

Abstract
NMDAR activity in the hypothalamic paraventricular nucleus (PVN) is increased and critically involved in heightened sympathetic vasomotor tone in hypertension. Calcium/calmodulin-dependent protein kinase II (CaMKII) binds to and modulates NMDAR activity. In this study, we determined the role of CaMKII in regulating NMDAR activity of PVN presympathetic neurons in male spontaneously hypertensive rats (SHRs). NMDAR-mediated EPSCs and puff NMDA-elicited currents were recorded in spinally projecting PVN neurons in SHRs and male Wistar-Kyoto (WKY) rats. The basal amplitude of evoked NMDAR-EPSCs and puff NMDA currents in retrogradely labeled PVN neurons were significantly higher in SHRs than in WKY rats. The CaMKII inhibitor autocamtide-2-related inhibitory peptide (AIP) normalized the increased amplitude of NMDAR-EPSCs and puff NMDA currents in labeled PVN neurons in SHRs but had no effect in WKY rats. Treatment with AIP also normalized the higher frequency of NMDAR-mediated miniature EPSCs of PVN neurons in SHRs. CaMKII-mediated phosphorylation level of GluN2B serine 1303 (S1303) in the PVN, but not in the hippocampus and frontal cortex, was significantly higher in SHRs than in WKY rats. Lowering blood pressure with celiac ganglionectomy in SHRs did not alter the increased level of phosphorylated GluN2B S1303 in the PVN. In addition, microinjection of AIP into the PVN significantly reduced arterial blood pressure and lumbar sympathetic nerve discharges in SHRs. Our findings suggest that CaMKII activity is increased in the PVN and contributes to potentiated presynaptic and postsynaptic NMDAR activity to elevate sympathetic vasomotor tone in hypertension.
SIGNIFICANCE STATEMENT Heightened sympathetic vasomotor tone is a major contributor to the development of hypertension. Although glutamate NMDA receptor (NMDAR)-mediated excitatory drive in the hypothalamus plays a critical role in increased sympathetic output in hypertension, the molecular mechanism involved in potentiated NMDAR activity of hypothalamic presympathetic neurons remains unclear. Here we show that the activity of calcium/calmodulin-dependent protein kinase II (CaMKII) is increased and plays a key role in the potentiated presynaptic and postsynaptic NMDAR activity of hypothalamic presympathetic neurons in hypertension. Also, the inhibition of CaMKII in the hypothalamus reduces elevated blood pressure and sympathetic nerve discharges in hypertension. This new knowledge extends our understanding of the mechanism of synaptic plasticity in the hypothalamus and suggests new strategies to treat neurogenic hypertension.
Keywords: autonomic nervous system, hypertension, hypothalamus, sympathetic nerve activity, synaptic transmission
Introduction
Heightened sympathetic vasomotor tone is one of major mechanisms contributing to the development of neurogenic hypertension (Judy et al., 1976; Allen, 2002; Li and Pan, 2007b). The presympathetic neurons in the paraventricular nucleus (PVN) of the hypothalamus can regulate sympathetic outflow through projections to the rostral ventrolateral medulla and the intermediolateral cell column in the spinal cord (Ranson et al., 1998). The PVN is a critical brain region involved in the development and maintenance of hypertension in several animal models such as salt-sensitive hypertension, angiotensin II-induced hypertension (Chen et al., 2010), obesity-related hypertension (Zhou et al., 2015), and spontaneously hypertensive rats (SHRs; Li and Pan, 2007a,b). However, the molecular mechanism underlying the hyperactivity of PVN presympathetic neurons in hypertension is not fully known.
Enhanced glutamatergic input, especially the NMDAR activity, in the PVN is a major source of the excitatory drive to PVN presympathetic neurons, which in turn increases sympathetic vasomotor tone in SHRs (Li and Pan, 2007b; Li et al., 2008). Both presynaptic and postsynaptic NMDAR activity are potentiated in the PVN in SHRs and contribute to the hyperactivity of PVN presympathetic neurons (Li et al., 2008; Ye et al., 2011; Qiao et al., 2017). The NMDAR activity and the phosphorylation level in the PVN is tightly regulated by many protein kinases, such as casein kinase 2 (CK2; Ye et al., 2011, 2012) and Src kinases (Qiao et al., 2017). In addition, calcium/calmodulin-dependent protein kinase II (CaMKII), a synapse-enriched serine/threonine protein kinase (Kelly et al., 1984; Hell, 2014), is abundantly expressed in the brain including the hypothalamus (Erondu and Kennedy, 1985; Wang et al., 2013). CaMKII has four isoforms, including α, β, γ, and δ (Hell, 2014), and each can be activated by cytoplasmic Ca2+ rises (Sanhueza et al., 2011; Barcomb et al., 2016). Through binding to GluN2B subunit of NMDARs, CaMKII is closely involved in synaptic NMDAR plasticity. In this regard, the interaction of CaMKII with NMDARs is critical to the maintenance of synaptic strength in the hippocampus during long-term potentiation (Sanhueza et al., 2011; Halt et al., 2012; Barcomb et al., 2016). However, despite the close association between CaMKII and NMDARs, little is known about whether CaMKII plays a role in the enhanced synaptic NMDAR activity in the PVN in hypertension. In the present study, we used in vitro and in vivo approaches to determine the role of CaMKII in regulating the synaptic NMDAR activity of PVN presympathetic neurons and sympathetic vasomotor tone in SHRs.
Materials and Methods
Animal models.
Male Wistar-Kyoto (WKY) rats and SHRs (age, 13 weeks; Harlan) were used in this study. SHRs are commonly used as an animal model of essential hypertension, and elevated sympathetic outflow is involved in the development and maintenance of hypertension in SHRs (Judy et al., 1976; Allen, 2002; Li and Pan, 2007b). The surgical procedures and experimental protocols were approved by the Institutional Animal Care and Use Committee of The University of Texas MD Anderson Cancer Center and conformed to the National Institutes of Health guidelines for the ethical use of animals.
Retrograde labeling of spinally projecting PVN neurons.
The spinally projecting PVN neurons were identified by fluorescent microspheres injected into the intermediolateral cell column of spinal cord (Li et al., 2014, 2015). Briefly, the rats were anesthetized by 2–3% isoflurane in O2, and laminectomy was performed to expose the spinal cord at the T2–T4 level. FluoSpheres (0.04 μm; Invitrogen) were pressure ejected bilaterally into the intermediolateral region of the thoracic spinal cord in five or six separate 50 nl injections (Nanojector II, Drummond Scientific) through a glass pipette (Li et al., 2008). The injected rats were subcutaneously injected with enrofloxacin (5 mg/kg/d for 3 d) and buprenorphine (0.5 mg/kg every 12 h for 3 d) and allowed to recover for 3–7 d before being used for brain slice recordings.
Electrophysiological recordings in hypothalamic slices.
The brain slices were prepared as we described previously (Li et al., 2008; Ye et al., 2011). Briefly, the anesthetized rats were decapitated, and the brains were quickly removed and placed in ice-cold artificial CSF (aCSF) saturated by a mixture of 95% O2 and 5% CO2) containing the following (in mm): 124.0 NaCl, 3.0 KCl, 1.3 MgSO4, 2.4 CaCl2, 1.4 NaH2PO4, 10.0 glucose, and 26.0 NaHCO3. The coronal brain slices (300 μm thick) containing PVNs were sectioned by a vibrating microtome (VT1000, Leica Biosystems). The slices were then incubated in aCSF continuously gassed with a mixture of 95% O2 and 5% CO2 at 34°C for at least 1 h before the electrophysiological recording.
FluoSphere-labeled PVN neurons were recorded under whole-cell patch-clamp configuration. The hypothalamic slices were continuously perfused with aCSF (saturated by 95% O2 and 5% CO2; speed, 3.0 ml/min) at 34°C maintained by an in-line solution heater. Labeled PVN neurons were identified on an upright microscope (BX51WI, Olympus) with a combination of epifluorescence illumination and differential interference contrast optics. The resistance of the pipette recording electrode was 3–7 MΩ when filled with an internal solution containing the following (in mm): 110.0 Cs2SO4, 2.0 MgCl2, 0.1 CaCl2, 10.0 HEPES, 1.1 EGTA, 0.3 Na2-GTP, and 2.0 Mg-ATP, adjusted to pH 7.25 with 1.0 m CsOH (270–290 mOsm). The lidocaine N-ethyl bromide (QX-314; 10.0 mm), a sodium channel blocker was also added into the internal solution. The signal was processed through a Multiclamp 700B Amplifier (Molecular Devices), filtered at 1–2 kHz, and digitized at 10 kHz using a Digidata 1440 digitizer (Molecular Devices). The junction potential was calculated according to the composition of the internal and external solutions, and corrected off-line. The electrical stimulation (0.2 ms, 0.5 mA, 0.1 Hz) was delivered via a bipolar stimulating electrode to evoke the EPSCs (Li et al., 2008, 2014). AMPA receptor (AMPAR)-EPSCs were recorded at a holding potential of −60 mV in the presence of 10 μm gabazine, and NMDAR-EPSCs were recorded at a holding potential of +40 mV in the presence of 10 μm gabazine and 20 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX). Because the NMDAR channel is typically blocked by Mg2+ at a negative holding potential (Nowak et al., 1984), the NMDAR-EPSCs were recorded at a holding potential of +40 mV to remove the Mg2+ block of NMDAR channels. In contrast, the AMPAR-EPSCs recorded at a holding potential of −60 mV is large and can be readily recorded (Li et al., 2008). We have shown that the amplitude of AMPAR-EPSCs recorded in labeled PVN neurons at a holding potential of −60 mV is similar in WKY rats and SHRs. In this study, AMPAR-EPSCs were used to normalize the NMDAR-EPSCs elicited by the same amount of synaptic glutamate release in SHRs.
The miniature EPSCs (mEPSCs) were recorded at a holding potential of −60 mV in the presence of 10.0 μm gabazine and 1.0 μm tetrodotoxin (TTX). The postsynaptic NMDAR currents were recorded by puff NMDA directly to the labeled neuron at a holding potential of −60 mV (Li et al., 2008; Qiao et al., 2017). The NMDA current was recorded at 0.1 mm Mg2+, 10 μm glycine, and 1 μm TTX (Li et al., 2008; Ye et al., 2011). Gabazine, AP-5, autocamtide-2-related inhibitory peptide (AIP), CNQX, and QX-314 were obtained from Abcam.
In vivo recording of arterial blood pressure and lumbar sympathetic nerve activity.
Rats were anesthetized with a mixture of α-chloralose (60–75 mg/kg) and urethane (800 mg/kg) and mechanically ventilated after trachea cannulation. The arterial blood pressure (ABP) of left femoral artery was monitored, and heart rate (HR) was extracted from the pulsatile pressure wave. A small branch of the left lumbar postganglionic sympathetic nerve was isolated and cut distally (Li and Pan, 2007b). A stainless steel recording electrode was used to record the nerve activity, which was amplified, bandpass filtered (100–3000 Hz; model P511, Grass Instruments), and monitored via an audio amplifier (Grass Instruments). The lumbar sympathetic nerve activity (LSNA) and ABP were recorded by using a 1401-PLUS analog-to-digital converter and Spike2 system (Cambridge Electronic Design). At the end of each experiment, the electrical background noise was measured at 5 min after the rats were killed by an overdose of sodium pentobarbital (100 mg/kg, i.v.) and subtracted from the integrated LSNA values (Li and Pan, 2007a,b). The percentage change in LSNA from baseline was calculated.
PVN microinjection.
The rat head was fixed on a stereotactic apparatus, and the microinjection in the PVN was performed using the following coordinates: 1.6–2.0 mm caudal to the bregma; 0.5 mm lateral to the midline; and 7.0–7.5 mm ventral to the dura (Li and Pan, 2007a,b). The drug was microinjected into PVN through a glass microinjection pipette (tip diameter, 20–30 μm) by using a calibrated microinjection system (Nanojector II, Drummond Scientific). The depressor responses (at least 10 mmHg) to microinjection of 5.0 nmol GABA (20 nl, 250 mm) was used to select the injection site in the PVN. The injection solution contained 2% FluoSpheres microspheres (0.04 μm; Invitrogen) to estimate the drug diffusion site (Li and Pan, 2007a, b). At the end of the experiment, the rat brain was removed and fixed in 10% buffered formalin solution. Frozen coronal sections (40 μm thick) were cut, and rhodamine-labeled fluorescent regions were identified and plotted on standardized sections from the atlas of Paxinos and Watson (1999). The rats were excluded from analysis if the microinjection site was outside the PVN.
Real-time PCR.
Total RNA were extracted from the brain tissues using TRIsure (BIO-38032, Bioline). A RevertAid RT Reverse Transcription Kit (K1619, Thermo Fisher Scientific) was used for reverse transcription of the mRNA (500 ng) to cDNA. Two microliters of five-time diluted cDNA were added to a 20 μl SYBR green real-time PCR system (A25780, Thermo Fisher Scientific). The following primers were used for amplification. CAMKIIα forward primer: AGGATGAAGACACCAAAGTGC; CAMKIIα reverse primer: GGTTCAAAGGC-TGTCATTCC; CAMKIIβ forward primer: ACCTCATTTGAGCCTGA-AGC; CAMKIIβ reverse primer: CAGGATAGTGGTGTGGATCG; GAPDH forward primer: GACATGCCGCCTGGAGAAAC; and GAPDH reverse primer: AGCCCAGGATGCCCTTTAGT. The mean values of CaMKIIα and CaMKIIβ mRNA in WKY rats were considered to be 1.
Western immunoblotting.
PVN tissues (∼0.5 mm in diameter) were obtained using micro-punch methods (Ye et al., 2011; Li et al., 2014). Total proteins were extracted with a radioimmunoprecipitation assay buffer in the presence of a mixture of protease inhibitors. The protein concentration was determined by Bradford protein assay. Protein samples were loaded and run on 12% SDS-PAGE gels and were then transferred to a polyvinylidene difluoride membrane (Millipore). The protein blots were probed with the following antibodies: anti-phosphorylated GluN2BSer1303, anti-CaMKIIα, anti-CaMKIIβ, or anti-GAPAH. All antibodies were purchased from Abcam. The protein bands were detected by ECL kit (GE Healthcare), and quantified using ImageJ software. The mean values of phosphorylated GluN2BSer1303, CaMKIIα, and CaMKIIβ subunits in WKY rats were considered to be 1.
Celiac ganglionectomy and blood pressure measurement with telemetry.
The arterial blood pressure in SHRs was measured by a Millar telemetry system. Rats were anesthetized with 2% isoflurane, and the Millar catheter connected to the transmitter was inserted into the aorta below the bifurcation of the renal arteries. The transmitter was implanted into the abdominal cavity and attached with a suture to the peritoneum. After the surgery, rats were systemically administered buprenorphine (0.3 mg/kg, s.c.) and penicillin for 3 d. The blood pressure signals were monitored in free-moving rats through the receiver, and the data were collected every 2 d and analyzed with a data acquisition system (LabChart, AD Instruments).
After recording stable baseline ABP, the rats were anesthetized again with 2% isoflurane. The celiac ganglion area was exposed through a midline laparotomy and identified within the area near the superior mesenteric artery and celiac artery (Hamer and Santer, 1981). For rats undergoing celiac ganglionectomy (CGx), the celiac plexus and all visible nerves were dissected and removed as completely as possible. In sham control rats, the celiac ganglion plexus was exposed but not disrupted surgically.
Experimental design and statistical analysis.
For electrophysiological recording experiments, only one neuron was recorded in each brain slice, and at least four rats were used for each recording protocol. Data are presented as the mean ± SEM. The mEPSCs were analyzed off-line using a peak detection program (MiniAnalysis, Synaptosoft). The software pClamp 10 was used to determine the peak amplitude of evoked EPSCs and puff NMDA currents. The LSNA, ABP, and HR were analyzed using the Spike2 software program. LSNA was rectified and integrated off-line after subtracting the background noise. Control values were obtained by averaging the signal over a 60 s period immediately before each treatment. Response values after each intervention were averaged over 30 s when the maximal responses occurred. For comparisons of two groups, statistical significance was tested using Student's t test. For comparisons of more than two groups, repeated-measures ANOVA with Dunnett's post hoc test or one-way ANOVA with Tukey's post hoc test was performed to compare responses between experimental groups (GraphPad Prism 6, GraphPad Software). p < 0.05 was considered to be statistically significant.
Results
CaMKII contributes to enhanced postsynaptic NMDAR activity of PVN presympathetic neurons in SHRs
CaMKII is highly expressed in the hypothalamus (Wang et al., 2013). We first determined whether CaMKII is involved in regulating synaptic NMDAR activity of spinally projecting PVN neurons in SHRs. The AIP is a highly specific and potent inhibitor of CaMKII. It has been shown that 10 μm AIP completely blocks CaMII activity but has no effects on other kinases including PKC (Ishida et al., 1995). To determine the effect of AIP (10 μm) on evoked AMPAR-EPSCs and NMDAR-EPSCs in labeled spinally projecting PVN neurons, we incubated the hypothalamic slices from WKY rats and SHRs with either vehicle (aCSF) or AIP (10 μm) for 60 min before electrophysiological recording. The amplitude of evoked NMDAR-EPSCs of labeled PVN neurons was significantly larger in SHRs than in WKY rats in vehicle-treated slices (p < 0.0001, F(3,29) = 24.23; Fig. 1A,B). AIP treatment normalized the amplitude of evoked NMDAR-EPSCs of labeled PVN neurons in SHRs. However, treatment with AIP had no effect on the amplitude of evoked NMDAR-EPSCs of labeled PVN neurons in WKY rats (Fig. 1A,B). AIP treatment did not significantly alter the amplitude of evoked AMPAR-EPSCs of labeled PVN neurons in WKY rats or SHRs. Furthermore, the ratio of NMDAR-EPSCs to AMPAR-EPSCs in vehicle-treated slices from SHRs was significantly higher than that from WKY rats (p < 0.0001, F(3,29) = 29.72; Fig. 1C). AIP treatment normalized the ratio of NMDAR-EPSCs to AMPAR-EPSCs in SHRs to the level in WKY rats (Fig. 1C), but it had no effect on the ratio of NMDAR-EPSCs to AMPAR-EPSCs in WKY rats.
Figure 1.

CaMKII is involved in increased synaptic NMDAR activity of spinally projecting PVN neurons in SHRs. A, Representative current traces of evoked AMPAR-EPSCs (at a holding potential of −60 mV) and NMDAR-EPSCs (at a holding potential of +40 mV) recorded in labeled PVN neurons from WKY rats and SHRs treated with vehicle or 10 μm AIP. B, Summary data of evoked AMPAR-EPSCs and NMDAR-EPSCs in labeled PVN neurons recorded from WKY rats and SHRs treated with vehicle (n = 7 in the SHR group; n = 9 in the WKY group) or AIP (n = 9 in the SHR group; n = 8 in the WKY group). C, Group data show the ratio of NMDAR-EPSCs to AMPAR-EPSCs in labeled PVN neurons recorded from WKY rats or SHRs treated with vehicle or AIP. Data are presented as the mean ± SEM. One-way ANOVA with Tukey's post hoc test was used to analyze the difference in the NMDAR-EPSCs and the ratio of NMDAR-EPSCs to AMPAR-EPSCs among the four groups. *p < 0.05 compared with the values in the vehicle group in WKY rats. #p < 0.05 compared with the values in the SHR vehicle group.
To directly determine the role of CaMKII in the regulation of postsynaptic NMDAR activity, we examined the effect of AIP on NMDAR currents elicited by puff application of NMDA (100 μm) directly to labeled PVN neurons. Puff application of NMDA induced significantly larger currents in SHRs (n = 8) than in WKY rats (n = 9; p < 0.0001, F(3,30) = 17.27; Fig. 2) in vehicle-treated slices. Treatment with AIP (10 μm, 1 h) restored the NMDA currents of labeled PVN neurons in SHRs to the levels in WKY rats (Fig. 2). These results suggest that increased CaMKII activity contributes to increased postsynaptic NMDAR activity of PVN presympathetic neurons in SHRs.
Figure 2.

CaMKII contributes to the increased postsynaptic NMDAR activity of spinally projecting PVN neurons in SHRs. A, B, Original current traces (A) and summary data (B) show the effect of 10 μm AIP on puff-NMDA (100 μm)-elicited currents of labeled PVN neurons recorded from WKY rats (vehicle, n = 9; AIP, n = 9) and SHRs (vehicle, n = 8; AIP, n = 9). Data are presented as the mean ± SEM. One-way ANOVA with Tukey's post hoc test was used to analyze the difference in the NMDAR currents among the four groups. *p < 0.05 compared with the respective values in WKY rats; #p < 0.05 compared with the SHR vehicle group.
CaMKII is involved in tonic activation of presynaptic NMDAR of PVN presympathetic neurons in SHRs
The presynaptic NMDARs in the PVN are normally silent but become tonically activated in SHRs (Li et al., 2008; Ye et al., 2011). To determine whether CaMKII contributes to increased presynaptic NMDAR activity in the PVN in SHRs, we recorded mEPSCs of labeled PVN neurons, which reflect quantal release of glutamate from presynaptic terminals (Wuarin and Dudek, 1993; Sulzer and Pothos, 2000). We included an NMDAR channel blocker, MK-801 (1.0 mm), in the pipette solution to block postsynaptic NMDAR activity in these experiments (Zhou et al., 2010; Qiao et al., 2017). The baseline frequency of mEPSCs was significantly higher in SHRs than in WKY rats (p = 0.0004, F(5,45) = 9.120; Fig. 3), whereas the baseline amplitude of mEPSCs did not differ significantly between the two groups (Fig. 3). To confirm that the increase in the frequency of mEPSCs was mediated by NMDARs in SHRs, we blocked NMDARs by bath application of a specific NMDAR antagonist, AP-5 (50 μm). AP-5 application readily normalized the frequency of mEPSCs of labeled PVN neurons in SHRs to the level observed in WKY rats, but it had no effect in WKY rats (Fig. 3A,B,E). AIP (10 μm, 1 h) treatment significantly reduced the baseline frequency of mEPSCs of labeled PVN neurons in SHRs without changing their amplitude (p = 0.047, F(5,48) = 4.599; Fig. 3). However, AIP treatment had no effect on the frequency or amplitude of mEPSCs in WKY rats. After treatment with AIP, bath application AP-5 (50 μm) no longer had any effect on the frequency of mEPSCs of labeled PVN neurons in brain slices from SHRs (Fig. 3). These data suggest that increased CaMKII activity is involved in tonic activation of presynaptic NMDARs of PVN presympathetic neurons in SHRs.
Figure 3.

CaMKII increases presynaptic NMDAR activity of spinally projecting PVN neurons in SHRs. A–D, Original traces (A, C) and cumulative probability analysis (B, D) show the effect of bath application of AP-5 (50 μm) on mEPSCs of labeled PVN neurons recorded from WKY rats (A, B) and SHRs (C, D) that were treated with vehicle or 10 μm AIP. E, F, Summary data show the effects of AIP and AP-5 on the frequency and amplitude of mEPSCs in labeled PVN neurons of WKY rats (vehicle, n = 8; AIP, n = 8) and SHRs (vehicle, n = 9; AIP, n = 9). Data are presented as the mean ± SEM. *p < 0.05 compared with the respective value in WKY rats; #p < 0.05 compared with the baseline in the SHR vehicle group.
Converging role of CK2 and CaMKII in increased synaptic NMDAR activity of PVN presympathetic neurons in SHRs
CK2, a constitutively active protein serine/threonine kinase, plays a role in increased NMDAR activity of PVN presympathetic neurons in SHRs (Ye et al., 2011). Activated CaMKII promotes the CK2-mediated phosphorylation of GluN2B at S1480 sites to control the synaptic expression of NMDARs (Sanz-Clemente et al., 2013). We thus determined the possible cross talk between CK2 and CaMKII in the PVN in SHRs. 5,6-Dichloro-1-β-d-ribofuranosylbenzimidazole (DRB) is a selective CK2 inhibitor (Raaf et al., 2008). We have shown that treatment with DRB (100 μm) alone can reverse the increased frequency of mEPSCs and the amplitude of puff NMDAR currents in spinally projecting PVN neurons in SHRs (Ye et al., 2011). Treatment with AIP (10 μm) alone and AIP plus DRB (100 μm) similarly reduced the amplitude of NMDA puff-elicited currents of labeled PVN neurons in SHRs (p = 0.265, t(16) = 1.155, n = 10; Fig. 4A,B). The amplitude of puff NMDAR currents of labeled PVN neurons did not significantly differ between SHR brain slices treated with AIP alone and with AIP plus DRB (Fig. 4A,B).
Figure 4.
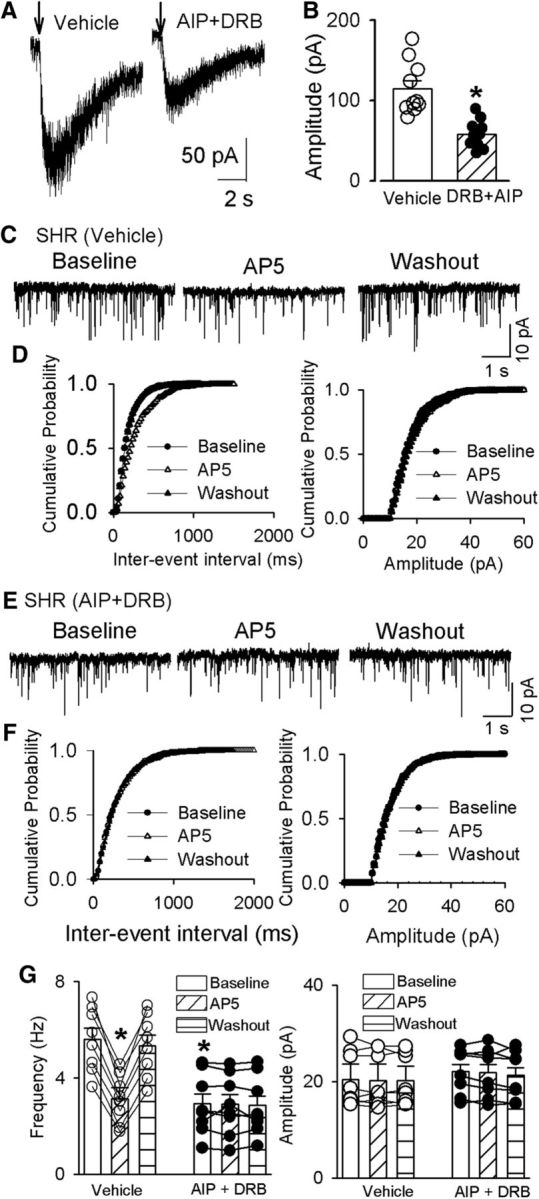
CaMKII and CK2 contributes to enhanced synaptic NMDAR activity of spinally projecting PVN neurons in SHRs. A, B, Original current traces (A) and summary data (B) show the effect of AIP (10 μm) plus DRB (100 μm) on puff NMDA (100 μm)-elicited currents of labeled PVN neurons recorded from SHRs (vehicle, n = 7; AIP plus DRB, n = 8). C–F, Original traces (C, E) and cumulative probability analyses (D, F) show that DRB plus AIP decreased the basal frequency of mEPSCs of labeled PVN neurons recorded from SHRs (C, E) and eliminated the effect of AP-5 (50 μm) on the frequency of mEPSCs (E, F). G, Summary data show the effects of AIP plus DRB and AP-5 on the frequency and amplitude of mEPSCs in labeled PVN neurons of SHRs (vehicle, n = 10; AIP plus DRB, n = 10). Data are presented as the mean ± SEM. *p < 0.05 compared with the baseline in the SHR vehicle group.
Furthermore, treatment with AIP alone and AIP plus DRB produced a similar reduction in the baseline frequency of mEPSCs in labeled PVN neurons in brain slices from SHRs (p = 0.0002, F(5,45) = 9.818; Fig. 4E–G). Also, after treatment with AIP plus DRB, AP-5 no longer had any effect on the frequency of mEPSCs in labeled PVN neurons in SHRs. These results suggest that CK2 and CaMKII have an overlapping role in increased presynaptic and postsynaptic NMDAR activity of PVN presympathetic neurons in hypertension.
CaMKII-dependent phosphorylation of NMDARs in the PVN is increased in SHRs
Because CaMKII directly phosphorylates the NMDAR subunit GluN2B on serine 1303 (S1303; Omkumar et al., 1996), we next determined CaMKII-mediated phosphorylated GluN2B S1303 as a measure of CaMKII activity in the PVN. Western immunoblotting showed a single band of phosphorylated GluN2B S1303, and the phosphorylated GluN2B S1303 protein level in the PVN was significantly greater in SHRs than in WKY rats (p = 0.0353, t(10) = 2.433; Fig. 5A,B). In contrast, phosphorylated GluN2B S1303 protein levels in the hippocampus and frontal cortex did not differ significantly between WKY rats and SHRs (Fig. 5A,B). The mRNA and protein levels of CaMKII-α and CaMKII-β, two of the major isoforms of CaMKII in the PVN, did not significantly differ between WKY rats and SHRs (Fig. 5E,F).
Figure 5.
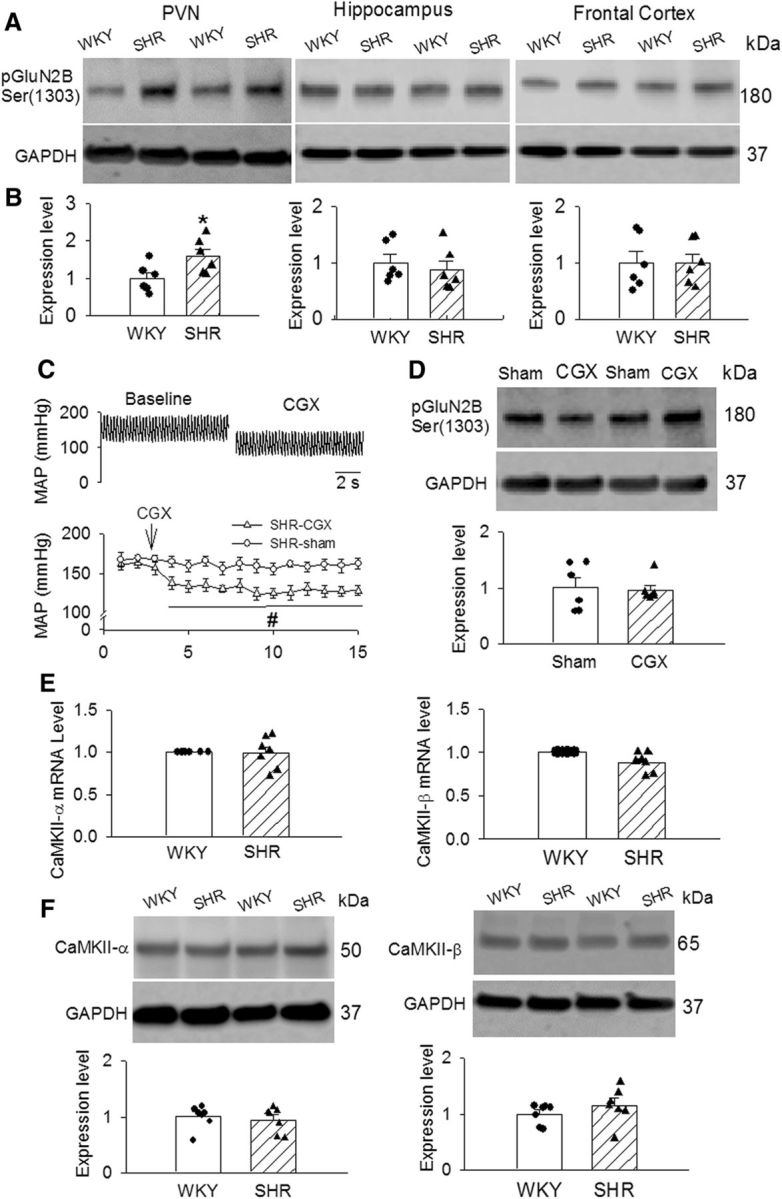
CaMKII-mediated phosphorylation of NMDARs in PVNs in WKY rats and SHRs. A, B, Representative gel images (A) and summary data (B) show the phosphorylated GluN2B (Ser1303) levels (normalized to GAPDH) in the PVN, hippocampus, and frontal cortex in WKY rats and SHRs (n = 6 rats in each group). C, Original ABP traces and group data show the effect of CGx and sham surgery on the mean ABP in SHRs (n = 6 rats in each group). D, Representative images and summary data show the phosphorylated GluN2B (Ser1303) levels (normalized to GAPDH) in the PVN in SHRs subjected to CGx or sham surgery (n = 6 rats in each group). E, Summary data show the mRNA levels of CaMKII-α and CaMKII-β subunits in PVN tissues from WKY and SHR (n = 7 rats in each group). F, Representative gel images and summary data show that CaMKII-α and CaMKII-β protein levels in the PVN tissue in WKY rats and SHRs (n = 6 rats in each group). Data are presented as the mean ± SEM. *p < 0.05 compared with the WKY group; #p < 0.05 compared with the sham group.
To determine whether the higher CaMKII activity in the PVN of SHRs was an adaptive response to high ABP, we performed CGx to lower ABP and then measured phosphorylated GluN2B S1303 levels in the PVNs of SHRs. ABP was monitored by radiotelemetry in free-moving SHRs subjected to sham surgery and CGx. CGx caused a significant reduction in ABP in SHRs compared with those subjected to sham surgery. CGx-induced decreases in ABP in SHRs occurred within 5–7 d after CGx surgery and lasted for at least 2 weeks (Fig. 5C). Immunoblotting showed that the phosphorylated GluN2B S1303 protein level in the PVNs of SHRs did not differ significantly between rats undergoing CGx and those undergoing sham surgery (Fig. 5D). These data suggest that increased CaMKII activity in the PVN is independent of high ABP in SHRs.
CaMKII-dependent NMDAR activity in the PVN contributes to heightened sympathetic vasomotor tone in SHRs
In addition, we determined the role of CaMKII-dependent NMDAR activity in the PVN in controlling sympathetic vasomotor tone in SHRs. We did not perform AIP injection in WKY rats because blocking NMDARs in the PVN has no effect on ABP or sympathetic nerve activity in normotensive WKY rats (Li and Pan, 2007b). Microinjection of AIP (50 pmol, 50 nl) bilaterally into the PVN significantly decreased the LSNA and mean ABP of SHRs (mean ABP: p < 0.0001, F(2,21) = 106.8; LSNA: p < 0.0001, F(2,21) = 151.3; n = 8; Fig. 6D,F). The LSNA and mean ABP started to decrease at a mean time of 5.1 ± 0.6 min after AIP injection, and this effect lasted for 26.3 ± 2.8 min. In SHRs receiving AIP microinjection, subsequent microinjection of the NMDAR antagonist AP-5 (1.0 nmol, 50 nl; Li and Pan, 2007b; Ye et al., 2011; Qiao et al., 2017) into the PVN failed to further decrease LSNA, ABP, and HR (Fig. 6D,F; n = 8). In contrast, AP-5 microinjection into the PVN significantly decreased LSNA and ABP in the SHRs subjected to a prior vehicle microinjection (Fig. 6C,E,F; n = 7). These data suggest that CaMKII-mediated NMDAR activity in the PVN contributes to elevated sympathetic vasomotor tone in SHRs.
Figure 6.

CaMKII-dependent NMDAR activity in the PVN maintains elevated sympathetic vasomotor tone in SHRs. A, B, A representative image (A) and schematic drawing (B) show the microinjection sites for vehicle plus AP-5 (●) and AIP plus AP-5 (○) in the PVN in SHRs. C, D, Representative recording traces show the effect of vehicle (C) or AIP (D) followed by AP-5 bilateral injections into the PVNs on ABP, LSNA, and HR in SHRs. E–G, Summary data show changes in mean ABP (E), LSNA (F), and HR (G) in response to AP-5 after microinjection of vehicle (n = 6) or AIP (n = 7) into the PVN in SHRs. Data are presented as the mean ± SEM. *p < 0.05 compared with the respective baseline. 3V, Third ventricle; AH, anterior hypothalamus.
Discussion
Our study showed that CaMKII is differentially involved in regulating synaptic NMDAR activity of PVN presympathetic neurons in normotensive and hypertensive conditions. We found that inhibition of CaMKII activity with AIP normalized the increased amplitude of evoked NMDAR-EPSCs and puff NMDA-induced currents in spinally projecting PVN neurons in SHRs but had no effect on NMDAR-EPSCs and puff NMDA currents in normotensive control rats. Although CaMKII plays a crucial role in the long-term potentiation in the CA1 field of the hippocampus through the phosphorylation of AMPA receptors (Barria et al., 1997), we found that the inhibition of CaMKII had no effect on the amplitude of mEPSCs or evoked AMPAR-EPSCs in PVN neurons in SHRs or WKY rats. It is unlikely that CaMKII is involved in the regulation of AMPAR activity PVN presympathetic neurons in SHRs. Presynaptic NMDARs in the hypothalamus are not functionally active under physiological conditions, but they become tonically activated and promote synaptic glutamate release to PVN presympathetic neurons in hypertension (Li et al., 2008; Ye et al., 2011). In addition to the expression on postsynaptic soma (Erondu and Kennedy, 1985; Wang et al., 2013), CaMKII is also expressed at nerve terminals (Kelly et al., 1984). We found that CaMKII inhibition normalized the increased NMDAR-mediated baseline frequency of mEPSCs of PVN neurons in SHRs but had no effect on the mEPSC frequency in normotensive WKY rats. This differential effect of CaMKII inhibition on synaptic NMDAR activity is probably due to the low basal level of CaMKII activity in the PVN under the normotensive condition. Our findings suggest that increased CaMKII activity plays a major role in potentiated activity of NMDARs, at both presynaptic and postsynaptic sites, in the PVN in hypertension.
NMDARs are present as tetramers typically composed of obligatory GluN1 and modulatory GluN2 subunits (Traynelis et al., 2010). The GluN2 subunits have a long intracellular C terminus, which is involved in protein–protein interactions and post-translational modifications (Bard and Groc, 2011). Activated CaMKII can bind to the GluN2B C terminus to phosphorylate S1303 (Omkumar et al., 1996; Strack and Colbran, 1998; Leonard et al., 1999). We found in this study that the phosphorylation level of GluN2B S1303 in the PVN was significantly increased in SHRs compared with WKY rats. However, the phosphorylation levels of GluN2B S1303 levels in the hippocampus and frontal cortex did not significantly differ between WKY rats and SHRs, suggesting that the increased CaMKII activity is not a general phenomenon throughout all brain regions in hypertension. We also found that the phosphorylation level of GluN2B S1303 in the PVN was not altered by lowering ABP in SHRs with CGx. This finding suggests that increased CaMKII activity in the PVN is not due to increased ABP but may instead serve as a primary contributor during hypertension development in SHRs. We recognize that the PVN is a heterogeneous nucleus containing many types of cells and that PVN presympathetic neurons may represent a small population of PVN neurons. It should be noted that injecting a retrograde dye to a small region of the spinal cord can only label a small portion of PVN presympathetic neurons in the PVN. Although immunoblotting data showed an increase in CaMKII-mediated GluN2B phosphorylation level in the PVN tissue in SHRs, it is less likely that the phosphorylated GluN2B involves only PVN presympathetic neurons. Thus, CaMKII-induced phosphorylation of GluN2B S1303 in the PVN may occur in cells other than presympathetic neurons and could play a role in other neuroendocrine and psychiatric disorders in SHRs (Yabuki et al., 2014). Although the expression levels of CaMKIIα/β in the PVN were not significantly different between SHRs and WKY rats, CaMKII can remain constitutively active after dissociation from its activator Ca2+/CaM (Miller et al., 1988; Schworer et al., 1988; Thiel et al., 1988; Lou and Schulman, 1989). It is likely that CaMKII is activated in response to NMDAR-mediated Ca2+ entry and in turn triggers the autophosphorylation within its regulatory domain (e.g., Thr286; Bayer et al., 2001; Shonesy et al., 2014). Furthermore, activated CaMKIIα can translocate to the postsynaptic density by binding to residues 1290–1309 within the GluN2B intracellular C terminus (Strack and Colbran, 1998; Leonard et al., 1999). This interaction between CaMKII and NMDARs may be critical for the phosphorylation of other substrates within the postsynaptic density (Strack et al., 2000; Bayer et al., 2001; Leonard et al., 2002).
NMDARs can be phosphorylated at different phosphorylation sites by CK2 and CaMKII (Ye et al., 2011, 2012; Sanz-Clemente et al., 2013; Barcomb et al., 2016). The NMDAR activity depends largely on the balance of the phosphorylation levels of NMDAR subunits. CaMKII and CK2 are both serine/threonine kinase and are critically involved in the phosphorylation of GluN2B (Ye et al., 2012; Sanz-Clemente et al., 2013). We found that the inhibition of CK2 with DRB did not cause further reduction in puff NMDA currents and NMDAR-mediated mEPSCs in PVN presympathetic neurons in SHRs after the inhibition of CaMKII activity. These data suggest that CK2 and CaMKII may have an overlapping role in enhanced synaptic NMDAR activity of PVN presympathetic neurons in SHRs through different phosphorylation sites. In addition, NMDAR phosphorylation may involve a coordinated interaction between CK2 and CaMKII kinases. In this regard, disruption of the CaMKII binding site on GluN2B S1303 leads to CK2-dependent phosphorylation of GluN2B (S1480). Also, activated CaMKII may bind to CK2 and promotes the coupling of CK2 to GluN2B (Sanz-Clemente et al., 2013). Thus, the overall activity of NMDARs in the PVN likely depends on their phosphorylation levels controlled by both CK2 and CaMKII.
The sympathetic vasomotor tone is tightly regulated by PVN presympathetic neurons (Allen, 2002; Li and Pan, 2007b; Li et al., 2008). Because CaMKII inhibition normalized the increased synaptic NMDAR activity of PVN presympathetic neurons in SHRs, we examined the role of CaMKII in controlling elevated sympathetic outflow in SHRs. Consistent with findings in brain slice recordings, we found that the microinjection of the CaMKII inhibitor AIP into the PVN significantly decreased LSNA and ABP in SHRs. Importantly, subsequent microinjection of the NMDAR antagonist AP-5 largely reduced LSNA and ABP in vehicle-injected SHRs, but not in AIP-injected SHRs. These in vivo data suggest that CaMKII-mediated NMDAR activity in the PVN critically contributes to the elevated sympathetic outflow in SHRs. Nevertheless, we cannot exclude other CaMKII phosphorylation targets in the PVN that may be involved in the maintaining sympathetic vasomotor tone in hypertension.
In conclusion, our study provides new information that increased CaMII activity in the PVN plays a pivotal role in the augmented presynaptic and postsynaptic NMDAR activity of PVN presympathetic neurons and elevated sympathetic vasomotor tone in SHRs. CaMKII may be a potential target for treating patients with neurogenic hypertension. It would be of interest to know whether the identified mechanism of NMDAR regulation by CaMKII in SHRs applies to other hypertension models. However, it is not clear whether presynaptic and postsynaptic NMDAR activity of PVN presympathetic neurons are altered in other hypertension models such as renal hypertension, salt-sensitive hypertension, and obesity-related hypertension. After the glutamatergic synaptic plasticity is defined, further studies are needed to determine the dynamic changes in various CaMKII isoforms in the PVN in other animal models of hypertension and to examine the long-term effect of CaMKII inhibitors in conscious hypertensive animals.
Footnotes
This work was supported by the National Institutes of Health (Grants HL-131161 and MH-096086) and the N.G. and Helen T. Hawkins Endowment (to H.-L.P.).
The authors declare no competing financial interests.
References
- Allen AM. (2002) Inhibition of the hypothalamic paraventricular nucleus in spontaneously hypertensive rats dramatically reduces sympathetic vasomotor tone. Hypertension 39:275–280. 10.1161/hy0202.104272 [DOI] [PubMed] [Google Scholar]
- Barcomb K, Hell JW, Benke TA, Bayer KU (2016) The CaMKII/GluN2B protein interaction maintains synaptic strength. J Biol Chem 291:16082–16089. 10.1074/jbc.M116.734822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard L, Groc L (2011) Glutamate receptor dynamics and protein interaction: lessons from the NMDA receptor. Mol Cell Neurosci 48:298–307. 10.1016/j.mcn.2011.05.009 [DOI] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR (1997) Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science 276:2042–2045. 10.1126/science.276.5321.2042 [DOI] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H (2001) Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411:801–805. 10.1038/35081080 [DOI] [PubMed] [Google Scholar]
- Chen QH, Andrade MA, Calderon AS, Toney GM (2010) Hypertension induced by angiotensin II and a high salt diet involves reduced SK current and increased excitability of RVLM projecting PVN neurons. J Neurophysiol 104:2329–2337. 10.1152/jn.01013.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erondu NE, Kennedy MB (1985) Regional distribution of type II Ca2+/calmodulin-dependent protein kinase in rat brain. J Neurosci 5:3270–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, Hell JW (2012) CaMKII binding to GluN2B is critical during memory consolidation. EMBO J 31:1203–1216. 10.1038/emboj.2011.482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamer DW, Santer RM (1981) Anatomy and blood supply of the coeliac-superior mesenteric ganglion complex of the rat. Anat Embryol (Berl) 162:353–362. 10.1007/BF00299978 [DOI] [PubMed] [Google Scholar]
- Hell JW. (2014) CaMKII: claiming center stage in postsynaptic function and organization. Neuron 81:249–265. 10.1016/j.neuron.2013.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida A, Kameshita I, Okuno S, Kitani T, Fujisawa H (1995) A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem Biophys Res Commun 212:806–812. 10.1006/bbrc.1995.2040 [DOI] [PubMed] [Google Scholar]
- Judy WV, Watanabe AM, Henry DP, Besch HR Jr, Murphy WR, Hockel GM (1976) Sympathetic nerve activity: role in regulation of blood pressure in the spontaenously hypertensive rat. Circ Res 38:21–29. 10.1161/01.RES.38.6.21 [DOI] [PubMed] [Google Scholar]
- Kelly PT, McGuinness TL, Greengard P (1984) Evidence that the major postsynaptic density protein is a component of a Ca2+/calmodulin-dependent protein kinase. Proc Natl Acad Sci U S A 81:945–949. 10.1073/pnas.81.3.945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AS, Lim IA, Hemsworth DE, Horne MC, Hell JW (1999) Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A 96:3239–3244. 10.1073/pnas.96.6.3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AS, Bayer KU, Merrill MA, Lim IA, Shea MA, Schulman H, Hell JW (2002) Regulation of calcium/calmodulin-dependent protein kinase II docking to N-methyl-D-aspartate receptors by calcium/calmodulin and alpha-actinin. J Biol Chem 277:48441–48448. 10.1074/jbc.M205164200 [DOI] [PubMed] [Google Scholar]
- Li DP, Pan HL (2007a) Role of gamma-aminobutyric acid (GABA)A and GABAB receptors in paraventricular nucleus in control of sympathetic vasomotor tone in hypertension. J Pharmacol Exp Ther 320:615–626. 10.1124/jpet.106.109538 [DOI] [PubMed] [Google Scholar]
- Li DP, Pan HL (2007b) Glutamatergic inputs in the hypothalamic paraventricular nucleus maintain sympathetic vasomotor tone in hypertension. Hypertension 49:916–925. 10.1161/01.HYP.0000259666.99449.74 [DOI] [PubMed] [Google Scholar]
- Li DP, Yang Q, Pan HM, Pan HL (2008) Pre- and postsynaptic plasticity underlying augmented glutamatergic inputs to hypothalamic presympathetic neurons in spontaneously hypertensive rats. J Physiol 586:1637–1647. 10.1113/jphysiol.2007.149732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Zhu LH, Pachuau J, Lee HA, Pan HL (2014) mGluR5 upregulation increases excitability of hypothalamic presympathetic neurons through NMDA receptor trafficking in spontaneously hypertensive rats. J Neurosci 34:4309–4317. 10.1523/JNEUROSCI.4295-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Zhou JJ, Pan HL (2015) Endogenous casein kinase-1 modulates NMDA receptor activity of hypothalamic presympathetic neurons and sympathetic outflow in hypertension. J Physiol 593:4439–4452. 10.1113/JP270831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou LL, Schulman H (1989) Distinct autophosphorylation sites sequentially produce autonomy and inhibition of the multifunctional Ca2+/calmodulin-dependent protein kinase. J Neurosci 9:2020–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SG, Patton BL, Kennedy MB (1988) Sequences of autophosphorylation sites in neuronal type II CaM kinase that control Ca2(+)-independent activity. Neuron 1:593–604. 10.1016/0896-6273(88)90109-2 [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A (1984) Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307:462–465. 10.1038/307462a0 [DOI] [PubMed] [Google Scholar]
- Omkumar RV, Kiely MJ, Rosenstein AJ, Min KT, Kennedy MB (1996) Identification of a phosphorylation site for calcium/calmodulindependent protein kinase II in the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem 271:31670–31678. 10.1074/jbc.271.49.31670 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1999) The rat brain in stereotaxic coordinates, Ed 4 San Diego: Academic. [DOI] [PubMed] [Google Scholar]
- Qiao X, Zhou JJ, Li DP, Pan HL (2017) Src kinases regulate glutamatergic input to hypothalamic presympathetic neurons and sympathetic outflow in hypertension. Hypertension 69:154–162. 10.1161/HYPERTENSIONAHA.116.07947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaf J, Brunstein E, Issinger OG, Niefind K (2008) The CK2 alpha/CK2 beta interface of human protein kinase CK2 harbors a binding pocket for small molecules. Chem Biol 15:111–117. 10.1016/j.chembiol.2007.12.012 [DOI] [PubMed] [Google Scholar]
- Ranson RN, Motawei K, Pyner S, Coote JH (1998) The paraventricular nucleus of the hypothalamus sends efferents to the spinal cord of the rat that closely appose sympathetic preganglionic neurones projecting to the stellate ganglion. Exp Brain Res 120:164–172. 10.1007/s002210050390 [DOI] [PubMed] [Google Scholar]
- Sanhueza M, Fernandez-Villalobos G, Stein IS, Kasumova G, Zhang P, Bayer KU, Otmakhov N, Hell JW, Lisman J (2011) Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J Neurosci 31:9170–9178. 10.1523/JNEUROSCI.1250-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Clemente A, Gray JA, Ogilvie KA, Nicoll RA, Roche KW (2013) Activated CaMKII couples GluN2B and casein kinase 2 to control synaptic NMDA receptors. Cell Rep 3:607–614. 10.1016/j.celrep.2013.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schworer CM, Colbran RJ, Keefer JR, Soderling TR (1988) Ca2+/calmodulin-dependent protein kinase II. Identification of a regulatory autophosphorylation site adjacent to the inhibitory and calmodulin-binding domains. J Biol Chem 263:13486–13489. [PubMed] [Google Scholar]
- Shonesy BC, Jalan-Sakrikar N, Cavener VS, Colbran RJ (2014) CaMKII: a molecular substrate for synaptic plasticity and memory. Prog Mol Biol Transl Sci 122:61–87. 10.1016/B978-0-12-420170-5.00003-9 [DOI] [PubMed] [Google Scholar]
- Strack S, Colbran RJ (1998) Autophosphorylation-dependent targeting of calcium/calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem 273:20689–20692. 10.1074/jbc.273.33.20689 [DOI] [PubMed] [Google Scholar]
- Strack S, McNeill RB, Colbran RJ (2000) Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem 275:23798–23806. 10.1074/jbc.M001471200 [DOI] [PubMed] [Google Scholar]
- Sulzer D, Pothos EN (2000) Regulation of quantal size by presynaptic mechanisms. Rev Neurosci 11:159–212. [DOI] [PubMed] [Google Scholar]
- Thiel G, Czernik AJ, Gorelick F, Nairn AC, Greengard P (1988) Ca2+/calmodulin-dependent protein kinase II: identification of threonine-286 as the autophosphorylation site in the alpha subunit associated with the generation of Ca2+-independent activity. Proc Natl Acad Sci U S A 85:6337–6341. 10.1073/pnas.85.17.6337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62:405–496. 10.1124/pr.109.002451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang C, Szábo G, Sun QQ (2013) Distribution of CaMKIIalpha expression in the brain in vivo, studied by CaMKIIalpha-GFP mice. Brain Res 1518:9–25. 10.1016/j.brainres.2013.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuarin JP, Dudek FE (1993) Patch-clamp analysis of spontaneous synaptic currents in supraoptic neuroendocrine cells of the rat hypothalamus. J Neurosci 13:2323–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabuki Y, Shioda N, Maeda T, Hiraide S, Togashi H, Fukunaga K (2014) Aberrant CaMKII activity in the medial prefrontal cortex is associated with cognitive dysfunction in ADHD model rats. Brain Res 1557:90–100. 10.1016/j.brainres.2014.02.025 [DOI] [PubMed] [Google Scholar]
- Ye ZY, Li DP, Li L, Pan HL (2011) Protein kinase CK2 increases glutamatergic input in the hypothalamus and sympathetic vasomotor tone in hypertension. J Neurosci 31:8271–8279. 10.1523/JNEUROSCI.1147-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZY, Li L, Li DP, Pan HL (2012) Casein kinase 2-mediated synaptic GluN2A up-regulation increases N-methyl-D-aspartate receptor activity and excitability of hypothalamic neurons in hypertension. J Biol Chem 287:17438–17446. 10.1074/jbc.M111.331165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HY, Chen SR, Chen H, Pan HL (2010) Opioid-induced long-term potentiation in the spinal cord is a presynaptic event. J Neurosci 30:4460–4466. 10.1523/JNEUROSCI.5857-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JJ, Yuan F, Zhang Y, Li DP (2015) Upregulation of orexin receptor in paraventricular nucleus promotes sympathetic outflow in obese Zucker rats. Neuropharmacology 99:481–490. 10.1016/j.neuropharm.2015.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]