

Abstract
Objective
The objective of this study was to compare the DNA methylation profile in the longissimus dorsi muscle between Small Tailed Han and Dorper×Small Tailed Han crossbred sheep which were known to exhibit significant difference in meat-production.
Methods
Six samples (three in each group) were subjected to the methylated DNA immunoprecipitation sequencing (MeDIP-seq) and subsequent bioinformatics analyses to detect differentially methylated regions (DMRs) between the two groups.
Results
23.08 Gb clean data from six samples were generated and 808 DMRs were identified in gene body or their neighboring up/downstream regions. Compared with Small Tailed Han sheep, we observed a tendency toward a global loss of DNA methylation in these DMRs in the crossbred group. Gene ontology enrichment analysis found several gene sets which were hypo-methylated in gene-body region, including nucleoside binding, motor activity, phospholipid binding and cell junction. Numerous genes were found to be differentially methylated between the two groups with several genes significantly differentially methylated, including transforming growth factor beta 3 (TGFB3), acyl-CoA synthetase long chain family member 1 (ACSL1), ryanodine receptor 1 (RYR1), acyl-CoA oxidase 2 (ACOX2), peroxisome proliferator activated receptor-gamma2 (PPARG2), netrin 1 (NTN1), ras and rab interactor 2 (RIN2), microtubule associated protein RP/EB family member 1 (MAPRE1), ADAM metallopeptidase with thrombospondin type 1 motif 2 (ADAMTS2), myomesin 1 (MYOM1), zinc finger, DHHC type containing 13 (ZDHHC13), and SH3 and PX domains 2B (SH3PXD2B). The real-time quantitative polymerase chain reaction validation showed that the 12 genes are differentially expressed between the two groups.
Conclusion
In the current study, a tendency to a global loss of DNA methylation in these DMRs in the crossbred group was found. Twelve genes, TGFB3, ACSL1, RYR1, ACOX2, PPARG2, NTN1, RIN2, MAPRE1, ADAMTS2, MYOM1, ZDHHC13, and SH3PXD2B, were found to be differentially methylated between the two groups by gene ontology enrichment analysis. There are differences in the expression of 12 genes, of which ACSL1, RIN2, and ADAMTS2 have a negative correlation with methylation levels and the data suggest that DNA methylation levels in DMRs of the 3 genes may have an influence on the expression. These results will serve as a valuable resource for DNA methylation investigations on screening candidate genes which might be related to meat production in sheep.
Keywords: DNA Methylation, Small Tailed Han Sheep, Dorper×Small Tailed Han Crossbred Sheep, Methylated DNA Immunoprecipitation Sequencing (MeDIP-seq), Longissimus Dorsi Muscle
INTRODUCTION
China is the largest mutton producer and consumer in the world. However, China does not have good indigenous commercial mutton sheep breeds, and hybridization of local breeds with known commercial mutton sheep breeds has been widely used.
The Small Tailed Han sheep is a Chinese indigenous breed that is famous for its precociousness and prolificacy [1]. The Dorper sheep, which is originated from South Africa, is well known for its hardiness, early maturity, and rapid growth [2]. Recently, the Dorper sheep has been imported into China as a meat sire breed to improve growth performance and carcass traits of the local Small Tailed Han sheep. The Dorper×Small Tailed Han crossbred sheep has therefore become a widely reared breed for mutton production in northern China. Compared with Small Tailed Han, the Dorper×Small Tailed Han crossbred sheep exhibited higher carcass weight, net meat weight, and greater dressing percentage. However, the underlying molecular mechanism for the growth and meat production differences remains unclear.
Currently, extensive genetic studies have identified many genetic polymorphisms affecting growth in sheep [3,4]. However, polymorphisms or quantitative trait loci cannot provide adequate explanations for them. Recently, DNA methylation, an important epigenetic mechanism in eukaryotes, has received considerable attention because of its potential effect on complex traits. It is believed that DNA methylation regulates many biological processes, including gene expression, genomic imprinting, and X chromosome inactivation [5–7]. Using the methylated DNA immunoprecipitation-sequencing (MeDIP-seq) technology, methylation studies have been extensively conducted in animals [8–10].
In this study, we aimed to survey the genome-wide DNA methylation pattern in the longissimus dorsi muscle (LDM) to identify methylated genes which contributed to the differences in growth and carcass traits between Small Tailed Han and Dorper×Small Tailed Han crossbred sheep. Our data revealed the DNA methylome of the two breeds, identified differentially methylated genes between breeds and genes related to meat production.
MATERIALS AND METHODS
Animal care
This study was approved by the Animal Ethics Committee of Jilin Academy of Agricultural Sciences. Sampling was carried out according to the “Guidelines on Ethical Treatment of Experimental Animals” (2006) established by the Ministry of Science and Technology, China.
Sample collection
Animals used in this study were from two breeds: Dorper×Small Tailed Han crossbred (F1, T) and Small Tailed Han sheep (C). The animals were all weaned at 4 months of age and raised from weaning to slaughter on a diet following the National Research Council (NRC) standard. The animals were raised in semi-confinement in the central region of Jilin province, China from March, 2015. The ewes in both groups were raised under same conditions on feed and water ad libitum and were humanely sacrificed at 280 days. Longissimus dorsi muscle tissues from six ewes (three in each group) were collected and snap frozen in liquid nitrogen until DNA or RNA extraction. DNA from these tissues was extracted with standard phenol chloroform method. Total RNA was isolated from each sample with RnaEx Total RNA Isolation Solution (GK3006, GENEray, Shanghai, China) according to the manufacturer’s instructions.
The methylated DNA immunoprecipitation-sequencing
MeDIP DNA libraries were prepared for a total of six samples (three in each group) following the protocol as previously described [11]. Briefly, DNA was fragmented to approximately 100 to 500 bp using a Bioruptor sonicator (Diagenode Inc., Denville, NJ, USA). Sequencing libraries were constructed with the Paired-End DNA Sample Prep kit (Illumina Inc., San Diego, CA, USA) following the manufacturer’s instructions. Adaptor-ligated DNA was immunoprecipitated by a monoclonal anti-methylcytidine antibody (Diagenode, USA). Quantitative real-time polymerase chain reaction (qRT-PCR) analysis was performed to validate the quality of immunoprecipitated fragments. DNA fragments of 200 to 300 bp were excised from the gel and purified using a gel extraction kit (Qiagen, Inc., Valencia, CA, USA). The extracted fragments were quantified using the Agilent 2100 Analyzer (Agilent Technologies, Palo Alto, CA, USA). Enriched fragments were amplified by adaptor-mediated PCR. DNA libraries were subjected to paired-end sequencing with a 50 bp read length using the Illumina HiSeq 2000 platform (Illumina, USA).
Bioinformatics analysis
Raw sequencing data were first processed to filter out low-quality reads containing adaptors or low-quality bases. The clean data were then aligned to the Ovis aries reference genome (oviAri3) using the Burrows-Wheeler Alignment software [12]. Uniquely mapped reads were retained for subsequent analyses.
Genome-wide methylation peak scanning was conducted using the model-based analysis of ChIP-Seq (MACS) [13]. The distribution of peaks in different regions of the sheep genome in each sample, including the promoter, 5′-untranslated region (UTR), 3′-UTR, exons, introns, downstream (2 kbp), CpG islands, and repeats, were analyzed. A CGI was defined using the following three criteria: i) greater than 200 bp in length; ii) GC content ≥50%; and iii) CpG observed/expected ratio ≥0.6. Gene information was downloaded from the public File Transfer Protocol site of Ensembl (ftp://ftp.ensembl.org/pub/release-75/gtf/ovis_aries/Ovis_aries.Oar_v3.1.75.gtf.gz).
To identify differentially methylated region (DMRs) in the samples, their peaks were merged and the differences in the number of reads within those peaks between the two groups were analyzed using the t-test (p<0.01).
All genes containing DMRs were used for subsequent gene ontology and Kyoto encyclopedia of genes and genomes pathway enrichment analyses using the database for annotation, visualization and integrated discovery (DAVID) web server (http://david.abcc.ncifcrf.gov/). Genes were mapped to their respective human orthologs and then submitted to DAVID for enrichment analysis, and the significant threshold was set as Benjamini-corrected p value <0.05.
Bisulphite sequencing PCR validation
Genomic DNA from the T and C sheep was treated with bisulphite sodium using the QIAGEN DNA Methylation Kit (cat: 59824). The bisulphite-treated DNA was used for PCR. PCR primers used are listed in Supplementary Table S2. PCR was performed using the following program: 95°C for 4 min; 40 cycles of 94°C for 30 s, 55°C for 40 s and 72°C for 40 s; and 72°C for 5 min. The PCR products were detected by gel electrophoresis and cloned into the pTG19-T vector (Generay, Shanhai, China). Ten positive clones for each gene per sample were randomly selected for sequencing (BGI, ShenZhen, China). The final sequence results were processed by BiQ Analyzer (v2.0).
Real-time quantitative PCR validation
Total RNA was quantified by the NanoDrop ND-2000 (Thermo Scientific, Wilmington, DE, USA) and the RNA integrity was assessed using agarose gel electrophoresis. SYBR Green PCR amplification was performed on ABI7500 (Applied Biosystems Inc., Foster City, CA, USA). The real-time quantitative PCR reaction contained 10 μL of Power qPCR PreMix (GK8020, GENEray, Shanghai, China), 20 ng of diluted cDNA, and 5 μM of each primer (Supplementary Table Table S3) contributing a total volume of 20 μL. Cycling conditions were as follows: 95°C for 10 min, 95°C for 10 s, 60°C for 34 s, 95°C for 15 s for 40 cycles, followed by melt analysis from 60°C to 95°C. After completion of the reaction, the data analysis was based on the obtained sample Ct value. 2−ΔCt method was used to analysis outputting data. β-Action was used as an internal reference gene.
RESULTS
Characteristics of Dorper×Small Tailed Han crossbred (F1) and Small Tailed Han sheep
As shown in Table 1, in general, the mean carcass weight of the crossbred sheep (30.53±2.60) was significantly higher than that of the Small Tailed Han sheep (24.33±1.86). The net meat weight, dressing percentage and meat percentage were also higher in the crossbred sheep than in the Small Tailed Han sheep. In addition, although not statistically significant, the shear force of the crossbred group was lower than that of the Small Tailed Han sheep, indicating that meat of the crossbred group was tenderer.
Table 1.
Characteristics of the Dorper×Small Tailed Han crossbred (F1) and Small Tailed Han sheep
| Small Tailed Han | Dorper×Small Tailed Han crossbred | |
|---|---|---|
| Weaning weight (kg) | 22.86±4.12 | 22.54±4.20 |
| Final weight (kg) | 47.89±8.81 | 56.57±9.38* |
| Cooked meat percentage (%) | 57.98±1.36 | 58.10±0.82 |
| Shear force (kgf) | 3.38±0.44 | 3.07±0.50 |
| Carcass weight (kg) | 24.33±1.86 | 30.53±2.60* |
| Net meat weight (kg) | 18.30±0.85 | 24.24±2.72* |
| Dressing percentage (%) | 49.01±1.64 | 52.62±2.30* |
| Meat percentage (%) | 38.36±1.95 | 42.06±2.48* |
p<0.05.
Landscape of the DNA methylomes
In the present study, as shown in Table 2, 23.08 Gb clean data were generated from six samples by MeDIP-seq (~3.85 Gb per sample). Approximately 93.65% of the clean reads were mapped to the reference genome, and approximately 56.54% of reads were uniquely mapped. MeDIP-Seq reads were detected in most chromosomal regions (chromosomes 1–26, and chromosome X) in each group. Figure 1 shows the distribution of MeDIP-seq reads in different CG density regions. The densities of 10 to 15 CpGs had the highest percentage of reads in both groups.
Table 2.
Summary of sequence read alignments to the reference genome and genes of MeDIP-Seq data for the Small Tailed Han (C) and Dorper×Small Tailed Han crossbred (T) libraries
| Sample | Total reads | Mapped reads | Unique mapped reads | Genome mean depth |
|---|---|---|---|---|
| C13 | 35965704 | 33407122 | 21829729 | 2.5× |
| C14 | 30590924 | 28575092 | 16517504 | 2.3× |
| C7 | 40647652 | 37889408 | 25692821 | 2.7× |
| T10 | 40073504 | 37651517 | 21456357 | 2.4× |
| T5 | 43973672 | 41451188 | 24290732 | 2.6× |
| T8 | 39585158 | 37221630 | 20727942 | 2.5× |
Figure 1.

The distribution of the methylated DNA immunoprecipitation-sequencing (MeDIP-seq) reads in different CG density regions.
To further reveal the genome-wide methylation pattern, MeDIP-Seq reads distribution were analyzed in 2 kb region upstream of the transcription start sites (TSS), 5′-UTRs, coding DNA sequence (CDS), introns, 3′-UTRs, 2 kb region downstream of the transcription termination site (TTS) and repeats (Figure 2). The results showed that uniquely mapped reads were mainly present in repeat elements (32.49% in Small Tailed Han and 34.36% in the crossbred group). This was probably because the total length of the repeats is much longer than that of other elements. Among all DNA elements (TSS, CDS, intron, 5′UTR, 3′UTR, and TTS), gene body (CDS, intron, 5′UTR, and 3′ UTR) had a higher level of DNA methylation than the flanking regions of genes.
Figure 2.

MeDIP-Seq reads distribution in 2 kb region upstream of the transcription start sites (TSS), 5′-untranslated regions (UTRs), coding DNA sequence (CDS), introns, 3′-UTRs, 2 kb region downstream of the TTS and repeats.
The uniquely mapped reads were further used to detect the peaks (methylation-enriched regions). As shown in Supplementary Table S1, an average of 106,061 and 105,400 peaks were detected in the Small Tailed Han and the crossbred group, respectively. We further analyzed the peak distribution in different components of the genome. Figure 3 shows the CpGs numbers in peaks. Most of the peaks (approximately 22%) had 10 to 15 CpG sites. Analysis of peak distribution in different components of the genome showed that the peaks were mainly in in the introns and the CDS (Figure 4).
Figure 3.

The distribution of CpGs numbers in peaks.
Figure 4.

Peak distribution in different components of the genome.
The peaks of the two groups of samples were merged as candidates (DMRs). We used t-test to detect the DMRs between the two groups. The results showed that there were a total of 42 promoters of differentially methylated genes between the Small Tailed Han and the crossbred group, of which 34 were down methylated and 8 were up methylated in the LDM in the crossbred group compared to the Small Tailed Han (Table 3; Figure 5). In other words, the DNA methylation level in the LDM was decreased in the crossbred group compared to the Small Tailed Han group. In addition, 746 DMRs were detected in gene bodies or downstream of the gene, suggesting that more DMRs were in gene body regions rather than promoters, and the DMRs were more prone to be hypomethylated in the crossbred group, which indicated that global methylation loss might be related to the changes of meat quality.
Table 3.
Numbers of genes showing differential methylation in different gene regions
| Small Tailed Han vs crossbred | Differentially methylated gene | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Upstream 2 kb | 5′-UTR | CDS | Intron | 3′-UTR | Downstream 2 kb | |
| Hyper-methylation | 26 | 2 | 90 | 87 | 4 | 3 |
| Hypo-methylation | 64 | 2 | 181 | 305 | 9 | 15 |
UTR, untranslated region; CDS, coding DNA sequence.
Figure 5.
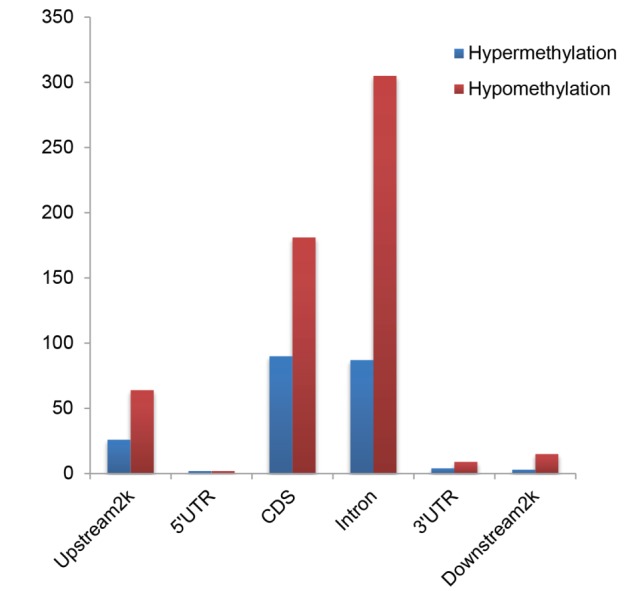
Distribution of the Hypermethylated and Hypomethylated genes.
Functional enrichment analysis for genes with DMRs
To examine the potential functions of the genes which showed differential methylation status, we performed an enrichment analysis for genes with DMRs in gene body and flanking regions (2 kb). DMRs on chromosome X were excluded from this analysis since DNA methylation concentrates on the X chromosome due to X chromosome inactivation. After multiple testing corrections, as shown in Table 4, all genes with DMR in their flanking region or gene-body were enriched in nucleoside binding (p = 0.0048), motor activity (p = 0.0050), ATP binding (p = 0.0074), dynein complex (p = 0.0085), vesicle-mediated transport (p = 0.0165) and cytoskeleton (p = 0.0167). Hypermethylated genes with DMRs in their promoters and gene-body were not significantly enriched in any pathways or gene ontology items which might be due to the relative small number of these genes. As shown in Table 4, gene-body hypo-methylated genes were significantly enriched for the molecular function of nucleoside binding (p = 0.0245), motor activity (p = 0.0248), phospholipid binding (p = 0.0385), and cell junction (p = 0.0395).
Table 4.
Gene ontology analysis of all DMR related genes and gene-body hypomethylated genes
| Category | Term | Description | Gene count | p value |
|---|---|---|---|---|
| All DMR related genes | ||||
| Molecular function | GO:0001882 | Nucleoside binding | 94 | 0.0048 |
| Molecular function | GO:0003774 | Motor activity | 18 | 0.0050 |
| Molecular function | GO:0032559 | Adenyl ribonucleotide binding | 89 | 0.0061 |
| Molecular function | GO:0001883 | Purine nucleoside binding | 94 | 0.0073 |
| Molecular function | GO:0005524 | ATP binding | 89 | 0.0074 |
| Cellular component | GO:0030286 | Dynein complex | 9 | 0.0085 |
| Molecular function | GO:0030554 | Adenyl nucleotide binding | 94 | 0.0121 |
| Biological process | GO:0016192 | Vesicle-mediated transport | 45 | 0.0165 |
| Cellular component | GO:0005856 | Cytoskeleton | 84 | 0.0167 |
| Gene-body hypomethylated genes | ||||
| Molecular function | GO:0001882 | Nucleoside binding | 63 | 0.0245 |
| Molecular function | GO:0003774 | Motor activity | 13 | 0.0248 |
| Molecular function | GO:0001883 | Purine nucleoside binding | 63 | 0.0303 |
| Molecular function | GO:0032559 | Adenyl ribonucleotide binding | 60 | 0.0371 |
| Molecular function | GO:0005543 | Phospholipid binding | 14 | 0.0385 |
| Cellular component | GO:0030054 | Cell junction | 30 | 0.0395 |
| Molecular function | GO:0030554 | Adenyl nucleotide binding | 63 | 0.0396 |
DMR, differentially methylated regions.
Genes involved in production
To further explore the potential roles of genes involved in production, we selected genes with DMRs in their flanking regions and gene bodies which are also known to be associated with sheep meat production. As shown in Table 5 [4,14–19], seven methylated genes identified in the present study are previously reported to be involved in carcass traits in animals.
Table 5.
Known meat production related genes included in the list of genes with differentially methylated regions (DMRs) in their flanking region and gene bodies
| Differentially methylated regions (DMR) | Previous published studies for DMR related genes | |||||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| Chromosome | Start | End | Type | Gene elements | Gene | Animal | Phenotype | Reference |
| 14 | 47508715 | 47510424 | hypomethylation | Exon | RYR1 | Pig | Daily weight gain | Kadarmideen [14] |
| 19 | 56580593 | 56581899 | hypomethylation | Exon | PPARG2 | Cattle | Carcass traits | Fan et al [15] |
| 7 | 84215883 | 84217928 | hypermethylation | Promoter | TGFB3 | Chicken | Growth traits | Jin et al [16] |
| 5 | 1961358 | 1961794 | hypermethylation | Intron | ADAMTS2 | Sheep | Post-weaning gain | Zhang et al [4] |
| 5 | 1961358 | 1961794 | hypermethylation | Intron | ADAMTS2 | Cattle | fat content of muscle | Lee et al [17] |
| 11 | 28039026 | 28040412 | hypomethylation | Intron | NTN1 | Sheep | Post-weaning gain | Zhang et al [4] |
| 16 | 4132670 | 4134204 | hypomethylation | Exon | SH3PXD2B | Mice | Postnatal growth | Mao et al [18] |
| 16 | 4134519 | 4135224 | hypomethylation | Intron | SH3PXD2B | Mice | Postnatal growth | Mao et al [18] |
| 26 | 13938540 | 13939276 | hypermethylation | Exon | ACSL1 | Cattle | Fatty acids of skeletal muscle | Widmann et al [19] |
RYR1, ryanodine receptor 1; PPARG2, peroxisome proliferator activated receptor-gamma 2; TGFB3, transforming growth factor beta 3; ADAMTS2, ADAM metallopeptidase with thrombospondin type 1 motif 2; NTN1, netrin 1; SH3PXD2B, SH3 and PX domains 2B; ACSL1, acyl-CoA synthetase long chain family member 1.
Validation of MeDIP-seq results by bisulphite sequencing PCR
To validate MeDIP-seq data, the methylation level of transforming growth factor beta 3 (TGFB3) promoter region was examined by bisulphite sequencing PCR (BSP). As shown in Figure 6, the methylated region and unmethylated region showed good consistency between the MeDIP-seq results and BSP results. To further confirm the reliability of DMRs analysis, the methylation level of four randomly selected DMRs in gene acyl-CoA oxidase 2 (ACOX2) intron, acyl-CoA synthetase long chain family member 1 (ACSL1) CDS region, and upstream regions of ras and rab interactor 2 (RIN2) and zinc finger, DHHC type containing 13 (ZDHHC13) were analyzed by BSP. DMRs in upstream region of RIN2 showed significant difference between the two groups (p<0.05, t-test). Although no significant difference, the BSP results of other three DMRs showed similar methylation changes as MeDIP-seq data (Figure 7).
Figure 6.

Bisulphite sequencing polymerase chain reaction (BSP) validation in the transforming growth factor beta 3 (TGFB3) promoter region. The BSP were performed in the TGFB3 promoter region (Chr7:84219421–84219679) among all six sequencing samples (C7, C13, C14, T5, T8, T10) BSP results.
Figure 7.

Bisulphite sequencing PCR (BSP) validation for differential methylated regions (DMRs). For the BSP, the y axes were the mean methylation percentage of three sequencing samples for each group in the DMR. For the methylated DNA immunoprecipitation-sequencing (MeDIP-seq), the y axes indicated mean normalized methylation level in the DMR. The group C and group T is the Small Tailed Han and Dorper×Small Tailed Han crossbred sheep. The star (*) indicated the significant difference (p<0.05, t-test). Subfigures a–d were the DMRs in gene ACOX2 intron region (chr19:42869604–42870470), ACSL1 CDS region (chr26:13938540–13939276), RIN2 upstream region (chr13:38607690–38608423), and ZDHHC13 upstream region (chr21:25146996–25148088), respectively.
Validation of differentially expressed for differentially methylated genes
To validation of differentially expressed for differentially methylated genes between groups, we confirmed twelve differentially methylated genes in MeDIP-seq by real-time quantitative PCR, including TGFB3, ACSL1, ryanodine receptor 1 (RYR1), ACOX2, peroxisome proliferator activated receptor-gamma2 (PPARG2), netrin 1 (NTN1), RIN2, microtubule associated protein RP/EB family member 1 (MAPRE1), ADAM metallopeptidase with thrombospondin type 1 motif 2 (ADAMTS2), myomesin 1 (MYOM1), ZDHHC13, and SH3 and PX domains 2B (SH3PXD2B). The results of real-time quantitative PCR demonstrated these genes are differentially expressed, thus validating the MeDIP-seq data (Figure 8). There were differences in the expression of each gene between the two populations. Two genes (TGFB3, ACSL1) are significantly different (p<0.05). Seven genes (RYR1, ACOX2, PPARG2, MAPRE1, ADAMTS2, MYOM1, and ZDHHC13) are significantly different (p<0.01). The others showed no significant difference. The methylation levels between the two groups are shown in Figure 9. TGFB3 and ACSL1 were significantly different between the two groups (p<0.05) and RYR1, ACOX2, PPARG2, MAPRE1, ADAMTS2, MYOM1 and ZDHHC13 were highly significantly different (p<0.01). Of which, ACSL1, RIN2, and ADAMTS2 were found have a negative correlation between the expression levels of genes and methylation levels in DMRs.
Figure 8.

Relative expression levels of 12 differently methylated genes. Data represent means±standard error of the mean (n = 3). * p≤0.05; ** p≤0.01. The Group C and Group T is the Small Tailed Han and Dorper×Small Tailed Han crossbred sheep, respectively.
Figure 9.

Differential methylated modification of genes. There were significant differences in the methylation levels of each gene between two groups. Data represent means±standard error of the mean (n = 3). ** p≤0.01. The Group C and Group T is the Small Tailed Han and Dorper×Small Tailed Han crossbred sheep, respectively.
DISCUSSION
Although global DNA methylation surveys have been performed on LDM tissues of sheep [20], the present study is the first to systematically compare the genome-wide LDM methylation profiles of Dorper×Small Tailed Han crossbred and Small Tailed Han sheep. We aimed to identify methylated genes associated with meat production.
Reads distribution analysis of our study found that uniquely mapped reads were enriched in the repeats and the gene body regions, which was consistent with previous reports in sheep using the RRBS [20], suggesting that MeDIP-seq is a cost-effective approach for analyses of the sheep DNA methylome. A similar methylation pattern was also observed in other animals and plants, such as chicken [21], bovine [22], pig [10], and Arabidopsis thaliana [23]. It is most likely that this methylation pattern is a mechanism that is conserved among different species.
Compared with Small Tailed Han sheep, we observed a tendency toward a global loss of DNA methylation in the crossbred group. In other words, the DNA methylation level in the LDM was decreased in the crossbred group compared to the Small Tailed Han group. Gene ontology enrichment analysis for genes hypomethylated in the gene-body regions found several important gene sets, such as motor activity. Currently, the relationship between gene-body hypomethylation and gene expression is still unclear. Previous studies on breast cancer showed that global DNA hypomethylation is coupled to gene silencing [24]. Therefore, it might be possible that gene-body hypomethylation in the crossbred group caused down regulation of certain genes and lower motor activity, leading to lower shear values and better meat tenderness. Consistent with this result, previous study reported that broilers reared in low stocking density showed greater motor activity and higher shear values [25].
This study compared methylation differences and expression differences of 12 genes, including TGFB3, ACSL1, RYR1, ACOX2, PPARG2, NTN1, RIN2, MAPRE1, ADAMTS2, MYOM1, ZDHHC13, and SH3PXD2B. Three genes, whose expressions are negatively correlated with DNA methylation are ACSL1, RIN2, and ADAMTS2. The result suggest that the differential expression of these three genes between two groups may be caused by the differences of DNA methylation, and other differential expression genes between the two groups may be due to reasons other than DNA methylation [26].
The expression regulation of ACSL1, RIN2, and ADAMTS2 may be affected by their methylation, and then affect the biological function of meat performance. ACSLs could transform fatty acid to ester acyl coenzyme A. The result indicates that ACSLs not only play a key role in the synthesis of triglycerides, phospholipids and cholesterol, but also affect the metabolism of fatty acid. ACSL1 can be used as a candidate gene for affecting fatty acids in bovine skeletal muscle, and can affect the composition of fat in beef [19]. RIN2 can connect three GTPases, R-Ras, Rab5, and Rac1, to promote endothelial cell adhesion through the regulation of integrin internalization and Rac1 activation [27], but there were no reports of an effect on animal production. ADAMTS2 has an important effect on the biosynthesis of collagen, it is presumed that the expression of the gene will affect the deposition of intramuscular fat. The recent study of Korean cattle showed that the expression levels of ADAMTS2 and ADAMTS4 have a significant impact in fat content of muscle tissue [17]. Thus further confirming that ADAMTS2 can indirectly regulate the deposition of intramuscular fat by affecting collagen synthesis.
DMR related genes are genes known to be involved in meat production. Current data also identified some DMR related genes which were reported to be involved in meat production. Most of these genes are from studies on the association between phenotypes and DNA polymorphisms. For example, two genes (ADAMTS2 and NTN1) have been reported to be related with post-weaning gain in sheep in a genome-wide association studies [4]. RYR1 is well known as a meat quality gene and T allele is associated with reduced meat quality [28]. RYR1 polymorphisms were also related to daily weight gain in pig as reported by Kadarmideen et al [14].
Differential mythelations in intron of ACOX2, MYOM1, SH3PXD2B, and NTN1, and exon of RYR1, and upstream of MAPRE1 were also identified in our results. However, further studies are needed to confirm their regulatory roles in the higher carcass weight observed in the crossbred group.
Furthermore, TGFB3 with promoter hyper-methylation was detected in the crossbred group and has been reported to be related with growth in chicken [16]. Transforming growth factor β3 is known as a cytokine that is involved in cell differentiation, embryogenesis, and development [29]. Differential expression and methylation of this gene have been related to leg muscle development in chicken [30]. Similar to RYR1, PPARG2 is another gene with exon hypo-methylation. Polymorphisms of PPARG2 gene were reported to be associated with meat quality and production in cattle [15]. PPARG2 encodes PPAR-gamma protein which is a regulator of adipocyte differentiation. In addition, PPARG2 has been implicated in numerous diseases including obesity [31]. Further investigations are needed to confirm the contribution from these genes to the difference between the crossbred group and Small Tailed Han group.
In summary, we provided a comprehensive analysis of genome-wide DNA methylation patterns in LDM of Dorper×Small Tailed Han crossbred and Small Tailed Han sheep. We identified remarkable DNA methylation changes between the two groups, such as a tendency toward hypomethylation in gene bodies in the LDM of the crossbred sheep. Furthermore, we identified numerous genes which might be potentially involved in the difference between the two groups. Several genes were highlighted due to their known association with growth and production, including TGFB3, ACSL1, RYR1, ACOX2, PPARG2, NTN1, RIN2, MAPRE1, ADAMTS2, MYOM1, ZDHHC13, and SH3PXD2B. There are differences in the expression of 9 genes, of which ACSL1, RIN2, and ADAMTS2 may be due to DNA methylation. These results will serve as a valuable resource for DNA methylation investigations on screening candidate genes which might be related to meat production in sheep.
Supplementary Data
ACKNOWLEDGMENTS
This study is supported by the National Sheep Industry Technology System (CARS-39), the science and technology development project of Jilin Province (20150204023NY).
Footnotes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.
REFERENCES
- 1.Cheng P. FAO Animal Production and Health Paper. Rome, Italy: Food and Agriculture Organization of the United Nations; 1985. Livestock breeds of China; p. 46. [Google Scholar]
- 2.Cloete SWP, Snyman MA, Herselman MJ. Productive performance of Dorper sheep. Small Rumin Res. 2000;36:119–35. doi: 10.1016/s0921-4488(99)00156-x. [DOI] [PubMed] [Google Scholar]
- 3.Walling GA, Visscher PM, Wilson AD, et al. Mapping of quantitative trait loci for growth and carcass traits in commercial sheep populations. J Anim Sci. 2004;82:2234–45. doi: 10.2527/2004.8282234x. [DOI] [PubMed] [Google Scholar]
- 4.Zhang L, Liu J, Zhao F, et al. Genome-wide association studies for growth and meat production traits in sheep. PLoS One. 2013;8:e66569. doi: 10.1371/journal.pone.0066569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Courtier B, Heard E, Avner P. Xce haplotypes show modified methylation in a region of the active X chromosome lying 3′ to Xist. Proc Natl Acad Sci USA. 1995;92:3531–5. doi: 10.1073/pnas.92.8.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sasaki H, Allen ND, Surani MA. DNA methylation and genomic imprinting in mammals. In: Jost JP, Saluz HP, editors. DNA methylation. Vol. 64. Basel, Switzerland: Birkhäuser. EXS; 1993. pp. 469–86. [DOI] [PubMed] [Google Scholar]
- 7.Siegfried Z, Eden S, Mendelsohn M, et al. DNA methylation represses transcription in vivo . Nat Genet. 1999;22:203–6. doi: 10.1038/9727. [DOI] [PubMed] [Google Scholar]
- 8.Gim JA, Hong CP, Kim DS, et al. Genome-wide analysis of DNA methylation before-and after exercise in the thoroughbred horse with MeDIP-Seq. Mol Cells. 2015;38:210–20. doi: 10.14348/molcells.2015.2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu Y, Xu H, Li Z, et al. Comparison of the genome-wide DNA methylation profiles between fast-growing and slow-growing broilers. PLoS One. 2013;8:e56411. doi: 10.1371/journal.pone.0056411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin L, Jiang Z, Xia Y, et al. Genome-wide DNA methylation changes in skeletal muscle between young and middle-aged pigs. BMC Genomics. 2014;15:653. doi: 10.1186/1471-2164-15-653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li N, Ye M, Li Y, et al. Whole genome DNA methylation analysis based on high throughput sequencing technology. Methods. 2010;52:203–12. doi: 10.1016/j.ymeth.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kadarmideen HN. Biochemical, ECF18R, and RYR1 gene polymorphisms and their associations with osteochondral diseases and production traits in pigs. Biochem Genet. 2008;46:41–53. doi: 10.1007/s10528-007-9127-5. [DOI] [PubMed] [Google Scholar]
- 15.Fan YY, Fu GW, Fu CZ, Zan LS, Tian WQ. A missense mutant of the PPAR-gamma gene associated with carcass and meat quality traits in Chinese cattle breeds. Genet Mol Res. 2012;11:3781–8. doi: 10.4238/2012.August.17.4. [DOI] [PubMed] [Google Scholar]
- 16.Jin S, Chen S, Li H, et al. Polymorphisms in the transforming growth factor beta3 gene and their associations with feed efficiency in chickens. Poult Sci. 2013;92:1745–9. doi: 10.3382/ps.2013-03018. [DOI] [PubMed] [Google Scholar]
- 17.Lee SH, Gondro C, Werf J, et al. Use of a bovine genome array to identify new biological pathways for beef marbling in Hanwoo (Korean Cattle) BMC Genomics. 2010;11:623. doi: 10.1186/1471-2164-11-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mao M, Thedens DR, Chang B, et al. The podosomal-adaptor protein SH3PXD2B is essential for normal postnatal development. Mamm Genome. 2009;20:462–75. doi: 10.1007/s00335-009-9210-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Widmann P, Nuernberg K, Kuehn C, et al. Association of an ACSL1 gene variant with polyunsaturated fatty acids in bovine skeletal muscle. BMC Genet. 2011;12:96. doi: 10.1186/1471-2156-12-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Couldrey C, Brauning R, Bracegirdle J, et al. Genome-wide DNA methylation patterns and transcription analysis in sheep muscle. PLoS One. 2014;9:e101853. doi: 10.1371/journal.pone.0101853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Q, Li N, Hu X, et al. Genome-wide mapping of DNA methylation in chicken. PLoS One. 2011;6:e19428. doi: 10.1371/journal.pone.0019428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang YZ, Sun JJ, Zhang LZ, et al. Genome-wide DNA methylation profiles and their relationships with mRNA and the microRNA transcriptome in bovine muscle tissue (Bos taurine) Sci Rep. 2014;4:6546. doi: 10.1038/srep06546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2007;39:61–9. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]
- 24.Hon GC, Hawkins RD, Caballero OL, et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012;22:246–58. doi: 10.1101/gr.125872.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farmer LJ, Perry GC, Lewis PD, et al. Responses of two genotypes of chicken to the diets and stocking densities of conventional UK and Label Rouge production systems-II. Sensory attributes. Meat Sci. 1997;47:77–93. doi: 10.1016/s0309-1740(97)00040-5. [DOI] [PubMed] [Google Scholar]
- 26.Song F, Smith JF, Kimura MT, et al. Association of tissue-specific differentially methylated regions (TDMs) with differential gene expression. Proc Natl Acad Sci USA. 2005;102:3336–41. doi: 10.1073/pnas.0408436102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fernandezborja M. A tale of three GTPases and a RIN in endothelial cell adhesion. Cell Res. 2012;22:1426. doi: 10.1038/cr.2012.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stratz P, Wellmann R, Preuss S, Wimmers K, Bennewitz J. Genome-wide association analysis for growth, muscularity and meat quality in Pietrain pigs. Anim Genet. 2014;45:350–6. doi: 10.1111/age.12133. [DOI] [PubMed] [Google Scholar]
- 29.Sanders EJ, Wride MA. Roles for growth and differentiation factors in avian embryonic development. Poult Sci. 1997;76:111–7. doi: 10.1093/ps/76.1.111. [DOI] [PubMed] [Google Scholar]
- 30.Lu Y, Chen S, Yang N. Expression and methylation of FGF2, TGF-beta and their downstream mediators during different developmental stages of leg muscles in chicken. PLoS One. 2013;8:e79495. doi: 10.1371/journal.pone.0079495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shao X, Wang M, Wei X, et al. Peroxisome Proliferator-Activated Receptor-gamma: master regulator of adipogenesis and obesity. Curr Stem Cell Res Ther. 2015;11:282–9. doi: 10.2174/1574888x10666150528144905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.