

Abstract
Single‐stranded DNA (ssDNA) regions form as an intermediate in many DNA‐associated transactions. Multiple cellular proteins interact with ssDNA via the oligonucleotide/oligosaccharide‐binding (OB) fold domain. The heterotrimeric, multi‐OB fold domain‐containing Replication Protein A (RPA) complex has an essential genome maintenance role, protecting ssDNA regions from nucleolytic degradation and providing a recruitment platform for proteins involved in responses to replication stress and DNA damage. Here, we identify the uncharacterized protein RADX (CXorf57) as an ssDNA‐binding factor in human cells. RADX binds ssDNA via an N‐terminal OB fold cluster, which mediates its recruitment to sites of replication stress. Deregulation of RADX expression and ssDNA binding leads to enhanced replication fork stalling and degradation, and we provide evidence that a balanced interplay between RADX and RPA ssDNA‐binding activities is critical for avoiding these defects. Our findings establish RADX as an important component of cellular pathways that promote DNA replication integrity under basal and stressful conditions by means of multiple ssDNA‐binding proteins.
Keywords: DNA replication, genome integrity, replication protein A, replication stress, single‐stranded DNA
Subject Categories: DNA Replication, Repair & Recombination
Introduction
Accurate DNA replication during each cell cycle is vital for faithful transmission of genetic information and for cell and organism fitness 1, 2. However, DNA replication fidelity is challenged by endogenous and exogenous genotoxic sources that jeopardize the integrity and progression of the replication machinery. The slowing or stalling of replication forks, a deleterious condition generally referred to as replication stress, is a major driver of genomic instability that may lead to cancer and other severe pathologies 3, 4, 5. The ATR kinase is a master organizer of the response to replication stress, phosphorylating numerous effector proteins to set in motion a multipronged protective response to such insults that prevents the detrimental breakdown of replication forks 6, 7, 8. Extended stretches of single‐stranded DNA (ssDNA), generated by uncoupling of replicative DNA helicase and polymerase activities upon insults that hinder polymerase progression, serve as a central trigger of replication stress responses 3, 9. Exposed ssDNA regions are rapidly coated by the ssDNA‐binding heterotrimeric Replication Protein A (RPA) complex, promoting recruitment of ATR via its obligate RPA‐binding partner ATRIP and subsequent stimulation of its kinase activity by activators including TopBP1 and ETAA1 10, 11, 12, 13, 14. RPA has strong, sub‐nanomolar ssDNA‐binding affinity by virtue of four oligonucleotide/oligosaccharide‐binding (OB) fold domains, three of which are found in RPA1, the largest subunit within the RPA complex 15, 16. RPA has essential functions during both normal DNA replication and responses to genotoxic stress, shielding naked ssDNA regions from nucleolytic processing and providing a recruitment platform for numerous RPA‐binding effector proteins in DNA replication and DNA damage responses 16.
It is becoming increasingly clear that a major function of the ATR‐dependent response to replication stress is to prevent irreversible fork collapse by limiting global origin firing and, consequently, the overall amount of ssDNA generated 8. While RPA is in considerable excess of the amount needed to globally coat ssDNA exposed during unperturbed replication, unscheduled ssDNA generation induced by DNA replication inhibitors such as hydroxyurea (HU) can effectively exhaust the cellular RPA pool, leaving some replication forks unprotected and susceptible to degradation by nucleases 17. Inhibiting ATR kinase activity exacerbates such irreversible replication “catastrophe” by disabling the suppression of new origin firing. The notion that RPA is rate‐limiting for fork stability during replication stress suggests that its availability and dynamic interaction with, and turnover from, ssDNA must be carefully controlled in accordance with the status of the genome to ensure faithful DNA replication and chromosomal stability; however, the underlying mechanisms are not well understood.
Besides RPA, eukaryotic cells encode a range of other OB fold‐containing ssDNA‐binding factors with important roles in DNA replication and genome maintenance pathways 18. These include SSB1/NABP2, which facilitates DNA double‐strand break signaling and repair, and POT1 and TPP1, components of the telomere‐protecting shelterin complex 18. Another illustrative example is BRCA2, an essential mediator of homologous recombination that promotes the exchange of ssDNA‐bound RPA with the recombinase RAD51 19. Recent studies revealed that BRCA2 and RAD51 also have key roles in protecting nascent DNA at stalled replication forks from degradation by the MRE11 nuclease 20, 21, 22. Thus, it transpires that a complex, and in all likelihood highly regulated, interplay between ssDNA and manifold ssDNA‐binding proteins operates to safeguard replication fork stability and genetic integrity in different chromosomal contexts and in response to a wide range of perturbations.
In this study, we discovered that the uncharacterized human protein RADX (CXorf57) is a hitherto unrecognized OB fold domain‐containing factor that interacts with ssDNA regions at DNA replication sites. Through its OB fold‐dependent ssDNA‐binding ability and a functional interplay with RPA, RADX plays an important role in supporting the stability of replication forks during normal S phase and in response to replication stress. Our findings establish RADX as a new member of cellular ssDNA‐binding factors promoting DNA replication integrity in human cells.
Results and Discussion
RADX (CXorf57) is an ssDNA‐binding protein
In bioinformatic screens for prospective new genome stability maintenance factors, we noted that the uncharacterized human protein CXorf57 contains three potential N‐terminal OB folds, a domain organization similar to that of the RPA1 subunit within the RPA complex (Fig 1A) 18. Human CXorf57 has been recently found to be among proteins showing enrichment at stalled replication forks and in RPA pull‐downs 23, 24, and the CXorf57 locus is a common integration site for the B‐cell lymphoma‐inducing avian leukosis virus 25. These observations suggested that CXorf57, which we refer to here as RADX, might have a role in DNA replication and/or genome stability maintenance pathways, and we therefore explored its cellular function. In silico modeling of RADX structure by the Phyre2 protein modeling suite 26 predicted high‐confidence similarity of the region comprising OB folds 2 and 3 in RADX to OB folds within RPA1 and other proteins that bind ssDNA but not RNA (Figs 1A and B, and EV1A). We therefore asked whether RADX is a DNA‐binding protein. Using immobilized biotin‐labeled DNA oligos, we found that stably expressed wild‐type GFP‐RADX was efficiently retrieved in ssDNA pull‐downs like RPA1 (Fig 1C). Deletion of the entire OB fold region (ΔOB) abolished the ssDNA‐binding activity of RADX (Fig 1A and C). Importantly, specific point mutations within the RADX OB2 domain (*OB) predicted to diminish its ssDNA‐binding ability based on alignment with mutations in the DBD‐A OB fold of RPA1 (K263A/E277A) that cause a 100‐fold reduction in ssDNA‐binding affinity 27 substantially reduced RADX interaction with ssDNA (Figs 1A and C, and EV1A). RADX bound ssDNA with high affinity, comparable to that of RPA (Fig 1D). Moreover, endogenous RADX interacted with ssDNA but not double‐stranded DNA (dsDNA) (Fig 1E). These findings suggest that the association between RADX and ssDNA is direct and mediated by the OB fold region. Interestingly, RADX expression was not cell cycle‐regulated but varied substantially among human cell lines, in a manner correlating with its mRNA levels (Figs 1F and EV1B–D).
Figure 1. RADX (CXorf57) is an ssDNA‐binding protein.
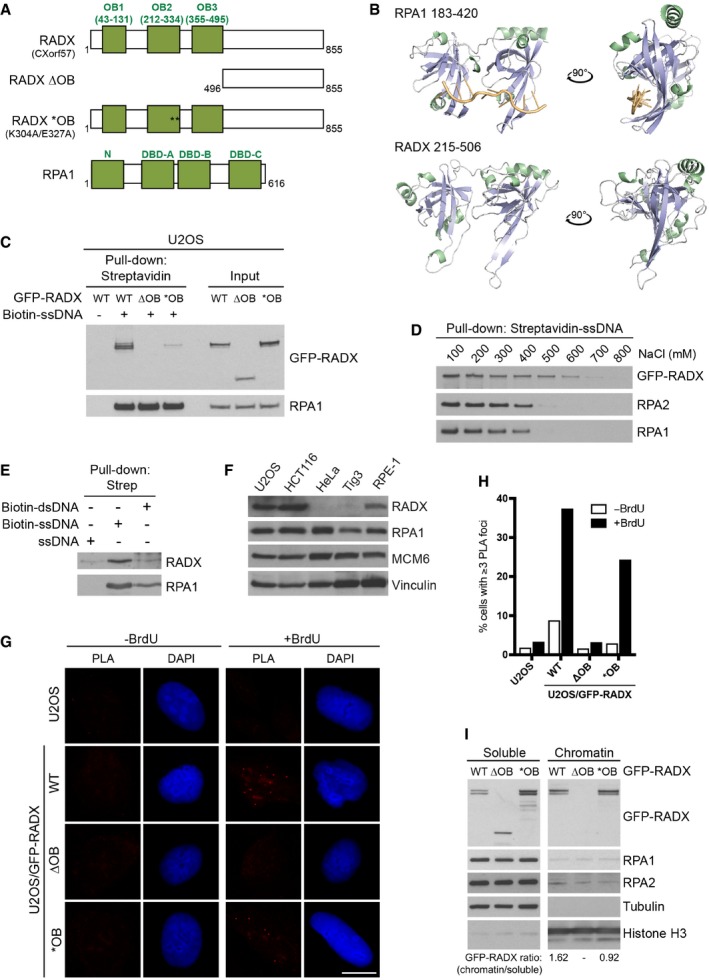
- Domain organization of human RADX and RPA1, showing location of OB folds, including three DNA‐binding domains (DBDs) in RPA1, and RADX mutants used in this study (see also Fig EV1A).
- Predicted in silico folding of OB folds 2 and 3 of human RADX, modeled by Phyre2, showing similarity to the structure of the region encompassing DBD‐A and DBD‐B in human RPA1 (Fig 1A).
- RADX interacts with ssDNA. Extracts of U2OS cells stably expressing GFP‐RADX WT or mutants were incubated with biotin‐coupled ssDNA oligo immobilized on streptavidin beads, washed extensively, and immunoblotted with GFP and RPA1 antibodies.
- ssDNA‐bound streptavidin beads incubated with cell extracts as in (C) were washed with buffer containing indicated salt concentrations prior to immunoblotting.
- Lysates of untransfected HCT116 cells were incubated with immobilized ssDNA or dsDNA probes as in (C) and immunoblotted with RADX and RPA1 antibodies.
- Immunoblot analysis of RADX expression in human cell lines. See also Fig EV1B.
- Representative images from in situ proximity ligation assays (PLAs) in BrdU‐labeled U2OS and U2OS/GFP‐RADX cell lines (Fig EV1E and F) using GFP and BrdU antibodies under native conditions. Scale bar, 10 μm.
- Quantification of data in (G) by quantitative image‐based cytometry (QIBC) (n ≥ 3,000 cells per condition; data from a representative experiment are shown). See also Fig EV1G.
- Soluble and chromatin‐enriched fractions of U2OS cell lines stably expressing GFP‐RADX alleles were immunoblotted with indicated antibodies.
Figure EV1. RADX expression, localization, and alignment with RPA1.
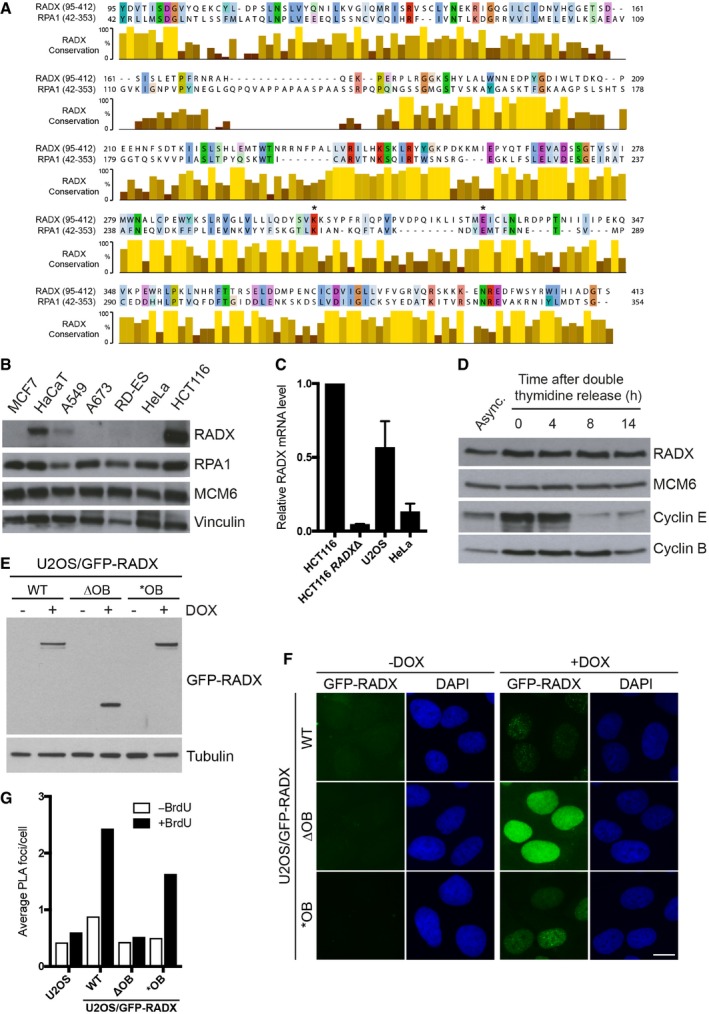
- Alignment of the regions comprising OB‐folds 2 and 3 in human RADX and the ssDNA‐binding DBD‐A and DBD‐B OB‐folds in human RPA1 (Fig 1A). The positions of the K263 and E277 residues in RPA1 and the corresponding K304 and E327 residues in RADX, mutated in RADX *OB, are indicated by asterisks.
- Immunoblot analysis of RADX expression in the indicated human cell lines.
- qPCR analysis of RADX mRNA levels in indicated cell lines relative to HCT116 WT cells. Primers to ubiquitin were used as a control for normalization (mean ± SEM; n = 3 independent experiments).
- U2OS cells were synchronized in early S phase by double thymidine block, released into fresh medium for the indicated times, and analyzed by immunoblotting with indicated antibodies.
- U2OS cell lines stably reconstituted with GFP‐RADX alleles were treated or not with doxycycline (DOX) to induce the transgenes and immunoblotted with GFP and tubulin antibodies.
- Cells in (E) were fixed with paraformaldehyde and analyzed by microscopy to visualize GFP‐RADX localization. Scale bar, 10 μm.
- Quantification of PLAs (Fig 1G) in BrdU‐labeled U2OS and U2OS/GFP‐RADX cell lines using GFP and BrdU antibodies under native conditions (n ≥ 3,000 cells per condition; data from a representative experiment are shown).
To test whether RADX interacts with ssDNA in cells, we performed proximity ligation assays (PLAs) 28 using BrdU‐labeled cell lines stably expressing wild‐type (WT) or mutant forms of GFP‐RADX at similar levels (Fig EV1E and F) to probe for proximity between the transgenes and BrdU under native conditions, where the BrdU epitope is exclusively accessible in ssDNA 29. Consistent with RADX associating with ssDNA regions via the OB fold domain, we observed specific PLA signals in nuclear foci between BrdU and GFP‐RADX WT, but not GFP‐RADX ΔOB, in a subset of cells, while a smaller proportion of cells expressing GFP‐RADX *OB displayed PLA foci (Figs 1G and H, and EV1G). Likewise, RADX WT but not RADX ΔOB associated with chromatin, whereas the *OB mutant showed reduced chromatin binding, as judged by its relative distribution between chromatin‐enriched and soluble fractions (Fig 1I).
OB fold‐dependent recruitment of RADX to genotoxic stress sites
Given the ssDNA‐binding ability of RADX, we asked whether it is recruited to sites of replication stress, which typically harbor extended ssDNA regions. While endogenous RADX was not stably associated with chromatin in unperturbed U2OS cells, it could be detected in chromatin fractions upon hydroxyurea (HU)‐induced replication fork stalling, albeit primarily upon prolonged replication stress that induces extensive ssDNA formation (Figs 2A and B, and EV2A). In the presence of ATR inhibitor, which dramatically increases ssDNA generation and ultimately causes DNA breakage upon HU‐induced replication stress 17, RADX chromatin accumulation was accelerated along with RPA (Figs 2B and EV2A). Stably expressed GFP‐RADX predominantly localized to the nucleus, displaying a pronounced puncta‐like pattern in unstressed cells that required the OB region but did not co‐localize with replication foci or a range of other nuclear compartments (Fig EV2B and C; our unpublished observations). However, we found that WT RADX rapidly accumulated at sites of microlaser irradiation‐inflicted DNA damage in a subset of cells, co‐localizing with γ‐H2AX and PCNA, while the ΔOB mutant showed no detectable recruitment (Fig 2C and D). Consistent with its residual ssDNA‐binding affinity, RADX *OB also accumulated at DNA damage sites (Fig 2C). To probe for recruitment of RADX to ssDNA regions upon replication stress, we used PLAs to assay for GFP‐RADX proximity to RPA, which accumulates strongly at these sites. In line with our earlier findings, GFP‐RADX WT and *OB, but not RADX ΔOB, gave rise to PLA signals with RPA in nuclear foci (Figs 2E and F, and EV2D). Importantly, increasing ssDNA formation by treatment with HU or camptothecin (CPT) markedly enhanced PLA foci specifically in S phase (EdU‐positive) cells (Fig 2E and F). We conclude that RADX is recruited to ssDNA regions in response to replication stress via its OB fold region.
Figure 2. Recruitment of RADX to sites of replication stress.
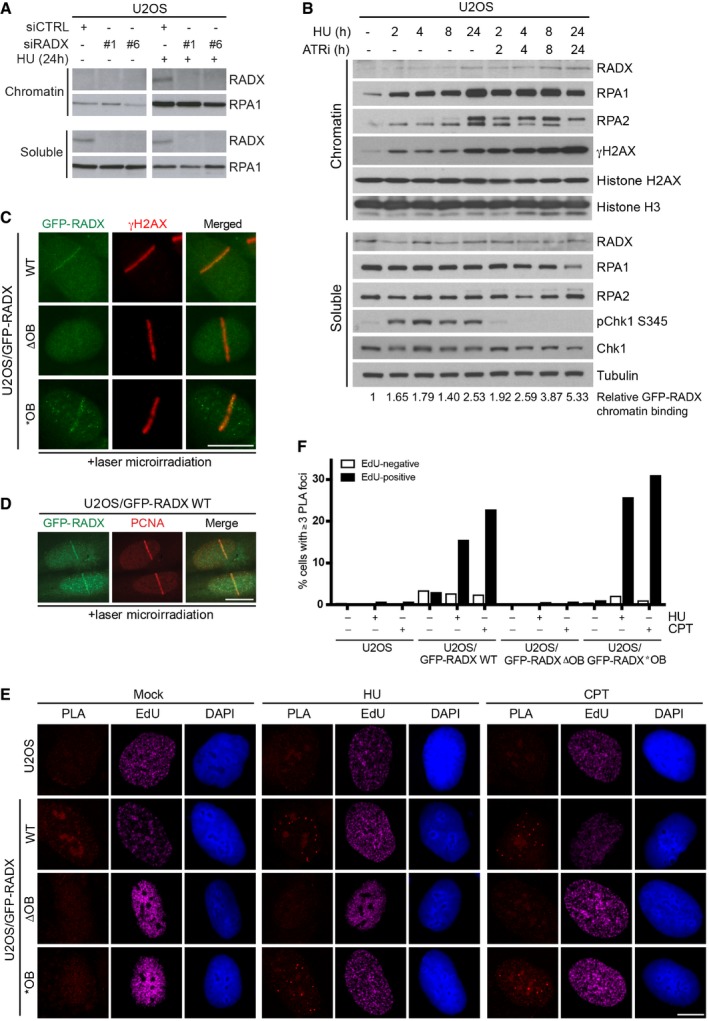
- Immunoblot analysis of chromatin fractions of U2OS cells transfected with indicated siRNAs and treated or not with HU.
- U2OS cells treated with HU in the presence or absence of ATR inhibitor (ATRi) were fractionated and immunoblotted with indicated antibodies. Relative RADX levels on chromatin were quantified and normalized to histone H3. See also Fig EV2A.
- Representative images of U2OS cells stably expressing indicated GFP‐RADX alleles that were subjected to laser microirradiation, fixed with methanol/acetone immediately afterwards, and immunostained with γ‐H2AX antibody.
- As in (C), except cells were fixed with paraformaldehyde 20 min after DNA damage infliction and immunostained with PCNA antibody.
- Representative images from PLAs with GFP and RPA2 antibodies in U2OS and U2OS/GFP‐RADX cell lines labeled with EdU for 30 min and then fixed or exposed to HU or CPT for 4 h before fixation.
- Quantification of data in (E) by QIBC (n ≥ 3,000 cells per condition; data from a representative experiment are shown). See also Fig EV2D.
Figure EV2. RADX recruitment to replication stress sites.
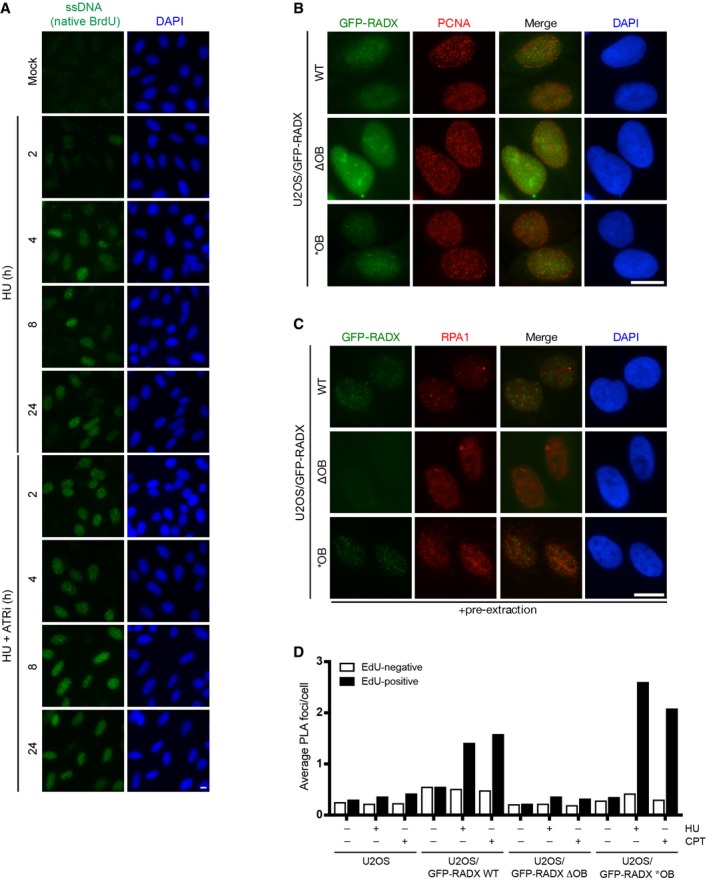
- U2OS cells labeled with BrdU and treated with HU in the presence or absence of ATR inhibitor (ATRi) were fixed and immunostained under native conditions. Representative images are shown.
- U2OS/GFP‐RADX cell lines treated with doxycycline to induce the transgenes were fixed with methanol/acetone and immunostained with PCNA antibody.
- As in (B), except cells were pre‐extracted prior to fixation with paraformaldehyde and immunostained with RPA1 antibody.
- Quantification of PLAs (Fig 2E) with GFP and RPA2 antibodies in U2OS and U2OS/GFP‐RADX cell lines labeled with EdU for 30 min and subsequently left untreated or exposed to HU or CPT for 4 h (n ≥ 3,000 cells per condition; data from a representative experiment are shown).
Loss of RADX deregulates the DNA replication machinery
The findings above suggested that RADX might promote DNA replication integrity. To test this, we performed DNA fiber assays to analyze the impact of RADX loss by siRNAs or CRISPR/Cas9‐based targeted knockout (Fig 3A and B) on the status of individual replication forks. Depletion of RADX in HCT116 cells did not markedly affect overall cell cycle distribution and had little impact on checkpoint signaling in response to replication stress (Fig EV3A and B). However, we found that RADX knockdown by means of different siRNAs markedly reduced fork elongation rates in otherwise unstressed cells (Fig 3C). This was accompanied by a substantial decrease in the symmetry of bidirectional replication forks (Fig 3D), suggesting that the reduced fork speeds in RADX‐depleted cells are due, at least in part, to an increased rate of fork pausing and stalling events. Indeed, the proportion of stalled forks was more than doubled in RADX knockdown cells (Fig 3E). Further demonstrating marked deregulation of normal DNA replication integrity, RADX depletion caused a robust increase in new origin usage (Fig 3F), possibly a compensatory effect for the diminished replication fork speeds and excessive fork stalling resulting from RADX loss 30. Importantly, the strong impact of RADX siRNAs on replication fork status was closely mirrored by CRISPR/Cas9‐mediated RADX knockout (RADXΔ) (Fig 3A–F), suggesting it was a specific consequence of RADX loss. Moreover, while siRNA‐mediated RADX depletion in U2OS cells essentially mirrored the effects seen in HCT116 cells, both of which express RADX at high levels, it only mildly reduced fork elongation rates in HeLa cells where RADX expression is low (Figs 1F and EV3C–F).
Figure 3. RADX promotes DNA replication integrity.
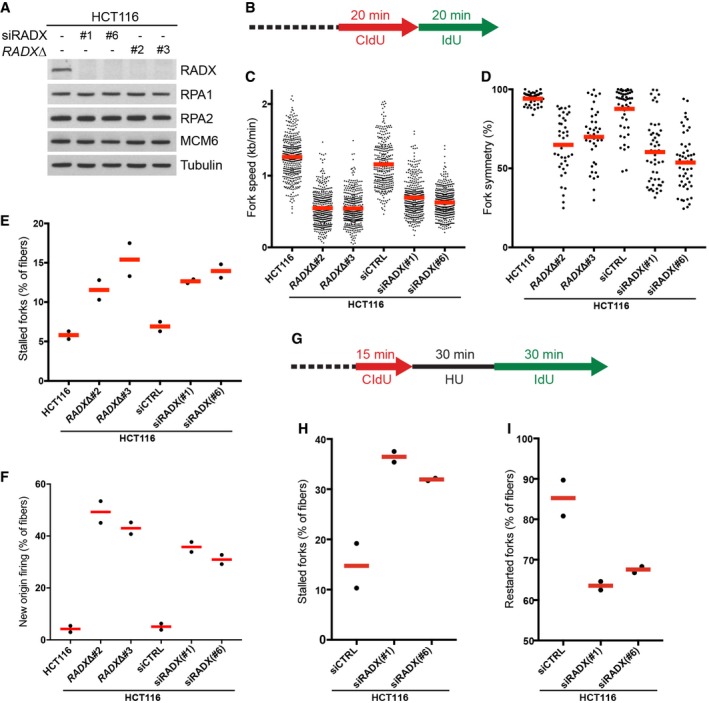
- Immunoblot analysis of HCT116 WT cells transfected with indicated RADX siRNAs and HCT116 RADX knockout cell lines (RADXΔ).
- Schematic for DNA fiber assays in (C–E).
- Cells in (A) were labeled with consecutive pulses of CldU and IdU as shown in (B). Replication fork speeds were calculated as length of labeled track divided by pulse time (bars, mean; n = 400 fibers, pooled from two independent experiments, per condition).
- Bidirectional replication fork symmetry was calculated as percentage of shorter divided by longer tracks from (C). Concordance is 100%, representing equal rates of bidirectional elongation for both daughter forks (bars, mean; n = 50 bidirectional forks, pooled from two independent experiments, per condition).
- Proportion of stalled forks (CldU‐only tracks) among DNA fibers from (C) (bars, mean; 200 fibers analyzed per condition; n = 2 independent experiments).
- Proportion of new origins (IdU‐only tracks) among DNA fibers in (C) (bars, mean; 20 fields of view quantified per condition; n = 2 independent experiments).
- Schematic for DNA fiber assays in (H) and (I).
- Proportion of stalled forks (CldU‐only tracks) among DNA fibers from cells labeled as in (G) (bars, mean; 200 fibers analyzed per condition; n = 2 independent experiments).
- Proportion of restarted forks (CldU‐ and IdU‐positive tracks) among DNA fibers in (G) (bars, mean; 200 fibers analyzed per condition; n = 2 independent experiments).
Figure EV3. Impact of RADX loss on cell cycle distribution, checkpoint signaling, and replication fork integrity.

- Quantitative image analysis of asynchronously growing HCT116 WT cells transfected with indicated RADX siRNAs (left) and HCT116 RADX knockout cell lines (RADXΔ) (right) labeled with EdU and stained with DAPI (n ≥ 3,000 cells per condition). Proportion of cells in different cell cycle phases is indicated.
- HCT116 WT or RADXΔ cells were treated with HU or CPT for the indicated times, collected, and immunoblotted with indicated antibodies.
- U2OS cells transfected with indicated siRNAs were labeled with consecutive pulses of CldU and IdU as shown in Fig 3B. Replication fork speeds were calculated as length of labeled track divided by pulse time (bars, mean; n = 200 fibers, pooled from two independent experiments, per condition).
- Fork symmetry was calculated as the percentage of shorter divided by longer tracks from (C). Concordance is 100%, representing fully bidirectional replication and equal rates of elongation for both daughter forks (bars, mean; n = 50 bidirectional forks, pooled from two independent experiments, per condition).
- Proportion of new origins in U2OS cells treated as in (C) was determined (bars, mean; 20 fields of view quantified per condition; n = 2 independent experiments).
- HeLa cells transfected with control (CTRL) or RADX siRNA were labeled with consecutive pulses of CldU and IdU as shown in Fig 3B. Replication fork speeds were calculated as length of labeled track divided by pulse time (bars, mean; n = 400 fibers, pooled from two independent experiments, per condition).
- U2OS cells transfected with indicated siRNAs were labeled with CldU, then incubated with HU, washed, and labeled with IdU as shown in Fig 3G. The proportion of new origins was determined (bars, mean; 20 fields of view quantified per condition; n = 2 independent experiments).
In addition to its impact on DNA replication in unperturbed cells, depletion of RADX led to a defective response to acute replication stress induced by short treatment with HU, evidenced by an increased proportion of stalled replication forks and a concomitant reduction in the rate of replication fork restart (Figs 3G–I and EV3G). Together, these findings demonstrate an important role of RADX in promoting DNA replication integrity under both unchallenged and stressful conditions.
The balance between RADX and RPA ssDNA‐binding activities is critical for DNA replication integrity
To understand how RADX supports DNA replication integrity, we asked whether its ssDNA‐binding ability is needed for this involvement. Indeed, using GFP‐RADX‐expressing cell lines (Fig EV1E and F), we found that the altered fork elongation and origin firing patterns resulting from depletion of endogenous RADX could be fully corrected by GFP‐RADX WT but not RADX ΔOB, while the RADX *OB mutant only modestly ameliorated these defects (Fig 4A–D). The compromised functionality of RADX *OB in supporting DNA replication integrity might alter the dynamics of its association with ssDNA in cells, potentially explaining why this mutant shows enhanced interaction with replication stress sites despite its diminished ssDNA‐binding affinity in vitro (Figs 1C and 2E and F). Increasing the expression of RADX WT, but not the ΔOB and *OB mutants, without concomitant depletion of endogenous RADX markedly reduced fork speeds in unstressed cells (Fig 4E), suggesting that a carefully controlled level of RADX ssDNA‐binding activity is important for DNA replication integrity. One possible underlying mechanism is that RADX has an antagonistic relationship with RPA for interaction with ssDNA regions at DNA replication sites that must be properly balanced to ensure intact fork progression. This hypothesis predicted that like RADX, deregulated expression of RPA might negatively impact normal DNA replication dynamics. Consistent with this idea, in cells where the abundance of the RPA complex was mildly elevated through stable ectopic expression of all three RPA subunits at near‐endogenous levels (“Super‐RPA”) 17, replication fork speeds were reduced to an extent comparable to that seen in cells lacking RADX (Figs 4E and EV4A). Depletion of RADX aggravated the fork progression defect arising from elevated RPA expression (Fig 4E). Mild depletion of RPA to an extent that does not impair overall DNA replication capacity 17 also reduced fork elongation rates, but ameliorated the fork speed, fork symmetry, and origin firing defects in a RADXΔ background (Figs 4F–H and EV4B). Interestingly, knockdown of RAD51 essentially phenocopied these effects (Figs 4F–H and EV4B). These findings suggest that via its affinity for ssDNA, RADX might counteract excessive associations of ssDNA‐binding factors including RPA and RAD51 with replication forks to facilitate proper replisome dynamics. However, using iPOND 23 and other assays, we did not observe pronounced alterations in RPA and RAD51 interactions with replication forks and chromatin upon RADX loss (Fig EV4C and data not shown), suggesting that any competition between RADX and these proteins for ssDNA binding may be highly dynamic in nature.
Figure 4. The balance between RADX and RPA ssDNA‐binding activities is critical for DNA replication integrity.
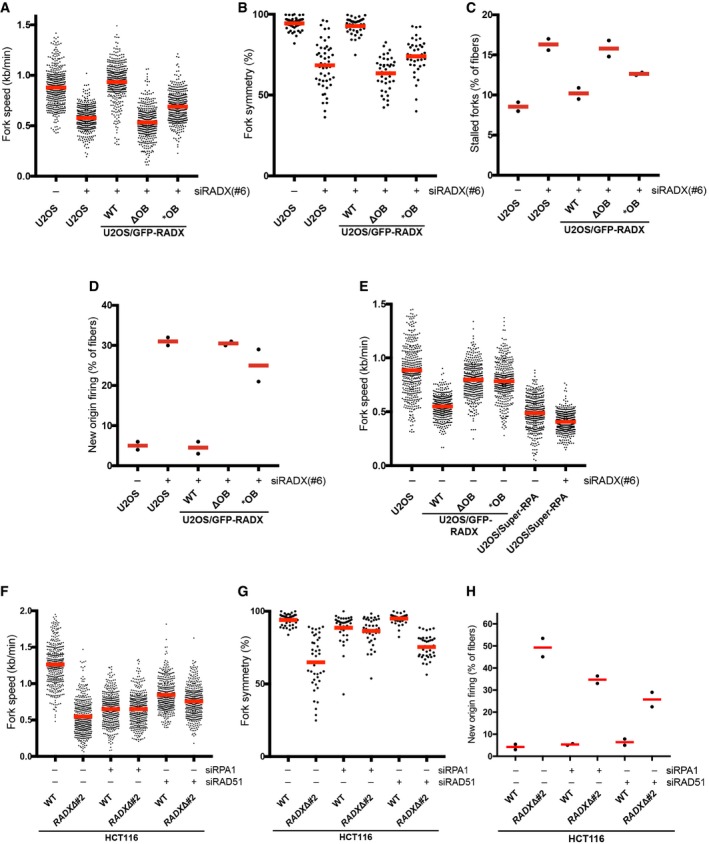
- Replication fork speeds in U2OS or U2OS/GFP‐RADX cells transfected with control (−) or RADX(#6) siRNA targeting the 3′UTR and labeled with CldU and IdU as in Fig 3B (bars, mean; n = 400 fibers, pooled from two independent experiments, per condition).
- Fork symmetry in cells treated as in (A) (bars, mean; n = 50 bidirectional forks, pooled from two independent experiments, per condition).
- Proportion of stalled forks (CldU‐only tracks) among DNA fibers in (A) (bars, mean; 200 fibers analyzed per condition; n = 2 independent experiments).
- Proportion of new origins (IdU‐only tracks) among DNA fibers in (A) (bars, mean; 20 fields of view quantified per condition; n = 2 independent experiments).
- Parental U2OS and derivative lines stably expressing GFP‐RADX alleles or all three RPA isoforms at near‐endogenous levels (U2OS/Super‐RPA) (Fig EV4A) were transfected with control (−) or RADX siRNA. Replication fork speeds were determined as in (A) (bars, mean; n = 400 fibers, pooled from two independent experiments, per condition).
- Fork symmetry among DNA fibers in (F) (bars, mean; n = 50 bidirectional forks, pooled from two independent experiments, per condition).
- New origin firing among DNA fibers in (F) (bars, mean; 20 fields of view quantified per condition; n = 2 independent experiments).
Figure EV4. RPA and RAD51 expression levels and association with replication forks under different experimental conditions.
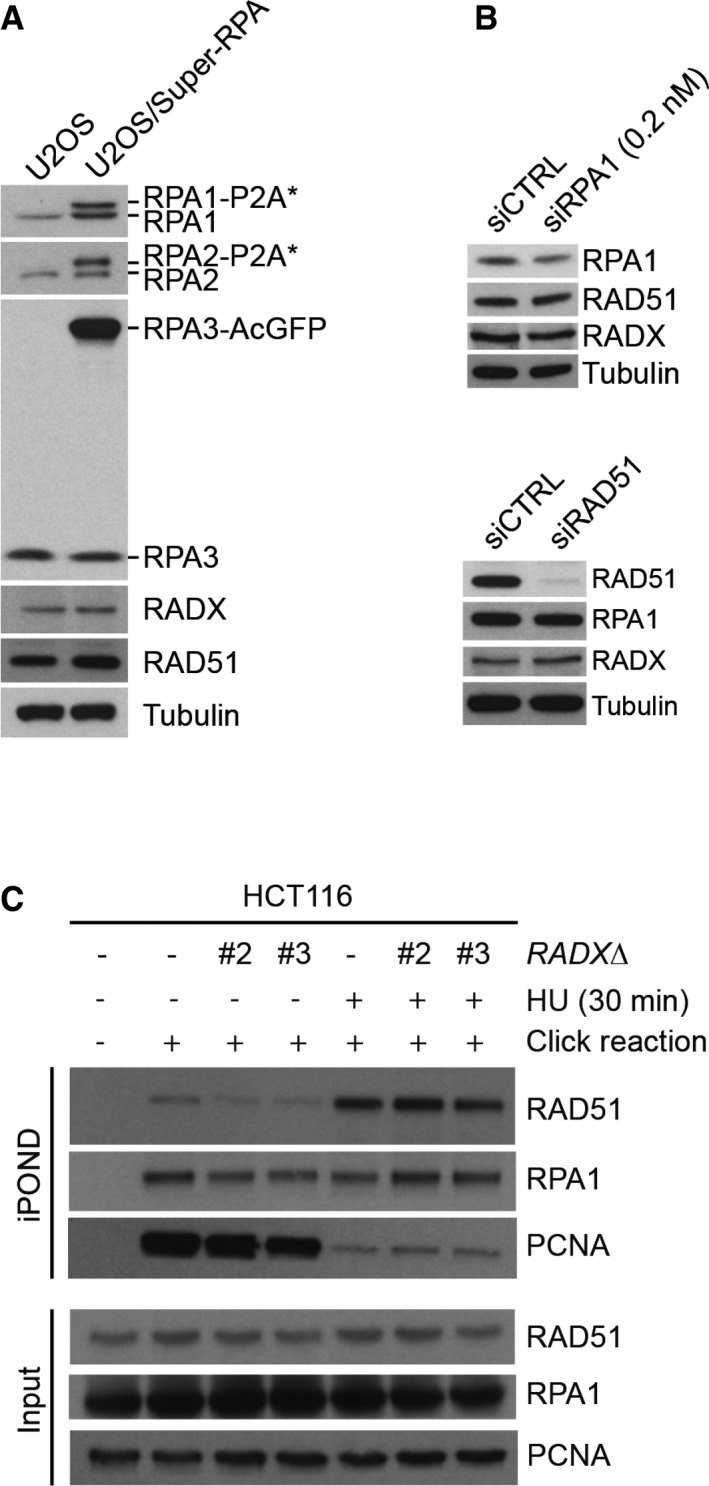
- Immunoblot analysis of total extracts of U2OS/Super‐RPA cells, using indicated antibodies. P2A, 2A peptide.
- Immunoblot analysis of RPA and RAD51 siRNA knockdown efficiency.
- Immunoblot analysis of levels of RAD51, RPA1, and PCNA associated with replication forks isolated by iPOND and in total cell extracts in HCT116 WT and RADXΔ cell lines.
RADX suppresses replication fork degradation and collapse
Replication fork components including BRCA2 and RAD51 have critical roles in preventing degradation of nascent DNA strands at stalled forks 20, 21. To test whether impaired replication fork protection might be an underlying cause of deregulated DNA replication integrity induced by RADX deficiency, we monitored the stability of newly replicated DNA tracts under HU‐induced replication stress (Fig 5A). In these assays, RADX knockdown led to a pronounced degree of fork degradation during prolonged HU treatment (Fig 5B). Overexpression of WT RADX similarly impaired fork protection, while the impact of RADX OB fold mutants was comparatively milder (Fig 5C). This suggests that altered replication fork dynamics resulting from deregulated RADX expression and ssDNA binding may, at least partially, be a consequence of impaired fork protection. Depletion of RADX also led to a markedly elevated rate of HU‐induced replication catastrophe, characterized by full chromatin loading (and thus exhaustion) of the cellular RPA pool and concomitant DNA breakage demarcated by H2AX hyperphosphorylation 17 (Figs 5D and EV5A). This defect was alleviated by expression of RADX WT but not the *OB mutant (Figs 5D and EV5A). Strikingly, elevated levels of RPA also mitigated the replication catastrophe phenotype caused by RADX loss (Fig 5E). At least two non‐mutually exclusive scenarios may account for these observations. First, consistent with an antagonistic relationship between RADX and RPA for ssDNA binding, RADX interaction with ssDNA might enhance RPA mobility at replication forks to increase the effective pool of RPA available to bind and protect extended ssDNA regions generated upon replication stress, thereby rendering cells less prone to fork breakage. Second, RADX may itself have a role in shielding unprotected ssDNA tracts from nuclease‐mediated degradation, supported by the observation that RADX primarily associates with chromatin upon persistent replication stress. In line with these findings, RADX loss caused mild but significant hypersensitivity to replication stress‐inducing agents (Fig 5F and G) similar to the effect of selectively abrogating the fork protection function of BRCA2 21, and this defect could be fully corrected by elevated RPA expression (Fig 5H).
Figure 5. RADX promotes replication fork stability and cell survival upon replication stress.
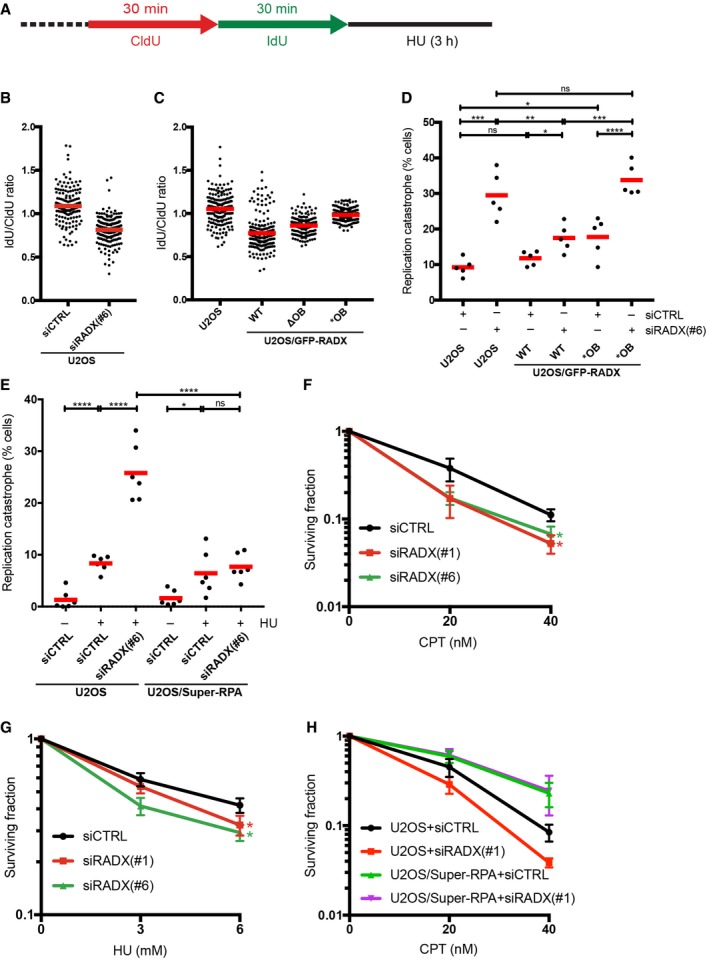
- Schematic for replication fork protection assays in (B) and (C).
- Replication fork degradation (IdU/CldU ratio) in U2OS cells transfected with indicated siRNAs and processed as in (A) (bars, mean; n = 200 fibers, pooled from two independent experiments, per condition).
- As in (B), except fork degradation was analyzed in U2OS/GFP‐RADX cell lines.
- U2OS or U2OS/GFP‐RADX cells transfected with indicated siRNAs and exposed to HU for 4 h were co‐immunostained with RPA1 and γ‐H2AX antibodies and analyzed by QIBC. Proportion of cells displaying maximal RPA chromatin loading accompanied by H2AX hyperphosphorylation (Fig EV5A), reflecting replication catastrophe 17, is indicated (bars, mean; n = 5 independent experiments (≥ 3,000 cells analyzed per condition); *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, unpaired t‐test).
- As in (D), but using U2OS or U2OS/Super‐RPA cells exposed or not to HU (n = 6 independent experiments). See also Fig EV5B.
- Clonogenic survival of U2OS cells transfected with indicated siRNAs and subjected to different CPT doses for 24 h (mean ± SEM; n = 3 independent experiments). LQ (linear quadratic) model was fitted to the fractional survival data, using non‐linear least square method. Overlap between confidence intervals of the fitting coefficients was used to evaluate the statistical difference between cell survival after siRADX and siCTRL treatment (*No overlap of 95% confidence interval with siCTRL).
- As in (F), except cells were treated with indicated doses of HU (mean ± SEM; n = 3 independent experiments).
- As in (F), using U2OS and U2OS/Super‐RPA cells (mean ± SEM; n = 3 independent experiments).
Figure EV5. Impact of altered RADX functionality on HU‐induced replication catastrophe.
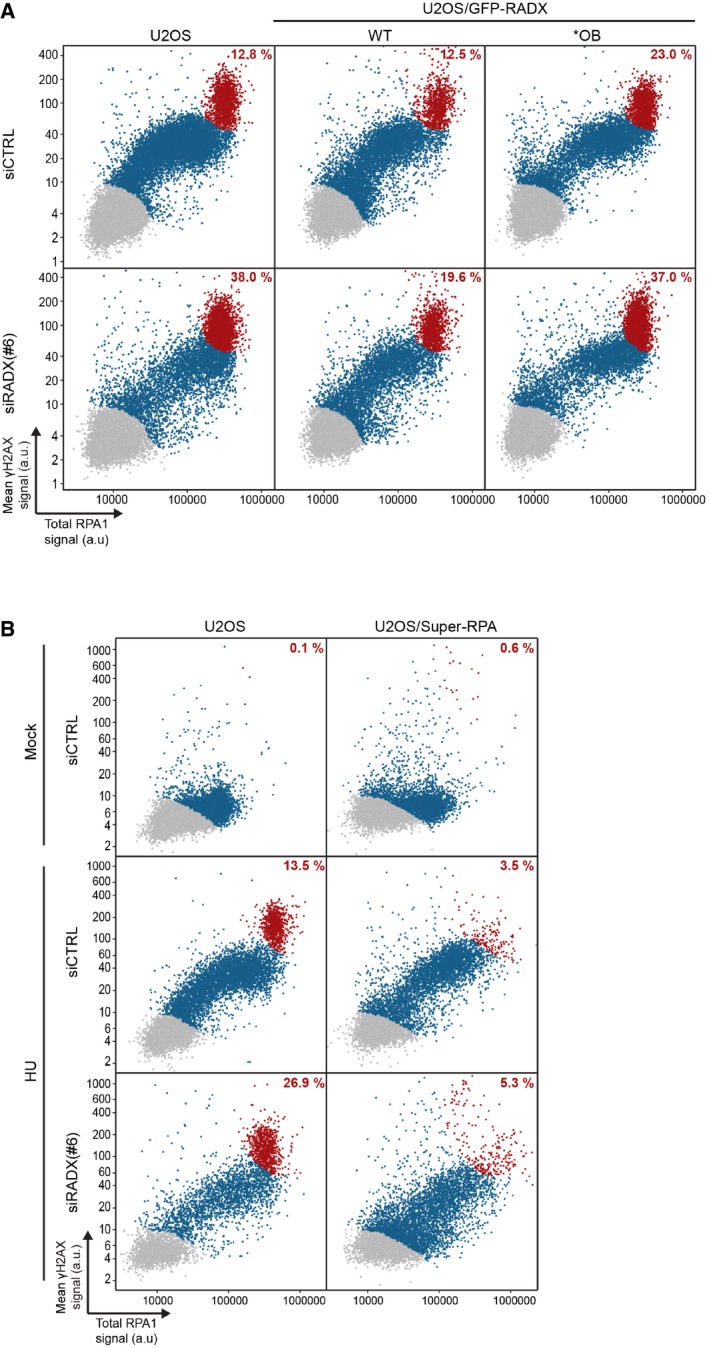
- U2OS or derivative U2OS/GFP‐RADX cell lines transfected with indicated siRNAs and exposed to HU for 4 h were co‐immunostained with RPA1 and γ‐H2AX antibodies and subjected to quantitative image analysis of total RPA1 and mean γ‐H2AX signal intensities in individual cells (each representing a single dot). Cells displaying maximal RPA chromatin loading accompanied by extensive DNA breakage (characterized by H2AX hyperphosphorylation) reflecting irreversible global replication catastrophe are indicated in red. Cells in blue show RPA1 and/or γ‐H2AX signals above background levels. Proportion of cells showing replication catastrophe among all cells displaying RPA1 and/or γ‐H2AX signals above background levels (blue and red) is indicated (n ≥ 3,000 cells per condition; data from a representative experiment are shown). See also Fig 5D.
- As in (A), but using U2OS or U2OS/Super‐RPA cells exposed or not to HU. Proportion of cells displaying replication catastrophe is indicated (n ≥ 3,000 cells per condition; data from a representative experiment are shown). See also Fig 5E.
Collectively, our work establishes RADX as a novel cellular ssDNA‐binding protein with an important role in promoting replication fork stability, adding RADX to a growing list of OB fold domain‐containing factors functioning in DNA‐associated transactions and genome stability maintenance. Similar findings were recently reported by Cortez and colleagues, who demonstrated a role of RADX in antagonizing RAD51 accumulation at replication forks to promote genome integrity 31. Our study is consistent with this work and additionally suggests that the interplay between RADX and RPA is also important for fork stability. The fork protection function of RADX largely correlates with its ssDNA‐binding affinity, which might help to facilitate optimally balanced fork interactions of ssDNA‐ and RPA‐binding factors including RAD51 and SMARCAL1 that centrally influence fork remodeling and protection after replication stress 20, 32, 33. Additional mechanisms may also contribute to the role of RADX in promoting DNA replication and replication stress responses. For instance, while we have not observed pronounced interaction of RADX with RPA, RAD51, and associated genome stability regulators (our unpublished observations), RADX might recruit other factors to ssDNA regions; indeed, OB fold proteins often form part of multisubunit complexes 18. Identifying cellular binding partners of RADX will be important to address this possibility. The highly variable RADX expression pattern among different cancer cell lines raises the possibility that alteration of RADX abundance could be a mechanism to mitigate the harmful consequences of chronic replication stress. Future efforts to illuminate the mechanistic basis of how RADX facilitates replication fork integrity and stability through interplay with cellular binding partners and other ssDNA‐binding proteins, and how this impacts chromosomal stability in health and disease, are clearly warranted.
Materials and Methods
Plasmids and siRNAs
Full‐length RADX cDNA was cloned into pCR8/GW/TOPO (Invitrogen) using TOPO TA Cloning kit (Invitrogen). Point mutations and deletions in RADX (*OB: K304A/E327A; ΔOB: deletion of amino acids 1–495) were introduced with the QuikChange Site‐Directed Mutagenesis Kit (Stratagene), according to the manufacturer's protocol. Using Gateway LR Clonase (Invitrogen) RADX WT, *OB and ΔOB cDNAs were inserted into the destination vector pcDNA4/TO/GFP for doxycycline‐inducible expression. Plasmids for generation of HCT116 ΔRADX cells using CRISPR/Cas9 were generated as described 34 using the pX459 plasmid (Addgene #62988) for Cas9 and gRNA delivery. Briefly, gRNA sequences were ordered as complementary primers, mixed in a 1:1 ratio, and annealed. Subsequently, pX459 was digested with BbsI and the gRNA introduced using a normal ligation reaction according to the manufacturer's instructions (New England Biolabs). The following sequences were used: RADX sgRNA #2 (forward): 5′‐CACCGACATCATAGCAATAAAGGGG‐3′; RADX sgRNA #2 (reverse): 5′‐AAACCCCCTTTATTGCTATGATGTC‐3′; RADX sgRNA #3 (forward): 5′‐CACCGTTAACAGACAAGCAACCTG‐3′; RADX sgRNA #3 (reverse): 5′‐AAACCAGGTTGCTTGTCTGTTAAC‐3′.
Plasmid DNA and siRNA transfections were performed using FuGENE HD Transfection Reagent (Promega) and Lipofectamine RNAiMAX (Invitrogen), respectively, according to the manufacturers’ protocols. All siRNAs were used at a final concentration of 50 nM, unless otherwise indicated. The following siRNA oligonucleotides were used:
Non‐targeting control (CTRL): 5′‐GGGAUACCUAGACGUUCUA‐3′;
RADX(#1): 5′‐GCTTGAACTCTCTCGTATA‐3′; RADX(#6) (targeting the 3′UTR): 5′‐GGUUCGAAUUUCUCAGUAU‐3′; RAD51: 5′‐GUAGAGAAGUGGAGCGUAA‐3′. siRNA to RPA1 was described previously 17.
Cell culture, drug treatment, and colony survival assays
All cell lines used in this study were obtained from ATCC, cultured in DMEM containing 10% FBS, and regularly tested for mycoplasma infection. The cell lines were not authenticated. To generate U2OS cell lines inducibly expressing GFP‐RADX WT, ∆OB, or *OB alleles, U2OS cells were co‐transfected with pcDNA4/TO/GFP‐RADX constructs and pcDNA6/TR (Invitrogen) and positive clones were selected by incubation in medium containing blasticidin S (Invitrogen) and zeocin (Invitrogen) for 14 days. To generate HCT116 RADXΔ cell lines, parental cells were transfected with pX459‐sgRADX #2 or #3 (gRNAs targeting unique sequences within the RADX locus) and selected briefly with puromycin during clonal selection. Clones were screened for RADX expression by immunoblotting. The RADXΔ#2 and RADXΔ#3 cell lines were derived independently with RADX gRNA #2 and #3, respectively. A U2OS derivative cell line stably expressing RPA1, RPA2, and RPA3 at near‐endogenous levels from a single transcript (“Super‐RPA”) 17 was a kind gift from Dr. Luis Toledo (Center for Chromosome Stability, University of Copenhagen, Denmark).
Unless otherwise indicated, the following drug concentrations were used: camptothecin (1 μM, Sigma‐Aldrich), hydroxyurea (2 mM, Sigma‐Aldrich), ATR inhibitor (AZ20, 10 μM, Sigma‐Aldrich), and doxycycline (1 μg/ml, Sigma‐Aldrich). For colony formation assays, cells were seeded in duplicates (6‐cm dishes) per condition and allowed to adhere for a minimum of 16 h. Cells were then treated with indicated concentrations of HU or CPT for 24 h, washed extensively and incubated in drug‐free medium for 7–8 days. Plates were then washed once in PBS, left to dry, and stained with cell staining solution (0.5% w/v crystal violet, 25% v/v methanol). Finally, the plates were washed three times in deionized water. Colonies were counted manually and surviving fraction calculated as: colonies/(seeded colonies × plating efficiency).
Immunoblotting, cell fractionation, and antibodies
For immunoblotting, which was done as described 35, cells were lysed in EBC buffer (50 mM Tris, pH 7.5; 150 mM NaCl; 1 mM EDTA; 0.5% NP40; 1 mM DTT) or RIPA buffer (50 mM Tris, pH 8.0; 150 mM NaCl; 1% NP40; 0.1% SDS; 0.1% sodium deoxycholate) supplemented with protease and phosphatase inhibitors. For chromatin fractionation, cells were lysed in Buffer 1 (100 mM NaCl; 300 mM sucrose; 3 mM MgCl2; 10 mM PIPES, pH 6.8; 1 mM EGTA; 0.2% Triton X‐100) containing protease, phosphatase, and DUB inhibitors and incubated on ice for 5 min. After centrifugation, the pellet was washed in Buffer 1 and resuspended in Buffer 2 (50 mM Tris–HCl, pH 7.5; 150 mM NaCl; 5 mM EDTA; 1% Triton X‐100; 0.1% SDS) containing protease, phosphatase, and DUB inhibitors. Lysates were then incubated 10 min on ice and sonicated. Isolation of replication fork‐associated proteins by iPOND was done as described 23.
Antibodies used in this study included: GFP (sc‐9996 (Clone B2), Santa Cruz (1:1,000 dilution); sc‐8334, Santa Cruz (1:5,000)), RPA1 (Ab79398, Abcam (1:1,000)), RPA2 (ab76420, Abcam (1:1,000); NA19L (Clone Ab‐3), RPA34‐20, Calbiochem (1:1,000)), RPA2‐pSer4/Ser8 (A300‐245A, Bethyl (1:1,000)), RPA3 (ab588‐100, Abcam (1:1,000)), γ‐H2AX (05‐636 (Clone JBW301), Millipore (1:500)), histone H2AX (2595S, Cell Signaling (1:1,000)), BrdU (RPN202, GE Healthcare Life Sciences (1:5,000)), MCM6 (C‐20, sc‐9843, Santa Cruz (1:1,000)), RAD51 (PC130, Ab‐1, Millipore (1:500)), Chk1 (sc‐8408 (Clone G4), Santa Cruz (1:1,000)), Chk1 pSer345 (2348, Cell Signaling (1:1,000)), Chk2 pT68 (2661S, Cell Signaling (1:1,000)), Chk2 (sc‐9064, Santa Cruz (1:200)), ATM pS1981 (4526, Cell Signaling (1:500)), ATM (ab78 (clone 2C1), Abcam (1:1,000)), tubulin alpha (T9026, Sigma‐Aldrich (1:5,000)), histone H3 (ab1791, Abcam (1:5,000)), PCNA (sc‐56, Santa Cruz (1:500)), cyclin B (610220, BD Biosciences (1:1,000)), cyclin E (sc‐247, Santa Cruz (1:1,000), vinculin (V9131, Sigma (1:10,000)). Polyclonal sheep antibody to RADX was raised against full‐length recombinant human RADX, purified from bacteria. Quantification of immunoblots was done with ImageJ software.
ssDNA‐binding assays and RADX OB fold modeling
For DNA immobilization on beads, a 5′‐biotinylated ssDNA oligo (ss90‐1‐Biotin) or reverse complemented non‐biotinylated (negative control) ssDNA oligo (ss90‐2) was incubated with streptavidin‐coated magnetic beads (10 pmol per condition, DynaBeads M‐280 streptavidin) in modified EBC buffer (50 mM Tris, pH 7.5; 150 mM NaCl; 0.5% NP40; 1 mM DTT) at room temperature for 30 min and subsequently washed four times in modified EBC. For dsDNA pull‐downs, ss90‐1 and ss90‐2 were annealed prior to incubation with beads. For analysis of RADX binding to DNA‐conjugated beads, U2OS cells stably expressing GFP‐RADX alleles were lysed in modified EBC buffer supplemented with protease inhibitors. Extracts were sonicated and centrifuged at 16,000 g. Cleared cell extracts were pre‐incubated with unconjugated streptavidin beads at 4°C for 1 h to reduce unspecific binding. Pre‐cleared extracts were then incubated with ssDNA‐conjugated beads for 30 min at room temperature and subsequently washed five times in modified EBC buffer (or modified EBC containing increasing NaCl concentrations). ssDNA‐bound proteins were eluted by boiling beads in Laemmli sample buffer for 10 min and analyzed by immunoblotting. DNA oligo sequences:
ss90‐1: 5′‐Biotin‐ATCGCATTGGCATTGGCAATGCGATACGACTGATCGAGGGTACTCAGCTAGCTGATTCCGATCGGCTTATTCCGTGTACATACATCGGAT‐3′; ss90‐2: 5′‐ ATCCGATGTATGTACACGGAATAAGCCGATCGGAATCAGCTAGCTGAGTACCCTCGATCAGTCGTATCGCATTGCCAATGCCAATGCGAT‐3′.
In silico modeling of RADX OB fold domain structure, based on solved OB structures and secondary structure prediction, was performed using the Phyre2 protein structure prediction server (http://www.sbg.bio.ic.ac.uk/phyre2) 26. Homology to ssDNA‐binding OB folds in RPA1, BRCA2, POT1, and SSB1 was detected. Based on best model fit, a putative RADX structure of OB folds 2 and 3 (residues 215–506) was exported and compared to a published structure of RPA1 DBD‐A and DBD‐B (residues 183–420) bound to ssDNA (PDB code: 4GOP).
Immunofluorescence, proximity ligation assays (PLAs), and high‐content imaging analysis
Cells were pre‐extracted in PBS containing 0.2% Triton X‐100 for 2 min on ice, before fixation with 4% formaldehyde for 10 min. If cells were not pre‐extracted, they were subjected to a permeabilization step with PBS containing 0.2% Triton X‐100 for 5 min and incubated with primary antibodies diluted in DMEM for 1 h at room temperature. Following staining with secondary antibodies (Alexa Fluor; Life Technologies) diluted in DMEM for 30 min at room temperature, coverslips were mounted in Vectashield mounting medium (Vector Laboratories) containing nuclear stain DAPI. For EdU staining, cells were treated with EdU (10 μM) for 30 min before fixation and then stained using the Click‐iT® Plus EdU Alexa Fluor 647 Imaging Kit (Invitrogen) according to the manufacturer's instructions. In situ proximity ligation assay (PLA) was performed according to the manufacturer's guidelines (Duolink). Laser microirradiation was performed as described 36. Confocal images were acquired with an LSM880 confocal microscope (Carl Zeiss Microimaging Inc.) mounted on Zeiss AxioObserver.Z1 equipped with a Plan Apochromat 40×/1.3 NA oil immersion objective, using standard settings. Image acquisition and analysis were carried out with ZEN2.1 software. Raw images were exported as TIFF files, and if adjustments in image contrast and brightness were applied, identical settings were used on all images of a given experiment. Quantitative image‐based cytometry (QIBC) was performed as described 17. Briefly, cells were pre‐extracted or not, fixed, and stained as described above. Nuclear DNA was counterstained with DAPI (Molecular Probes) alongside incubation with secondary antibodies. Cells were mounted onto glass slides using ProLong® Gold Antifade (Invitrogen, Molecular Probes). Images were acquired with an Olympus IX‐81 wide‐field microscope equipped with an MT20 Illumination system and a digital monochrome Hamamatsu C9100 CCD camera. Olympus UPLSAPO 10×/0.4 NA, 20×/0.75 NA objectives were used. Automated and unbiased image analysis was carried out with the ScanR analysis software. Unless otherwise stated, between 1,000 and 5,000 cells were analyzed per condition. Data were exported and processed using Spotfire (Tibco) software.
DNA fiber assays
Exponentially growing cells (1 × 106) were labeled with consecutive pulses of CldU (25 μM) and IdU (250 μM) for 20 min (HCT116) or 25 min (U2OS and HeLa). Cells were then trypsinized and resuspended in PBS. Cell suspension (2 μl) was spotted onto Superfrost glass slides and lysed in buffer containing 200 mM Tris–HCl, pH 5.5; 50 mM EDTA; 0.5% SDS for 2 min. Slides were tilted at an angle to allow the DNA to run slowly down the slide. Slides were air‐dried before fixing in 3:1 methanol:acetic acid. DNA fiber spreads were denatured with 2.5 M HCl for 80 min before blocking in 2% BSA‐PBS with 0.1% Tween for 15 min. Slides were then incubated with rat anti‐BrdU (Abcam, ab6326) for 1 h at 1:100 to detect CldU. Slides were washed in PBS‐Tween and PBS before antibody cross‐linking in 4% formaldehyde for 10 min. Slides were then incubated with Alexa Fluor 594 goat anti‐rat antibody (Thermo Fisher) for 1 h at 1:100. Following similar washes, slides were incubated with mouse anti‐BrdU (BD Bioscience, #347580) at 1:500 overnight at 4°C to detect IdU. Slides were washed and incubated with Alexa Fluor 488 goat anti‐mouse antibody (Thermo Fisher) for 1 h at 1:100. After washing, the slides were air‐dried and mounted with 50 μl of Vectashield mounting medium (Vector Laboratories). Track lengths were measured using ImageJ software. For HU treatment, cells were incubated with CldU (25 μM) for 15 min, followed by 2 mM HU for 30 min and finally IdU (250 μM) for 30 min before trypsinization and slide preparation as above. For fork protection assays, cells were incubated with CldU (25 μM) for 25 min, followed by IdU (250 μM) for 25 min and finally HU (5 mM) for 3 h before trypsinization and slide preparation as above.
Quantification of mRNA levels by quantitative PCR
RNA was purified from cells using the RNeasy kit (Qiagen). cDNA was generated by reverse transcription PCR (iScript™ cDNA Synthesis Kit, Bio‐Rad), according to the manufacturer's protocol. Real‐time quantitative PCR was performed using the Stratagene Mx3005P System and Brilliant III Ultra‐Fast SYBR® Green qPCR Master Mix (Agilent). Ubiquitin mRNA level was used as a control for normalization. For amplification of the indicated cDNAs, the following primers were used: RADX (forward): 5′‐ATGATGTGACGATCTCAGATGGG‐3′; RADX (reverse): 5′‐CCCCTGGCCTATCCTTTTCTC‐3′; ubiquitin (forward) 5′‐CACTTGGTCCTGCGCTTGA‐3′; ubiquitin (reverse) 5′‐CAATTGGGAATGCAACAACTTTAT‐3′.
Statistics and reproducibility
For all experiments, samples were not randomized and the investigators were not blinded to the group allocation during experiments and outcome assessment. No samples were excluded from the analysis and no statistical method was used to predetermine sample size. Statistical tests used are described in the figure legends. All experiments shown in this study were repeated independently at least twice with similar results. Data from representative experiments are shown in Figs 1C–I, 2A–F, 3A, EV1B and D–G, EV2A–D, EV3A and B, EV4A–C, and EV5A and B.
Author contributions
LS, TH, SH, PH, and CG performed experiments. LS, TH, SH, PH, and NM designed experiments and analyzed and discussed the data. NM wrote the manuscript with comments from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Luis Toledo for providing reagents, Anna Bizard (Center for Chromosome Stability) for help with experiments, and Gopal Karemore (Novo Nordisk Foundation Center for Protein Research) for assistance with and helpful advice on statistical analyses. This work was supported by grants from The Novo Nordisk Foundation (grants no. NNF14CC0001, NNF12OC0002114, and NNF16CC0020906), European Research Council (ERC, grant agreement no. 616236 (DDRegulation)), Danish Cancer Society (grant no. R146‐RP11394), Danish Council for Independent Research, and Danish National Research Foundation (grant no. DNRF115).
EMBO Reports (2017) 18: 1991–2003
References
- 1. Jackson SP, Bartek J (2009) The DNA‐damage response in human biology and disease. Nature 461: 1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hoeijmakers JH (2001) Genome maintenance mechanisms for preventing cancer. Nature 411: 366–374 [DOI] [PubMed] [Google Scholar]
- 3. Zeman MK, Cimprich KA (2014) Causes and consequences of replication stress. Nat Cell Biol 16: 2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Branzei D, Foiani M (2010) Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 11: 208–219 [DOI] [PubMed] [Google Scholar]
- 6. Marechal A, Zou L (2013) DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5: a012716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blackford AN, Jackson SP (2017) ATM, ATR, and DNA‐PK: the trinity at the heart of the DNA damage response. Mol Cell 66: 801–817 [DOI] [PubMed] [Google Scholar]
- 8. Toledo L, Neelsen KJ, Lukas J (2017) Replication catastrophe: when a checkpoint fails because of exhaustion. Mol Cell 66: 735–749 [DOI] [PubMed] [Google Scholar]
- 9. Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA (2005) Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR‐dependent checkpoint. Genes Dev 19: 1040–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA‐ssDNA complexes. Science 300: 1542–1548 [DOI] [PubMed] [Google Scholar]
- 11. Cortez D, Guntuku S, Qin J, Elledge SJ (2001) ATR and ATRIP: partners in checkpoint signaling. Science 294: 1713–1716 [DOI] [PubMed] [Google Scholar]
- 12. Kumagai A, Lee J, Yoo HY, Dunphy WG (2006) TopBP1 activates the ATR‐ATRIP complex. Cell 124: 943–955 [DOI] [PubMed] [Google Scholar]
- 13. Haahr P, Hoffmann S, Tollenaere MA, Ho T, Toledo LI, Mann M, Bekker‐Jensen S, Raschle M, Mailand N (2016) Activation of the ATR kinase by the RPA‐binding protein ETAA1. Nat Cell Biol 18: 1196–1207 [DOI] [PubMed] [Google Scholar]
- 14. Bass TE, Luzwick JW, Kavanaugh G, Carroll C, Dungrawala H, Glick GG, Feldkamp MD, Putney R, Chazin WJ, Cortez D (2016) ETAA1 acts at stalled replication forks to maintain genome integrity. Nat Cell Biol 18: 1185–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim C, Paulus BF, Wold MS (1994) Interactions of human replication protein A with oligonucleotides. Biochemistry 33: 14197–14206 [DOI] [PubMed] [Google Scholar]
- 16. Marechal A, Zou L (2015) RPA‐coated single‐stranded DNA as a platform for post‐translational modifications in the DNA damage response. Cell Res 25: 9–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker‐Jensen S, Mailand N, Bartek J, Lukas J (2013) ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155: 1088–1103 [DOI] [PubMed] [Google Scholar]
- 18. Flynn RL, Zou L (2010) Oligonucleotide/oligosaccharide‐binding fold proteins: a growing family of genome guardians. Crit Rev Biochem Mol Biol 45: 266–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moynahan ME, Jasin M (2010) Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol 11: 196–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V (2010) Rad51 protects nascent DNA from Mre11‐dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17: 1305–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M (2011) Double‐strand break repair‐independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145: 529–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ et al (2016) Replication fork stability confers chemoresistance in BRCA‐deficient cells. Nature 535: 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, Cortez D (2015) The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol Cell 59: 998–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wan L, Huang J (2014) The PSO4 protein complex associates with replication protein A (RPA) and modulates the activation of ataxia telangiectasia‐mutated and Rad3‐related (ATR). J Biol Chem 289: 6619–6626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Justice JFT, Morgan RW, Beemon KL (2015) Common viral integration sites identified in avian Leukosis virus‐induced B‐cell lymphomas. MBio 6: e01863–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10: 845–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haring SJ, Mason AC, Binz SK, Wold MS (2008) Cellular functions of human RPA1. Multiple roles of domains in replication, repair, and checkpoints. J Biol Chem 283: 19095–19111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gustafsdottir SM, Ostman A, Landegren U (2002) Protein detection using proximity‐dependent DNA ligation assays. Nat Biotechnol 20: 473–477 [DOI] [PubMed] [Google Scholar]
- 29. Groth A, Corpet A, Cook AJ, Roche D, Bartek J, Lukas J, Almouzni G (2007) Regulation of replication fork progression through histone supply and demand. Science 318: 1928–1931 [DOI] [PubMed] [Google Scholar]
- 30. Techer H, Koundrioukoff S, Nicolas A, Debatisse M (2017) The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat Rev Genet 18: 535–550 [DOI] [PubMed] [Google Scholar]
- 31. Dungrawala H, Bhat KP, Le Meur R, Chazin WJ, Ding X, Sharan SK, Wessel SR, Sathe AA, Zhao R, Cortez D (2017) RADX promotes genome stability and modulates chemosensitivity by regulating RAD51 at replication forks. Mol Cell 67: 374–386. e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Betous R, Mason AC, Rambo RP, Bansbach CE, Badu‐Nkansah A, Sirbu BM, Eichman BF, Cortez D (2012) SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev 26: 151–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M (2015) Rad51‐mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208: 563–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Poulsen M, Lukas C, Lukas J, Bekker‐Jensen S, Mailand N (2012) Human RNF169 is a negative regulator of the ubiquitin‐dependent response to DNA double‐strand breaks. J Cell Biol 197: 189–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mosbech A, Gibbs‐Seymour I, Kagias K, Thorslund T, Beli P, Povlsen L, Nielsen SV, Smedegaard S, Sedgwick G, Lukas C et al (2012) DVC1 (C1orf124) is a DNA damage‐targeting p97 adaptor that promotes ubiquitin‐dependent responses to replication blocks. Nat Struct Mol Biol 19: 1084–1092 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File