nat Rev Clin Oncol. 2011;8:677–688. doi: 10.1038/nrclinonc.2011.141. [DOI] [PubMed] [Google Scholar]
- 53.Bulte JW, Douglas T, Witwer B, Zhang SC, Strable E, Lewis BK, Zywicke H, Miller B, van Gelderen P, Moskowitz BM, Duncan ID, Frank JA. Magnetodendrimers allow endosomal magnetic labeling and in vivo tracking of stem cells. Nat Biotechnol. 2001;19:1141–1147. doi: 10.1038/nbt1201-1141. [DOI] [PubMed] [Google Scholar]
- 54.Aime S, Carrera C, Delli Castelli D, Geninatti Crich S, Terreno E. Tunable imaging of cells labeled with MRI-PARACEST agents. Angew Chem Int Ed Engl. 2005;44:1813–1815. doi: 10.1002/anie.200462566. [DOI] [PubMed] [Google Scholar]
- 55.Partlow KC, Chen J, Brant JA, Neubauer AM, Meyerrose TE, Creer MH, Nolta JA, Caruthers SD, Lanza GM, Wickline SA. 19F magnetic resonance imaging for stem/progenitor cell tracking with multiple unique perfluorocarbon nanobeacons. FASEB J. 2007;21:1647–1654. doi: 10.1096/fj.06-6505com. [DOI] [PubMed] [Google Scholar]
- 56.Smith CA, de la Fuente J, Pelaz B, Furlani EP, Mullin M, Berry CC. The effect of static magnetic fields and tat peptides on cellular and nuclear uptake of magnetic nanoparticles. Biomaterials. 2010;31:4392–4400. doi: 10.1016/j.biomaterials.2010.01.096. [DOI] [PubMed] [Google Scholar]
- 57.Vilekar P, Awasthi V, Lagisetty P, King C, Shankar N, Awasthi S. In vivo trafficking and immunostimulatory potential of an intranasallyadministered primary dendritic cell-based vaccine. BMC Immunol. 2010;11:60. doi: 10.1186/1471-2172-11-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ruiz-de-Angulo A, Zabaleta A, Gomez-Vallejo V, Llop J, Mareque-Rivas JC. Microdosed lipidCoated (67)Ga-magnetite enhances antigenspecific immunity by image tracked delivery of antigen and CpG to lymph nodes. ACS Nano. 2016;10:1602–1618. doi: 10.1021/acsnano.5b07253. [DOI] [PubMed] [Google Scholar]
- 59.Shevtsov MA, Nikolaev BP, Yakovleva LY, Parr MA, Marchenko YY, Eliseev I, Yudenko A, Dobrodumov AV, Zlobina O, Zhakhov A, Ischenko AM, Pitkin E, Multhoff G. 70-kDa heat shock protein coated magnetic nanocarriers as a nanovaccine for induction of anti-tumor immune response in experimental glioma. J Control Release. 2015;220:329–340. doi: 10.1016/j.jconrel.2015.10.051. [DOI] [PubMed] [Google Scholar]
- 60.Kim HS, Woo J, Lee JH, Joo HJ, Choi Y, Kim H, Moon WK, Kim SJ. In vivo tracking of dendritic cell using MRI reporter gene, Ferritin. PLoS One. 2015;10:e0125291. doi: 10.1371/journal.pone.0125291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu Y, Wu C, Zhu W, Xia C, Wang D, Zhang H, Wu J, Lin G, Wu B, Gong Q, Song B, Ai H. Superparamagnetic MRI probes for in vivo tracking of dendritic cell migration with a clinical 3 T scanner. Biomaterials. 2015;58:63–71. doi: 10.1016/j.biomaterials.2015.04.016. [DOI] [PubMed] [Google Scholar]
- 62.Crisci E, Fraile L, Novellas R, Espada Y, Cabezon R, Martinez J, Cordoba L, Barcena J, Benitez-Ribas D, Montoya M. In vivo tracking and immunological properties of pulsed porcine monocyte-derived dendritic cells. Mol Immunol. 2015;63:343–354. doi: 10.1016/j.molimm.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 63.de Chickera S, Willert C, Mallet C, Foley R, Foster P, Dekaban GA. Cellular MRI as a suitable, sensitive non-invasive modality for correlating in vivo migratory efficiencies of different dendritic cell populations with subsequent immunological outcomes. Int Immunol. 2012;24:29–41. doi: 10.1093/intimm/dxr095. [DOI] [PubMed] [Google Scholar]
- 64.de Chickera SN, Snir J, Willert C, Rohani R, Foley R, Foster PJ, Dekaban GA. Labelling dendritic cells with SPIO has implications for their subsequent in vivo migration as assessed with cellular MRI. Contrast Media Mol Imaging. 2011;6:314–327. doi: 10.1002/cmmi.433. [DOI] [PubMed] [Google Scholar]
- 65.Dekaban GA, Snir J, Shrum B, de Chickera S, Willert C, Merill M, Said EA, Sekaly RP, Foster PJ, O’Connell PJ. Semiquantitation of mouse dendritic cell migration in vivo using cellular MRI. J Immunother. 2009;32:240–251. doi: 10.1097/CJI.0b013e318197b2a0. [DOI] [PubMed] [Google Scholar]
- 66.Zhang XZ, De Chickera SN, Willert C, Economopoulos V, Noad J, Rohani R, Wang AY, Levings MK, Scheid E, Foley R, Foster PJ, Dekaban GA. Cellular magnetic resonance imaging of monocyte-derived dendritic cell migration from healthy donors and cancer patients as assessed in a scid mouse model. Cytotherapy. 2011;13:1234–1248. doi: 10.3109/14653249.2011.605349. [DOI] [PubMed] [Google Scholar]
- 67.Zhang ZL, Li WG, Procissi D, Li KG, Sheu AY, Gordon AC, Guo Y, Khazaie K, Huan Y, Han GH, Larson AC. Antigen-loaded dendritic cell migration: MR imaging in a pancreatic carcinoma model. Radiology. 2015;274:192–200. doi: 10.1148/radiol.14132172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ferguson PM, Slocombe A, Tilley RD, Hermans IF. Using magnetic resonance imaging to evaluate dendritic cell-based vaccination. PLoS One. 2013:8. doi: 10.1371/journal.pone.0065318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martelli C, Borelli M, Ottobrini L, Rainone V, Degrassi A, Russo M, Gianelli U, Bosari S, Fiorini C, Trabattoni D, Clerici M, Lucignani G. In vivo imaging of lymph node migration of MNPand (111)In-labeled dendritic cells in a transgenic mouse model of breast cancer (MMTVRas) Mol Imaging Biol. 2012;14:183–196. doi: 10.1007/s11307-011-0496-0. [DOI] [PubMed] [Google Scholar]
- 70.Mackay PS, Kremers GJ, Kobukai S, Cobb JG, Kuley A, Rosenthal SJ, Koktysh DS, Gore JC, Pham W. Multimodal imaging of dendritic cells using a novel hybrid magneto-optical nanoprobe. Nanomedicine. 2011;7:489–496. doi: 10.1016/j.nano.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mou YB, Hou YY, Chen BA, Hua ZC, Zhang Y, Xie H, Xia GH, Wang ZY, Huang XF, Han W, Ni YH, Hu QG. In vivo migration of dendritic cells labeled with synthetic superparamagnetic iron oxide. Int J Nanomedicine. 2011;6:2633–2640. doi: 10.2147/IJN.S24307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Long CM, van Laarhoven HW, Bulte JW, Levitsky HI. Magnetovaccination as a novel method to assess and quantify dendritic cell tumor antigen capture and delivery to lymph nodes. Cancer Research. 2009;69:3180–3187. doi: 10.1158/0008-5472.CAN-08-3691. [DOI] [PMC free article] [PubMed] [Google Scholar]