

Abstract
Calcitonin gene-related peptide (CGRP), the most abundant neuropeptide in primary afferent sensory neurons, is strongly implicated in the pathophysiology of migraine headache, but its role in migraine is still equivocal. As a new approach to migraine treatment, humanized anti-CGRP monoclonal antibodies (CGRP-mAbs) were developed to reduce the availability of CGRP, and were found effective in reducing the frequency of chronic and episodic migraine. We recently tested the effect of fremanezumab (TEV-48125), a CGRP-mAb, on the activity of second-order trigeminovascular dorsal horn neurons that receive peripheral input from the cranial dura, and found a selective inhibition of high-threshold but not wide-dynamic range class of neurons. To investigate the basis for this selective inhibitory effect, and further explore the mechanism of action of CGRP-mAbs, we tested the effect of fremanezumab on the cortical spreading depression-evoked activation of mechanosensitive primary afferent meningeal nociceptors that innervate the cranial dura, using single-unit recording in the trigeminal ganglion of anesthetized male rats. Fremanezumab pretreatment selectively inhibited the responsiveness of Aδ neurons, but not C-fiber neurons, as reflected in a decrease in the percentage of neurons that showed activation by cortical spreading depression. These findings identify Aδ meningeal nociceptors as a likely site of action of fremanezumab in the prevention of headache. The selectivity in its peripheral inhibitory action may partly account for fremanezumab's selective inhibition of high-threshold, as a result of a predominant A-δ input to high-threshold neurons, but not wide dynamic-range dorsal horn neurons, and why it may not be effective in all migraine patients.
SIGNIFICANCE STATEMENT Recently, we reported that humanized CGRP monoclonal antibodies (CGRP-mAbs) prevent activation and sensitization of high-threshold (HT) but not wide-dynamic range trigeminovascular neurons by cortical spreading depression (CSD). In the current paper, we report that CGRP-mAbs prevent the activation of Aδ but not C-type meningeal nociceptors by CSD. This is the first identification of an anti-migraine drug that appears to be selective for Aδ-fibers (peripherally) and HT neurons (centrally). As the main CGRP-mAb site of action appears to be situated outside the brain, we conclude that the initiation of the headache phase of migraine depends on activation of meningeal nociceptors, and that for selected patients, activation of the Aδ-HT pain pathway may be sufficient for the generation of headache perception.
Keywords: calcitonin gene-related peptide, cortical spreading depression, headache, migraine, pain, trigeminovascular
Introduction
Calcitonin gene-related peptide (CGRP) is the most abundant neuropeptide present in primary afferent sensory neurons in dorsal root and trigeminal ganglia (O'Connor and van der Kooy, 1988; McCarthy and Lawson, 1990; Price and Flores, 2007). A large body of evidence in both man and animals supports an important role for CGRP in the pathophysiology of migraine headache (Hansen and Ashina, 2014; Karsan and Goadsby, 2015; Russo, 2015). The mechanism of CGRP's involvement in migraine is not well understood, but one line of research has focused on its role in the physiology of neurons that supply the sensory innervation of the cranial meninges, a sensory pathway that has been critically implicated in the generation of migraine headache (Moskowitz, 1984; Strassman et al., 1996; Burstein et al., 2015). CGRP is present in a substantial subpopulation of the meningeal sensory neurons (O'Connor and van der Kooy, 1988; Tsai et al., 1988; Uddman et al., 1989; Keller and Marfurt, 1991; Messlinger et al., 1993; Edvinsson et al., 2001), and can be released from both the peripheral endings in the meninges (Zagami et al., 1990; Kurosawa et al., 1995; Eltorp et al., 2000) and the central endings in the medullary and upper cervical dorsal horn (Storer et al., 2004; Fischer et al., 2005). Peripherally, CGRP produces meningeal vasodilatation (McCulloch et al., 1986; Uddman et al., 1986; Edvinsson et al., 1987, 1998; Kurosawa et al., 1995; Williamson et al., 1997), and thus contributes to meningeal neurogenic inflammation. The actions of CGRP as a neuromodulator when released from the central terminals of meningeal sensory neurons has not yet been well elucidated, but there is evidence from studies at spinal levels that CGRP contributes to sensitization of dorsal horn neurons (Sun et al., 2004a,b; Han et al., 2005; Bird et al., 2006; Adwanikar et al., 2007). Among the most well documented effects of the anti-migraine agent sumatriptan is inhibition of neuropeptide release from both the peripheral and central terminals of meningeal sensory neurons (Messlinger et al., 1998; Durham and Russo, 1999; Eltorp et al., 2000).
As a new approach to migraine treatment, humanized CGRP monoclonal antibodies (CGRP-mAbs) have been developed as therapeutic agents, and were found to reduce the frequency of attacks in chronic migraine patients (Dodick et al., 2014a,b; Bigal et al., 2015a,b; Sun et al., 2016). Recently, we examined the effect of fremanezumab, a CGRP-mAb, on the central neurons in the meningeal sensory pathway, and found a selective inhibition of the high-threshold (HT) but not the wide-dynamic range (WDR) class of dorsal horn neurons (Melo-Carrillo et al., 2017). This selective action is presumed to be through a peripheral site of action, since the antibodies do not cross the blood–brain barrier. Such a selective inhibition of HT neurons has not been reported previously, and it is not known what type of peripheral action could produce such a selective central inhibition. To investigate this question, we examined the effect of fremanezumab on the evoked activity of mechanosensitive primary afferent neurons in the trigeminal ganglion that innervate the cranial dura. Neuronal activation was evoked by cortical spreading depression (CSD), a cortical event that has been implicated in the initiation of migraine attacks (Bolay and Moskowitz, 2005) and that is effective in activating both first- and second-order neurons in the meningeal sensory pathway (Zhang et al., 2010, 2011).
Materials and Methods
Surgical preparation.
Experiments were approved by the Beth Israel Deaconess Medical Center and Harvard Medical School standing committees on animal care, and were in accordance with the U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male Sprague-Dawley rats (225–325 g) were anesthetized with urethane (1.5 g/kg, i.p.), and treated with atropine (0.4 ml, i.p.) to reduce intraoral secretions. The femoral vein was cannulated for drug infusion, an endotracheal tube was implanted, and the rat was mounted in a stereotaxic apparatus. Core temperature was maintained at 37°C using a feedback controlled heating pad, and pO2 was maintained >98% by O2 inhalation through the endotracheal tube. A craniotomy was made to expose the left transverse sinus, to allow electrical and mechanical stimulation of dural primary afferent nociceptors. A small burr hole was drilled in the calvarium on the left side, at ∼1 mm rostral to bregma and ∼1.5–2 mm lateral, to allow insertion of a glass micropipette for recording of electrocorticogram activity. A second burr hole was drilled ∼5 mm caudal to the first one (∼5 mm rostral to the transverse sinus), for induction of cortical spreading depression by pinprick (brief insertion of a metal microelectrode into the cortex). The dura exposed by these burr holes was not removed. All of the exposed dura was kept moist using a modified synthetic interstitial fluid (in mm: 135 NaCl, 5 KCl, 1 MgCl2, 5 CaCl2, 10 glucose, and 10 HEPES, pH 7.2). For neural recording from the left trigeminal ganglion, a craniotomy was made on the right (contralateral) side to allow the microelectrodes to be advanced through the contralateral cortex to reach the ganglion, using an angled approach. This was done to avoid damage to the ipsilateral cortex. The craniotomy was made to allow electrode insertions into the right cortex covering an area of 1–3 mm caudal to bregma, and 1.5–3 mm lateral. The dura covering this area of cortex was removed to allow microelectrode insertion.
Neural recording.
A glass micropipette filled with 0.9% saline (∼1 MΩ, 7 μm tip) was inserted through the rostral burr hole 500 μm below the cortical surface for electrocorticogram recording. A platinum-coated tungsten microelectrode (impedance 150–300 kΩ) was advanced into the trigeminal ganglion for single-unit recording. To reach the ganglion via a contralateral approach, the electrode was angled medially 21° and was advanced through the contralateral cortex (see above for coordinates). Dural afferent neurons in the ganglion were identified by their constant latency response to single shock stimulation delivered to the dura overlying the ipsilateral transverse sinus with a bipolar stimulating electrode (0.5 ms pulse, 5 mA, 0.75 Hz). The response latency was used to calculate conduction velocity (c.v.), based on a conduction distance to the trigeminal ganglion of 12 mm. Neurons were classified as either C-units (c.v. ≤1.5 ms) or Aδ-units (c.v. 1.5–5 m/s). Neurons with conduction velocity >5 m/s were not studied. Spike 2 software (CED) was used for acquisition and waveform discrimination of the electrically evoked spikes, and for off-line analysis.
Mechanical receptive fields of dural afferents were initially identified by probing the dura with blunt forceps and von Frey hairs. Only neurons for which a mechanical receptive field could be identified were selected for study.
Experimental paradigm.
Ongoing discharge activity was recorded continuously throughout the experiment. After recording of baseline spontaneous activity an intravenous infusion was made of either the humanized monoclonal anti-CGRP antibody fremanezumab (TEVA Pharmaceutical Industries) or the corresponding human IgG2 isotype control antibody (isotype-conAb), both were given at a dose of 30 mg/kg in saline. Four hours after drug administration, a single wave of CSD was induced by briefly inserting a metal microelectrode into the cortex through the caudal burr hole, and spontaneous activity was recorded continuously for another 60 min.
Data analysis.
The 30 min period before CSD induction (3.5 h after drug administration) was used as the baseline for determination of CSD responses. A neuron was considered to have a response to CSD if its firing rate increased above baseline by 2 SD for a period at least 10 min. Calculation of response magnitudes were based on two 10 min periods where mean firing rates were highest during the two 30 min periods before (baseline firing rate) and after occurrence of CSD (post-CSD firing rate). The latency from CSD to the onset of response, as well as response duration were calculated for each neuron.
Baseline firing rates were compared between (1) the isotype (control) and CGRP-mAb groups, and (2) Aδ and C fibers using the nonparametric Mann–Whitney analysis. Firing rates before and after CSD were compared using the nonparametric Wilcoxon analysis. Latency and duration of CSD responses were compared based on Group (isotype/CGRP-mAb) and neuron type (Aδ/C), comprising four subgroups, using the nonparametric Kruskal–Wallis test. The association between responsiveness to CSD (yes/no) and Group (isotype/CGRP-mAb) per neuron type (Aδ/C) were tested using χ2 tests. All analyses were performed using Statistical Package for the Social Sciences (SPSS v22, IBM). Continuous variables are presented as median (interquartile range: 25th–75th percentile). Statistical significance was set at p < 0.05.
Results
Single-unit recordings were obtained from 19 Aδ- and 30 C-class meningeal nociceptors in the trigeminal ganglion that were identified by their response to electrical and mechanical stimulation of the dura overlying the ipsilateral transverse sinus.
The effect of CSD on neuronal discharge was tested following intravenous infusion of either CGRP-mAb (n = 10 Aδ and 14 C-fibers) or the corresponding isotype antibody (n = 9 Aδ- and 16 C-fibers). CSD was induced by pinprick 4 h after the drug infusion. Before CSD, neurons displayed firing rates of 0.37 (1.47) [median (IQR)] in CGRP-MAb-treated animals, and 0.45 (0.69) [median (IQR)] in the isotype-treated animals (Z = −0.13, p = 0.897). There was no significant difference in the baseline firing rates between CGRP-MAb-treated Aδ neurons 0.08 (0.95) [median (IQR)] and the isotype-treated Aδ neurons 0.79 (1.37) [median (IQR); Z = −1.72, p = 0.095]. Similarly, the CGRP-mAb-treated C-fibers 0.46 (1.64) [median (IQR)] were comparable to the isotype-treated group 0.37 (0.51) [median (IQR); Z = −1.43, p = 0.154].
Following CSD, according to the aforementioned criteria, an increase in firing rate was observed in 10/24 (41%) neurons in CGRP-MAb-treated animals and 13/25 (52%) neurons in isotype-treated animals (Table 1).
Table 1.
Summary of results
| Activation by CSD | Neurons (N = 49) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Isotype (n = 25) |
CGRP-mAb (n = 24) |
|||||||
| Aδ (n = 9) |
C (n = 16) |
Aδ (n = 10) |
C (n = 14) |
|||||
| Yes (n = 6) | No (n = 3) | Yes (n = 7) | No (n = 9) | Yes (n = 2) | No (n = 8) | Yes (n = 8) | No (n = 6) | |
| Baseline, mean spikes/s | 0.79 (1.4) | 0.49 (0.8) | 0.53 (0.6) | 0.30 (0.5) | 0.03 (0) | 0.44 (1.0) | 0.32 (1.0) | 1.70 (1.9) |
| Post-CSD, mean spikes/s | 1.29 (2.1)* | 0.33 (2.0) | 1.10 (2.0)* | 0.29 (0.5) | 0.35 (0.1) | 0.44 (1.1) | 1.07 (2.0)* | 0.96 (1.8) |
| Latency, min | 6 (7.7) | NA | 5 (4.0) | NA | 5, 8a | NA | 5 (3.7) | NA |
| Duration, min | 30 (26.2) | NA | 60 (30.0) | NA | 20, 60a | NA | 60 (7.5) | NA |
Data are represented as median (IQR). NA, Not applicable.
*Statistically significant difference compared to baseline (p < 0.05).
aAnalyses were not carried out due to small sample (n = 2); results are displayed per neuron (n = 2).
CSD effects on Aδ-fibers
Isotype-treated group
In the isotype-treated group (Fig. 1; Table 1), CSD activated 6/9 (66%) Aδ-meningeal nociceptors; i.e., the firing rate of each of these neurons increased by >2 SD after occurrence of CSD as compared with their baseline firing (Fig. 1A–C). These neurons demonstrated a baseline firing rate of 0.79 (1.42)[median (IQR)] that increased to 1.29 (2.16)[median (IQR); Z = −2.20, p = 0.028] after occurrence of CSD (Fig. 1B). Their activation started 6.0 (7.75) [median (IQR)] min after occurrence of CSD (range: 2–18 min; Fig. 1D) and lasted for 30.0 (26.25) [median (IQR)] min (range: 15–60 min; Fig. 1E). No significant change was observed in the activity level of the remaining 3 neurons after CSD occurrence (baseline: 0.49 (0.83)[median (IQR)]; post-CSD: 0.33 (0.93) [median (IQR)]; Z = −1.60, p = 0.109; Fig. 1F–H).
Figure 1.
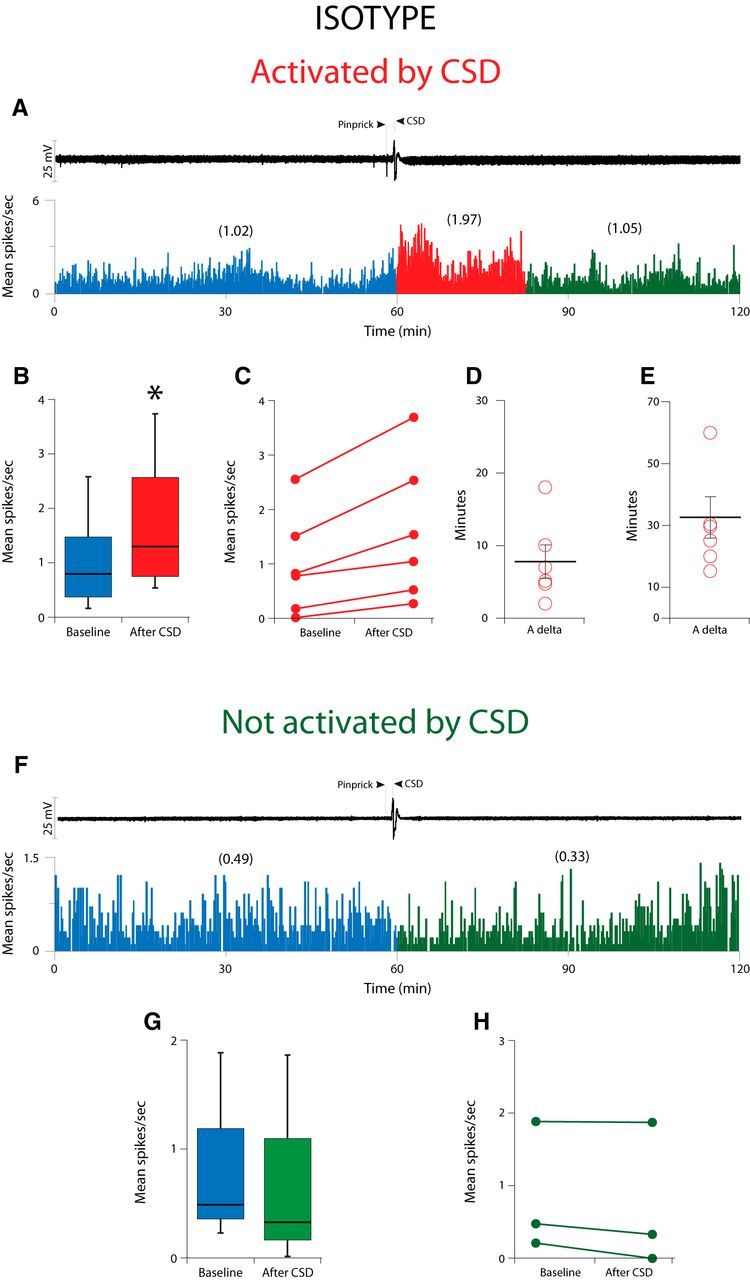
Effect of CSD on activity of Aδ meningeal primary afferent nociceptors in animals treated with isotype. A, CSD recording (top trace) and plot of firing rate (bottom trace) for one Aδ neuron that was activated by CSD. B, Here and in the next 3 figures, box plots of neuronal firing rate before and after CSD illustrate the median and interquartile range of 25th–75th percentile. C, Plot of baseline and post-CSD firing rates (see Materials and Methods) for the isotype-treated Aδ neurons that were activated by CSD (n = 6). D, E, Plots of the latency to the onset of activation (D) and the duration of activation (E) for the neurons shown in B. F, Example of one Aδ-fiber that was not activated by CSD. G, Box plots of neuronal firing rate before and after CSD. H, Plot as in B, for the isotype-treated Aδ neurons that were not activated by CSD (n = 3). Asterisks in Figs. 1B, 3B, and 4B indicate statistically significant difference (p < 0.05).
CGRP-mAb-treated group
In contrast, in the CGRP-mAb-treated group (Fig. 2; Table 1), CSD activated only 2/10 (20%) Aδ-meningeal nociceptors (Fig. 2A,B). These neurons demonstrated a baseline firing rate of 0.03 (0.0)[median (IQR)] that increased to 0.35 (0.18)[median (IQR); Z = −1.34, p = 0.180] after occurrence of CSD. Similar to the isotype-treated group, activation latencies (8 and 5 min) and duration (20 and 60 min) for these two neurons were within the same range (Fig. 2C,D). The activity level of the majority of the Aδ-units (8/10) which were studied in this CGRP-mAb-treated group remained unchanged after the occurrence of CSD (baseline: 0.44 (1.0)[median (IQR)]; post-CSD: 0.44 (1.14) [median (IQR)]; Z = −0.98, p = 0.326; Fig. 2E–G).
Figure 2.
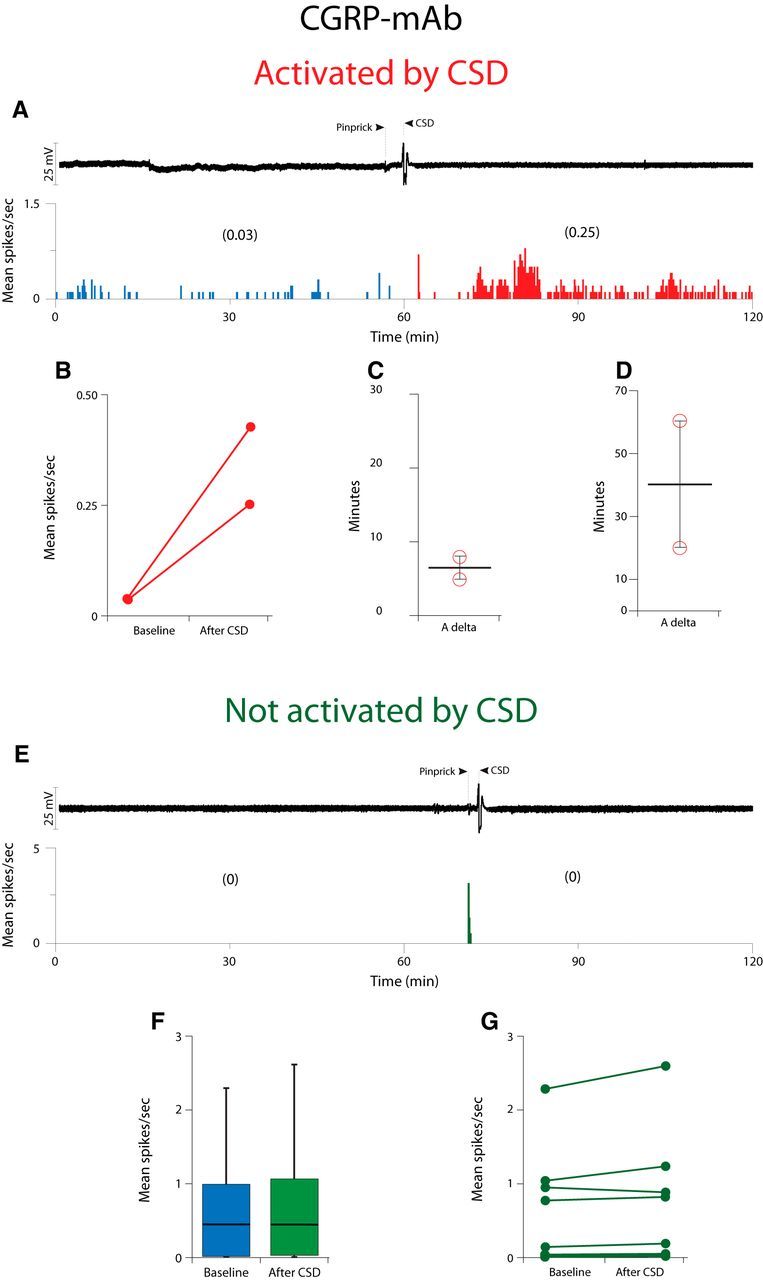
Effect of CSD on activity of Aδ meningeal primary afferent nociceptors in animals treated with CGRP-mAb. A, CSD recording (top trace) and plot of firing rate (bottom trace) for one Aδ neuron that was activated by CSD. B, Plot of baseline and post-CSD firing rates for the CGRP-mAb-treated Aδ neurons that were activated by CSD (n = 2). C, D, Plots of the latency to the onset of activation (C) and the duration of activation (D) for the neurons shown in B. E, Example of one Aδ fiber that was not activated by CSD. F, Box plots of neuronal firing rate before and after CSD. G, Plot as in B, for the mAb-treated Aδ neurons that were not activated by CSD (n = 8).
Isotype versus CGRP-mAb
There was a significant association between responsiveness to CSD and Group such that of a total of eight CSD activated neurons, 6 (75%) were treated with isotype and only 2 (25%) were treated with CGRP-mAb. Additionally, of 9 isotype-treated neurons, 6 (66.6%) were and 3 (33.3%) were not activated by CSD. As to the CGRP-mAb-treated group, of 10 neurons, 2 (20%) were and 8 (80%) were not activated by CSD (χ(1)2 = 4.23, p = 0.040)].
CSD effects on C-fibers
Isotype-treated group
In the isotype-treated group (Fig. 3; Table 1), CSD activated 7/16 (43%) C-type meningeal nociceptors; i.e., the firing rate of each of these neurons increased by >2 SD after occurrence of CSD compared with their baseline firing (Fig. 3A–C). These neurons demonstrated a baseline firing rate of 0.53 (0.61)[median (IQR)] that increased to 1.1 (2.0)[median (IQR); Z = −2.36, p = 0.018] after occurrence of CSD (Fig. 3B). Their activation started 5.0 (4.0) [median (IQR)] min after occurrence of CSD (range: 3–25 min; Fig. 3D) and lasted for 60.0 (30.0) [median (IQR)] min (range:15–60 min; Fig. 3E). No significant change was observed in the activity level of the remaining nine neurons after the CSD (baseline: 0.3 (0.56)[median (IQR)]; post-CSD: 0.29 (0.53)[median (IQR)]; Z = −0.98, p = 0.327; (Fig. 3F–H).
Figure 3.
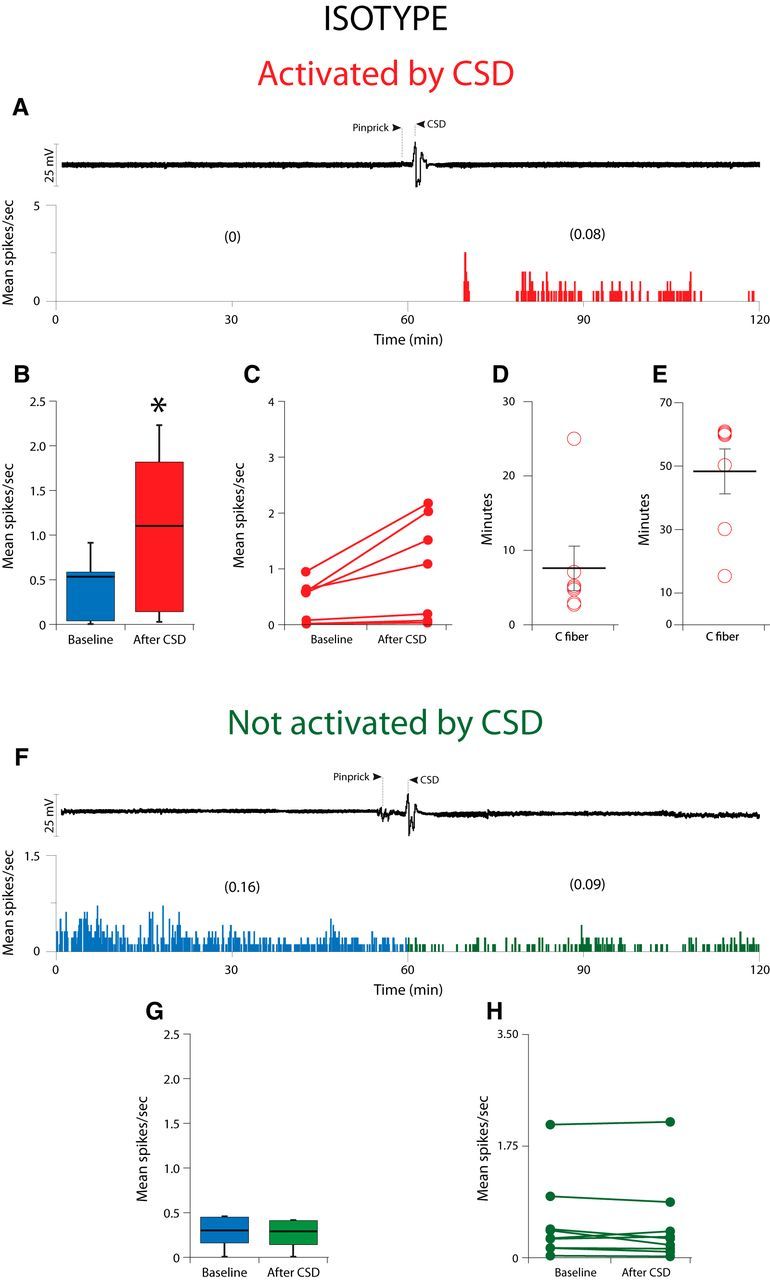
Effect of CSD on activity of C-fiber meningeal primary afferent nociceptors in animals treated with isotype. A, CSD recording (top trace) and plot of firing rate (bottom trace) for one C-fiber neuron that was activated by CSD. B, Box plots of neuronal firing rate before and after CSD. C, Plot of baseline and post-CSD firing rates for the isotype-treated C-fiber neurons that were activated by CSD (n = 7). D, E, Plots of the latency to the onset of activation (D) and the duration of activation (E) for the neurons shown in B. F, Example of one C-fiber neuron that was not activated by CSD. G, Box plots of neuronal firing rate before and after CSD. H, Plot as in B, for the isotype-treated C-fiber neurons that were not activated by CSD (n = 9).
CGRP-mAb-treated group
In the CGRP-mAb-treated group (Fig. 4; Table 1), CSD activated 8/14 (57%) C-type meningeal nociceptors (Fig. 4A–C). These neurons demonstrated a baseline firing rate of 0.32 (1.04)[median (IQR)] that increased to 1.07 (2.07)[median (IQR); Z = −2.52, p = 0.012] after occurrence of CSD (Fig. 4B). As in the isotype-treated group, their activation started 5.0 (3.75)[median (IQR)] min after occurrence of CSD (range: 2–10 min; Fig. 4D) and lasted for 60 (7.5)[median (IQR)] min (range: 20–60 min; Fig. 4E). No significant change was observed in the activity level of the remaining six neurons by the CSD (baseline: 1.70 (1.98)[median (IQR)]; post-CSD 0.96 (1.82) [median (IQR)]; Z = −1.57, p = 0.116; Fig. 4F–H).
Figure 4.
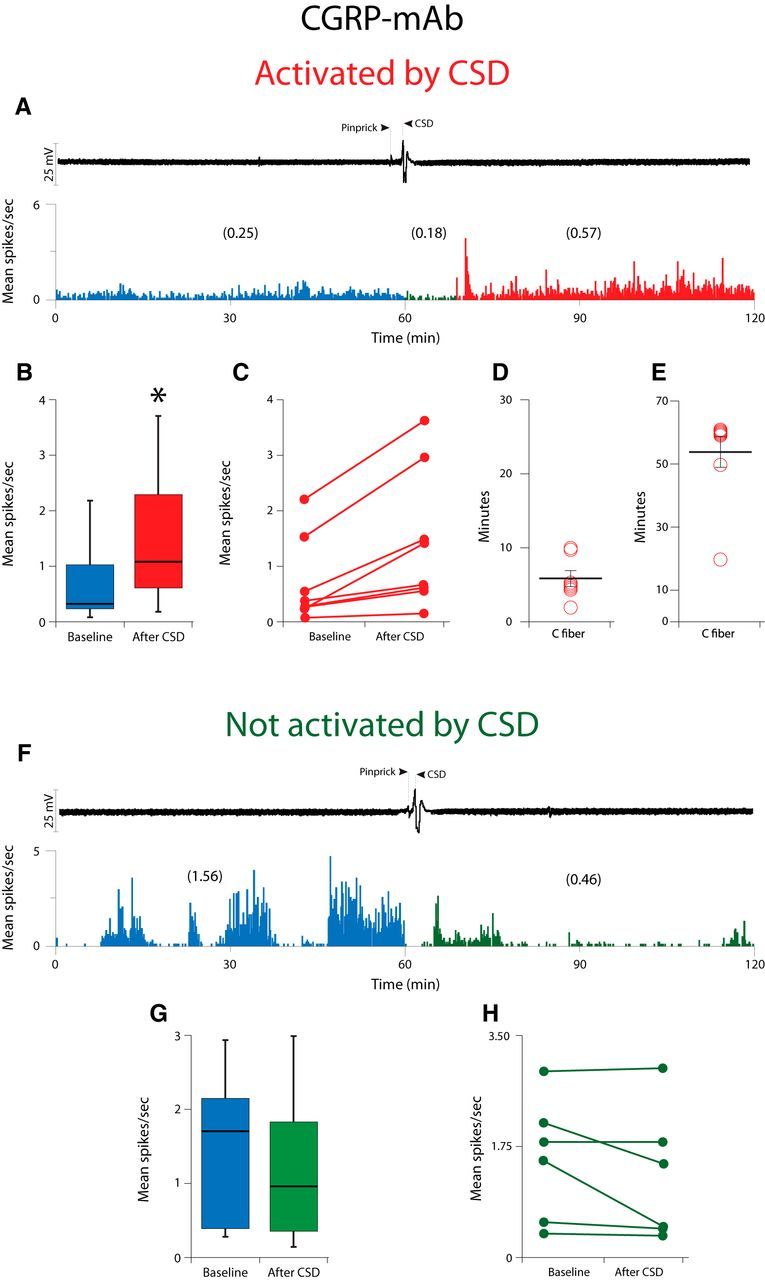
Effect of CSD on activity of C-fiber meningeal primary afferent nociceptors in animals treated with CGRP-mAb. A, CSD recording (top trace) and plot of firing rate (bottom trace) for one C-fiber neuron that was activated by CSD. B, Box plots of neuronal firing rate before and after CSD. C, Plot of baseline and post-CSD firing rates for the CGRP-mAb-treated C-fiber neurons that were activated by CSD (n = 8). D, E, Plots of the latency to the onset of activation (D) and the duration of activation (E) for the neurons shown in B. F, Example of one Aδ fiber that was not activated by CSD. G, Box plots of neuronal firing rate before and after CSD. H, Plot as in B, for the mAb-treated Aδ neurons that were not activated by CSD (n = 6).
Isotype versus CGRP-mAb
No significant association was found between responsiveness to CSD and Group. Namely, of 15 CSD-activated neurons, 7 (46.7%) were treated with isotype and 8 (53.3%) were treated with CGRP-mAb. Additionally, of a total of 16 isotype-treated neurons, 7 (43.8%) were and 9 (56.3%) were not activated by CSD. As to the CGRP-mAb-treated group, of a total of 14 neurons, 8 (57.1%) were and 6 (42.9%) were not activated by CSD (χ(1)2 = 0.54, p = 0.464).
Discussion
This is the first study to test the effects of a CGRP-mAb on the response properties of peripheral trigeminovascular neurons. It is also the first study that identifies a molecule that prevents CSD from activating thinly myelinated Aδ but not unmyelinated C type meningeal nociceptors. This selectivity may be critical for the unique prophylactic profile of this class of drugs in migraine therapy. Mechanistically, the findings define Aδ meningeal nociceptors as a likely anti-nociceptive site of action of fremanezumab, which is critical for its ability to prevent the headache. The findings may also explain fremanezumab's selective inhibition of HT but not WDR trigeminovascular neurons in the dorsal horn (Melo-Carrillo et al., 2017). Clinically, fremanezumab's selective inhibition of peripheral Aδ meningeal nociceptors and central HT trigeminovascular neurons may also explain how CGRP-mAbs prevent the headaches, and equally important, why it may not work for everybody (Bigal et al., 2015a,b).
By far, the most important finding of this study is that fremanezumab prevented the activation of Aδ but not C-type meningeal nociceptors by CSD. Mechanistically, the most obvious explanation for this selectivity is that the calcitonin receptor-like receptor (CLR) and/or the receptor activity-modifying protein 1 (RAMP1) are expressed exclusively in the Aδ- but not C-fibers. In support of this explanation, Eftekhari et al. (2013) showed that in the rat and human dura, CGRP is expressed in C- but not A-fibers whereas CLR and RAMP1 are expressed in the A- but not C-fibers. Our electrophysiological/functional findings suggest that most dural CLR- and RAMP1-positive A-fibers are thinly myelinated Aδ-fibers; a conclusion supported by anatomical studies showing that almost all A-fibers in the dura are Aδ (Strassman et al., 2004). According to this scenario, the Aδ activation by CSD involves increased CGRP release in the dura, most likely by the CGRP-positive C-fibers, whereas the prevention of their activation by fremanezumab is achieved by the sequestration of the secreted CGRP molecules (Fig. 5, steps 2–3).
Figure 5.

Proposed mechanisms for prevention of migraine by CGRP-mAbs. CSD induces brief constriction, brief dilatation, and prolonged constriction of pial arteries (Step 1), as well as immediate and delayed activation of C-fiber meningeal nociceptors containing CGRP (Step 2). Upon their CGRP-independent activation, meningeal C-fibers release CGRP in the dura and by doing so; mediate a CGRP-dependent activation of the nearby Aδ-fibers (Step 3). Once activated, C-fiber meningeal nociceptors converge on and activate WDR neurons in the spinal trigeminal nucleus, whereas Aδ-fibers converge on and activate both WDR and HT neurons (Step 4) that eventually transmit the nociceptive signals from the dura to the thalamus (Step 5). The absence of CGRP receptors from the meningeal C-fibers renders the activation of the C-WDR pathway CGRP-independent (red), and thus, unresponsive to the CGRP-mAb. In contrast, the presence of CGRP receptors on meningeal Aδ-fibers renders the activation of the Aδ-HT pathway CGRP-dependent (blue), and thus, responsive to the CGRP-mAb.
Another explanation for the selective inhibition of the Aδ-fibers by fremanezumab is that the mechanosensitivity of the Aδ meningeal nociceptors is greater than the mechanosensitivity of the C-fibers (Levy and Strassman, 2002), in which case, the CSD-induced cerebral vasodilatation should have a greater effect on the activation of the Aδ-fibers because the vasodilatation-associated stretching of the wall of the blood vessels is a mechanical stimulus. According to this scenario, the blockade of CGRP action by fremanezumab might have a greater effect on Aδ-fibers than C-fibers, because CGRP receptors are present on cerebral vascular smooth muscle cells (Eftekhari et al., 2013; Miller et al., 2016), and CSD-induced cerebral vasodilatation (Bolay et al., 2002; Ayata and Lauritzen, 2015) is partly dependent on CGRP (Colonna et al., 1994; Reuter et al., 1998).
Recently, we showed that fremanezumab prevents CSD-induced activation and sensitization of HT but not WDR trigeminovascular neurons, and proposed that in migraine patients whose headaches are reduced by the CGRP-mAb, the headaches are initiated and/or maintained by the HT rather than the WDR neurons (Melo-Carrillo et al., 2017). When we wrote this paper, we did not know that fremanezumab prevents the activation of Aδ- but not C-type meningeal nociceptors. The current study suggests that the selective inhibition of (central) HT trigeminovascular neurons by fremanezumab is secondary to its selective inhibition of the (peripheral) Aδ meningeal nociceptors (Fig. 5, step 4). The functional and anatomical relationship between Aδ nociceptors and HT trigeminal neurons has been recognized for quite some time. In the primate, HT neurons in the spinal trigeminal nucleus, defined by their responses to noxious pinch and pinprick but not to innocuous brush, exhibit exclusive input from Aδ fibers (Price et al., 1976). In humans, Aδ fibers, especially the mechanosensitive and mechanoheat-sensitive type I which are also capsaicin-insensitive, are the only type of nociceptors that mediate the perception of pinprick (Magerl et al., 2001). In agreement with these, it is now well established that central branches of Aδ but not C-type nociceptors terminate in the two laminae (I and V) where we recorded all of our HT neurons (Bráz and Basbaum, 2009).
The functional network and the physiological events that may explain fremanezumab's selective inhibition of meningeal Aδ- (but not C-) fibers and HT (but not WDR) trigeminovascular neurons' (Melo-Carrillo et al., 2017) responses to CSD is summarized in Figure 5. Scientifically, these findings may be the first to allow us to distinguish between HT and WDR neurons' contribution to the perception of pain at different times after their activation (e.g., early vs late) and under different conditions (e.g., acute, chronic, neuropathic, inflammatory). Clinically, these findings force us to modify a notion that dominated the understanding of the different stages of allodynia, hyperalgesia, and central sensitization, the role they play in the chronification of pain and migraine, and their impact on treatment. Regarding the Aδ-HT versus C-WDR pathways, the complete or nearly complete prevention of headache by fremanezumab suggests that the headache perception of some patients is mediated by the neuronal Aδ-HT pathway alone, and conversely, the complete or near complete lack of effect (i.e., no or insignificant reduction in migraine days) suggests that the headache perception of some patients could be mediated by the neuronal C-WDR pathway alone. Regarding allodynia, hyperalgesia and central sensitization, a synthesis of all currently available clinical data on the treatment outcome of the four CGRP-mAbs under development indicates clearly that a class of drugs that is most likely to block the flow of nociceptive signals from the peripheral meninges to the spinal trigeminal nucleus, is capable of reducing migraine in patients whose attacks were frequent enough before treatment as to classify them chronic migraineurs. Until now, we thought that the transition from acute to chronic migraine follows, at least in part, the transition from early stage central sensitization that depends on pain signals that come from peripheral nociceptors to late stage central sensitization that is independent of the pain signals from the nociceptors, and accordingly, proposed that late-phase central sensitization and allodynia explain the inferior efficacy of late treatment with drugs that lack the ability to reverse the sensitization(Burstein and Jakubowski, 2004; Burstein et al., 2004; Levy et al., 2004). Our current and recent (Melo-Carrillo et al., 2017) studies call for a revision of this concept. Fremanezumab's ability to reduce migraine in patients (i.e., the responders) who are likely to exhibit signs of late-phase, already established, allodynia and central sensitization (Lipton et al., 2017) suggests that their ongoing allodynia and central sensitization continue to depend on the pain signals they receive from the meningeal nociceptors. Conversely, fremanezumab's inability to reduce migraine in this patient population (i.e., the nonresponders) suggests that their ongoing allodynia and central sensitization progress into the established phase whereby the activity of their central trigeminovascular neurons is in fact independent of the pain signals they receive from the meningeal nociceptors.
In summary, our findings provide direct evidence for the assertion that the prophylactic effect of CGRP-mAbs is achieved mainly through their ability to prevent the activation of peripheral trigeminovascular neurons of the Aδ type by events that lead to cerebral release of CGRP, such as during migraine headache (Goadsby et al., 1990). Because CGRP-mAb therapy is effective in the prevention of new headache attacks, it is reasonable to conclude that the activation of Aδ-fibers in the dura, which leads to activation of HT neurons in the spinal trigeminal nucleus, is responsible and sufficient for the initiation of the headache.
One caveat regarding the present findings is that the sample included only male rats. Our prior study on fremanezumab effects on the central neurons of the trigeminovascular pathway in the dorsal horn did include both male and female rats, and found no significant difference between males and females in the effects of fremanezumab on neuronal activity. Nonetheless, it remains to be determined in future studies whether the drug has effects on the meningeal primary afferent neurons in females, and whether the effects in females are similar to those found in the males, as was the case for the central neurons.
Footnotes
This work was supported by a grant from TEVA Pharmaceutical Industries; NIH Grants R37-NS079678, RO1 NS069847, and RO1 NS094198 (R.B.); and a grant from R. Chemers Neustein (A.S.).
Commercial interest: TEVA Pharmaceutical Industries holds the patent for treating episodic and chronic migraine with Fremanezumab and funded parts of the study. J.S. is an employee of TEVA and R.B. is a consultant to TEVA.
References
- Adwanikar H, Ji G, Li W, Doods H, Willis WD, Neugebauer V (2007) Spinal CGRP1 receptors contribute to supraspinally organized pain behavior and pain-related sensitization of amygdala neurons. Pain 132:53–66. 10.1016/j.pain.2007.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayata C, Lauritzen M (2015) Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 95:953–993. 10.1152/physrev.00027.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigal ME, Dodick DW, Rapoport AM, Silberstein SD, Ma Y, Yang R, Loupe PS, Burstein R, Newman LC, Lipton RB (2015a) Safety, tolerability, and efficacy of TEV-48125 for preventive treatment of high-frequency episodic migraine: a multicentre, randomised, double-blind, placebo-controlled, phase 2b study. Lancet Neurol 14:1081–1090. 10.1016/S1474-4422(15)00249-5 [DOI] [PubMed] [Google Scholar]
- Bigal ME, Edvinsson L, Rapoport AM, Lipton RB, Spierings EL, Diener HC, Burstein R, Loupe PS, Ma Y, Yang R, Silberstein SD (2015b) Safety, tolerability, and efficacy of TEV-48125 for preventive treatment of chronic migraine: a multicentre, randomised, double-blind, placebo-controlled, phase 2b study. Lancet Neurol 14:1091–1100. 10.1016/S1474-4422(15)00245-8 [DOI] [PubMed] [Google Scholar]
- Bird GC, Han JS, Fu Y, Adwanikar H, Willis WD, Neugebauer V (2006) Pain-related synaptic plasticity in spinal dorsal horn neurons: role of CGRP. Mol Pain 2:31. 10.1186/1744-8069-2-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolay H, Moskowitz MA (2005) The emerging importance of cortical spreading depression in migraine headache. Rev Neurol 161:655–657. 10.1016/S0035-3787(05)85108-2 [DOI] [PubMed] [Google Scholar]
- Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA (2002) Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med 8:136–142. 10.1038/nm0202-136 [DOI] [PubMed] [Google Scholar]
- Bráz JM, Basbaum AI (2009) Triggering genetically-expressed transneuronal tracers by peripheral axotomy reveals convergent and segregated sensory neuron-spinal cord connectivity. Neuroscience 163:1220–1232. 10.1016/j.neuroscience.2009.07.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein R, Jakubowski M (2004) Analgesic triptan action in an animal model of intracranial pain: a race against the development of central sensitization. Ann Neurol 55:27–36. 10.1002/ana.10785 [DOI] [PubMed] [Google Scholar]
- Burstein R, Collins B, Jakubowski M (2004) Defeating migraine pain with triptans: a race against the development of cutaneous allodynia. Ann Neurol 55:19–26. 10.1002/ana.10786 [DOI] [PubMed] [Google Scholar]
- Burstein R, Noseda R, Borsook D (2015) Migraine: multiple processes, complex pathophysiology. J Neurosci 35:6619–6629. 10.1523/JNEUROSCI.0373-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna DM, Meng W, Deal DD, Busija DW (1994) Calcitonin gene-related peptide promotes cerebrovascular dilation during cortical spreading depression in rabbits. Am J Physiol 266:H1095–H1102. [DOI] [PubMed] [Google Scholar]
- Dodick DW, Goadsby PJ, Spierings EL, Scherer JC, Sweeney SP, Grayzel DS (2014a) Safety and efficacy of LY2951742, a monoclonal antibody to calcitonin gene-related peptide, for the prevention of migraine: a phase 2, randomised, double-blind, placebo-controlled study. Lancet Neurol 13:885–892. 10.1016/S1474-4422(14)70128-0 [DOI] [PubMed] [Google Scholar]
- Dodick DW, Goadsby PJ, Silberstein SD, Lipton RB, Olesen J, Ashina M, Wilks K, Kudrow D, Kroll R, Kohrman B, Bargar R, Hirman J, Smith J; the ALD403 study investigators (2014b) Safety and efficacy of ALD403, an antibody to calcitonin gene-related peptide, for the prevention of frequent episodic migraine: a randomised, double-blind, placebo-controlled, exploratory phase 2 trial. Lancet Neurol 13:1100–1107. 10.1016/S1474-4422(14)70209-1 [DOI] [PubMed] [Google Scholar]
- Durham PL, Russo AF (1999) Regulation of calcitonin gene-related peptide secretion by a serotonergic antimigraine drug. J Neurosci 19:3423–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvinsson L, Ekman R, Jansen I, McCulloch J, Uddman R (1987) Calcitonin gene-related peptide and cerebral blood vessels: distribution and vasomotor effects. J Cereb Blood Flow Metab 7:720–728. 10.1038/jcbfm.1987.126 [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Mulder H, Goadsby PJ, Uddman R (1998) Calcitonin gene-related peptide and nitric oxide in the trigeminal ganglion: cerebral vasodilatation from trigeminal nerve stimulation involves mainly calcitonin gene-related peptide. J Auton Nerv Syst 70:15–22. 10.1016/S0165-1838(98)00033-2 [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Elsås T, Suzuki N, Shimizu T, Lee TJ (2001) Origin and co-localization of nitric oxide synthase, CGRP, PACAP, and VIP in the cerebral circulation of the rat. Microsc Res Tech 53:221–228. 10.1002/jemt.1086 [DOI] [PubMed] [Google Scholar]
- Eftekhari S, Warfvinge K, Blixt FW, Edvinsson L (2013) Differentiation of nerve fibers storing CGRP and CGRP receptors in the peripheral trigeminovascular system. J Pain 14:1289–1303. 10.1016/j.jpain.2013.03.010 [DOI] [PubMed] [Google Scholar]
- Eltorp CT, Jansen-Olesen I, Hansen AJ (2000) Release of calcitonin gene-related peptide (CGRP) from guinea pig dura mater in vitro is inhibited by sumatriptan but unaffected by nitric oxide. Cephalalgia 20:838–844. 10.1046/j.1468-2982.2000.00131.x [DOI] [PubMed] [Google Scholar]
- Fischer MJ, Koulchitsky S, Messlinger K (2005) The nonpeptide calcitonin gene-related peptide receptor antagonist BIBN4096BS lowers the activity of neurons with meningeal input in the rat spinal trigeminal nucleus. J Neurosci 25:5877–5883. 10.1523/JNEUROSCI.0869-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goadsby PJ, Edvinsson L, Ekman R (1990) Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol 28:183–187. 10.1002/ana.410280213 [DOI] [PubMed] [Google Scholar]
- Han JS, Li W, Neugebauer V (2005) Critical role of calcitonin gene-related peptide 1 receptors in the amygdala in synaptic plasticity and pain behavior. J Neurosci 25:10717–10728. 10.1523/JNEUROSCI.4112-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JM, Ashina M (2014) Calcitonin gene-related peptide and migraine with aura: a systematic review. Cephalalgia 34:695–707. 10.1177/0333102413520084 [DOI] [PubMed] [Google Scholar]
- Karsan N, Goadsby PJ (2015) Calcitonin gene-related peptide and migraine. Curr Opin Neurol 28:250–254. 10.1097/WCO.0000000000000191 [DOI] [PubMed] [Google Scholar]
- Keller JT, Marfurt CF (1991) Peptidergic and serotoninergic innervation of the rat dura mater. J Comp Neurol 309:515–534. 10.1002/cne.903090408 [DOI] [PubMed] [Google Scholar]
- Kurosawa M, Messlinger K, Pawlak M, Schmidt RF (1995) Increase of meningeal blood flow after electrical stimulation of rat dura mater encephali: mediation by calcitonin gene-related peptide. Br J Pharmacol 114:1397–1402. 10.1111/j.1476-5381.1995.tb13361.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Strassman AM (2002) Mechanical response properties of A and C primary afferent neurons innervating the rat intracranial dura. J Neurophysiol 88:3021–3031. 10.1152/jn.00029.2002 [DOI] [PubMed] [Google Scholar]
- Levy D, Jakubowski M, Burstein R (2004) Disruption of communication between peripheral and central trigeminovascular neurons mediates the antimigraine action of 5HT 1B/1D receptor agonists. Proc Natl Acad Sci U S A 101:4274–4279. 10.1073/pnas.0306147101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton RB, Munjal S, Buse DC, Bennett A, Fanning KM, Burstein R, Reed ML (2017) Allodynia is associated with initial and sustained response to acute migraine treatment: results from the American migraine prevalence and prevention study. Headache 57:1026–1040. 10.1111/head.13115 [DOI] [PubMed] [Google Scholar]
- Magerl W, Fuchs PN, Meyer RA, Treede RD (2001) Roles of capsaicin-insensitive nociceptors in cutaneous pain and secondary hyperalgesia. Brain 124:1754–1764. 10.1093/brain/124.9.1754 [DOI] [PubMed] [Google Scholar]
- McCarthy PW, Lawson SN (1990) Cell type and conduction velocity of rat primary sensory neurons with calcitonin gene-related peptide-like immunoreactivity. Neuroscience 34:623–632. 10.1016/0306-4522(90)90169-5 [DOI] [PubMed] [Google Scholar]
- McCulloch J, Uddman R, Kingman TA, Edvinsson L (1986) Calcitonin gene-related peptide: functional role in cerebrovascular regulation. Proc Natl Acad Sci U S A 83:5731–5735. 10.1073/pnas.83.15.5731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo-Carrillo A, Noseda R, Nir R, Schain AJ, Stratton J, Strassman AM, Burstein R (2017) Selective inhibition of trigeminovascular neurons by fremanezumab: a humanized monoclonal anti-CGRP antibody. J Neurosci 37:7149–7163. 10.1523/JNEUROSCI.0576-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messlinger K, Hanesch U, Baumgärtel M, Trost B, Schmidt RF (1993) Innervation of the dura mater encephali of cat and rat: ultrastructure and calcitonin gene-related peptide-like and substance P-like immunoreactivity. Anat Embryol 188:219–237. [DOI] [PubMed] [Google Scholar]
- Messlinger K, Ebersberger A, Schaible HG (1998) Release of immunoreactive substance P in the brain stem upon stimulation of the cranial dura mater with low pH: inhibition by the serotonin (5-HT1) receptor agonist CP 93,129. Br J Pharmacol 125:1726–1732. 10.1038/sj.bjp.0702247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S, Liu H, Warfvinge K, Shi L, Dovlatyan M, Xu C, Edvinsson L (2016) Immunohistochemical localization of the calcitonin gene-related peptide binding site in the primate trigeminovascular system using functional antagonist antibodies. Neuroscience 328:165–183. 10.1016/j.neuroscience.2016.04.046 [DOI] [PubMed] [Google Scholar]
- Moskowitz MA. (1984) The neurobiology of vascular head pain. Ann Neurol 16:157–168. 10.1002/ana.410160202 [DOI] [PubMed] [Google Scholar]
- O'Connor TP, van der Kooy D (1988) Enrichment of a vasoactive neuropeptide (calcitonin gene related peptide) in the trigeminal sensory projection to the intracranial arteries. J Neurosci 8:2468–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DD, Dubner R, Hu JW (1976) Trigeminothalamic neurons in nucleus caudalis responsive to tactile, thermal, and nociceptive stimulation of monkey's face. J Neurophysiol 39:936–953. [DOI] [PubMed] [Google Scholar]
- Price TJ, Flores CM (2007) Critical evaluation of the colocalization between calcitonin gene-related peptide, substance p, transient receptor potential vanilloid subfamily type 1 immunoreactivities, and isolectin b(4) binding in primary afferent neurons of the rat and mouse. J Pain 8:263–272. 10.1016/j.jpain.2006.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter U, Weber JR, Gold L, Arnold G, Wolf T, Dreier J, Lindauer U, Dirnagl U (1998) Perivascular nerves contribute to cortical spreading depression-associated hyperemia in rats. Am J Physiol 274:H1979–H1987. [DOI] [PubMed] [Google Scholar]
- Russo AF. (2015) Calcitonin gene-related peptide (CGRP): a new target for migraine. Annu Rev Pharmacol Toxicol 55:533–552. 10.1146/annurev-pharmtox-010814-124701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storer RJ, Akerman S, Goadsby PJ (2004) Calcitonin gene-related peptide (CGRP) modulates nociceptive trigeminovascular transmission in the cat. Br J Pharmacol 142:1171–1181. 10.1038/sj.bjp.0705807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strassman AM, Raymond SA, Burstein R (1996) Sensitization of meningeal sensory neurons and the origin of headaches. Nature 384:560–564. 10.1038/384560a0 [DOI] [PubMed] [Google Scholar]
- Strassman AM, Weissner W, Williams M, Ali S, Levy D (2004) Axon diameters and intradural trajectories of the dural innervation in the rat. J Comp Neurol 473:364–376. 10.1002/cne.20106 [DOI] [PubMed] [Google Scholar]
- Sun H, Dodick DW, Silberstein S, Goadsby PJ, Reuter U, Ashina M, Saper J, Cady R, Chon Y, Dietrich J, Lenz R (2016) Safety and efficacy of AMG 334 for prevention of episodic migraine: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol 15:382–390. 10.1016/S1474-4422(16)00019-3 [DOI] [PubMed] [Google Scholar]
- Sun RQ, Lawand NB, Lin Q, Willis WD (2004a) Role of calcitonin gene-related peptide in the sensitization of dorsal horn neurons to mechanical stimulation after intradermal injection of capsaicin. J Neurophysiol 92:320–326. 10.1152/jn.00086.2004 [DOI] [PubMed] [Google Scholar]
- Sun RQ, Tu YJ, Lawand NB, Yan JY, Lin Q, Willis WD (2004b) Calcitonin gene-related peptide receptor activation produces PKA- and PKC-dependent mechanical hyperalgesia and central sensitization. J Neurophysiol 92:2859–2866. 10.1152/jn.00339.2004 [DOI] [PubMed] [Google Scholar]
- Tsai SH, Tew JM, McLean JH, Shipley MT (1988) Cerebral arterial innervation by nerve fibers containing calcitonin gene-related peptide (CGRP): I. Distribution and origin of CGRP perivascular innervation in the rat. J Comp Neurol 271:435–444. 10.1002/cne.902710310 [DOI] [PubMed] [Google Scholar]
- Uddman R, Edvinsson L, Ekblad E, Hakanson R, Sundler F (1986) Calcitonin gene-related peptide (CGRP): perivascular distribution and vasodilatory effects. Regul Pept 15:1–23. 10.1016/0167-0115(86)90071-6 [DOI] [PubMed] [Google Scholar]
- Uddman R, Hara H, Edvinsson L (1989) Neuronal pathways to the rat middle meningeal artery revealed by retrograde tracing and immunocytochemistry. J Auton Nerv Syst 26:69–75. 10.1016/0165-1838(89)90109-4 [DOI] [PubMed] [Google Scholar]
- Williamson DJ, Hargreaves RJ, Hill RG, Shepheard SL (1997) Intravital microscope studies on the effects of neurokinin agonists and calcitonin gene-related peptide on dural vessel diameter in the anaesthetized rat. Cephalalgia 17:518–524. 10.1046/j.1468-2982.1997.1704518.x [DOI] [PubMed] [Google Scholar]
- Zagami AS, Goadsby PJ, Edvinsson L (1990) Stimulation of the superior sagittal sinus in the cat causes release of vasoactive peptides. Neuropeptides 16:69–75. 10.1016/0143-4179(90)90114-E [DOI] [PubMed] [Google Scholar]
- Zhang X, Levy D, Noseda R, Kainz V, Jakubowski M, Burstein R (2010) Activation of meningeal nociceptors by cortical spreading depression: implications for migraine with aura. J Neurosci 30:8807–8814. 10.1523/JNEUROSCI.0511-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Levy D, Kainz V, Noseda R, Jakubowski M, Burstein R (2011) Activation of central trigeminovascular neurons by cortical spreading depression. Ann Neurol 69:855–865. 10.1002/ana.22329 [DOI] [PMC free article] [PubMed] [Google Scholar]