

Abstract
Most type VI secretion systems (T6SSs) described to date are protein delivery apparatuses that mediate bactericidal activities. Several T6SSs were also reported to mediate virulence activities, although only few anti‐eukaryotic effectors have been described. Here, we identify three T6SSs in the marine bacterium Vibrio proteolyticus and show that T6SS1 mediates bactericidal activities under warm marine‐like conditions. Using comparative proteomics, we find nine potential T6SS1 effectors, five of which belong to the polymorphic MIX‐effector class. Remarkably, in addition to six predicted bactericidal effectors, the T6SS1 secretome includes three putative anti‐eukaryotic effectors. One of these is a MIX‐effector containing a cytotoxic necrotizing factor 1 domain. We demonstrate that T6SS1 can use this MIX‐effector to target phagocytic cells, resulting in morphological changes and actin cytoskeleton rearrangements. In conclusion, the V. proteolyticus T6SS1, a system homologous to one found in pathogenic vibrios, uses a suite of polymorphic effectors that target both bacteria and eukaryotic neighbors.
Keywords: bacterial competition, CNF1, Vibrio, virulence
Subject Categories: Microbiology, Virology & Host Pathogen Interaction
Introduction
A common strategy used by bacteria to manipulate neighbors is to secrete proteins, termed toxins or effectors, via specialized protein secretion systems 1. Whereas some secretion systems secrete toxins to the extracellular space, others can deliver them directly into neighboring cells 1. The type VI secretion system (T6SS) is a contact‐dependent protein delivery apparatus found in many Gram‐negative bacteria 2, 3. This system is composed of a tail‐tube made of stacked hexameric rings of a protein called Hcp 3, which are capped by a spike complex made of a VgrG trimer 4 and a PAAR repeat‐containing protein 5, 6. The tail‐tube is decorated with effector proteins that mediate the activity of the T6SS 7. Upon activation, a sheath‐like structure engulfing the tail‐tube contracts and propels the tail‐tube, with the effectors that decorate it, out of the bacterial cell and into an adjacent recipient, where effectors are deployed 6, 8. T6SSs are unique as they can directly deliver effectors into either bacterial or eukaryotic cells 9, 10 and thus play a role in interbacterial competition or virulence, respectively 7.
T6SS effectors come in two flavors: as domains fused to tail‐tube components, or as proteins that bind to these tail‐tube components either directly 11, 12 or via adapter proteins 13, 14, 15, 16. As many T6SSs described to date mediate antibacterial activities, most effectors that have been characterized are bactericidal and carry various activities such as lipases 17, 18, nucleases 19, peptidoglycan hydrolases 20, pore‐forming 21, and NAD(P)+ glycohydrolases 22. To prevent self‐intoxication, these bactericidal effectors are encoded adjacent to cognate immunity genes 20, 23. Several T6SSs were shown to target eukaryotic cells, yet compared to the number of bactericidal effectors, few anti‐eukaryotic effectors have been described to date 24. These include toxin domains fused to “evolved VgrGs” 4, 25, pore‐forming VasX 21, 26, PldB phospholipase 27, TecA deamidase 28, EvpP 29, and effectors secreted by the Francisella T6SS 30.
We recently uncovered a widespread, polymorphic class of T6SS effectors that share an N‐terminal domain named MIX (Marker for type sIX effectors) and variable C‐terminal toxin domains 31. MIX‐effectors group into five clans named MIX I–V 31. Members of MIX V clan are horizontally shared between marine bacteria and are suggested to enhance their fitness 32. In a recent report, we determined the secretome of Vibrio proteolyticus (hereafter referred to as Vpr), a marine bacterium that has been previously isolated from diseased corals 33, 34. We identified a functional hemolysin that is cytotoxic toward mammalian cells 35. The Vpr secretome also included tail‐tube components of T6SSs and MIX‐effectors, indicating the presence of functional T6SSs.
Here, we set out to characterize the T6SSs of Vpr and determine their effector repertoires and activities. We found that of the three T6SSs encoded by Vpr, T6SS1 mediated antibacterial activities. Using comparative proteomics analysis to identify the T6SS1 effector repertoire, combined with functional assays, we found that in addition to antibacterial MIX‐ and non‐MIX‐effectors, T6SS1 also secrets MIX‐effectors with anti‐eukaryotic activities. Thus, Vpr employs T6SS1 and a suite of MIX‐effectors to manipulate both bacterial and eukaryotic neighbors.
Results and Discussion
Vpr encodes three T6SSs and seven MIX‐effectors
Genomic analysis of the Vpr strain ATCC 15338 (also called NBRC 13287) revealed three T6SSs encoded on three main gene clusters, each including the conserved core T6SS components and accessory genes 36 (Fig 1). The T6SS1 cluster (vpr01s_06_00050–vpr01s_6_00300) is homologous to the antibacterial Vibrio parahaemolyticus T6SS1 and Vibrio alginolyticus T6SS1, which we previously characterized 31, 32. It contains two putative transcription regulators (vpr01s_06_00080 and vpr01s_06_00230) 37 and a MIX‐effector/immunity (E/I) pair (vpr01s_06_00050/vpr01s_06_00060) 31. However, it is missing the gene encoding a PAAR repeat‐containing protein at the end of the cluster. The T6SS2 cluster (vpr01s_08_01740–vpr01s_08_01940) is similar to the T6SS3 cluster (vpr01s_01_04280–vpr01s_01_05020) in terms of gene content and organization, with two differences: (i) the genes encoding VgrG2‐PAAR2 (vpr01s_08_01940–vpr01s_08_01920) are transcribed in the same direction as the rest of the cluster, whereas their counterparts in T6SS3 (vpr01s_01_05000–vpr01s_01_05020) are inverted; (ii) the genes between clpV and fha differ between the clusters (vpr01s_08_01820–vpr01s_08_01800 in T6SS2; vpr01s_01_04890–vpr01s_01_04880 in T6SS3) (Fig 1). Neither the T6SS2 nor the T6SS3 clusters appear to encode effectors.
Figure 1. T6SS clusters and modules identified in silico in Vpr .
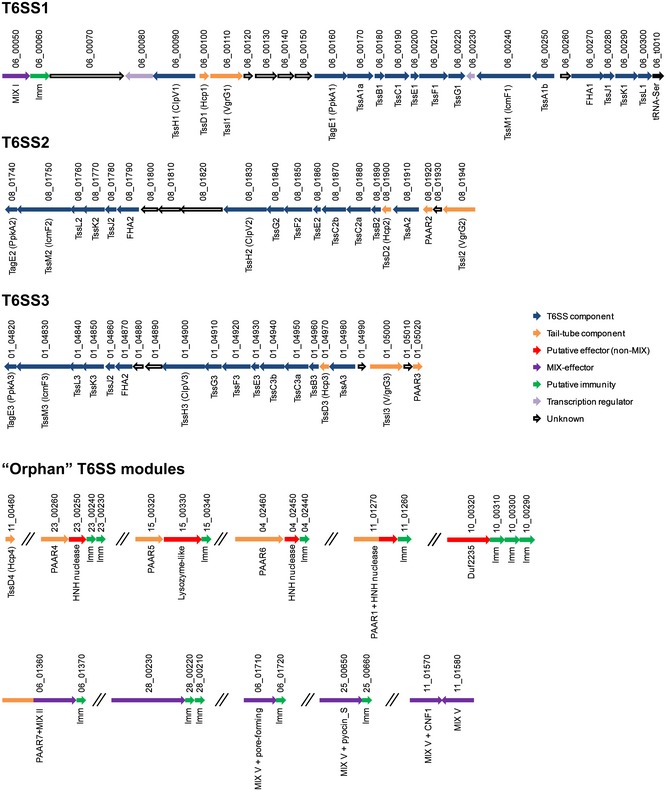
- Genes are represented by arrows indicating direction of translation. Locus tags (vpr01s_xx_xxxxx) shown above. Encoded proteins and known domains shown below.
The Vpr genome also contains “orphan” T6SS modules and effectors found outside the main T6SS clusters. An “orphan” hcp4 (vpr01s_11_00460) encodes a protein 99% identical to Hcp1. Five “orphan” modules encode PAAR repeat‐containing proteins which are either found upstream of putative antibacterial E/I pairs with predicted nuclease or lysozyme‐like activities (vpr01s_23_00260, vpr01s_15_00320, and vpr01s_04_02460) or are fused to a putative C‐terminal effector domain (vpr01s_01_01270 and vpr01s_06_01360). There are also six “orphan” MIX‐effectors, in addition to the MIX‐effector found in the T6SS1 cluster. vpr01s_06_01360 is a MIX II clan member that is fused to an N‐terminal PAAR domain mentioned above. vpr01s_28_00230 contains two MIX domains belonging to clans IV and V. vpr01s_06_01710 and vpr01s_25_00650 belong to the MIX V clan and carry putative antibacterial pore‐forming (amino acid 502–584 similar to TcdA/TcdB pore‐forming toxin domain with 46% probability, according to HHpred 38) and Pyocin_S nuclease (according to NCBI conserved domains) toxin domains, respectively. Interestingly, two “orphan” MIX‐effectors belonging to the MIX V clan appear to be anti‐eukaryotic rather than antibacterial. vpr01s_11_01570 contains a virulence CNF1 (cytotoxic necrotizing factor 1) domain, whereas vpr01s_11_01580 does not contain a known C‐terminal domain but is not part of an E/I pair as it has no adjacent immunity gene. An additional putative antibacterial effector with a DUF2235 lipase domain 18 is encoded by vpr01s_10_00320 followed by three homologous putative immunity genes (vpr01s_10_00310–vpr01s_10_00290) (Fig 1). Interestingly, we found no “orphan” vgrG genes.
Vpr kills competing bacteria under warm marine‐like conditions
To characterize the Vpr T6SSs, we first set out to identify conditions that activate them. As our analysis revealed several T6SS effectors with predicted bactericidal activities, we reasoned that at least one of the three Vpr T6SSs will mediate antibacterial toxicity, which we can monitor as an indicator of T6SS activity. Therefore, we performed bacterial competition assays and monitored the viability of Escherichia coli prey (which is commonly used as a Gram‐negative prey in bacterial competition assays) when incubated together with Vpr under different temperature (23, 30, or 37°C) and salinity conditions (1% or 3% NaCl). We found that Vpr bactericidal activity against E. coli prey was most prominent at 30°C on media containing 3% NaCl, as under these conditions we observed the largest decrease in E. coli prey viability (Fig 2). Thus, Vpr kills competing bacteria preferentially under conditions that are found in marine environments during summer months.
Figure 2. Vpr is bactericidal under warm marine‐like conditions.
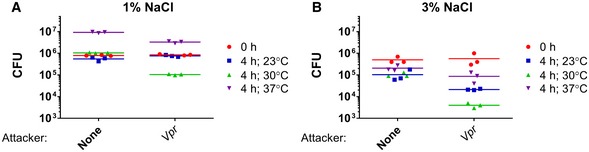
-
A, BViability counts of Escherichia coli prey before (0 h) and after (4 h) co‐culture with wild‐type Vpr or with no attacker (none) at indicated temperatures on media containing 1% (A) or 3% (B) NaCl.
Vpr bactericidal activity is mediated by T6SS1
To determine whether the bactericidal activity of Vpr is mediated by either of the T6SSs, we generated strains deleted for each of the three vgrG genes that correspond to the three T6SS clusters. We then tested the ability of these mutants to kill E. coli prey. Inactivation of T6SS1 (ΔvgrG1) resulted in complete loss of bactericidal activity, whereas inactivation of T6SS2 (ΔvgrG2) or T6SS3 (ΔvgrG3) had no effect (Fig 3A). These results suggested that T6SS1 was responsible for the Vpr‐mediated bactericidal activity seen in Fig 2. Indeed, the expression and secretion pattern of the secreted T6SS1 tail‐tube spike component VgrG1 agreed with the level of Vpr bactericidal activity under the same conditions and peaked at 30°C in media containing 3% NaCl (Appendix Fig S1). Attempts to rescue the bactericidal activity of ΔvgrG1 by exogenous expression of VgrG1 from a plasmid were unsuccessful, possibly due to a polar effect of vgrG1 deletion on downstream T6SS1 components (Appendix Fig S2). Therefore, we generated another T6SS1 mutant in which we deleted the gene encoding the essential T6SS baseplate component TssG1 (ΔtssG1) 39. As with ΔvgrG1, the ΔtssG1 mutant lost bactericidal activity against E. coli (Fig 3B) as well as against V. parahaemolyticus, a marine bacterium that is found in the natural habitat of Vpr (Fig 3C). Exogenous expression of TssG1 from a plasmid rescued the bactericidal activity and the secretion of the spike component VgrG1 (Fig 3B–D). Notably, deletion of tssG1 had no adverse effects on Vpr growth (Appendix Fig S3). Thus, T6SS1 mediates the bactericidal activity of Vpr under warm marine‐like conditions.
Figure 3. T6SS1 mediates Vpr bactericidal activity.
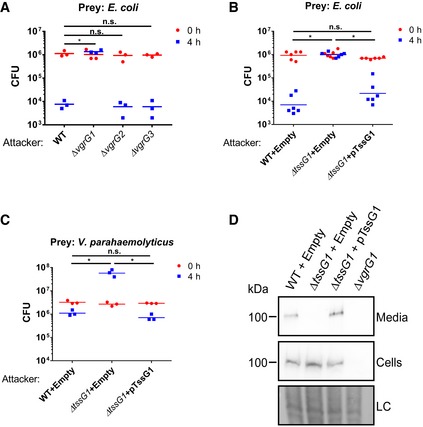
-
A–CViability counts of Escherichia coli (A, B) and Vibrio parahaemolyticus POR1/Δhcp1 (C) prey before (0 h) and after (4 h) co‐culture with indicated Vpr strains on media containing 3% NaCl at 30°C. Asterisks mark statistical significance between samples at 4‐h timepoint by unpaired, two‐tailed Student's t‐test (*P < 0.05); n.s., no significant difference; WT, wild‐type.
-
DExpression (Cells) and secretion (Media) of VgrG1 were detected by immunoblot using specific antibodies against VgrG1. Loading control (LC) is shown for total protein lysate.
The Vpr T6SS1 secretome includes multiple tail‐tube components and putative antibacterial and anti‐eukaryotic effectors
Next, we set out to determine the Vpr T6SS1 effectors that mediate the bactericidal activity. To this end, we used mass spectrometry to analyze and compare the secretomes of Vpr wild‐type (T6SS1+) and ΔtssG1 (T6SS1−) grown under T6SS1‐inducing conditions (i.e., 30°C in media containing 3% NaCl). We identified 27 proteins that were predominantly found in the secretome of the wild‐type Vpr (ratio of wild‐type/ΔtssG1 > 5) (Table 1 and Dataset EV1). Of these 27 proteins, 15 are predicted to be T6SS1 components and effectors (Fig 4), as they (i) are tail‐tube components; (ii) contain MIX domains; (iii) are homologs of known T6SS effectors; (iv) contain predicted toxin domains; (v) do not contain a canonical secretion signal; (vi) are in an operon with T6SS tail‐tube components. As expected, the tail‐tube components Hcp1 and VgrG1, encoded within the T6SS1 gene cluster, were secreted in a T6SS1‐dependent manner. Interestingly, the “orphan” Hcp4 and two “orphan” PAAR proteins (PAAR4 and PAAR6) were also identified as T6SS1 components 5.
Table 1.
T6SS1‐dependent Vpr secretome
| Function/Role | Uniprot | Locus [vpr01s_] | Description | Domains | Length [aa] | Signal peptidea |
|---|---|---|---|---|---|---|
| T6SS | ||||||
| Tail‐tube components | U3BN98 | 06_00100 | Hcp1 | Hcp | 172 | No |
| U3BB23 | 06_00110 | VgrG1 | VgrG | 692 | No | |
| U3BK42 | 11_00460 | Hcp4 | Hcp | 172 | No | |
| U3BHI2 | 23_00260 | PAAR4 | DUF4150 (PAAR) | 503 | No | |
| U2ZG25 | 04_02460 | PAAR6 | DUF4150 (PAAR) | 942 | No | |
| Antibacterial effectors | U2ZJC6 | 10_00320 | Lipase | DUF2235 | 750 | No |
| U2ZYX6 | 04_02450 | Nuclease | HNH/ENDO VII | 256 | No | |
| U3A2L0 | 08_01700 | Pore‐forming | TcdA/B pore‐forming domain‐like (TM) | 356 | No | |
| U3BK37 | 06_00050 | MIX‐effector | MIX I | 555 | No | |
| U2ZN70 | 25_00650 | MIX‐effector nuclease | MIX V, Pyocin_S | 758 | No | |
| U3BSB9 | 28_00230 | MIX‐effector | MIX IV, LysM, MIX V (TM) | 1,402 | No | |
| Anti‐eukaryotic effectors | U3BEL6 | 11_01580 | MIX‐effector | MIX V | 591 | No |
| U3BNK6 | 11_01570 | MIX‐effector deamidase | MIX V, CNF1 | 603 | No | |
| U3A3W8 | 11_00400 | Txp40 homolog | 90 | No | ||
| Unknown | U2ZZY6 | 06_00070 | VP1390 homolog | OmpA_C‐like | 1,539 | No |
| Non‐T6SS | ||||||
| Flagella | U3B8Q8 | 03_01380 | Flagellin | Flagellin_N, Flagellin_C | 288 | No |
| U2ZXS3 | 03_01390 | Flagellar hook‐associated protein 2 | FliD_N, FliD_C | 445 | No | |
| U3B8S7 | 03_01530 | Flagellar hook‐associated protein 1 | FlgK | 457 | No | |
| U3BHW0 | 03_01570 | Flagellar basal body rod | FlgG | 261 | No | |
| U2ZXU5 | 03_01590 | Flagellar hook | FlgE | 398 | No | |
| U3BHV5 | 03_01520 | Flagellar hook‐associated protein | FlgL | 299 | No | |
| U2ZYS5 | 03_01510 | Flagellin and related hook‐associated protein | FlgL | 347 | No | |
| Siderophore/heme transport | U3BJL7 | 05_01130 | Putative ferric siderophore receptor | TonB‐dependent receptor | 663 | Yes |
| U3A4E3 | 17_00240 | Ferrioxamine B receptor | TonB‐dependent receptor | 710 | Yes | |
| U2ZCG7 | 01_01640 | Putative heme/hemoglobin receptor | TonB‐dependent receptor | 711 | Yes | |
| Other | U3BAK7 | 05_01090 | Hemolysin‐type calcium‐binding region homolog | WD‐like beta propeller repeat | 463 | Yes |
| U3A3U4 | 11_00150 | Protease | Peptidase_M28 | 596 | Yes | |
TM, transmembrane.
As determined by SignalP 4.1.
Figure 4. The Vpr T6SS1 secretome.
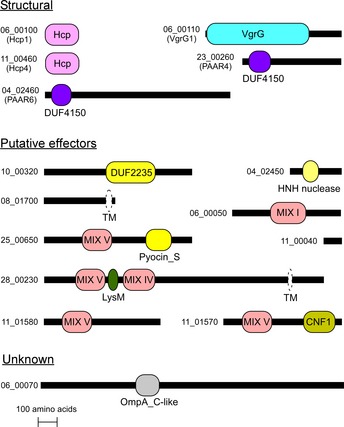
- Schematic representation of putative Vpr T6SS1‐secreted proteins identified by comparative proteomics (see Table 1). Horizontal black bars are relative to protein length. MIX domains and domains identified in NCBI CDD are illustrated in color; domain names are inside or below the rectangles. Transmembrane helices (TM) identified in UniProt are illustrated as white ovals with dashed perimeters. DUF4150 belong to PAAR‐like superfamily; DUF2235 is a phospholipase.
We also detected nine potential effectors in the T6SS1 secretome, including five MIX‐effectors (Table 1 and Fig 4). Six are predicted antibacterial effectors with various toxin domains and downstream small open reading frames that likely encode cognate immunity proteins (Figs 1 and 4, and Appendix Fig S4). Vpr01s_10_00320 contains a DUF2235 lipase domain; Vpr01s_04_02450 is a HNH‐family nuclease (99% probability according to HHpred 38 analysis) encoded downstream of PAAR6 (Fig 1); Vpr01s_08_01700 contains a C‐terminal transmembrane region in a domain similar to pore‐forming toxins (37% probability according to HHpred 38 analysis) (Table 1 and Fig 4) and is encoded close to the T6SS2 cluster (Appendix Fig S4). Three antibacterial effectors belong to the MIX‐effector class: Vpr01s_06_00050 is encoded within the T6SS1 cluster and is homologous to the antibacterial MIX‐effectors VP1388 and Va01565 from V. parahaemolyticus 31 and V. alginolyticus 32, respectively; Vpr01s_25_00650 belongs to the horizontally shared MIX V clan 32 and contains a Pyocin_S nuclease domain; Vpr01s_28_00230 contains both MIX IV and MIX V domains, as well as a C‐terminal region homologous to that of the Vibrio cholerae pore‐forming effector VasX 21 (Table 1 and Fig 4).
Surprisingly, this bactericidal T6SS1 also secreted three effectors with putative anti‐eukaryotic activities, which are not part of bicistronic units that encode potential immunity proteins (Figs 1 and 4). Vpr01s_11_01570 (named Vpr01570 hereafter) belongs to the horizontally shared MIX V clan 32 and contains a CNF1 deamidase domain that is predicted to target Rho GTPases 40, 41 (Appendix Fig S5). Vpr01s_11_01580 is encoded next to Vpr01570 and contains a MIX V domain (Fig 1). Its C‐terminus is homologous to proteins encoded by bacteria of the genera Vibrio, Morganella, Pseudomonas, and Yersinia. Some of these homologs are annotated as cytotoxic and others contain Rhs repeats, which were suggested to be T6SS1 effectors 42. Vpr01s_11_00400 is a small protein homologous to the C‐terminus of the insecticidal toxin Txp40 43 (Appendix Figs S4 and S6).
Another T6SS1‐secreted protein is Vpr01s_06_00070. It is encoded downstream of the MIX‐E/I pair Vpr01s_06_00050/Vpr01s_06_00060 and is homologous to the V. parahaemolyticus VP1390, which we previously reported is secreted by the T6SS, yet its role remains unknown. Thus, the Vpr T6SS1 effector repertoire is diverse and includes multiple MIX‐effectors with both antibacterial and anti‐eukaryotic toxin domains. Notably, unlike the Vpr T6SS1, homologous systems in V. parahaemolyticus and V. alginolyticus secrete only one of each of the tail‐tube components Hcp, VgrG, and PAAR 31, 32. Thus, it appears that the Vpr T6SS1 acquired additional “orphan” Hcp and PAAR proteins which possibly allow for diverse tail‐tube combinations and enable secretion of a wide effector repertoire.
The remaining 12 proteins predominantly found in the wild‐type Vpr secretome are likely not direct substrates of T6SS1. These include flagellar components or proteins that contain a signal peptide and are associated with siderophore/heme transport, a protein containing a WD‐like beta propeller repeat, and a protease (Table 1). While we do not yet know what mechanism leads to this apparent down‐regulation of the flagellar machinery and other non‐T6SS proteins, others have previously reported similar findings. For example, the T6SS in Citrobacter was reported to modulate flagellar gene expression and secretion 44, as did the T6SS in Ralstonia 45. Therefore, it is possible that a common cross talk mechanism exists between T6SS activity and motility, and perhaps also between the T6SS and siderophore or heme transport.
The T6SS1 MIX‐effector Vpr01570 is a functional CNF1 toxin
Our comparative proteomics analysis revealed that T6SS1, which mediates bactericidal activities (Fig 3), also secretes effectors with predicted anti‐eukaryotic activities (Table 1). This led us to hypothesize that the Vpr T6SS1 can use its diverse effector repertoire to target both bacteria and eukaryotic neighbors. To determine whether T6SS1 can also manipulate eukaryotic cells, we focused our efforts on the T6SS1 MIX‐effector Vpr01570 that contains a CNF1 toxin domain and set out to monitor its functionality. CNF1 toxins are deamidases that target and activate Rho GTPases 40, 41. As Rho GTPases are master regulators of the actin cytoskeleton, CNF1 toxins induce actin cytoskeleton rearrangements inside eukaryotic cells, which are readily visualized by microscopy 41.
We recently reported that Vpr induces cell death in human epithelial HeLa cells and in RAW 264.7 murine macrophages 35. This cytotoxicity was mediated by a secreted pore‐forming hemolysin named VPRH 35. Strangely, however, unmasking this VPRH‐mediated cytotoxicity by infecting HeLa cells with a Δvprh mutant did not reveal any observable actin cytoskeleton rearrangements 35. This result implied that the CNF1 MIX‐effector is either not a functional toxin or is not delivered into HeLa cells during infection. We therefore tested whether Vpr01570 is a functional virulence toxin by exogenously expressing it in HeLa cells and in yeast. Expression of Vpr01570 fused to superfolder green fluorescent protein (sfGFP) induced dramatic actin cytoskeleton rearrangements in HeLa cells, including formation of actin stress fibers and ruffles which are hallmark phenotypes caused by CNF1 activity 40 (Fig EV1A). Vpr01570 was also toxic when expressed in yeast (Fig EV1B). These toxic effects were dependent on a functional CNF1 domain as they were not observed when a catalytic mutant (cysteine 450 to serine) (Appendix Fig S5) was expressed (Fig EV1A and B). Notably, expression of the Vpr01570 mutant form was detected in both HeLa cells and yeast, whereas the wild‐type form was detected only in HeLa cells, presumably due to its deleterious effect in yeast (Fig EV1C and D). Moreover, we confirmed that deletion of vprh did not impair T6SS1 activity as a Δvprh mutant remained bactericidal in a bacterial competition assay (Appendix Fig S7). Thus, Vpr01570 is a functional CNF1 toxin, yet Vpr does not use T6SS1 or Vpr01570 to manipulate the actin cytoskeleton during infection of HeLa cells.
Figure EV1. Vpr01570 is a functional anti‐eukaryotic toxin.
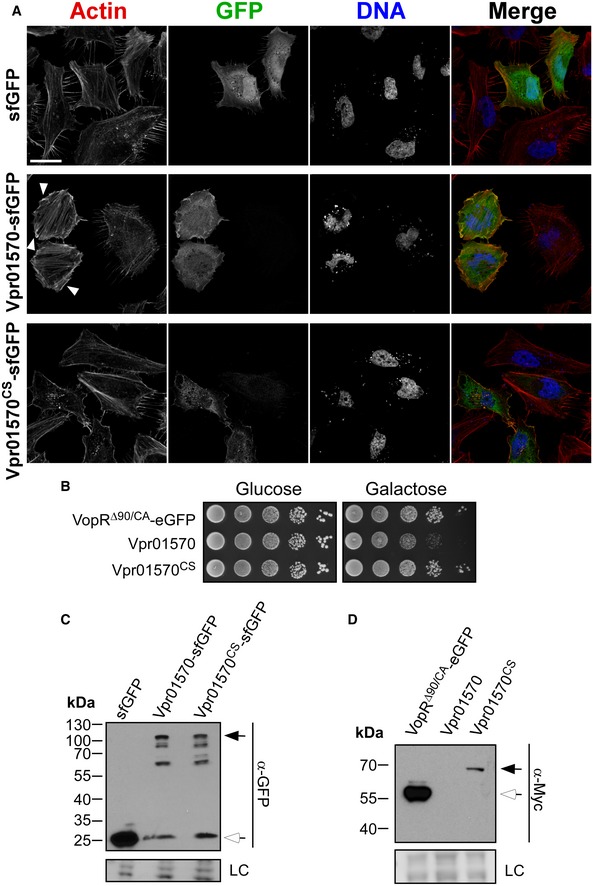
-
AConfocal micrograph of HeLa cells transfected with vectors expressing the indicated proteins. Cells were stained for F‐actin and DNA using rhodamine‐phalloidin (red) and Hoechst stain (blue), respectively. Scale bar = 30 μm; sfGFP, superfolder green fluorescent protein. Arrowheads mark actin ruffles.
-
BGrowth of BY4741 yeast containing vectors for galactose‐inducible expression of indicated proteins on repressing (glucose) and inducing (galactose) plates. Yeast were spotted in 10‐fold serial dilutions. VopRΔ90/CA is a truncated and catalytically inactive form of the Vibrio parahaemolyticus type III effector VopR and serves as a non‐toxic control.
-
C, DImmunoblot using anti‐GFP (C) or anti‐Myc (D) antibodies to verify expression of proteins used in (A) and (B), respectively. In (C), black arrow marks expected size of Vpr01570‐sfGFP fusions and white arrow marks expected size of sfGFP. In (D), black arrow marks the expected size of Vpr01570‐myc, and white arrow marks the expected size of VopRΔ90/CA‐eGFP fusion. LC, loading control.
Source data are available online for this figure.
Vpr induces hemolysin‐independent morphological changes in macrophages
Previous reports suggested that the anti‐eukaryotic activities of V.cholerae and Burkholderia pseudomallei T6SSs are activated upon internalization by phagocytic cells during infection 9, 46. As we did not observe T6SS1‐mediated morphological changes during infection of HeLa cells with the Δvprh mutant strain, we decided to test whether such effects will be detectable upon Vpr infection of phagocytic cells. To this end, we infected RAW 264.7 murine macrophages with Vpr. In agreement with our previous report 35, Vpr was cytotoxic to RAW 264.7 cells upon infection, and free nuclei from lysed cells were visible (Fig 5A). Remarkably, unmasking the VPRH‐mediated cytotoxicity by infecting macrophages with a Δvprh mutant revealed dramatic morphological changes that were not detected upon infection of HeLa cells. RAW 264.7 cells infected with Δvprh became flat, spread out, developed actin ruffles on the top side, and showed a pronounced and smooth actin border starting at 4 h post‐infection (Figs 5 and EV2). Notably, infection of macrophages with an avirulent derivative of the bacterium V. parahaemolyticus 47 did not induce similar phenotypes (Fig 5A), nor did incubation with lipopolysaccharide (LPS) (Fig EV2). These results suggested that the phenotypes observed during infection with the Vpr Δvprh mutant were induced by a Vpr determinant, and were not likely simply the result of macrophage activation by recognition of common pathogen‐associated molecular patterns 48.
Figure 5. The T6SS1 MIX‐effector Vpr01570 induces actin rearrangements in macrophages.
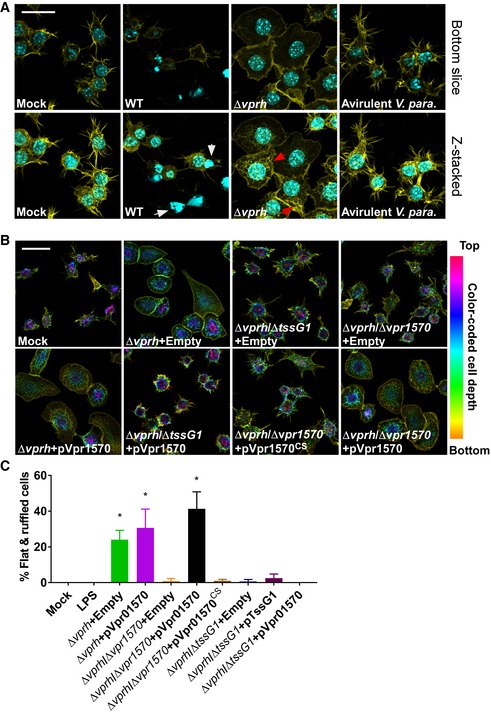
- Confocal micrograph of RAW 264.7 cells infected with indicated Vpr or avirulent Vibrio parahaeolyticus (V. para) strains for 4 h. Cells were stained for F‐actin and DNA using rhodamine‐phalloidin (yellow) and Hoechst stain (cyan), respectively. Top panels show bottom focal plane of the cells; lower panels show stacked z‐axis slices of the same field of view. White arrows mark free nuclei of lysed cells. Red arrows mark actin ruffles. Scale bar = 20 μm; WT, wild‐type.
- Confocal micrograph of RAW 264.7 cells infected with indicated Vpr strains containing either an empty vector (Empty) or arabinose‐inducible vectors for expression of wild‐type Vpr1570 (pVpr1570) or C450S mutant form (pVpr1570CS) for 4 h at MOI = 25. Cells were stained for F‐actin using rhodamine‐phalloidin. F‐actin is color‐coded to show cell depth in the z‐axis. Scale bar = 20 μm; WT, wild‐type.
- Quantification of percentage of infected cells presenting a flatten and ruffled morphology after 6 h infection with the indicated strains or addition of LPS (0.1 μg/ml). Results shown as mean percentage of cells in a field ± standard deviation (n = 3, minimum number of cells per field is 40; maximum number of cells per field is 87). Asterisks mark treatments that are significantly different from LPS treatment as determined by unpaired, two‐tailed Student's t‐test (*P < 0.05). Experiment was performed at least twice and a representative result is shown.
Figure EV2. Vpr‐induced morphological changes in macrophages begin 4 h post‐infection.
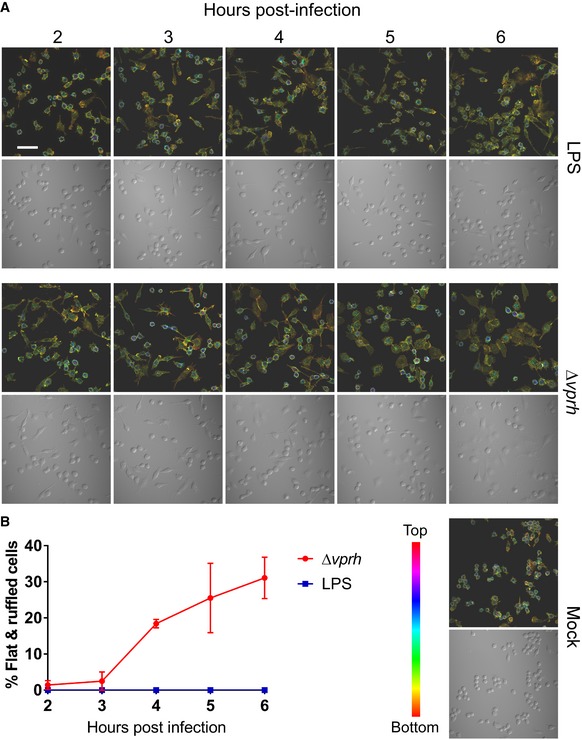
- Confocal micrograph of RAW 264.7 cells infected with Vpr Δvprh strain at MOI = 25 or treated with LPS (0.1 μg/ml) for the indicated times. Cells were stained for F‐actin using rhodamine‐phalloidin. Top panels show fluorescence images. F‐actin is color‐coded to show cell depth in the z‐axis. Bottom panels show DIC of same fields of view. Scale bar = 50 μm.
- Quantification of percentage of infected cells presenting a flatten and ruffled morphology during Vpr infection or LPS treatment as in (A). Results shown as mean percentage of cells in a field ± standard deviation (n = 3, minimum number of cells per field is 41; maximum number of cells per field is 95). Experiment was performed three times, and a representative result is shown.
Vpr uses the T6SS1 MIX‐effector Vpr01570 to manipulate actin in macrophages
Next, we tested whether the Vpr‐induced morphological changes in macrophages were mediated by T6SS1 activity. Inactivation of T6SS1 by deleting tssG1 abolished the actin rearrangements seen upon infection of RAW 264.7 cells with the Δvprh strain (Fig 5B and C). This result indicated that T6SS1 is required for the observed Vpr‐induced phenotypes. Furthermore, the ability of Vpr to induce T6SS1‐dependent actin rearrangements in macrophages but not in HeLa cells suggested that uptake of Vpr by a phagocytic cell may be a prerequisite for activation of T6SS1 during infection of eukaryotic cells. Surprisingly, exogenous expression of TssG1 from a plasmid did not restore the ability of the Δvprh/ΔtssG1 mutant to induce morphological changes in RAW 264.7 cells (Fig 5C). A possible explanation is that activation of T6SS1 during infection of macrophages requires precise regulation which is lost upon inducible overexpression of TssG1 from a plasmid.
Interestingly, actin reorganization phenotypes similar to those observed upon infection of RAW 264.7 cells with Vpr Δvprh were previously reported upon treating macrophages with the Yersinia CNFy toxin 49, which is homologous to the CNF1 toxin domain of the T6SS1 MIX‐effector Vpr01570 (Appendix Fig S5). Therefore, we asked whether Vpr01570 plays a role in the T6SS1‐mediated actin phenotypes in macrophages. Indeed, deletion of vpr01570 impaired the ability of Vpr Δvprh to induce the T6SS1‐mediated phenotypes in macrophages (Fig 5B and C). Exogenous expression of the wild‐type Vpr01570, but not of its catalytic mutant form (Vpr01570CS), rescued the mutant strain's ability to induce actin rearrangements. However, overexpression of Vpr1570 did not induce similar phenotypes when T6SS1 was inactive (Fig 5B and C). Importantly, deletion of vpr01570 did not impair T6SS1 functionality as the Δvprh/Δvpr01570 mutant retained bactericidal activity, nor did it affect bacterial growth (Appendix Fig S8). These results demonstrate that T6SS1 delivers the MIX‐effector Vpr01570 into macrophages where it employs its CNF1 toxin domain to induce morphological changes. Thus, Vpr can target both bacterial and eukaryotic neighbors with its T6SS1. This ability to use T6SS1 for delivery of polymorphic MIX‐effectors into both bacteria and potential eukaryotic predator or host cells must provide a fitness advantage as it allows Vpr to manipulate neighbors which are present in its natural marine habitat 33, 34.
Moreover, MIX‐effector‐secreting T6SSs homologous to the Vpr T6SS1 are present in many other marine bacteria, including emerging pathogens such as V. parahaemolyticus 37 and V. alginolyticus 32. We previously characterized MIX‐effectors with antibacterial activities 31, 32 and showed that effectors containing the MIX V domain are horizontally shared between marine bacteria and are used to diversify effector repertoires, thus enhancing competitive fitness 32. Based on our current results, we further propose that bacteria can also acquire virulence MIX‐effectors, which are evidently circulating in the Vibrio pan‐genome, and use them to diversify their virulence traits and range.
After validating that Vpr01570 is an anti‐eukaryotic MIX‐effector delivered by T6SS1, we set out to determine whether this effector is not only necessary but also sufficient to induce the T6SS1‐mediated morphological changes in macrophages. To this end, we transfected RAW 264.7 cells with an expression vector encoding Vpr01570 fused to sfGFP. Cells transfected with the wild‐type Vpr01570 presented pronounced actin ruffling at the top of the cells which was not apparent upon expression of the catalytic mutant form Vpr01570CS (Fig EV3). Vpr01570 also co‐localized with the actin ruffles (Fig EV3). However, Vpr01570‐expressing cells did not spread or flatten as cells infected with Vpr Δvprh did. These results suggested that whereas Vpr01570 is necessary and sufficient to induce actin ruffling in macrophages, it is not sufficient to cause cell spreading. Thus, it is possible that either additional effectors (of the T6SS1 or of other systems) or macrophage activation by recognition of pathogen‐associated molecular patterns play a role, together with Vpr01570, in the induction of the observed morphological changes during infection. Notably, the Vpr T6SS3 may also be active under the same conditions as T6SS1. This is supported by the presence of the T6SS3 tail‐tube proteins Hcp3 (Vpr01s_01_04970) and VgrG3 (Vpr01s_01_05000) in the wild‐type Vpr secretome (Dataset EV1). As our results do not support a role for T6SS3 in bacterial competition, it is possible that this system targets eukaryotic cells. Further studies are required to determine the role and effector repertoire of T6SS3, as well as the mechanisms of action and targets of the T6SS1 putative anti‐eukaryotic effectors Vpr01s_11_01580 and Vpr01s_11_00400.
Figure EV3. Vpr01570 is sufficient to induce actin ruffles in macrophages.
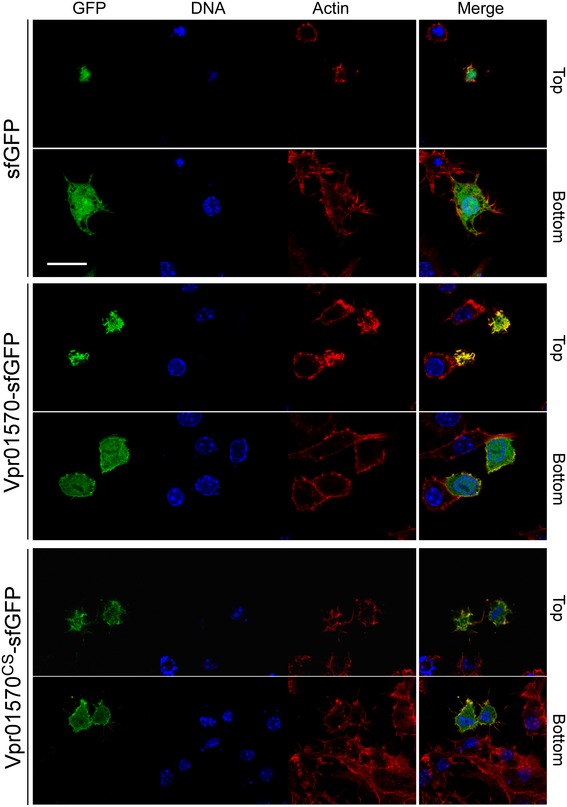
- Confocal micrograph of RAW 264.7 cells transfected with vectors expressing the indicated proteins. Cells were stained for F‐actin and DNA using rhodamine‐phalloidin (red) and Hoechst stain (blue), respectively. Top panels show a focal plane at the top of the cells; bottom panels show a focal plane located at the bottom of the cells. Scale bar = 20 μm; sfGFP, superfolder green fluorescent protein.
In conclusion, this study demonstrates that Vpr uses its T6SS1 to mediate both bactericidal and virulence activities by delivering a suite of antibacterial and anti‐eukaryotic effectors. The ability of the Vpr T6SS1 to deliver MIX‐effectors into both bacteria and eukaryotic cells makes it, together with polymorphic members of the MIX‐effector class, a lucrative candidate to use as a platform for biocontrol against pathogens from different kingdoms.
Materials and Methods
Bacterial strains, cell cultures, and yeast
Vibrio proteolyticus strain ATCC 15338 (also named NBRC 13287) (Vpr) and its derivative strains, as well as the V. parahaemolyticus RIMD 2210633 derivatives POR1/Δhcp1 (ΔtdhAS/Δhcp1) 37 and POR4 (ΔtdhAS/ΔvcrD1/ΔvcrD2) 47, were routinely grown in marine Luria‐Bertani (MLB) broth (Luria‐Bertani broth supplemented with NaCl to a final concentration of 3% w/v) at 30°C. To induce expression of genes from a plasmid, 0.1% (w/v) L‐arabinose was included in the medium. Escherichia coli strain DH5α were used for plasmid maintenance and amplification, and as prey in competition assays (see below). Escherichia coli strain S17‐1 (λ pir) were used for maintenance of pDM4 plasmids and mating (see below). Escherichia coli were routinely grown in 2× YT broth (1.6% w/v tryptone, 1% w/v yeast extract, 0.5% w/v NaCl) at 37°C. When necessary, media were supplemented with 30 μg/ml (for E. coli) or 250 μg/ml (for Vpr) kanamycin, or 25 μg/ml chloramphenicol. RAW 264.7 murine macrophages (ATCC TIB‐71) and human epithelial HeLa cells (ATCC CCL‐2) were purchased from ATCC and cultured in high‐glucose Dulbecco's modified Eagle's medium [DMEM (Gibco)] supplemented with 10% (v/v) fetal bovine serum, 1% (v/v) penicillin–streptomycin–glutamine, and 1% (v/v) sodium pyruvate (termed supplemented DMEM) and kept at 5% CO2 at 37°C. Cells have not been authenticated following receipt from ATCC. Saccharomyces cerevisiae strain BY4741 (MATa his3Δ0 leu2Δ0 met15Δ0 ura3Δ0) were grown in synthetic dropout media, as previously described 50.
Plasmid construction
For arabinose‐inducible expression in bacteria, the coding sequences (CDS) of VgrG1 (accession number GAD66994) and TssG1 (accession number GAD67005) were amplified from Vpr genomic DNA and inserted into the multiple cloning site (MCS) of the pBAD2 vector (KanR pBAD/Myc‐His in which the NcoI restriction site had been removed 37) in‐frame with the C‐terminal Myc‐6xHis tag (producing pVgrG1 and pTssG1, respectively). For arabinose‐inducible expression in bacteria, the CDS of Vpr01570 (accession number GAD68163), including the stop codon, was amplified from Vpr genomic DNA and inserted into the MCS of pMBAD (KanR pBAD/Myc‐His in which an N‐terminal c‐Myc tag was inserted in the NcoI and XhoI restriction sites 35), in‐frame with the N‐terminal c‐Myc tag (producing pVpr01570). To generate a mutant form of Vpr01570 with a mutation in cysteine 450 to serine (pVpr01570CS), site‐directed mutagenesis was performed on pVpr01570 using a pfu‐turbo DNA polymerase (Thermo Fisher Scientific) and the primers 5′‐GGAACATTGAGTGGCaGCACCATGGTCACGGG‐3′ and 5′‐CCCGTGACCATGGTGCtGCCACTCAATGTTCC‐3′, followed by DpnI treatment. The mutation was confirmed by nucleic acid sequencing.
For expression of Vpr01570 and Vpr01570CS sfGFP‐fusions in mammalian cells, the CDS of the wild‐type Vpr01570 and the C450S mutant form were amplified from pMBAD vectors (described above) and inserted into the MCS of the sfGFP‐N1 vector (Addgene) in‐frame with the C‐terminal sfGFP.
For galactose‐inducible expression of Vpr01570 and Vpr01570CS in yeast, the CDS of the wild‐type Vpr01570 and the C450S mutant form were amplified from pMBAD vectors (described above) and inserted into the MCS of the pGMU10 (Riken) expression vector in‐frame with the C‐terminal c‐Myc tag. The CDS of the V. parahaemolyticus type III secretion system effector VopR (vp1683), encoding amino acids 90–325 with a mutation in a catalytic cysteine 223 to alanine (VopRΔ90/CA), was inserted into the MCS of the pDGFP vector 51 in‐frame with the C‐terminal eGFP‐Myc tag, and used as a negative control for the yeast toxicity assay (see below).
Construction of deletion strains
For in‐frame deletions of tssG1(accession number GAD67005) and vpr01570 (accession number GAD68163), 1‐kb sequences directly upstream and downstream of each gene were cloned into pDM4, a CmrOriR6K suicide plasmid 52. These pDM4 constructs were inserted into V. proteolyticus via conjugation by S17‐1(λ pir) E. coli. Transconjugants were selected for on MLB agar containing chloramphenicol. The resulting transconjugants were plated onto MLB agar containing 15% (w/v) sucrose for counter‐selection and loss of the sacB‐containing pDM4. Deletions were confirmed by PCR. The generation of in‐frame deletions of vprh (accession number GAD67085), vgrG1 (accession number GAD66994), vgrG2 (accession number GAD67611), and vgrG3 (accession number GAD65726) were described previously 35.
Bacterial competition
Bacterial strains were grown overnight in MLB (Vpr and V. parahaemolyticus) or 2× YT (E. coli). Cultures were normalized to OD600 = 0.5, mixed at 4:1 ratio (attacker:prey) and spotted on LB or MLB plates, as previously described 37. Colony forming units (CFU) of the prey spotted at t = 0 h were determined by plating 10‐fold serial dilutions on selective media plates. Bacterial spots were harvested from assay plates after 4‐h incubation and the CFU of the surviving prey cells were determined. Assays were repeated at least twice with similar results, and results from a representative experiment are shown.
Protein secretion
Bacterial strains were grown overnight in MLB with appropriate antibiotics to maintain plasmids when necessary. 0.9 OD600 units were washed and re‐suspended in 20 ml borosilicate glass tubes containing 5 ml of LB or MLB media (to produce cultures of OD600 = 0.18) with appropriate antibiotics to maintain plasmids, and with 0.1% (w/v) L‐arabinose if induction of expression from a plasmid was required. Cultures were incubated with agitation (215 rpm) for 5 h at 23, 30, or 37°C, as indicated. For expression fractions (Cells) 1.0 OD600 units were collected and cell pellets were re‐suspended in 100 μl of Tris‐glycine SDS sample buffer ×2 (Novex, Life Sciences). Supernatants of 10 OD600 units were filtered and precipitated with deoxycholate and trichloroacetic acid 53. Precipitated proteins were pelleted and washed twice with cold acetone, prior to re‐suspension in 20 μl of 10 mM Tris–HCl pH = 8.0, followed by addition of 20 μl of 2× protein sample buffer. Expression and secretion samples were resolved on TGX stain‐free gels (Bio‐Rad) in SDS–polyacrylamide gel electrophoresis (SDS–PAGE), transferred onto PVDF membranes, and immunoblotted with custom‐made anti‐VgrG1 antibodies (polyclonal antibodies were produced in house (Thermo Scientific Pierce Protein Biology) with rabbits for peptide KDMSTKVLNNRYRDIGQDE). Loading of total protein lysate was visualized by analysis of trihalo compounds fluorescence of the immunoblot membrane (for Fig 3D) or Coomassie staining (for Appendix Fig S1).
Bacterial growth
Overnight‐grown cultures of Vpr were normalized to OD600 of 0.1 in 25 ml MLB in triplicates and grown at 30°C with agitation for 6 h. Growth was assessed by OD600 measurements at indicated time points. Experiments were performed at least twice with similar results. Results from a representative experiment are shown.
Tissue culture infection
RAW 264.7 cells were seeded at 3 × 105 cells/ml in 6‐well plates containing UV‐pretreated 22‐ by 22‐mm glass coverslips. Overnight Vibrio cultures were normalized to an OD600 of 0.1 in 5 ml MLB and grown for 2 h with agitation at 30°C. When necessary, 200 μg/ml kanamycin and 0.1% (w/v) L‐arabinose were added to maintain plasmids and induce expression. OD600 measurements of the cultures were taken and used to calculate the volume needed to make solutions of DMEM without antibiotics (termed infection medium) containing a multiplicity of infection (MOI) of 25 for each bacterial strain (MOI of 10 was used for the V. parahaemolyticus avirulent POR4 strain, corresponding to the same OD600 as MOI of 25 for Vpr strains). The infection solutions included 200 μg/ml kanamycin and 0.1% (w/v) L‐arabinose when necessary. RAW 264.7 cells were washed twice with 1 ml of unsupplemented DMEM, and then 2 ml infection medium with bacteria were added to each well. The plates were centrifuged at 200 × g for 5 min to synchronize infection before incubation at 37°C with 5% CO2 for the indicated duration. Following infection, cells were washed once with 1× PBS (pH 7.4) and fixed to the coverslips by treating with 1 ml of 3.2% paraformaldehyde solution at 4°C overnight. Experiments were performed at least twice with similar results, and results from a representative experiment are shown.
Tissue culture transfection
For transfections, HeLa cells were seeded either at 1 × 105 cells/ml (for immunoblots) or 5 × 104 cells/ml (for fluorescent microscopy), and RAW 264.7 cell were seeded at 5 × 105 cells/ml in 2 ml supplemented DMEM into 6‐well plates containing UV‐pretreated 22‐ by 22‐mm glass coverslips. Culture media was changed 2 h prior to transfection. Cells were treated with addition of 200 μl OptiMEM containing 3:1 ratio of Fugene HD reagent (Promega) to plasmid DNA and cultured at 37°C. For immuoblots, HeLa cells were collected 16 h post‐transfection and centrifuged at 1,000 rpm for 5 min. Pelleted cells underwent two washing steps in 1× PBS followed by centrifugation, then resuspended in 100 μl 2× protein sample buffer. Samples were resolved on SDS–polyacrylamide gel electrophoresis (SDS–PAGE), transferred onto PVDF membranes, and immunoblotted with anti‐GFP antibodies (Living Colors A.v. Monoclonal antibody JL‐8, Clontech). Equal loading of total protein lysate was confirmed by analysis of representative bands using Ponceau S staining of the immunoblot membrane. For microscopy, 7 h post‐transfection cells were washed once with 1× PBS (pH 7.4) and then treated with 1 ml of 3.2% paraformaldehyde solution for 15 min at room temperature to fix cells to coverslips. The cells were then treated accordingly to the following Microscopy protocol.
Microscopy
Fixed cells were treated with 0.1% Triton X‐100 for 10 min to permeabilize membranes. The cells were washed with 1 ml 1× PBS and were subsequently stained with 200 μl of a dye solution for 10 min, covered. The dye solution contained a 1:1,000 dilution of 1 mg/ml Hoechst 33342 (Molecular Probes) and a 1:100 dilution of 200 U/ml rhodamine‐phalloidin (Molecular Probes) to stain nuclei and F‐actin, respectively. Cells were treated three times to a 5‐min wash with 1 ml 1× PBS to reduce background staining. After the final wash, the coverslips were mounted onto glass slides using a drop of ProLong Gold antifade mounting solution (Molecular Probes), and sealed after sitting at room temperature for several hours. Slides were imaged using a Zeiss LSM 710 or LSM 800 confocal microscope, and images were converted using ImageJ (NIH).
Quantification of Vpr‐induced morphological changes
Confocal z‐stack images of F‐actin for infected RAW 264.7 cells were stacked into a color‐coded image using ImageJ temporal‐color‐coded hyperstacks, to allow for simultaneous assessment of cell height and F‐actin morphology. Cells that presented altered morphology and were flat, spread out, developed actin ruffles on the top side, and showed a pronounced and smooth actin border were identified, and their percentage out of the total number of cells visible in the field of view was calculated. For each treatment, three fields of view were processed per experiment. Three independent experiments were performed for each treatment with similar results. Results from a representative experiment are shown.
Yeast toxicity
pGMU10 and pDGFP expression vectors carrying Vpr01570 and VopR forms were transformed into the S. cerevisiae strain BY4741 by the lithium acetate transformation method 54. Transformed yeast were plated onto selective synthetic dropout media lacking uracil and supplemented with 2% glucose. Growth‐inhibition assays were performed as previously described 50. Briefly, yeast cultures were grown overnight at 30°C in liquid selective media containing 2% glucose, washed, and normalized to an OD600 of 1.0. Each strain was 10‐fold serially diluted and spotted (10 μl) onto repressing (2% glucose) or inducing (2% galactose and 1% raffinose) solid selective media. Yeast were then incubated at 30°C for 2 days. Validation of protein expression in yeast was performed for 2 OD units of cells as previously described 50. Proteins were detected using anti‐c‐Myc monoclonal antibodies (Santa Cruz, sc‐40).
Sequence analysis
T6SS clusters and “orphan” modules were identified by performing BLAST 55 searches using T6SS sequences from V. parahaemolyticus RIMD 2210633 37. MIX‐effectors sequences were identified previously 31. Domains within T6SS effectors were determined using the NCBI conserved domains database 56 or predicted using HHpred 38 and BLAST 55. Secretion signals were predicted using the SignalP 4.1 server 57. For multiple sequence alignments, protein sequences were aligned using T‐coffee 58. Alignments were visualized using Boxshade.
Mass spectrometry
For secretome analysis, overnight cultures of wild‐type or ΔtssG1 Vpr were normalized to an OD600 of 0.18 in 50 ml MLB media in triplicates and grown for 5 h at 30°C with agitation. Media were collected, and proteins were precipitated as previously described using deoxycholate and trichloroacetic acid 53. Protein samples were run 10 mm into the top of an SDS–PAGE gel, stained with Coomassie blue, and excised.
Overnight in‐gel digestion with trypsin (Promega) was performed after reduction and alkylation with DTT and iodoacetamide (Sigma‐Aldrich). The resulting samples were analyzed by tandem MS using an LTQ Orbitrap Fusion Lumos ETD Tribrid mass spectrometer (Thermo Fisher Scientific) coupled to an EASY nLC1200‐Nano liquid chromatography system (Thermo Fisher Scientific). Peptides were loaded onto a 75–μm i.d., 50‐cm long, column containing 2 μm C18 resin (Thermo Fisher Scientific) and eluted with a gradient: 0–5% B in 2 min, 5–30% in 65 min; 30–60% B in 10 min. Buffer A consisted of 2% (v/v) acetonitrile (ACN) and 0.1% formic acid in water. Buffer B consisted of 80% (v/v) ACN, 10% (v/v) trifluoroethanol, and 0.1% formic acid in water. The Lumos mass spectrometer acquired up to 20 high‐energy, collision‐induced dissociation fragment spectra for each full spectrum acquired.
Raw MS data files were converted to a peak list format and analyzed using the central proteomics facilities pipeline (CPFP), version 2.0.3 59, 60. Peptide identification was performed using the X!Tandem 61 and open MS search algorithm (OMSSA) 62 search engines against the V. proteolyticus NBRC 13287 protein database from UniProt. Fragment and precursor tolerances of 20 ppm and 0.5 Da were specified, and three missed cleavages were allowed. Carbamidomethylation of Cys was set as a fixed modification, with oxidation of Met set as a variable modification. Label‐free quantitation of proteins across samples was performed using SINQ normalized spectral index software 63. Protein identifications were filtered to an estimated 1% protein false discovery rate (FDR) using the concatenated target‐decoy method 64, and proteins needed to be present in all three wild‐type samples to be considered. Additional requirements of two unique peptide sequences per protein and two spectral counts per protein per wild‐type sample replicate were imposed, resulting in a final protein FDR < 1%. Two‐tailed, unequal variance Student t‐test was used to calculate P‐values. Samples in which no peptides were identified were set at value 0 for the analysis.
Statistical analysis
Unless otherwise stated, data are shown as mean ± standard deviation of the mean. For bacterial competition assays, data are presented as individual data points and their mean. Statistical significance (P < 0.05) was determined by two‐tailed, unpaired Student's t‐test.
Data availability
The data supporting the findings of this study are available within the paper and its supplementary information. The raw mass spectrometry data files and the corresponding peak lists were uploaded to the MassIVE dataset repository (https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=fa38fde1dc3e43e3abc675600e588838) and are available under MassIVE ID: MSV000080535.
Author contributions
DS, AR, and KO designed the experiments. AR, DS, NS, MSS, and JZ performed experiments. DS, AR, JZ, and KO analyzed the data. DS wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 3D
Acknowledgements
We thank the University of Texas Southwestern Medical Center Proteomics core for assistance with MS analysis. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant Agreement No. 714224) (D.S.), and by the NIH grant R01‐AI087808, the Welch Foundation grant I‐1561, and Once Upon a Time… (K.O.); K.O. is a Burroughs Welcome Investigator in Pathogenesis of Infectious Disease, a Beckman Young Investigator, and a W. W. Caruth, Jr., Biomedical Scholar, and has an Earl A. Forsythe Chair in Biomedical Science. D.S. is an Alon Fellow. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
EMBO Reports (2017) 18: 1978–1990
References
- 1. Costa TRD, Felisberto‐Rodrigues C, Meir A, Prevost MS, Redzej A, Trokter M, Waksman G (2015) Secretion systems in Gram‐negative bacteria: structural and mechanistic insights. Nat Rev Microbiol 13: 343–359 [DOI] [PubMed] [Google Scholar]
- 2. Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ (2006) Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci USA 103: 1528–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mougous JD, Cuff MEME, Raunser S, Shen A, Zhou M, Gifford CA, Goodman AL, Joachimiak G, Ordoñez CL, Lory S et al (2006) A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science 312: 1526–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pukatzki S, Ma AT, Revel AT, Sturtevant D, Mekalanos JJ (2007) Type VI secretion system translocates a phage tail spike‐like protein into target cells where it cross‐links actin. Proc Natl Acad Sci USA 104: 15508–15513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shneider MM, Buth SA, Ho BT, Basler M, Mekalanos JJ, Leiman PG (2013) PAAR‐repeat proteins sharpen and diversify the type VI secretion system spike. Nature 500: 350–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zoued A, Brunet YR, Durand E, Aschtgen M‐S, Logger L, Douzi B, Journet L, Cambillau C, Cascales E (2014) Architecture and assembly of the type VI secretion system. Biochim Biophys Acta 1843: 1664–1673 [DOI] [PubMed] [Google Scholar]
- 7. Alcoforado Diniz J, Liu YC, Coulthurst SJ (2015) Molecular weaponry: diverse effectors delivered by the type VI secretion system. Cell Microbiol 17: 1742–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Basler M, Pilhofer M, Henderson GP, Jensen GJ, Mekalanos JJ (2012) Type VI secretion requires a dynamic contractile phage tail‐like structure. Nature 483: 182–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ma AT, McAuley S, Pukatzki S, Mekalanos JJ (2009) Translocation of a Vibrio cholerae Type VI secretion effector requires bacterial endocytosis by host cells. Cell Host Microbe 5: 234–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vettiger A, Basler M (2016) Type VI secretion system substrates are transferred and reused among sister cells. Cell 167: 99–110.e12 [DOI] [PubMed] [Google Scholar]
- 11. Flaugnatti N, Le TTH, Canaan S, Aschtgen M‐S, Nguyen VS, Blangy S, Kellenberger C, Roussel A, Cambillau C, Cascales E et al (2016) A phospholipase A 1 antibacterial Type VI secretion effector interacts directly with the C‐terminal domain of the VgrG spike protein for delivery. Mol Microbiol 99: 1099–1118 [DOI] [PubMed] [Google Scholar]
- 12. Silverman J, Agnello D, Zheng H, Andrews B, Li M, Catalano C, Gonen T, Mougous J (2013) Haemolysin coregulated protein is an exported receptor and chaperone of type VI secretion substrates. Mol Cell 51: 584–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liang X, Moore R, Wilton M, Wong MJQ, Lam L, Dong TG (2015) Identification of divergent type VI secretion effectors using a conserved chaperone domain. Proc Natl Acad Sci USA 112: 9106–9111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Unterweger D, Kostiuk B, Otjengerdes R, Wilton A, Diaz‐Satizabal L, Pukatzki S (2015) Chimeric adaptor proteins translocate diverse type VI secretion system effectors in Vibrio cholerae . EMBO J 34: 2198–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alcoforado Diniz J, Coulthurst SJ (2015) Intraspecies competition in serratia marcescens is mediated by type VI‐secreted Rhs effectors and a conserved effector‐associated accessory protein. J Bacteriol 197: 2350–2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bondage DD, Lin J‐S, Ma L‐S, Kuo C‐H, Lai E‐M (2016) VgrG C terminus confers the type VI effector transport specificity and is required for binding with PAAR and adaptor–effector complex. Proc Natl Acad Sci USA 113: E3931–E3940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dong TG, Ho BT, Yoder‐Himes DR, Mekalanos JJ (2013) Identification of T6SS‐dependent effector and immunity proteins by Tn‐seq in Vibrio cholerae . Proc Natl Acad Sci USA 110: 2623–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Russell AB, LeRoux M, Hathazi K, Agnello DM, Ishikawa T, Wiggins PA, Wai SN, Mougous JD (2013) Diverse type VI secretion phospholipases are functionally plastic antibacterial effectors. Nature 496: 508–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ma LS, Hachani A, Lin JS, Filloux A, Lai EM (2014) Agrobacterium tumefaciens deploys a superfamily of type VI secretion DNase effectors as weapons for interbacterial competition in planta. Cell Host Microbe 16: 94–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Russell AB, Singh P, Brittnacher M, Bui NK, Hood RD, Carl MA, Agnello DM, Schwarz S, Goodlett DR, Vollmer W et al (2012) A widespread bacterial type VI secretion effector superfamily identified using a heuristic approach. Cell Host Microbe 11: 538–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miyata ST, Unterweger D, Rudko SP, Pukatzki S (2013) Dual expression profile of type VI secretion system immunity genes protects pandemic Vibrio cholerae . PLoS Pathog 9: e1003752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Whitney JC, Quentin D, Sawai S, LeRoux M, Harding BN, Ledvina HE, Tran BQ, Robinson H, Goo YA, Goodlett DR et al (2015) An interbacterial NAD(P)+ glycohydrolase toxin requires elongation factor Tu for delivery to target cells. Cell 163: 607–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Russell AB, Hood RD, Bui NK, LeRoux M, Vollmer W, Mougous JD (2011) Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475: 343–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hachani A, Wood TE, Filloux A (2016) Type VI secretion and anti‐host effectors. Curr Opin Microbiol 29: 81–93 [DOI] [PubMed] [Google Scholar]
- 25. Suarez G, Sierra JC, Erova TE, Sha J, Horneman AJ, Chopra AK (2010) A type VI secretion system effector protein, VgrG1, from Aeromonas hydrophila that induces host cell toxicity by ADP ribosylation of actin. J Bacteriol 192: 155–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miyata ST, Kitaoka M, Brooks TM, McAuley SB, Pukatzki S (2011) Vibrio cholerae requires the type VI secretion system virulence factor vasx to kill dictyostelium discoideum. Infect Immun 79: 2941–2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang F, Waterfield NR, Yang J, Yang G, Jin Q (2014) A Pseudomonas aeruginosa type VI secretion phospholipase D effector targets both prokaryotic and eukaryotic cells. Cell Host Microbe 15: 600–610 [DOI] [PubMed] [Google Scholar]
- 28. Aubert DF, Xu H, Yang J, Shi X, Gao W, Li L, Bisaro F, Chen S, Valvano MA, Shao F (2016) A Burkholderia type VI effector deamidates Rho GTPases to activate the pyrin inflammasome and trigger inflammation. Cell Host Microbe 19: 664–674 [DOI] [PubMed] [Google Scholar]
- 29. Chen H, Yang D, Han F, Tan J, Zhang L, Xiao J, Zhang Y (2017) The bacterial T6SS effector EvpP prevents NLRP3 inflammasome activation by inhibiting the Ca2+ – dependent MAPK‐Jnk pathway article the bacterial T6SS effector EvpP prevents NLRP3 inflammasome activation by inhibiting. Cell Host Microbe 21: 47–58 [DOI] [PubMed] [Google Scholar]
- 30. Eshraghi A, Kim J, Walls AC, Ledvina HE, Miller CN, Ramsey KM, Whitney JC, Radey MC, Peterson SB, Ruhland BR et al (2016) Secreted effectors encoded within and outside of the Francisella Pathogenicity Island promote intramacrophage growth. Cell Host Microbe 20: 573–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Salomon D, Kinch LN, Trudgian DC, Guo X, Klimko JA, Grishin NV, Mirzaei H, Orth K (2014) Marker for type VI secretion system effectors. Proc Natl Acad Sci USA 111: 9271–9276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Salomon D, Klimko JA, Trudgian DC, Kinch LN, Grishin NV, Mirzaei H, Orth K (2015) Type VI secretion system toxins horizontally shared between marine bacteria. PLoS Pathog 11: e1005128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Merkel JR, Traganza ED, Mukherjee BB, Griffin TB, Prescott JM (1964) Proteolytic activity and general characteristics of a marine bacterium, proteolytic activity and general characteristics of a marine. J Bacteriol 87: 1227–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cervino JM, Thompson FL, Gomez‐Gil B, Lorence EA, Goreau TJ, Hayes RL, Winiarski‐Cervino KB, Smith GW, Hughen K, Bartels E (2008) The Vibrio core group induces yellow band disease in Caribbean and Indo‐Pacific reef‐building corals. J Appl Microbiol 105: 1658–1671 [DOI] [PubMed] [Google Scholar]
- 35. Ray A, Kinch LN, de Souza Santos M, Grishin NV, Orth K, Salomon D (2016) Proteomics analysis reveals previously uncharacterized virulence factors in Vibrio proteolyticus . MBio 7: e01077–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boyer F, Fichant G, Berthod J, Vandenbrouck Y, Attree I (2009) Dissecting the bacterial type VI secretion system by a genome wide in silico analysis: what can be learned from available microbial genomic resources? BMC Genom 10: 104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Salomon D, Gonzalez H, Updegraff BL, Orth K (2013) Vibrio parahaemolyticus Type VI secretion system 1 is activated in marine conditions to target bacteria, and is differentially regulated from system 2. PLoS One 8: e61086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hildebrand A, Remmert M, Biegert A, Söding J (2009) Fast and accurate automatic structure prediction with HHpred. Proteins 77: 128–132 [DOI] [PubMed] [Google Scholar]
- 39. Brunet YR, Zoued A, Boyer F, Douzi B, Cascales E (2015) The type VI secretion TssEFGK‐VgrG phage‐like baseplate is recruited to the TssJLM membrane complex via multiple contacts and serves as assembly platform for tail tube/sheath polymerization. PLoS Genet 11: e1005545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lemonnier M, Landraud L, Lemichez E (2007) Rho GTPase‐activating bacterial toxins: from bacterial virulence regulation to eukaryotic cell biology. FEMS Microbiol Rev 31: 515–534 [DOI] [PubMed] [Google Scholar]
- 41. Aktories K (2015) Rho‐modifying bacterial protein toxins. Pathog Dis 73: ftv091 [DOI] [PubMed] [Google Scholar]
- 42. Koskiniemi S, Lamoureux JG, Nikolakakis KC, t'Kint de Roodenbeke C, Kaplan MD, Low DA, Hayes CS (2013) Rhs proteins from diverse bacteria mediate intercellular competition. Proc Natl Acad Sci USA 110: 7032–7037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brown SE, Cao AT, Dobson P, Hines ER, Akhurst RJ, East PD (2006) Txp40, a ubiquitous insecticidal toxin protein from xenorhabdus and photorhabdus bacteria. Appl Environ Microbiol 72: 1653–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu L, Hao S, Lan R, Wang G, Xiao D, Sun H, Xu J (2015) The type VI secretion system modulates flagellar gene expression and secretion in Citrobacter freundii and contributes to adhesion and cytotoxicity to host cells. Infect Immun 83: 2596–2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang L, Xu J, Xu J, Zhang H, He L, Feng J (2014) TssB is essential for virulence and required for type VI secretion system in Ralstonia solanacearum . Microb Pathog 74: 1–7 [DOI] [PubMed] [Google Scholar]
- 46. Shalom G, Shaw JG, Thomas MS (2007) In vivo expression technology identifies a type VI secretion system locus in Burkholderia pseudomallei that is induced upon invasion of macrophages. Microbiology 153: 2689–2699 [DOI] [PubMed] [Google Scholar]
- 47. Park K‐S, Ono T, Rokuda M, Jang M‐H, Okada K, Iida T, Honda T (2004) Functional characterization of two type III secretion systems of Vibrio parahaemolyticus . Infect Immun 72: 6659–6665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mogensen TH (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22: 240–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schweer J, Kulkarni D, Kochut A, Pezoldt J, Pisano F, Pils MC, Genth H, Huehn J, Dersch P (2013) The cytotoxic necrotizing factor of Yersinia pseudotuberculosis (CNF Y) enhances inflammation and Yop delivery during infection by activation of rho GTPases. PLoS Pathog 9: e1003746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Salomon D, Sessa G (2010) Identification of growth inhibition phenotypes induced by expression of bacterial type III effectors in yeast. J Vis Exp 37: 4–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Salomon D, Guo Y, Kinch LN, Grishin NV, Gardner KH, Orth K (2013) Effectors of animal and plant pathogens use a common domain to bind host phosphoinositides. Nat Commun 4: 2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. O'Toole R, Milton DL, Wolf‐Watz H (1996) Chemotactic motility is required for invasion of the host by the fish pathogen Vibrio anguillarum . Mol Microbiol 19: 625–637 [DOI] [PubMed] [Google Scholar]
- 53. Bensadoun A, Weinstein D (1976) Assay of proteins in the presence of interfering materials. Anal Biochem 70: 241–250 [DOI] [PubMed] [Google Scholar]
- 54. Agatep R, Kirkpatrick RD, Parchaliuk DL, Woods RA, Gietz RD (1998) Transformation of Saccharomyces cerevisiae by the lithium acetate/single‐stranded carrier DNA/polyethylene glycol protocol. Tech Tips Online 3: 133–137 [Google Scholar]
- 55. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410 [DOI] [PubMed] [Google Scholar]
- 56. Marchler‐Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR et al (2017) CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res 45: D200–D203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8: 785–786 [DOI] [PubMed] [Google Scholar]
- 58. Moretti S, Armougom F, Wallace IM, Higgins DG, Jongeneel CV, Notredame C (2007) The M‐Coffee web server: a meta‐method for computing multiple sequence alignments by combining alternative alignment methods. Nucleic Acids Res 35: W645–W648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Trudgian DC, Thomas B, McGowan SJ, Kessler BM, Salek M, Acuto O (2010) CPFP: a central proteomics facilities pipeline. Bioinformatics 26: 1131–1132 [DOI] [PubMed] [Google Scholar]
- 60. Trudgian DC, Mirzaei H (2012) Cloud CPFP: a shotgun proteomics data analysis pipeline using cloud and high performance computing. J Proteome Res 11: 6282–6290 [DOI] [PubMed] [Google Scholar]
- 61. Craig R, Beavis RC (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20: 1466–1467 [DOI] [PubMed] [Google Scholar]
- 62. Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH (2004) Open mass spectrometry search algorithm. J Proteome Res 3: 958–964 [DOI] [PubMed] [Google Scholar]
- 63. Trudgian DC, Ridlova G, Fischer R, Mackeen MM, Ternette N, Acuto O, Kessler BM, Thomas B (2011) Comparative evaluation of label‐free SINQ normalized spectral index quantitation in the central proteomics facilities pipeline. Proteomics 11: 2790–2797 [DOI] [PubMed] [Google Scholar]
- 64. Elias JE, Gygi SP (2007) Target‐decoy search strategy for increased confidence in large‐scale protein identifications by mass spectrometry. Nat Methods 4: 207–214 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 3D
Data Availability Statement
The data supporting the findings of this study are available within the paper and its supplementary information. The raw mass spectrometry data files and the corresponding peak lists were uploaded to the MassIVE dataset repository (https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=fa38fde1dc3e43e3abc675600e588838) and are available under MassIVE ID: MSV000080535.