

Abstract
While β‐catenin has been demonstrated as an essential molecule and therapeutic target for various cancer stem cells (CSCs) including those driven by MLL fusions, here we show that transcriptional memory from cells of origin predicts AML patient survival and allows β‐catenin‐independent transformation in MLL‐CSCs derived from hematopoietic stem cell (HSC)‐enriched LSK population but not myeloid–granulocyte progenitors. Mechanistically, β‐catenin regulates expression of downstream targets of a key transcriptional memory gene, Hoxa9 that is highly enriched in LSK‐derived MLL‐CSCs and helps sustain leukemic self‐renewal. Suppression of Hoxa9 sensitizes LSK‐derived MLL‐CSCs to β‐catenin inhibition resulting in abolishment of CSC transcriptional program and transformation ability. In addition, further molecular and functional analyses identified Prmt1 as a key common downstream mediator for β‐catenin/Hoxa9 functions in LSK‐derived MLL‐CSCs. Together, these findings not only uncover an unexpectedly important role of cells of origin transcriptional memory in regulating CSC self‐renewal, but also reveal a novel molecular network mediated by β‐catenin/Hoxa9/Prmt1 in governing leukemic self‐renewal.
Keywords: cells of origin, Hoxa9, MLL leukemia, Prmt1, Wnt/β‐catenin
Subject Categories: Cancer, Stem Cells, Transcription
Introduction
Self‐renewal is a critical feature of stem cells, but is diminished upon differentiation into their progenitors. During the differentiation process, gene expression programs responsible for self‐renewal are down‐regulated and frequently replaced by lineage‐specific transcriptional programs. Increasing evidence suggests that genes involved in promoting normal stem cell self‐renewal are commonly hijacked in cancer stem cells (CSCs), which are believed to sustain the disease and be responsible for relapse of various cancers including acute myeloid leukemia (AML; Zeisig et al, 2012; Fung et al, 2013). Among them, one of the most striking molecules is β‐catenin, which is required for leukemic stem cells (LSCs) driven by MLL‐fusion proteins or their downstream targets, Meis1/Hoxa9 (Wang et al, 2010; Yeung et al, 2010). Suppression of β‐catenin reversed MLL‐LSC to pre‐LSC stage (Yeung et al, 2010), and its complete inactivation prevented development of leukemia driven by MLL fusions or Meis1/Hoxa9 (Wang et al, 2010; Yeung et al, 2010). While β‐catenin is critical for embryonic and a number of somatic stem cells including fetal hematopoietic stem cells (HSCs; Zhao et al, 2007; Malhotra & Kincade, 2009), it is largely dispensable for adult HSCs, which can function normally when β‐catenin alone or even together with γ‐catenin is deleted (Cobas et al, 2004; Jeannet et al, 2008; Koch et al, 2008), highlighting the therapeutic potentials of targeting β‐catenin for eradication of LSCs.
In spite of the fundamental difference between stem cells and their progenitors in self‐renewal ability, we and others have shown that phenotypically and genetically indistinguishable cancers, including MLL‐rearranged leukemia, can arise from not only stem cells, but also their immediate downstream short‐lived progenitors with distinctive transcriptional programs (Cozzio et al, 2003; So et al, 2003; Huntly et al, 2004; Krivtsov et al, 2006; Visvader, 2011; Blanpain, 2013). Consistently, gene expression signatures associated with stem cells and progenitors correlate with different clinical outcomes in AML (Eppert et al, 2011; Krivtsov et al, 2013). MLL leukemia derived from mouse HSC‐enriched Lin−Sca‐1+c‐kit+ (LSK) populations can be more aggressive and less responsive to standard chemotherapy than those derived from granulocyte–myeloid progenitors (GMPs; Krivtsov et al, 2013). In line with this, a more recent study also reveals that HSC‐derived leukemia drives an invasive EMT‐related gene expression program, which may partly account for the aggressive nature of the disease (Stavropoulou et al, 2016). In spite of these recent evidences indicating the importance of cancer cells of origin in disease pathogenesis, we still do not know whether and how they may govern the utilization of molecular pathways for self‐renewal, which is a defining feature of CSC and has been a major focus for development of effective cancer therapeutics in the past decade.
Given the important function of β‐catenin in CSC biology, we carried out detailed functional biology and molecular studies examining β‐catenin requirement in MLL‐CSCs originated from different cells of origin. Here, we report that transcriptional memory from cells of origin that robustly predicts AML patient survival can govern and help to override the β‐catenin requirement in MLL‐CSCs. Mechanistically, we identify a novel transcriptional network mediated by Hoxa9/Prmt1 in sustaining leukemic self‐renewal in the absence of β‐catenin in HSC‐derived MLL‐CSCs. These findings reveal previously unrecognized functions and molecular networks from cancer cells of origin that allow override of β‐catenin‐dependent leukemic self‐renewal, adding a new dimension to the ongoing research efforts in developing effective therapeutics for eradication of CSCs.
Results
LSK‐ but not GMP‐derived MLL‐CSCs can override β‐catenin requirements for leukemic self‐renewal
To determine the functional requirement of β‐catenin in MLL‐CSCs derived from different cells of origin, we employed the previously described retroviral transduction/transformation assays (RTTA; Yeung & So, 2009; Zeisig & So, 2009) using HSC‐enriched Lin−Sca‐1+c‐Kit+ population (LSK), granulocyte/macrophage progenitors (GMPs), and control c‐Kit+ cells (mixed population consisting of mostly progenitors) from Ctnnb1 fl/fl CreER (Brault et al, 2001) conditional knockout mice (Fig 1A, Appendix Fig S1A and B). Consistent with previous findings (Yeung et al, 2010), β‐catenin was not required for MLL‐ENL in vitro transformation of c‐Kit+ cells (Appendix Fig S1C–E), but essential for in vivo development of CSCs (Appendix Fig S1F). Similarly, MLL‐ENL could transform LSK and GMPs independently of β‐catenin in vitro and formed compact colonies with early myeloid phenotypes (Fig 1B–D, Appendix Fig S1G and H). However, while β‐catenin deletion in GMP‐MLL‐ENL abolished its leukemogenic potentials in vivo (Fig 1E), β‐catenin deletion had little impact on LSK‐MLL‐ENL, which could still induce leukemia with indistinguishable phenotypes and largely similar latencies as compared with the wild‐type controls (Fig 1F–H). More importantly, LSK‐MLL‐ENL β‐catenin‐deficient cells could competently induce AML upon secondary transplant (Fig 1F–H, Appendix Fig S1I and J), which readout the self‐renewal property of CSCs and indicate the largely uncompromised CSC property in the absence of β‐catenin in LSK‐derived but not GMP‐derived MLL‐CSCs. The results could also be reproduced using a different MLL‐ENL construct carrying the minimal transformation domain (Slany et al, 1998) and MLL‐AF9 (Smith et al, 2011), and were not due to different expression levels of the MLL fusions in these populations (Appendix Fig S1K–M).
Figure 1. MLL‐ENL leukemic stem cells derived from LSK or GMP populations have contrasting functional requirements of β‐catenin for their initiation and maintenance of disease.
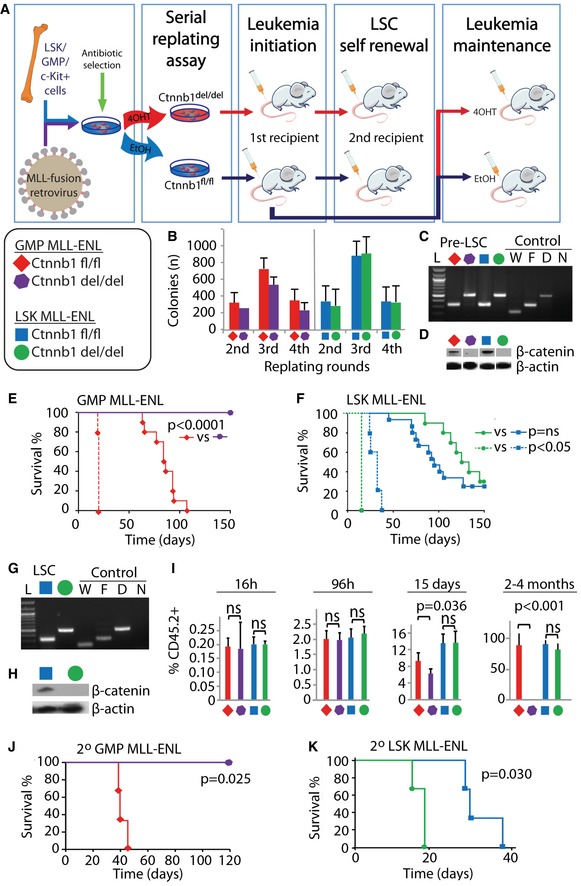
-
ASchematic overview of the experimental procedures. Keys and color codes in the legend box indicate the cells of origin and the β‐catenin status of MLL‐ENL‐transduced cells in the following experiments (B–K).
-
BColony numbers in serial replating assay of the different MLL‐ENL‐transduced cells. Data are represented as mean ± SD (n = 4).
-
CPCR validation of Ctnnb1 deletion on genomic DNA isolated from the indicated MLL‐ENL‐transduced GMP and LSK cells. L, 100‐bp ladder; W, wild‐type control; F, Ctnnb1‐floxed allele; D, Ctnnb1‐deleted allele; N, negative control.
-
DCell lysates from indicated MLL‐ENL‐transduced GMP and LSK cells after the fourth round of plating were blotted with anti‐β‐catenin (top) and anti‐actin (bottom) antibodies.
-
E, FKaplan–Meier survival curves of indicated MLL‐ENL‐transduced cells transplanted into primary recipient (solid lines) and secondary recipient mice (dotted lines). N = 10 mice per group were used in primary transplants (solid lines) in (E). N = 15 mice were used for Ctnnb1 fl/fl (blue solid line), and n = 10 mice were used for Ctnnb1 del/del (green solid line) in (F). For all secondary transplants (dotted lines), n = 5 mice were used per group.
-
GPCR validation of Ctnnb1 deletion on genomic DNA isolated from leukemic cells. L, 100‐bp ladder; W, wild‐type control; F, Ctnnb1‐floxed allele; D, Ctnnb1‐deleted allele; N, negative control.
-
HCell lysates from indicated leukemic mice were blotted with anti‐β‐catenin (top) and anti‐actin (bottom) antibodies.
-
IPercentage of CD45.2+ donor cells in the bone marrow of recipient mice at the indicated time points post‐transplantation of the indicated GMP‐MLL‐ENL‐ and LSK‐MLL‐ENL‐transduced cells. Data are represented as mean ± SD. N = 4, two‐tailed t‐test was performed.
-
J, KKaplan–Meier survival curves of secondary transplanted GMP‐MLL‐ENL (J) and LSK‐MLL‐ENL (K) primary leukemia bone marrow cells treated with DMSO as floxed controls or tamoxifen for β‐catenin deletion prior to secondary transplantation (n = 3 per group).
To gain further insights into the role of β‐catenin in disease development, we followed the in vivo kinetics of the MLL‐transformed cells derived from different cellular origins with or without β‐catenin. The results showed a similar percentage of engraftment across all samples of different cellular origins and genotypes at 16 and 96 h post‐transplant (Fig 1I), suggesting that β‐catenin deletion did not significantly affect homing and early in vivo proliferation abilities. In contrast to LSK‐derived MLL‐CSCs that continued to expand and induced leukemia in the absence of β‐catenin, the expansion of GMP‐MLL‐ENL Ctnnb1 del/del cells slowed down at 15 days and were gradually lost in vivo over a 4‐month period (Fig 1I), consistent with an impaired self‐renewal.
β‐Catenin is also not required for leukemia maintenance by LSK‐derived MLL‐CSCs
To explore the function of β‐catenin in the maintenance of leukemia derived from different origin‐specific CSCs, full‐blown leukemic cells harvested from primary leukemic mice carrying Ctnnb1‐floxed alleles were then treated with either EtOH or tamoxifen prior to transplantation into secondary recipients (Fig 1A). As expected, both EtOH‐treated LSK‐ and GMP‐derived MLL leukemic cells could competently induce leukemia. Inactivation of β‐catenin in GMP‐MLL‐ENL totally abolished their leukemogenic potential (Fig 1J), while β‐catenin‐deficient LSK‐MLL‐ENL still efficiently induced leukemia and even with a slightly shorter latency in secondary recipients (Fig 1K). In contrast to the absolute requirement of β‐catenin for development of various LSCs (Zhao et al, 2007; Wang et al, 2010; Yeung et al, 2010), the finding of largely dispensable function of β‐catenin in LSK‐derived MLL‐CSCs reveals an unexpected and previously unrecognized role of cells of origin in governing leukemic self‐renewal for both cancer initiation and maintenance.
Genomic variations do not account for contrasting β‐catenin dependence in MLL‐CSCs from different cells of origin
To assess whether the observed cell type‐specific differences could be explained by random genetic changes associated with particular cell types, nucleotide variations were called from all actively transcribed genes using RNA‐Seq. Among 23,766,084 high‐quality base pairs (depth ≥ 10 and quality ≥ 30), the vast majority of sites (23,747,763) were invariant in all our primary cell lines, and identical to the reference genome GRCm38, while a very small proportion, 4,663 SNPs (single nucleotide polymorphisms), were invariant and different from GRCm38. In‐depth variance analysis revealed that the difference between the LSK‐MLL‐ENL and GMP‐MLL‐ENL cells was not larger than between normal cells (Fig 2A, Dataset EV1A–C), and there were no fixed differences between the samples that could have caused the observed phenotypic differences. To further profile the non‐coding genomes, whole‐genome sequence analysis on LSK‐MLL‐ENL and GMP‐MLL‐ENL cells covering 918,583,518 high‐quality base pairs revealed 917,764,811 invariant sites, and a very small number of variants in the samples; 39,846 variants were different between the two biological replicates (mice), and only 17,225 were found different between the two cell types (Fig 2B, Dataset EV1C). Interestingly, the distribution of SNPs differing between cell types or mice was comparable for both comparisons in non‐coding and coding regions (Appendix Fig S2A). Consistently, SNPs in coding regions occurred in similar proportions in exon, intron, and UTR regions in both comparisons (Appendix Fig S2B). Moreover, the number of derived SNPs in GMP‐MLL‐ENL (9,309) was higher than those in LSK‐MLL‐ENL cells (7,916) that exhibited β‐catenin‐independent phenotypes (Dataset EV1C). Additionally, we examined and compared the copy number variances (CNVs) between the LSK‐MLL‐ENL and GMP‐MLL‐ENL genomes (Fig 2C and D). As a result, we observed very little CNVs in both genomes. There is only a very small genomic region of about 1 kb showing CNV between same cell types (i.e., LSK‐MLL‐ENL vs. GMP‐MLL‐ENL), whereas multiple chromosomal regions of about 50 kb exhibiting CNV were detected between samples (i.e., mouse Exp60 vs. mouse Exp69 in Dataset EV1D). Importantly, there is also no known coding gene in the 1‐kb CNV region shared between cell types (Fig 2D), consistently indicating insignificant genomic difference between LSK‐MLL‐ENL vs. GMP‐MLL‐ENL cells, which could account for their contrasting β‐catenin dependence. Together, these data reveal relatively few genomic variation in LSK‐MLL‐ENL compared with GMP‐MLL‐ENL and the controls, suggesting that non‐genomic influence from the cells of origin can be a key factor in governing the self‐renewal property of genetically and phenotypically indistinguishable cancers.
Figure 2. Cells of origin transcriptional memory predicts survival in AML patients.
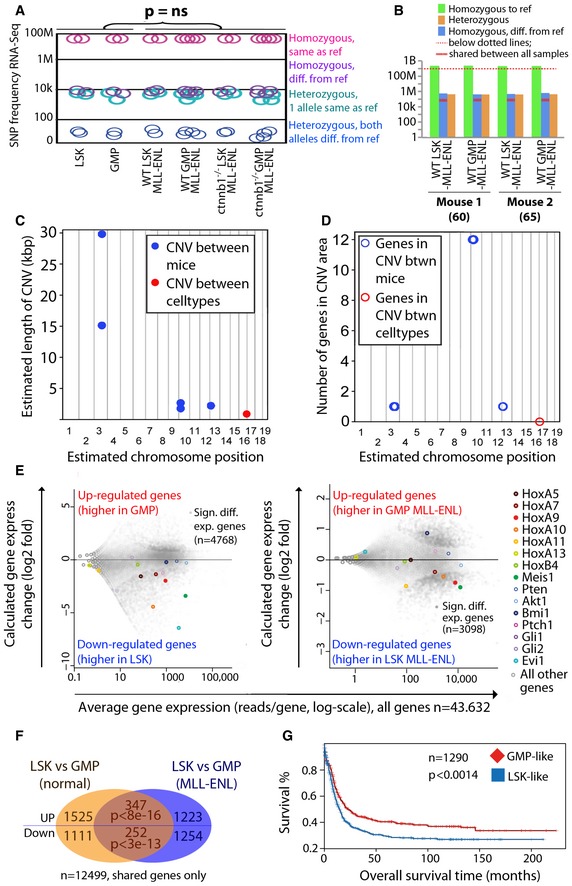
-
A, BNumber of identified genomic variants in indicated MLL‐ENL transformed cells using RNA‐Seq (A) and genomic sequencing (B). A two‐tailed t‐test was performed in (A). LSK, wild‐type LSK cells; GMP, wild‐type GMP cells.
-
C, DManhattan plots indicating estimated length of CNVs (C) or number of genes in CNV areas (D) on the y‐axis in the respective chromosomal positions shown in the x‐axis.
-
EMA‐plots showing the log2‐fold gene expression changes in the normal (left panel) and MLL‐ENL transformed (right panel) cells as indicated.
-
FTranscriptional memory signature; the overlap of differentially expressed genes in GMP‐LSK in normal vs. MLL‐ENL transformed cells is significantly enriched using a hypergeometric test.
-
GSurvival differences between patients clustered using transcriptional memory signatures with a log‐rank test.
Transcriptional memory from cells of origin governs self‐renewal pathways and predicts AML patient survival
As self‐renewal in normal stem cells is maintained by specific transcriptional programs, we hypothesized that the transcriptional memories from LSK and GMPs would be partially preserved even after transformation, resulting in transcriptional and functional differences observed in the respective CSCs (Zeisig et al, 2012). Thus, RNA‐seq analyses of normal LSK, GMPs, and their MLL‐ENL‐transformed counterparts were carried out. There were, as expected, large transcriptional differences between normal LSK and GMPs with 4,768 significantly differentially expressed genes, including Hox genes, Meis1 and Evi1 (Fig 2E, Appendix Fig S2C, Dataset EV2A and B), while overall gene expression differences between cells of different origin decreased after MLL‐ENL transformation (Fig 2E, Appendix Fig S2D). Nevertheless, a significantly larger than expected by chance number of genes remained differentially expressed between LSK and GMP even after transformation (Fig 2F, Appendix Fig S2C, Dataset EV2C), indicating the presence of “transcriptional memory” retained from the cells of origin. Toppgene functional annotation revealed genes associated with AML are consistently present in both signatures (Appendix Fig S2F–I, Dataset EV2D).
To further investigate the relevance of this cells of origin transcriptional memory gene signature in human leukemia, we employed it to stratify 1,290 human AML patients from multiple independent centers (Valk et al, 2004, Raponi et al, 2007, 2008; Metzeler et al, 2008; Wouters et al, 2009; Cancer Genome Atlas Research N, 2013; Dataset EV2E). AML patients with LSK‐transcriptional memory signature had much worse prognosis with a median survival 14.5 months as compared to patients with GMP‐transcriptional memory signature with median survival 22.7 months (Fig 2G), even though the two groups had similar WBC count (means = 40.3, 45.3, t‐test P = 0.30), age distributions (means = 48.0, 50,0, t‐test P = 0.07), and cytogenetic risk (cytogenetic risk (1/2/3) = 71/199/85, 82/165/66, chi‐square test P = 0.16). When compared with the previously identified human HSC signature (Eppert et al, 2011) and MLL leukemic‐GMP (LGMP) signatures from different cells of origin (Krivtsov et al, 2013), the current transcriptional memory signature represents a stronger predictor to stratify patients into different prognostic subgroups based on both resultant median survivals and P‐values (Appendix Fig S2J). Moreover, multivariate analyses consistently resulted in significant Cox proportional hazards ratios > 1; z‐score < 0.1 with both human HSC signature and transcriptional memory signature (Dataset EV2F). Together, these data indicate functional and pathological relevance of the newfound cells of origin transcriptional memory in governing human cancer biology beyond known cytogenetic/genetic risk factors.
Hoxa9 as a key transcriptional memory gene phenocopies β‐catenin function in development of origin‐specific MLL leukemia
Given the largely dispensable function of β‐catenin in LSK‐derived MLL‐CSCs, we hypothesize that some self‐renewal programs from normal stem cells may persist after transformation, and can sustain self‐renewal in the absence of β‐catenin. In the transcriptional memory signature, there were a small number of self‐renewal genes such as Hoxa9, Hoxa10, and Meis1 (Fig 2E and F), which are known downstream targets of MLL fusions (Milne et al, 2002; Zeisig et al, 2004; Huang et al, 2012), indicating that their degrees of activation are in part also determined by the cellular origins. Strikingly, RNA‐sequencing analysis on MLL‐ENL transformed cells upon β‐catenin inactivation revealed a specific up‐regulation of targets genes suppressed by Hoxa9/Meis1, suggesting a critical function of β‐catenin in regulation Hox/Meis1 axis for leukemic self‐renewal (Fig 3A, Dataset EV3A–C). Moreover, various stem cell‐related gene sets were positively enriched in β‐catenin‐deleted LSK‐MLL‐ENL cells as compared with β‐catenin‐deleted GMP‐MLL‐ENL (Fig 3B, Appendix Fig S3A, Dataset EV3C). β‐catenin‐deleted LSK‐MLL‐ENL not only expressed higher levels of Hoxa9 (Fig 3C and D) but also showed a negative enrichment for genes repressed by Hoxa9 (Fig 3E, Dataset EV3C). Together, the data consistently suggest a potential Hoxa9 complementation function in replacing β‐catenin in LSK‐derived MLL‐CSCs.
Figure 3. Key transcriptional memory gene Hoxa9 may help to overcome β‐catenin‐dependent transformation in LSK‐derived MLL‐CSCs.
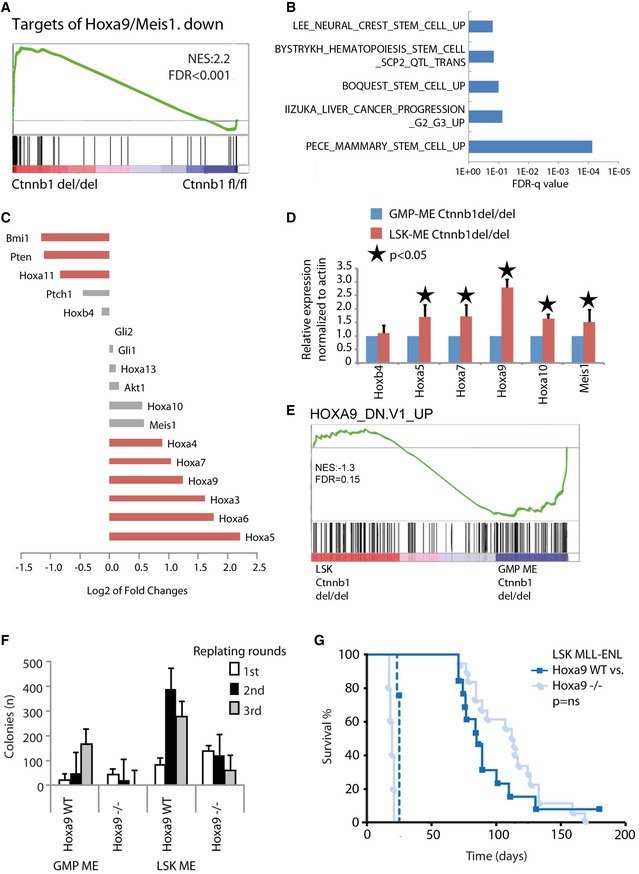
-
AGene set enrichment analysis (GSEA) shows “Targets of Hoxa9/Meis1, down” (Hess et al, 2006) for the indicated comparison.
-
BLog10‐fold FDR q‐values of the indicated gene sets positively enriched in β‐catenin‐deleted LSK‐MLL‐ENL compared to β‐catenin‐deleted GMP‐MLL‐ENL.
-
C–ERNA‐seq log2‐fold change of key self‐renewal genes (C), RT–qPCR validation of Hox/Meis1 expression represented as mean ± SD of three independent experiments. Two‐tailed t‐test was performed (D), and GSEA showing “Hoxa9_dn.v1_up” (Faber et al, 2009) for the β‐catenin‐deleted LSK‐MLL‐ENL to β‐catenin‐deleted GMP‐MLL‐ENL comparison (E).
-
FColony numbers in serial replating assay of indicated MLL‐ENL‐transduced cells. Data are represented as mean ± SD (n = 3).
-
GKaplan–Meier survival curve of mice transplanted with Hoxa9 −/− (n = 18) or WT LSK‐MLL‐ENL (n = 13) transformed cells (solid lines) and secondary recipient mice (n = 5 for Hoxa9 −/− and n = 4 for wt, dotted lines) as indicated. Comparisons between Hoxa9 WT and Hoxa9 −/− were not significantly different (ns).
Similar to β‐catenin, activation of Hoxa9 enhances self‐renewal, while its deletion does not have significant impact on HSCs (So et al, 2004; Lawrence et al, 2005; Smith et al, 2011), consistent with the existence of multiple complementary self‐renewal pathways in HSCs. We hypothesize whether there is indeed a functional complementation between Hoxa9 and β‐catenin, Hoxa9 requirement for MLL transformation may also be influenced by cells of origin. To address this issue, we used purified hematopoietic populations from Hoxa9‐knockout mice for RTTA. While MLL‐ENL could competently transform wild‐type LSK and GMPs in vitro, only LSK but not GMPs could be transformed by MLL‐ENL in the absence of Hoxa9 (Fig 3F, Appendix Fig S3B). More importantly, LSK‐MLL‐ENL Hoxa9 −/− similar to wild‐type LSK‐MLL‐ENL could induce serially transplantable leukemia in recipient mice (Fig 3G, Appendix Fig S3C), strongly suggesting that Hoxa9 requirement, similar to β‐catenin, is also largely determined by cancer cells of origin. These results not only assert the critical function of the newfound cells of origin transcription memory in governing the biology of the resultant disease, but also suggest that LSK‐MLL‐CSCs may be able to utilize the Hoxa9‐mediated self‐renewal pathways as a molecular mechanism to overcome targeted disruption of β‐catenin function.
Suppression of Hoxa9 abolishes β‐catenin‐independent transformation in LSK‐derived MLL‐CSCs
To gain further molecular insights into the functional interplay between β‐catenin and Hoxa9 in mediating self‐renewal in origin‐specific CSCs, we generated a novel compound Hoxa9 −/− Ctnnb1 fl/fl Rosa‐CreER Rosa‐YFP mouse by crossing Hoxa9 −/− mice (Smith et al, 2011) with Ctnnb1 fl/fl Rosa‐CreER Rosa‐YFP mice for RTTA and RNA‐sequencing analysis. While MLL‐ENL‐transduced LSK and GMPs isolated from compound Hoxa9 −/− Ctnnb1 fl/fl Rosa‐CreER Rosa‐YFP mice produced a similar number of first round colonies (Appendix Fig S4A), a further β‐catenin inactivation significantly compromised their transformation ability resulting in reduced number and size of colonies with early myeloid phenotypes (Fig 4A and B, Appendix Fig S4B–D).
Figure 4. Deletion of both β‐catenin and Hoxa9 abrogates leukemia development in LSK‐MLL‐ENL cells.
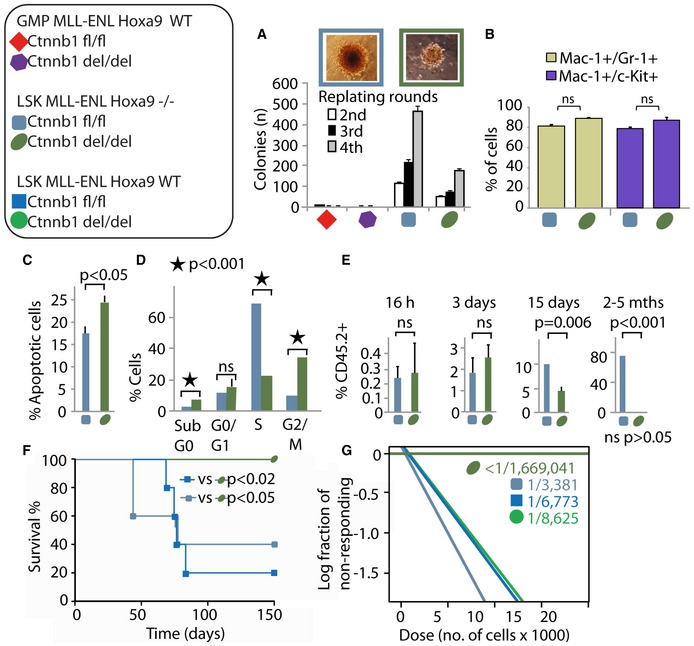
-
AColony numbers in serial replating assay of MLL‐ENL‐transduced cells. Images of typical 4th round colonies shown above. Data are represented as mean ± SD (n = 3).
-
BSummary of immunophenotypic analysis (Appendix Fig S4D) of LSK‐MLL‐ENL Hoxa9 −/− Ctnnb1 fl/fl with or without 72 h of tamoxifen treatment. Data are represented as mean ± SD. N = 3; two‐tailed t‐test was performed.
-
C–EPercentage of apoptotic cells (AnnexinV assay) (C), cells in the indicated cell cycle phases (BrdU assay) (D) and CD45.2+ donor cells in the bone marrow of recipient mice at the indicated time points post‐transplantation (E) of LSK‐MLL‐ENL‐transduced cells. Data are represented as mean ± SD. N = 4, two‐tailed t‐test was performed.
-
FKaplan–Meier survival curve of mice transplanted with LSK‐MLL‐ENL carrying different Hoxa9 and Ctnnb1 genotypes. N = 5 per genotype was used.
-
GSummary of the LSC frequencies obtained from the in vivo limiting dilution experiments (Appendix Fig S4E–H) using the LSK‐MLL‐ENL leukemic cells with different genotypes.
LSK‐MLL‐ENL Hoxa9 −/− Ctnnb1 −/− displayed a higher percentage of apoptosis (Fig 4C), and an increase in G2/M arrest at the expense of S‐phase (Fig 4D), which might help explain their reduced numbers and colony sizes (Fig 4A). Upon transplantation, both LSK‐MLL‐ENL Hoxa9 −/− Ctnnb1 fl/fl and Hoxa9 −/− Ctnnb1 del/del engrafted in comparable levels into the bone marrow and were able to proliferate and transiently expand (Fig 4E). However, inactivation of β‐catenin in LSK‐MLL‐ENL Hoxa9 −/− led to a gradual loss of their self‐renewal property and failed to induce leukemia (Fig 4E and F). Further in vivo limiting dilution analysis revealed similar frequency of CSCs found in wild‐type, Hoxa9 −/− , or Ctnnb1 −/− LSK‐MLL‐ENL, ranging from 1/3,381 to 1/8,625 (Fig 4G and Appendix Fig S4E–G), indicating a rather limited impact of single inactivation of Hoxa9 or β‐catenin on LSK‐derived MLL‐CSCs. In contrast, deletion of both proteins in LSK‐MLL‐ENL resulted in a drastic reduction of CSC frequency (estimated to be below 1/1,669,041; Fig 4G and Appendix Fig S4H), consistent with a critical functional crosstalk between Hoxa9 and β‐catenin, in which a high level of Hoxa9 expression allows β‐catenin‐independent transformation in LSK‐derived MLL‐CSCs.
β‐catenin and Hoxa9 co‐regulate Prmt1 in LSK‐MLL‐ENL cells
To gain further insights into the molecular pathways underlying β‐catenin‐ and Hoxa9‐mediated leukemic self‐renewal, we sought to define common gene sets that were deregulated upon the loss of leukemic self‐renewal (i.e., β‐catenin inactivation in Hoxa9 −/− LSK‐MLL‐ENL, Hoxa9 inactivation in Ctnnb1 −/− LSK‐MLL‐ENL cells). As a result, 38 gene sets were commonly up‐regulated (Fig 5A) and 33 were commonly down‐regulated (Fig 5B, Dataset EV4A and B) in compound Hoxa9 −/− Ctnnb1 −/− LSK‐MLL‐ENL cells. Consistent with the loss of leukemic self‐renewal, LSC_maintenance signatures (Somervaille et al, 2009) were inversely enriched. Moreover, the Hoxa9/Meis1 targets (Hess et al, 2006) were also inversely enriched, supporting the hypothesis that β‐catenin and Hoxa9 may co‐regulate common sets of genes critical for self‐renewal.
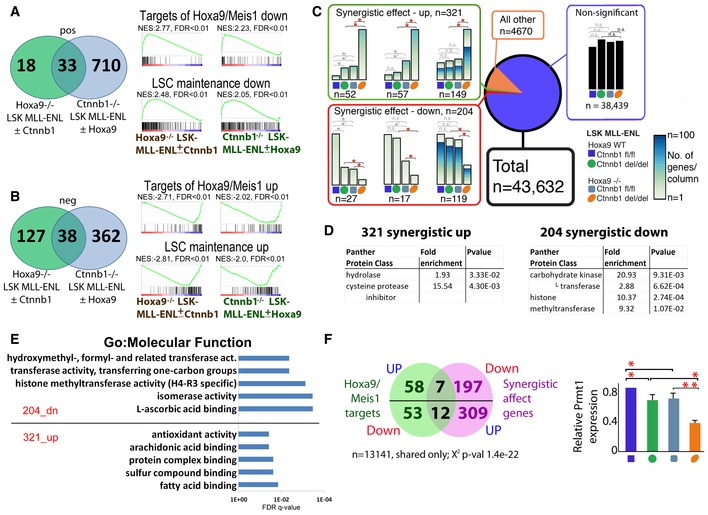
Figure 5. β‐catenin and Hoxa9 co‐regulate Prmt1 in LSK‐MLL‐ENL cells.
-
A, BVenn diagram showing the overlap of 33 commonly enriched up‐regulated (A) and 38 commonly enriched down‐regulated (B) highly significant (FDR < 0.05) gene sets between indicated comparisons (i.e., β‐catenin inactivation in Hoxa9 −/− LSK‐MLL‐ENL, Hoxa9 inactivation in Ctnnb1 −/− LSK‐MLL‐ENL cells), including “Hoxa9/Meis1_DN” (Hess et al, 2006), “LSC_mainentence_down” (Somervaille et al, 2009), “Hoxa9/Meis1_UP” (Hess et al, 2006), and “LSC_maintenance_up” (Somervaille et al, 2009) as indicated.
-
CA small set of genes is synergistically regulated by β‐catenin and Hoxa9 (the effect of knockout of both genes is smaller or larger than could have been predicted from their single knockouts). Example sets of genes synergistic up‐regulated (top), down‐regulated (bottom) and non‐significantly regulated (bottom right) are shown (all sets are in Appendix Fig S5). Keys and color code in the top corner indicate the origin and the β‐catenin and Hoxa9 status of MLL‐ENL‐transduced cells. Five differential expression comparisons were performed using DESeq2 with negative binomial GLM fitting and Wald statistics; comparisons are indicated with braces. All asterisks mark significant (P adjusted < 0.05) differential expression between the indicated conditions. Colored asterisks highlight differential expression which defines them as synergistically regulated genes. The height of the column represents the sum of normalized counts of all genes following the selected pattern of gene expression for a particular condition. The gradient fill of the columns indicates the number of genes represented in that column.
-
DPantherdb protein classes (http://pantherdb.org/) are shown for the 321_synergistic_up and 204_synergistic_down genes as indicated. P‐value was obtained after Bonferroni correction.
-
EThe FDR q‐values of the top 5 Toppgene GO:Molecular function (https://toppgene.cchmc.org/enrichment.jsp) are shown for the synergistic up and down‐regulated genes as indicated.
-
FStatistically significant directionality in the overlap of the synergistic genes with the Hox/Meis1_DN gene set (Dataset EV5B) including Prmt1, which has also been independently validated by RT–qPCR (right panel: n = 3, *P < 0.05, **P < 0.01, t‐test).
In order to specifically identify β‐catenin and Hoxa9 co‐regulated targets in LSK‐MLL‐ENL, we performed global quantitative expression analyses using RNA‐Seq and revealed a small fraction of genes ~1% (n = 525) showing a synergistic pattern in the double knockout compared to single knockouts alone (Fig 5C, Appendix Fig S5, Dataset EV4C). Different protein classes are present in the 321 synergistically up‐ and 204 synergistically down‐regulated gene lists. While hydrolase and cysteine protease inhibitors were enriched in the up‐regulated list, methyltransferases among others were enriched in the down‐regulated list (Fig 5D). These methyltransferases were arginine‐specific Prmt1, Prmt5, and Prmt7. Consistently, H4‐R3‐specific histone methyltransferase activity was among the enriched GO:Molecular functions (Fig 5E, Dataset EV4D), suggesting that arginine methylation may be co‐regulated by Hoxa9 and Ctnnb1. Indeed, when the targets of Hoxa9/Meis1 gene sets were overlapped with the synergistically up‐ and down‐regulated genes, 19 out of 111 (17%) genes from the Hoxa9/Meis1 targets (Fig 5F, Dataset EV4E), were synergistically regulated by β‐catenin and Hoxa9 in LSK‐MLL‐ENL (χ2, P = 1.4e‐22). Moreover, among them is again Prmt1, which had also been independently confirmed by RT–qPCR (Fig 5F). Strikingly, Prmt1 is a key epigenetic modifying enzyme known to be recruited by various fusion proteins involved in AML pathogenesis (Cheung et al, 2007, 2016; Shia et al, 2012), leading us to examine its role in mediating β‐catenin/Hoxa9 functions in LSK‐MLL‐ENL cells.
Prmt1 regulates similar and overlapping transcriptional programs mediated by β‐catenin in Hoxa9 −/− LSK‐MLL‐ENL cells
To investigate the transcriptional functions and potential molecular crosstalk between Prmt1 and β‐catenin, global transcriptional analyses by RNA sequencing were performed in Hoxa9 −/− LSK‐MLL‐ENL cells in the presence or absence of shRNA‐mediated Prmt1 knockdown using previously validated shRNAs (Cheung et al, 2007). As a result, we identified 1,416 differentially expressed genes, including 686 differentially up‐ and 730 differentially down‐regulated genes from two biological replicates upon Prmt1 inactivation (Fig 6A, Dataset EV5A). Similar transcriptomic analyses were then performed between Hoxa9 −/− LSK‐MLL‐ENL and Hoxa9 −/− Ctnnb1 −/− LSK‐MLL‐ENL, where we identified 342 differentially up‐regulated genes and 134 differentially down‐regulated genes from two biological replicates (Fig 6A). To assess whether Prmt1 and β‐catenin may regulate common transcriptional targets, we compared the differentially expressed genes from both analyses. As a result, we revealed similar and highly significant overlapping gene expression signatures associated with the loss of Prmt1 vs. β‐catenin in Hoxa9−/− LSK‐MLL‐ENL cells (Fig 6A, Dataset EV5A). While 24 genes were commonly down‐regulated by β‐catenin and Prmt1 (P < 2.4E‐7), 68 genes showed the opposite pattern (P < 2.7E‐22) upon their individual inactivation (Fig 6A). Functional annotation analysis revealed increased myeloid differentiation and apoptosis but reduced histone binding, chromatin silencing, and negative regulation of gene expression as dominant GO:Molecular functions and GO:Biological processes upon Prmt1 knockdown and β‐catenin knockout (Fig 6B). Strikingly, GSEA revealed that about 75% of the gene sets/pathways affected by Prmt1 knockdown were also regulated by β‐catenin knockout in Hoxa9 −/− LSK‐MLL‐ENL cells (Fig 6C, Dataset EV5B) including those involved in leukemic/normal stem cell functions and stemness (Fig 6D), consistent with the hypothesis that Prmt1 mediates β‐catenin functions in Hoxa9 −/− LSK‐MLL‐ENL cells. Together, these results strongly suggest a molecular and functional overlap between Prmt1 and β‐catenin in regulating critical transcriptional programs in LSK‐MLL‐ENL cells.
Figure 6. Prmt1 regulates similar and overlapping transcriptional programs mediated by β‐catenin in LSK‐MLL‐ENL cells.
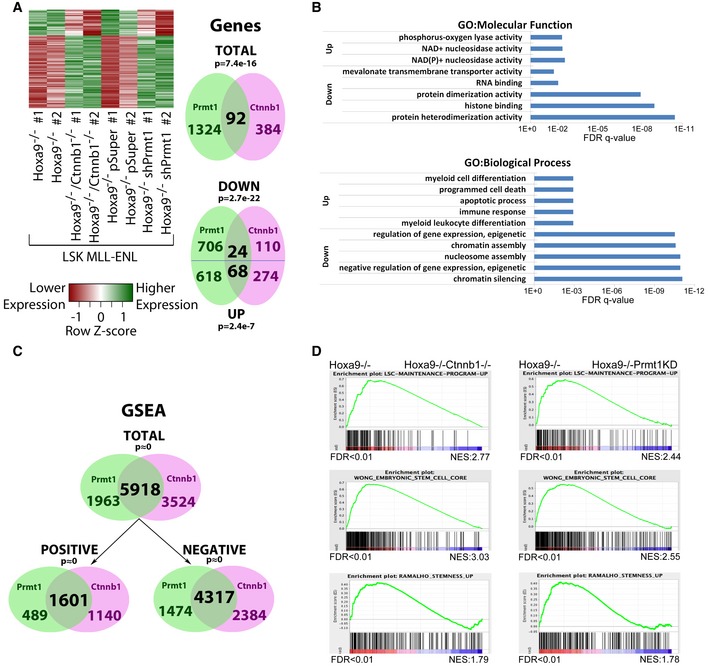
- Heatmap analysis and Venn diagram showing the commonly regulated genes after the loss of function of Prmt1 and β‐catenin in Hoxa9 −/− LSK‐MLL‐ENL cells.
- The FDR q‐values of the top GO:Molecular function (top panel) and GO:Biological process (bottom panel) (https://toppgene.cchmc.org/enrichment.jsp) for the indicated up‐ and down‐regulated genes is shown.
- Venn diagrams showing the total, positively and negatively regulated genesets identified by GSEA upon the loss of function of Prmt1 and β‐catenin in Hoxa9 −/− LSK‐MLL‐ENL cells.
- Examples of stem cell and stemness signatures which were commonly higher expressed in the single knockout Hoxa9 −/− LSK‐MLL‐ENL cells compared to Prmt1 down‐regulated or β‐catenin inactivated Hoxa9 −/− LSK‐MLL‐ENL cells.
Suppression of Prmt1 abolishes Hoxa9‐independent transformation in LSK‐derived MLL‐CSCs
To finally evaluate the biological function of Prmt1 as a critical mediator for β‐catenin/Hoxa9 functions, we first assessed its requirement in mediating Hoxa9 −/− LSK‐MLL‐ENL transformation. Prmt1 expression was independently down‐regulated in Hoxa9 −/− LSK‐MLL‐ENL by two different shRNAs, which also resulted in reduction of H4R3 asymmetric dimethylation mark (H4R3me2as) specifically conferred by Prmt1 (Appendix Fig S6A and B). Inhibition of Prmt1 expression compromised in vitro MLL‐ENL transformation of Hoxa9 −/− LSK cells (Fig 7A, Appendix Fig S6C) and mimic in vivo inactivation of β‐catenin in LSK‐MLL‐ENL. Similar to Hoxa9 −/− Ctnnb1 −/− LSK‐MLL‐ENL cells (Fig 4G), Hoxa9 −/−Prmt1 KD LSK‐MLL‐ENL cells were able to engraft and proliferate short term, but gradually lost their self‐renewal ability (Fig 7B). Crucially, Prmt1 knockdown suppressed oncogenic potential of Hoxa9 −/− LSK‐MLL‐ENL cells, which could otherwise induce leukemia within a month (Fig 7C).
Figure 7. Suppression of Prmt1 abolishes β‐catenin‐ or Hoxa9‐independent transformation in HSC‐derived MLL‐CSCs.
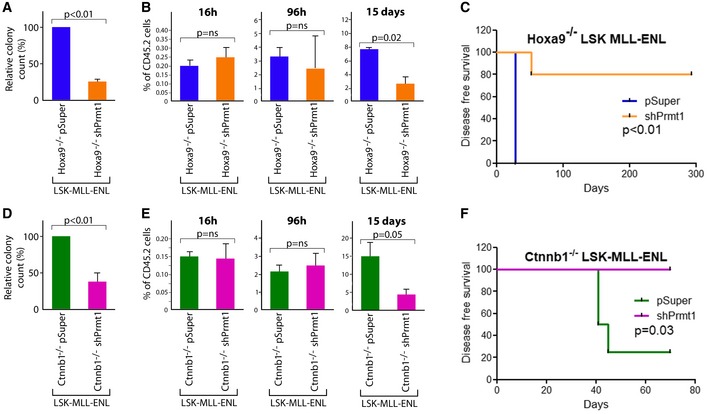
- The relative number of colonies from Hoxa9 −/− LSK‐MLL‐ENL leukemic cells with empty vector or shPRMT1 (n = 3, t‐test).
- The % of CD45.2+ donor cells in the bone marrow of recipient mice at the indicated time points post‐transplantation of Hoxa9 −/− LSK‐MLL‐ENL leukemic cells with empty vector or shPrmt1 (n = 4, t‐test).
- Kaplan–Meier survival curve of secondary recipient mice transplanted with Hoxa9 −/− LSK‐MLL‐ENL leukemic cells with vector control or shPrmt1 (n = 5/cohort, log‐rank test).
- The relative number of colonies from Ctnnb1 −/− LSK‐MLL‐ENL leukemic cells with empty vector or shPRMT1 (n = 3, t‐test).
- The % of CD45.2+ donor cells in the bone marrow of recipient mice at the indicated time points post‐transplantation of Ctnnb1 −/− LSK‐MLL‐ENL leukemic cells with empty vector or shPrmt1 (n = 4, t‐test).
- Kaplan–Meier survival curve of secondary recipient mice transplanted with Ctnnb1 −/− LSK‐MLL‐ENL leukemic cells with vector control or shPrmt1 (n = 4/cohort, log‐rank test).
To further determine whether Prmt1 as a key mediator for β‐catenin/Hoxa9 can also replace the function of Hoxa9, Prmt1 expression was suppressed in Ctnnb1 −/− LSK‐MLL‐ENL (Appendix Fig S6A and B). As a result, Prmt1 inhibition mimic Hoxa9 inactivation leading to suppression of colony formation ability of Ctnnb1 −/− LSK‐MLL‐ENL cells (Fig 7D), abolished their self‐renewal potentials (Fig 7E) and oncogenic ability in vivo (Fig 7F). Together with the comprehensive global gene expression network analyses, these results consistently indicate Prmt1 as a key player and novel downstream target mediating β‐catenin/Hoxa9 functions in LSK‐derived MLL‐CSCs.
Discussion
Self‐renewal as a defining feature of normal and cancer stem cells is tightly regulated by complex transcriptional networks. Most of the current targeted therapies and their intended clinical utility are developed without considering the cancer cells of origin, which can have distinctive self‐renewal and transcriptional properties. This traditional view has been challenged by the identification of phenotypically indistinguishable leukemia from different cells of origin (Cozzio et al, 2003; So et al, 2003; Huntly et al, 2004), which exhibit different responses to standard chemotherapy treatment (Krivtsov et al, 2013; George et al, 2016; Stavropoulou et al, 2016). However, until now, very little is known about impact of cells of origin on cancer self‐renewal and the molecular pathways underpinning this defining feature of CSCs. By performing comprehensive genomic and transcriptomic analyses on origin‐specific CSCs in combination with various in vitro and in vivo functional genomic assays, here we provide the experimental evidence for the presence of cells of origin transcriptional memory governing molecular pathways available for CSC self‐renewal, urging that both genetic mutations and transcriptional memory inherited from cells of origin determine the resultant CSC biology and heterogeneous responses to treatment. Key components of the canonical Wnt/β‐catenin signaling pathway are recurrently deregulated in various human cancers, and a number of inhibitor are in early‐phase clinical trials (Anastas & Moon, 2013). This is particularly relevant to leukemia as normal HSCs remain largely intact upon a complete inactivation of β‐catenin and targeting β‐catenin represents a promising venue for eradication of LSCs (Fung et al, 2013). However, our results reveal an added dimension of cancer heterogeneity conferred by cells of origin transcriptional memory and predict that pharmacological targeting of β‐catenin is unlikely to be effective in MLL leukemia originated from HSCs. We would like to point out that all the published literatures including our studies related to cancer cells of origin were performed using mouse cells, which could have different features from the human counterparts. Given the recent evidences from mouse models indicate the importance of the cells of origin in governing the resultant cancer biology, future relevant studies in human cell systems are paramount to give necessary insights and to improve our understanding of the cancer cell biology and designing effective therapeutics in the human diseases.
In contrast to its essential function in embryonic and other somatic stem cells, β‐catenin is dispensable for adult HSCs, suggesting the presence of residual canonical Wnt signaling for normal HSC function (Malhotra & Kincade, 2009) or an alternative molecule/pathway compensated for β‐catenin in adult hematopoiesis. Interestingly, most of the known molecules/pathways involved in canonical Wnt signaling predominately identified in epithelial cells or ES cells are not significantly affected upon β‐catenin deletion in MLL leukemia regardless of their cells of origin (Datasets EV2A and EV3C). In contrast, we identified a number of novel β‐catenin targets including those downstream of Meis1/Hoxa9 that are critical for HSC self‐renewal, consistently indicating co‐regulation of common self‐renewal pathways by β‐catenin and Hoxa9 in hematopoietic cells. Hoxa9 recently proposed as a key component of human LSC signature in AML (Jung et al, 2015) can mediate Bmi‐1‐independent leukemic self‐renewal (Smith et al, 2011) and resistance to PARPi treatment in AML (Esposito et al, 2015). In line with this finding, we and others have also reported the ability of β‐catenin or Hoxa9 in promoting HSC self‐renewal (Argiropoulos & Humphries, 2007; Malhotra & Kincade, 2009; Zeisig et al, 2012), but deletion of either one of them yields only mild hematopoietic phenotypes (Cobas et al, 2004; So et al, 2004; Lawrence et al, 2005; Jeannet et al, 2008; Koch et al, 2008; Smith et al, 2011), suggesting their overlapping function in HSC self‐renewal. This is further supported by identification of Prmt1 as a key common downstream target that mediates their transcriptional and self‐renewal functions. However, the lack of good‐quality ChIP‐grade antibodies against Hoxa9 and β‐catenin makes it unfeasible to reliably determine whether they might directly bind to Prmt1 regulatory regions. While the lack of known consensus binding sites of Hox or β‐catenin/Tcf by in silico analysis (data not shown) suggests an intermediate instead of direct regulation, future investigation is critical to gain further insights into any potential direct functional interactions and regulations. Nevertheless, our comprehensive transcriptomic analyses in combined with functional genomic studies have revealed the novel molecular networks mediated by β‐catenin/Hoxa9/Prmt1 in regulating leukemic self‐renewal in LSK‐derived MLL‐CSCs, and exemplify the intricate diversity in molecular pathways utilized by cancer cells to evade therapies, and underscores that simultaneous targeting of multiple self‐renewal pathways may be required for successful elimination of certain CSCs.
Intriguingly, activation of canonical Wnt/β‐catenin in MLL‐CSCs has also recently been identified as a major mechanism for development of resistance to pharmacological inhibition of BET (Fong et al, 2015; Rathert et al, 2015), a targeted therapy at its early clinical phase for MLL leukemia, further highlighting the importance of adequately targeting β‐catenin in the context of cells of origin for future targeted cancer therapies. Given the challenges involved in developing clinically effective inhibitors to β‐catenin (Fung et al, 2013), the identification of Prmt1 as a nexus for mediating leukemic self‐renewal in LSK‐MLL‐ENL transformed cells not only provides novel mechanistic insights into the downstream targets and molecular networks regulated by β‐catenin, but also suggests an alternative avenue for targeting β‐catenin in MLL‐CSCs.
Materials and Methods
Animals and transplantation studies
All experimental procedures were approved by King's College London ethics committees and conform to the UK Home Office regulations. For all in vivo experiments, mice were distributed into their respective groups randomly. Investigators were not blinded to the sample identity. Mice were considered leukemic when an engraftment of donor cell (> 30%) was detected in the bone marrow. Ctnnb1 fl/fl mice (Brault et al, 2001) were crossed with Rosa26‐CreER Rosa26‐YFP mice to generate Ctnnb1 fl/fl Rosa‐CreER RosaYFP mice. These mice were crossed with Hoxa9−/− knockout mice (So et al, 2004; Smith et al, 2011) to generate Hoxa9 −/− Ctnnb1 fl/fl Rosa‐CreER RosaYFP mice. Compound homozygous animals were used for experiments. C57BL/6 or SJL mice were given 11 Gy total body γ‐irradiation and injected via tail vein with test cells mixed with C57BL/6 or SJL bone marrow nuclear cells. Mice were culled when sign of sickness appeared. Survival curves were produced using GraphPad Prism software, and survival differences tested with the log‐rank test. For leukemia development experiments, up to 500,000 test cells mixed with 200,000 C57BL/6 or SJL bone marrow nuclear cells were transplanted. For primary transplants n = 5–18 mice/cohort and for secondary transplants n = 3–10 mice/cohort were used. For in vivo limiting dilution assays, varying numbers of cells from indicted populations were transplanted into n = 5 sublethally irradiated syngeneic mice/cohort and monitored for disease development. ELDA was used for statistical analysis (Hu & Smyth, 2009). For in vivo homing experiments, 5 million test cells were transplanted into n ≥ 4 (when studying genetic ablation) and n = 2 (when studying shPRMT1 knockdown) sublethally irradiated syngeneic mice/cohort/time point. Homing differences were statistically tested using a two‐tailed t‐test in Excel.
Hematopoietic stem and progenitor purification
Mouse femur and tibias were prepared and c‐Kit+ (CD117) cells isolated (Zeisig & So, 2009) using MACS (Miltenyi Biotech Technology, Germany). LSK and GMP populations were isolated as previously described (Yeung & So, 2009). Briefly, LSK (Lin−, Sca‐1+, c‐Kit+) and GMP (Lin−, Sca‐1−, c‐Kit+, CD34+, CD16/32lo) were isolated from lineage‐negative cells after lineage (Gr‐1, Mac‐1, B220, Ter119, CD3e, CD4, CD8) (Biolegend/eBiosciences) depletion using a BD FACS ARIA cell sorter. Post‐sort purity of > 97% was routinely achieved. For functional analysis, sorted LSK and GMP were plated in Methocult M3434 (Stem Cell Technologies, Canada), and after 7–10 days of incubation, different colony types were scored.
Retroviral transduction and transformation assays
Retroviral transduction and transformation assays (RTTA) were performed as previously described with some modifications (Zeisig & So, 2009). The MSCV‐MLL‐ENL construct has been described previously. The two independent shRNAs against murine Prmt1 have been reported previously (Cheung et al, 2007). Briefly, isolated c‐Kit+, LSK, or GMP cells were cultured overnight in RPMI + 10%FBS supplemented with 20 ng/ml SCF, 10 ng/ml IL3, and 10 ng/ml IL6 prior to viral transduction with virus particles carrying MLL‐ENL by centrifugation at 800 × g at 32°C for 2 h. Cells were plated in M3234 Methylcellulose medium supplemented with 20 ng/ml SCF, 10 ng/ml of each IL3, IL6, and GM‐CSF and appropriate antibiotic selection on the following day. Colonies were scored after 7 days and replated every 7 days. To induce the deletion of Ctnnb1, 20 nM tamoxifen (Sigma, USA) was added to the methylcellulose medium in the second round of plating and YFP positive were sorted after the second round of plating using a BD FACS ARIA and plated into the 3rd round. After the 4th round of plating, cells were cultured in R20/20 to establish cell lines as previously described (Yeung et al, 2010). Differences in colony numbers were statistically tested using a two‐tailed t‐test in Excel.
Phenotypic analysis
Immunophenotypic analysis was performed by FACS using fluorochrome‐conjugated monoclonal antibodies to murine c‐Kit (2B8 clone), Mac‐1 (M1/70), and Gr‐1 (RB6‐8C5) (eBiosciences). Staining was generally performed on ice for 15 min and washed once before analysis using a BD LSR II system. Differences in surface marker expression were statistically tested using a two‐tailed t‐test in Excel.
Western blot
Cell lysates from primary transformed cells or sorted leukemic cells were isolated and subjected to Western blot as described (Yeung et al, 2010).
Histone extraction and detection
Histone proteins were prepared by acid extraction (Abcam protocol, http://www.abcam.com/protocols/histone-extraction-protocol-for-western-blot). Briefly, cells were lysed for 10 minutes on ice in TEB buffer (PBS/0.5% Triton X‐100/protease inhibitors) at a cell density of 107 per ml. The nuclei were harvested by centrifugation and washed in half the volume of TEB. Pellets were resuspended in 0.2 N HCl at a density of 4 × 107 nuclei per ml, and histones were extracted overnight at 4C. Samples were centrifuged to pellet debris, and the supernatant containing the histones was transferred into a new Eppendorf tube. 10 μl of histones was mixed with 40 μl PBS and 50 μl 2× SDS loading buffer and incubated at 95°C for 8 min. 25 μl of the denatured histones were subjected to Western blot using a 10% Next gel (AMRESCO LLC). Antibodies against H4R3me2a (Active Motif) and Histone H3 (Abcam) were used.
Genotyping PCR
Genomic DNA was isolated, and β‐catenin genotyping PCR was performed as described previously (Brault et al, 2001). Primer sequences for Hoxa9 are available on request. Differences in gene expression were statistically tested using a two‐tailed t‐test in Microsoft Excel.
qRT–PCR
qRT–PCR was performed on StepOne qPCR machine (Applied Biosystems) using TaqMan or SYBR green chemistries. See Appendix Table S1 for the primer sequences used throughout the paper for validation.
Apoptosis and cell cycle analysis
MLL‐ENL‐transduced Hoxa9−/− Ctnnb1 fl/fl cells were treated for 72 h with or without tamoxifen and stained with AnnexinV for apoptosis. Cell cycle was analyzed using the BRDU Flow kit (BD Pharmingen) according to manufacturer's instructions. Cells were analyzed using a BD LSR II system (BD, USA), and differences were statistically tested using a two‐tailed t‐test in Excel.
RNA sequencing
300 ng to 1 μg of mirVANA (Ambion) isolated total RNA was used for RNA‐Seq library preparation using TruSeq Stranded Total RNA kit (Illumina) and sequenced on HIseq2000 platform (Illumina) as per manufacturers recommendations. All samples are listed in Dataset EV1A.
Whole‐genome sequencing
Genomic DNA was isolated with the QIAamp DNA Micro kit (Qiagen) according to the manufacturer's instructions, and 30 ng of genomic DNA was used as input for each library preparation. Whole‐genome sequencing libraries were generated using the tagmentation method as previously described (Wang et al, 2013), but without bisulfite treatment and with minor modifications. Briefly, genomic DNA was subjected to tagmentation with a hyperactive Tn5 transposase (Epizyme), which fragmented the DNA and appended sequencing adaptors in a single step. After PCR amplification of libraries, DNA fragments of 200–800 bp were double‐side selected using SPRI Ampure XP beads, with left–right ratios of 1.5–0.55. Purified libraries were subjected to 125‐bp paired‐end sequencing on an Illumina HiSeq2500 machine.
Bioinformatic analyses
Mapping and read counts
The FASTQ files were de‐tagged, and the quality of the FASTQ files inspected using FastQC v.0.11.2 (Andrews, 2015). Remaining adapters were trimmed using TRIMGalore! v.0.3.7 (Kreuger, 2015). Whole‐genome sequencing reads were mapped to the Ensembl mouse genome GRCm38 (Aken et al, 2016) using Bowtie2 v.2.2.5 (Langmead & Salzberg, 2012). RNA‐Seq reads were mapped to the Ensembl mouse genome using TopHat2 v2.0.13 (Kim et al, 2013), and reads were filtered for quality and counted using samtools v.0.1.18 and bedtools v2.23.0‐10‐g447cb97 (Li et al, 2009; Quinlan & Hall, 2010; Dataset EV1B).
SNP calling
SNP calling (for both RNA‐Seq and WGS) was performed using samtools v.0.1.18 mpileup and bcftools v.1.2 (using htslib v.1.2.1) (Li, 2011), SNPs were leniently filtered only for quality ≥ Q30, and custom scripts used to produce summary stats (Dataset EV1C). To summarize the regions in which SNPs fell, variant call files and Ensembl GTF annotation (Mus_musculus.GRCm38.82.gtf) were converted to bed format using custom scripts and compared using bedtools intersect v2.23.0. Bedtools complement v2.23.0 (Quinlan & Hall, 2010) was used to extract intronic and intergenic coordinates.
Gene differential expression
Differential expression was determined either using DESeq2 v.1.10.1 (Love et al, 2014), negative binomial GLM fitting and Wald statistics; or with limma v.3.26.9 (Ritchie et al, 2015) using voom to normalize read counts, and eBayes to determine differential expression (as detailed in Expanded View Datasets). Functional enrichment analysis was conducted using the GSEA software, using various Molecular Signature Databases (c2 set version 5.0, version 6.0 and all_gene_sets) (MSigDB) (Subramanian et al, 2005) appended with a custom‐made gene set for LSC stem cell maintenance (Somervaille et al, 2009; Dataset EV3B), comparing log2‐fold changes in gene expression as the ranking metric. For the GSEA analysis, human‐mouse gene orthologues were identified using MGI list of orthologous genes (Blake et al, 2014), and Ensembl bioMart used to transfer MGI IDs to Ensembl gene IDs (Kinsella et al, 2011). In order to find genes which showed a synergistic effect of simultaneous knockout of Cbnnt1 and Hoxa9, differential expression was tested with DESeq2 Negative Binomial GLM fitting and Wald statistics. The data were subjected to five DE analyses, which served to categorize the genes into 78 different classes. These comparisons are as follows: 1. LSK WT to Ctnnb1 KO, 2. LSK WT to HoxA9 KO, 3. Additive effect LSK WT to Ctnnb1 KO and Hoxa9 KO, 4. LSK Ctnnb1 KO to Ctnnb1:HoxA9 interaction, 5. LSK HoxA9 KO to Ctnnb1:HoxA9 interaction. For overlapping significantly differentially expressed genes, heatmaps were plotted using the function heatmap.2 from the R‐package gplots v3.0.1 (Warnes et al, 2016) with Z‐score scaling of rows and/or columns.
Survival analysis
Statistical analysis and data visualization were performed using R (R Core Team, 2014). Survival and Cox proportional hazards ratio analysis were conducted using R‐packages survival v.2.39.5 (Therneau, 2015) and survcomp v.1.20.0 (Schroder et al, 2011) on all patients which had complete survival and expression data from the following datasets: GSE1159 (n = 293), GSE12417; GPL570 (n = 79), GPL96 (n = 163), GSE14468 + GSE6891 (n = 602), GSE5122 (n = 58), GSE8970 (n = 34) available from the Gene Expression Omnibus database ncbi.nlm.nih.gov/geo/ and The Cancer Genome Atlas AML dataset (n = 183; Cancer Genome Atlas Research N, 2013). Summary data for patient cohorts are presented in Dataset EV2D. Samples were normalized using the GENENORM algorithm from the R‐package inSilicoMerging v.1.15.0 (Taminau et al, 2012) with prior DESeq2 Vst transformation of RNA‐Seq samples used in this study to derive the cell of origin‐specific signature (Love et al, 2014). All probe intensities were translated into human genes and averaged prior to merging. MLL patients in the datasets were identified from associated karyotyping and PCR diagnosis.
Data availability
All WGS has been submitted to the ENA study PRJEB14461. RNA‐Seq is available on ArrayExpress E‐MTAB‐3647.
Statistical analysis
All the experimental results were analyzed using two‐tailed Student's t‐test, chi‐square test, hypergeometric test, as indicated in the figure legends and Expanded View Datasets. Groups that were statistically compared shared a similar variance, as shown in the figures. P < 0.05 was considered as statistically significant. For GSEA analysis, FDR q‐values < 0.25 were considered as statistically significant (Subramanian et al, 2005). The log‐rank test was used to compare survival curves.
Author contributions
TS, BBZ, TKF, MM, C‐TT, PNIL, PV, JB, and AW performed experiments, BBZ contributed to writing the manuscript, MZ, QCH, CL, and BL contributed the bioinformatics analyses and editing the manuscript, and CWES contributed to overall research design, direction, and writing of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Review Process File
Source Data for Figure 1D
{kind=link}
Source Data for Figure 4B
{kind=link}
Acknowledgements
We would like to thank Sam Tung, Jayant Rane for assistance on bioinformatics analyses; Erica Tse for graphical assistance; Peter Parker and Ryan Driskell for critical advice on the manuscript. This work was supported mainly by a Cancer Research UK (CRUK) program grant A16753 and in part by a Bloodwise program grant to CWE So.
The EMBO Journal (2017) 36: 3139–3155
References
- Aken BL, Ayling S, Barrell D, Clarke L, Curwen V, Fairley S, Fernandez Banet J, Billis K, Garcia Giron C, Hourlier T, Howe K, Kahari A, Kokocinski F, Martin FJ, Murphy DN, Nag R, Ruffier M, Schuster M, Tang YA, Vogel JH et al (2016) The Ensembl gene annotation system. Database 2016: baw093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastas JN, Moon RT (2013) WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 13: 11–26 [DOI] [PubMed] [Google Scholar]
- Andrews S (2015). FastQC. v.0.11.2. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Argiropoulos B, Humphries RK (2007) Hox genes in hematopoiesis and leukemogenesis. Oncogene 26: 6766–6776 [DOI] [PubMed] [Google Scholar]
- Blake JA, Bult CJ, Eppig JT, Kadin JA, Richardson JE, Grp MGD (2014) The mouse genome database: integration of and access to knowledge about the laboratory mouse. Nucleic Acids Res 42: D810–D817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C (2013) Tracing the cellular origin of cancer. Nat Cell Biol 15: 126–134 [DOI] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R (2001) Inactivation of the beta‐catenin gene by Wnt1‐Cre‐mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128: 1253–1264 [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N (2013) Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368: 2059–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung N, Chan LC, Thompson A, Cleary ML, So CW (2007) Protein arginine‐methyltransferase‐dependent oncogenesis. Nat Cell Biol 9: 1208–1215 [DOI] [PubMed] [Google Scholar]
- Cheung N, Fung TK, Zeisig BB, Holmes K, Rane JK, Mowen KA, Finn MG, Lenhard B, Chan LC, So CW (2016) Targeting aberrant epigenetic networks mediated by PRMT1 and KDM4C in acute myeloid leukemia. Cancer Cell 29: 32–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobas M, Wilson A, Ernst B, Mancini SJ, MacDonald HR, Kemler R, Radtke F (2004) Beta‐catenin is dispensable for hematopoiesis and lymphopoiesis. J Exp Med 199: 221–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML, Weissman IL (2003) Similar MLL‐associated leukemias arising from self‐renewing stem cells and short‐lived myeloid progenitors. Genes Dev 17: 3029–3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, Metzeler KH, Poeppl A, Ling V, Beyene J, Canty AJ, Danska JS, Bohlander SK, Buske C, Minden MD, Golub TR, Jurisica I, Ebert BL, Dick JE (2011) Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med 17: 1086–1093 [DOI] [PubMed] [Google Scholar]
- Esposito MT, Zhao L, Fung TK, Rane JK, Wilson A, Martin N, Gil J, Leung AY, Ashworth A, So CW (2015) Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat Med 21: 1481–1490 [DOI] [PubMed] [Google Scholar]
- Faber J, Krivtsov AV, Stubbs MC, Wright R, Davis TN, van den Heuvel‐Eibrink M, Zwaan CM, Kung AL, Armstrong SA (2009) HOXA9 is required for survival in human MLL‐rearranged acute leukemias. Blood 113: 2375–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong CY, Gilan O, Lam EY, Rubin AF, Ftouni S, Tyler D, Stanley K, Sinha D, Yeh P, Morison J, Giotopoulos G, Lugo D, Jeffrey P, Lee SC, Carpenter C, Gregory R, Ramsay RG, Lane SW, Abdel‐Wahab O, Kouzarides T et al (2015) BET inhibitor resistance emerges from leukaemia stem cells. Nature 525: 538–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung TK, Leung AY, So CW (2013) The Wnt/beta‐catenin pathway as a potential target for drug resistant leukemic stem cells In Stem Cells and Cancer Stem Cells, Hayat MA. (ed.), Vol. 10, pp 163–172. Netherlands: Springer; [Google Scholar]
- George J, Uyar A, Young K, Kuffler L, Waldron‐Francis K, Marquez E, Ucar D, Trowbridge JJ (2016) Leukaemia cell of origin identified by chromatin landscape of bulk tumour cells. Nat Commun 7: 12166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess JL, Bittner CB, Zeisig DT, Bach C, Fuchs U, Borkhardt A, Frampton J, Slany RK (2006) c‐Myb is an essential downstream target for homeobox‐mediated transformation of hematopoietic cells. Blood 108: 297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Smyth GK (2009) ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 347: 70–78 [DOI] [PubMed] [Google Scholar]
- Huang Y, Sitwala K, Bronstein J, Sanders D, Dandekar M, Collins C, Robertson G, MacDonald J, Cezard T, Bilenky M, Thiessen N, Zhao Y, Zeng T, Hirst M, Hero A, Jones S, Hess JL (2012) Identification and characterization of Hoxa9 binding sites in hematopoietic cells. Blood 119: 388–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, Rowan R, Amaral S, Curley D, Williams IR, Akashi K, Gilliland DG (2004) MOZ‐TIF2, but not BCR‐ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 6: 587–596 [DOI] [PubMed] [Google Scholar]
- Jeannet G, Scheller M, Scarpellino L, Duboux S, Gardiol N, Back J, Kuttler F, Malanchi I, Birchmeier W, Leutz A, Huelsken J, Held W (2008) Long‐term, multilineage hematopoiesis occurs in the combined absence of beta‐catenin and gamma‐catenin. Blood 111: 142–149 [DOI] [PubMed] [Google Scholar]
- Jung N, Dai B, Gentles AJ, Majeti R, Feinberg AP (2015) An LSC epigenetic signature is largely mutation independent and implicates the HOXA cluster in AML pathogenesis. Nat Commun 6: 8489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella RJ, Kahari A, Haider S, Zamora J, Proctor G, Spudich G, Almeida‐King J, Staines D, Derwent P, Kerhornou A, Kersey P, Flicek P (2011) Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database 2011: bar030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch U, Wilson A, Cobas M, Kemler R, Macdonald HR, Radtke F (2008) Simultaneous loss of beta‐ and gamma‐catenin does not perturb hematopoiesis or lymphopoiesis. Blood 111: 160–164 [DOI] [PubMed] [Google Scholar]
- Kreuger F (2015) Trim Galore! 0.3.7. http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, Golub TR, Armstrong SA (2006) Transformation from committed progenitor to leukaemia stem cell initiated by MLL‐AF9. Nature 442: 818–822 [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Figueroa ME, Sinha AU, Stubbs MC, Feng Z, Valk PJ, Delwel R, Dohner K, Bullinger L, Kung AL, Melnick AM, Armstrong SA (2013) Cell of origin determines clinically relevant subtypes of MLL‐rearranged AML. Leukemia 27: 852–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence HJ, Christensen J, Fong S, Hu YL, Weissman I, Sauvageau G, Humphries RK, Largman C (2005) Loss of expression of the Hoxa‐9 homeobox gene impairs the proliferation and repopulating ability of hematopoietic stem cells. Blood 106: 3988–3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S (2009) The sequence alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27: 2987–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra S, Kincade PW (2009) Wnt‐related molecules and signaling pathway equilibrium in hematopoiesis. Cell Stem Cell 4: 27–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland MC, Heinecke A, Radmacher M, Marcucci G, Whitman SP, Maharry K, Paschka P, Larson RA, Berdel WE, Buchner T, Wormann B, Mansmann U, Hiddemann W, Bohlander SK, Buske C et al (2008) An 86‐probe‐set gene‐expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood 112: 4193–4201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL (2002) MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell 10: 1107–1117 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2014) A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; http://www.r-project.org/ [Google Scholar]
- Raponi M, Harousseau JL, Lancet JE, Lowenberg B, Stone R, Zhang Y, Rackoff W, Wang Y, Atkins D (2007) Identification of molecular predictors of response in a study of tipifarnib treatment in relapsed and refractory acute myelogenous leukemia. Clin Cancer Res 13: 2254–2260 [DOI] [PubMed] [Google Scholar]
- Raponi M, Lancet JE, Fan H, Dossey L, Lee G, Gojo I, Feldman EJ, Gotlib J, Morris LE, Greenberg PL, Wright JJ, Harousseau JL, Lowenberg B, Stone RM, De Porre P, Wang Y, Karp JE (2008) A 2‐gene classifier for predicting response to the farnesyltransferase inhibitor tipifarnib in acute myeloid leukemia. Blood 111: 2589–2596 [DOI] [PubMed] [Google Scholar]
- Rathert P, Roth M, Neumann T, Muerdter F, Roe JS, Muhar M, Deswal S, Cerny‐Reiterer S, Peter B, Jude J, Hoffmann T, Boryn LM, Axelsson E, Schweifer N, Tontsch‐Grunt U, Dow LE, Gianni D, Pearson M, Valent P, Stark A et al (2015) Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 525: 543–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 43: e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder MS, Culhane AC, Quackenbush J, Haibe‐Kains B (2011) survcomp: an R/Bioconductor package for performance assessment and comparison of survival models. Bioinformatics 27: 3206–3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shia WJ, Okumura AJ, Yan M, Sarkeshik A, Lo MC, Matsuura S, Komeno Y, Zhao X, Nimer SD, Yates JR III, Zhang DE (2012) PRMT1 interacts with AML1‐ETO to promote its transcriptional activation and progenitor cell proliferative potential. Blood 119: 4953–4962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slany RK, Lavau C, Cleary ML (1998) The oncogenic capacity of HRX‐ENL requires the transcriptional transactivation activity of ENL and the DNA binding motifs of HRX. Mol Cell Biol 18: 122–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LL, Yeung J, Zeisig BB, Popov N, Huijbers I, Barnes J, Wilson AJ, Taskesen E, Delwel R, Gil J, Van Lohuizen M, So CW (2011) Functional crosstalk between Bmi1 and MLL/Hoxa9 axis in establishment of normal hematopoietic and leukemic stem cells. Cell Stem Cell 8: 649–662 [DOI] [PubMed] [Google Scholar]
- So CW, Karsunky H, Passegue E, Cozzio A, Weissman IL, Cleary ML (2003) MLL‐GAS7 transforms multipotent hematopoietic progenitors and induces mixed lineage leukemias in mice. Cancer Cell 3: 161–171 [DOI] [PubMed] [Google Scholar]
- So CW, Karsunky H, Wong P, Weissman IL, Cleary ML (2004) Leukemic transformation of hematopoietic progenitors by MLL‐GAS7 in the absence of Hoxa7 or Hoxa9. Blood 103: 3192–3199 [DOI] [PubMed] [Google Scholar]
- Somervaille T, Matheny C, Spencer G, Iwasaki M, Rinn J, Witten D, Chang H, Shurtleff S, Downing J, Cleary M (2009) Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell 4: 129–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavropoulou V, Kaspar S, Brault L, Sanders MA, Juge S, Morettini S, Tzankov A, Iacovino M, Lau IJ, Milne TA, Royo H, Kyba M, Valk PJ, Peters AH, Schwaller J (2016) MLL‐AF9 expression in hematopoietic stem cells drives a highly invasive AML expressing EMT‐related genes linked to poor outcome. Cancer Cell 30: 43–58 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taminau J, Meganck S, Lazar C, Steenhoff D, Coletta A, Molter C, Duque R, de Schaetzen V, Weiss Solis DY, Bersini H, Nowe A (2012) Unlocking the potential of publicly available microarray data using inSilicoDb and inSilicoMerging R/Bioconductor packages. BMC Bioinformatics 13: 335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therneau T (2015) A Package for Survival Analysis in S. http://cran.r-project.org/package=survival
- Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Waalwijk van Barjesteh van Doorn‐Khosrovani S, Boer JM, Beverloo HB, Moorhouse MJ, van der Spek PJ, Lowenberg B, Delwel R (2004) Prognostically useful gene‐expression profiles in acute myeloid leukemia. N Engl J Med 350: 1617–1628 [DOI] [PubMed] [Google Scholar]
- Visvader JE (2011) Cells of origin in cancer. Nature 469: 314–322 [DOI] [PubMed] [Google Scholar]
- Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, Armstrong SA (2010) The Wnt/beta‐catenin pathway is required for the development of leukemia stem cells in AML. Science 327: 1650–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Gu L, Adey A, Radlwimmer B, Wang W, Hovestadt V, Bähr M, Wolf S, Shendure J, Eils R, Plass C, Weichenhan D (2013) Tagmentation‐based whole‐genome bisulfite sequencing. Nat Protoc 8: 2022–2032 [DOI] [PubMed] [Google Scholar]
- Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M, Venables B (2016) gplots: Various R Programming Tools for Plotting Data. https://cran.r-project.org/package=gplots
- Wouters BJ, Lowenberg B, Erpelinck‐Verschueren CA, van Putten WL, Valk PJ, Delwel R (2009) Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood 113: 3088–3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung J, So CW (2009) Identification and characterization of hematopoietic stem and progenitor cell populations in mouse bone marrow by flow cytometry. Methods Mol Biol 538: 301–315 [DOI] [PubMed] [Google Scholar]
- Yeung J, Esposito MT, Gandillet A, Zeisig BB, Griessinger E, Bonnet D, So CW (2010) beta‐Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 18: 606–618 [DOI] [PubMed] [Google Scholar]
- Zeisig BB, Milne T, Garcia‐Cuellar MP, Schreiner S, Martin ME, Fuchs U, Borkhardt A, Chanda SK, Walker J, Soden R, Hess JL, Slany RK (2004) Hoxa9 and Meis1 are key targets for MLL‐ENL‐mediated cellular immortalization. Mol Cell Biol 24: 617–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisig BB, So CW (2009) Retroviral/Lentiviral transduction and transformation assay. Methods Mol Biol 538: 207–229 [DOI] [PubMed] [Google Scholar]
- Zeisig BB, Kulasekararaj AG, Mufti GJ, So CW (2012) SnapShot: acute myeloid leukemia. Cancer Cell 22: 698.e1 [DOI] [PubMed] [Google Scholar]
- Zhao C, Blum J, Chen A, Kwon HY, Jung SH, Cook JM, Lagoo A, Reya T (2007) Loss of beta‐catenin impairs the renewal of normal and CML stem cells in vivo . Cancer Cell 12: 528–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Review Process File
Source Data for Figure 1D
Source Data for Figure 4B
Data Availability Statement
All WGS has been submitted to the ENA study PRJEB14461. RNA‐Seq is available on ArrayExpress E‐MTAB‐3647.