

Abstract
Endocytic processes are facilitated by both curvature‐generating BAR‐domain proteins and the coordinated polymerization of actin filaments. Under physiological conditions, the N‐BAR protein Bin1 has been shown to sense and curve membranes in a variety of cellular processes. Recent studies have identified Bin1 as a risk factor for Alzheimer's disease, although its possible pathological function in neurodegeneration is currently unknown. Here, we report that Bin1 not only shapes membranes, but is also directly involved in actin binding through its BAR domain. We observed a moderate actin bundling activity by human Bin1 and describe its ability to stabilize actin filaments against depolymerization. Moreover, Bin1 is also involved in stabilizing tau‐induced actin bundles, which are neuropathological hallmarks of Alzheimer's disease. We also provide evidence for this effect in vivo, where we observed that downregulation of Bin1 in a Drosophila model of tauopathy significantly reduces the appearance of tau‐induced actin inclusions. Together, these findings reveal the ability of Bin1 to modify actin dynamics and provide a possible mechanistic connection between Bin1 and tau‐induced pathobiological changes of the actin cytoskeleton.
Keywords: actin binding, Alzheimer's disease, genetic risk factor, N‐BAR protein Bin1, tau
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Neuroscience
Introduction
Plasma membrane rearrangements are crucial for many cellular processes such as endocytosis, trafficking, and motility. Membrane remodeling is achieved by an organized interplay between proteins that control plasma membrane curvature, membrane fission, and assembly of newly formed actin filaments 1. BAR (Bin‐amphiphysin/rvs) domain proteins are key players in regulating the shape of membranes by directly binding membrane lipids and inducing curvature 2, 3. This protein class also couples membrane rearrangements and actin dynamics via direct interactions with actin‐binding proteins 2, 4, thus orchestrating different morphogenetic processes. Recently, the BAR domain of two BAR proteins, pacsin2 and PICK1, has been identified to also directly interact with actin filaments, thereby adding an additional layer of regulation 5, 6.
Human Bin1 (hBin1, also called MYC box‐dependent interacting protein‐1 or bridging integrator‐1 or Amphiphysin 2) is a highly conserved membrane deforming protein. hBin1 has many different functions including tubulation of t‐tubules in muscle tissue, and regulation of clathrin‐mediated endocytosis and of endosome trafficking 7, 8, 9, 10, 11. Although hBin1 is broadly expressed, it is most abundant in muscle and brain tissue 12. Alternative splicing generates at least 10 transcripts in humans including ubiquitous and tissue‐specific isoforms 13. All isoforms of hBin1 contain a N‐terminal BAR domain, which through dimerization, induces membrane curvature 14, 15. A C‐terminal SH3 domain mediates the interaction of hBin1 with other proteins, such as endophilin, dynamin, and neuronal Wiskott–Aldrich syndrome protein (N‐WASP) for example 7, 16, 17. The neuron‐specific isoforms additionally contain a clathrin and AP2 (CLAP) binding domain 18 and have been implicated to function in clathrin‐mediated endocytosis and intracellular endosome trafficking 12. hBin1 belongs to a sub‐class of the BAR superfamily, called N‐BAR proteins, which contain an N‐terminal amphiphatic helix (H0) 14. Although the H0 helix is not required for membrane tubulation, it is important for hBin1 polymer formation at the membrane 19.
Interestingly, hBin1 was recently identified as a major risk factor in genomewide association studies (GWAS) of Alzheimer's disease (AD) 20. Different groups have reported altered hBin1 expression levels, even though the observations are conflicting 21, 22, 23, 24. Recent studies have associated hBin1 with two proteins in AD pathobiology: beta‐amyloid (Aβ) and tau. Aβ is generated from its precursor form in endosomes by a dual proteolytic cleavage 25. Since hBin1 is part of the endocytosis machinery, its depletion impairs the endocytic recycling of the first protease (β‐secretase) and thus may promote Aβ production 26, 27. Bin1 has also been shown to directly interact with tau and to modulate tau‐toxicity in Drosophila, although it is still controversial whether its loss enhances or mitigates tau‐toxicity 21, 28. Importantly, the underlying molecular mechanisms of hBin1‐mediated changes of tau neurotoxicity remain unclear.
In this study, we explore the mechanistic link between hBin1 and tau‐mediated pathology in AD. Tau aggregation leads to a plethora of different cellular impairments, like proteostasis disruption, mitochondrial dysfunction, and changes of the cytoskeleton 29, 30. The actin cytoskeleton is highly impaired in AD, with actin accumulating in aggregates 31. The presence of these actin‐rich inclusions correlates with the extent of tau pathology in AD patients 32, and tau can be detected in a subset of these rods 23. Given the association of hBin1 with N‐WASP and the involvement of BAR‐domain proteins in regulating actin dynamics, we speculated that hBin1 could be involved in tau pathology by directly affecting actin dynamics.
Here, we demonstrate that hBin1 is directly involved in modulating actin dynamics. hBin1 does not only stabilize filamentous actin and tau‐induced actin bundles in vitro; it also exhibits actin bundling activity itself. Furthermore, loss of Drosophila Bin1 (dBin1) leads to a decrease in actin accumulations in an in vivo tau Drosophila model. These novel observations provide a first mechanistic connection between Bin1 and pathological changes mediated by tau.
Results
hBin1 directly interacts with actin filaments
The BAR‐domain proteins PICK1 and pacsin2 have recently been shown to directly interact with actin filaments to execute their physiological functions 5, 6. To investigate whether the AD risk factor hBin1 is also able to directly bind to filamentous actin, we performed co‐sedimentation assays (Fig 1A). We analyzed the neuron‐specific isoform of hBin1 in this assay by incubating it with in vitro polymerized actin filaments for 30 min, before F‐actin filaments were separated from free G‐actin and unbound hBin1 via ultracentrifugation. hBin1 was detected only in the supernatant fraction in the absence of actin. However, upon incubation with F‐actin for 30 min, a proportion of hBin1 could also be observed in the pellet fraction, indicating an interaction of hBin1 with polymerized actin filaments (Fig 1B). Since some actin‐binding proteins also associate with G‐actin, we subsequently analyzed whether hBin1 can also bind to the monomeric form of actin. We incubated G‐actin coupled to Sepharose beads either with hBin1 or with the G‐actin‐binding protein profilin as a positive control. Both proteins could be pulled down with the G‐actin beads, indicating that hBin1 can bind to both F‐ and G‐actin (Fig EV1A). However, the binding of hBin1 to G‐actin was rather weak compared to profilin, suggesting that hBin1 preferentially binds to the filamentous form. Next, we aimed to visualize bound hBin1 on F‐actin filaments using negative stain electron microscopy (EM) (Fig 1C). Here, we observed F‐actin filaments partially decorated with an electron‐dense material, strongly supporting a direct interaction between hBin1 and F‐actin filaments. In order to verify that indeed hBin1 decorates actin filaments, we used dual‐color total internal reflection microscopy (TIRFM) with fluorescently labeled actin (actin 488) in the presence of fluorescently labeled hBin1 (hBin1‐SNAP‐549). This C‐terminally coupled hBin1‐SNAP was equally effective in F‐actin binding as untagged hBin1 in the co‐sedimentation assay (Fig EV1B), indicating no interfering effect of the SNAP‐tag for actin binding. Interestingly, hBin1 directly localized to actin filaments with hBin1 spots gradually accumulating on actin filaments until the filaments were decorated (Fig 1D), confirming our EM data. To further characterize the hBin1‐actin interaction, we increased the actin concentration systematically in our co‐sedimentation assays. Importantly, we observed a concentration‐dependent enrichment of hBin1 in the pellet fraction (Fig 1E). Under these experimental conditions, hBin1 bound F‐actin with a dissociation constant (K d) of 0.54 μM (Fig 1F). This dissociation constant is comparable with the K d of 0.3 μM of the BAR protein PICK1 6 and the K d of 1.9 μM of the BAR protein pacsin2 binding to F‐actin 5.
Figure 1. Identification of hBin1 as a new actin‐binding protein.
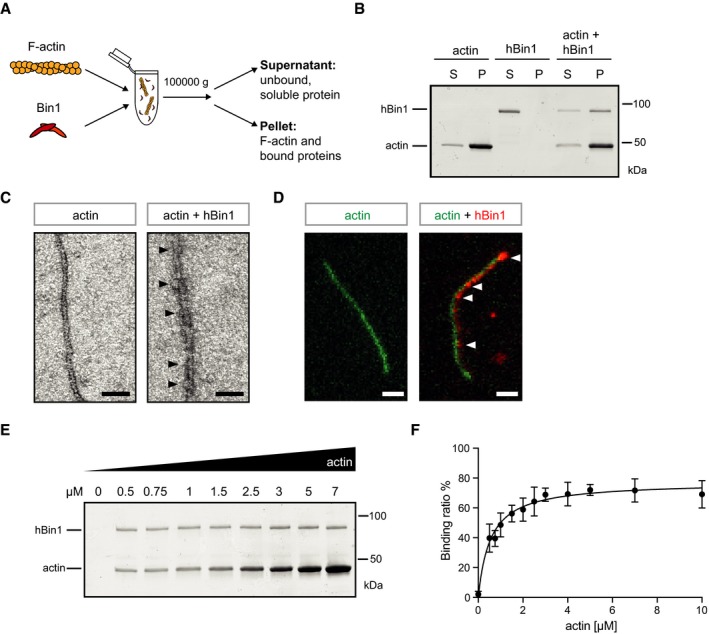
- Experimental setup to separate F‐actin and its binding partners (pellet) from unbound, soluble proteins (supernatant). F‐actin is incubated with Bin1 at RT for 30 min. Upon ultracentrifugation, F‐actin and proteins binding to it can be separated from monomeric G‐actin and unbound, soluble protein.
- hBin1 isoform 1 (1 μM) binds directly to F‐actin (4 μM). Co‐sedimentation assay as described in (A) was resolved by SDS–PAGE followed by Coomassie Blue staining. S, supernatant; P, pellet.
- Electron micrographs of negatively stained F‐Actin (0.4 μM) alone or together with hBin1 (isoform 1; 0.4 μM), scale bar = 50 nm. Black arrowheads point to heavily decorated parts of the actin filament.
- TIRFM image of actin‐488 filaments (green, 1 μM) in the presence or absence of hBin1‐SNAP594 (red, 250 nM), scale bar = 2 μm. White arrowheads point to hBin‐SNAP dots associating with the actin filament.
- Effects of increasing amounts of F‐actin on co‐sedimentation of hBin1 (isoform 1). F‐actin at indicated concentrations was incubated with 1 μM Bin1 and then subjected to a co‐sedimentation assay as described in (A). SDS–PAGE followed by a Coomassie Blue staining of the proteins found in the pellet fraction is shown.
- Binding of hBin1 (isoform 1) to F‐actin. Data represent mean values of four independent experiments similar to (E). Calculated K d of binding: 0.54 ± 0.07 μM. Error bars represent ± SD.
Figure EV1. Conserved binding between BAR domain and F‐actin within the Bin1 family.
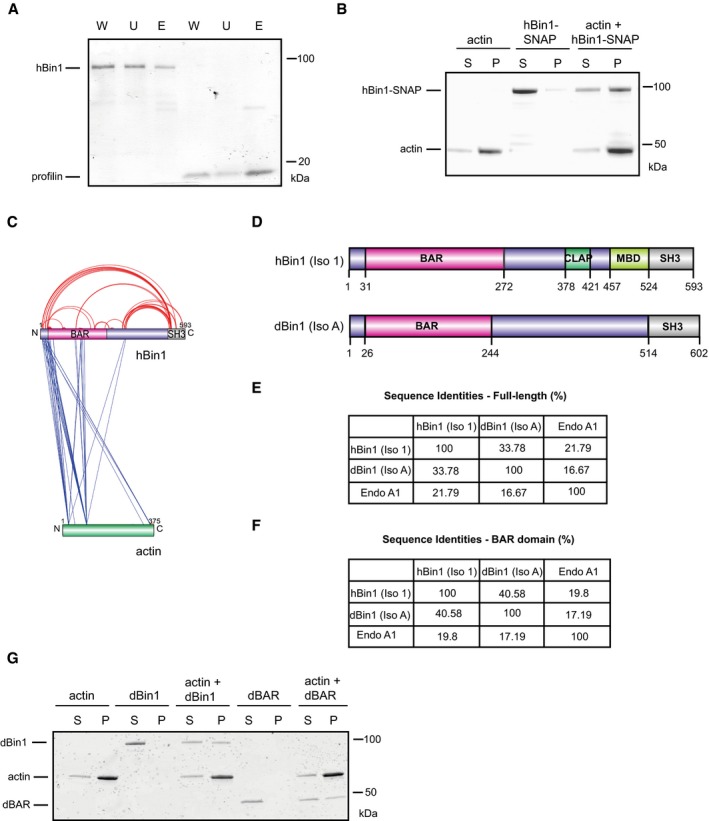
- hBin1 isoform 1 binds slightly to G‐actin. G‐actin‐coupled Sepharose beads were incubated with hBin1 and profilin (both 1.25 μM) and incubated for 1 h at RT before separating them from unbound protein via centrifugation at 6,000 g for 4 min. Fractions were resolved by SDS–PAGE followed by Coomassie Blue staining. W, whole input fraction; U, unbound fraction; E, eluted fraction.
- hBin1‐SNAP (isoform 1; 1 μM) binds directly to 4 μM F‐actin. Co‐sedimentation assay resolved by SDS–PAGE followed by Coomassie Blue staining.
- Xvis‐visualization of crosslink sites identified for the complex of hBin1 (isoform 1) with F‐actin. Only the crosslinks with a score of 80 and higher are displayed. Intra‐molecular crosslinks are indicated in red, whereas inter‐molecular crosslinks are indicated in blue.
- Domain overviews of hBin1 (isoform 1) and the Drosophila orthologue of Bin1 isoform A [dBin1 (isoform A)]. Both isoforms share the N‐terminal BAR domain and the C‐terminal SH3 domain, but differ in the middle domains.
- Sequence identities of the full‐length proteins hBin1 (isoform 1), dBin1 (isoform A), and the N‐BAR family member endophilin A1 (Endo A1) analyzed with Clustal omega. Gaps in sequences were not considered.
- Sequence identities of the BAR domains including the amphipathic helix of hBin1 (isoform 1), dBin1 (isoform A), and Endo A1 analyzed with Clustal omega. Gaps in sequences were not considered.
- dBin1 (isoform A) and its BAR domain (dBAR; both 1 μM) bind directly to 4 μM F‐actin. Co‐sedimentation assay resolved by SDS–PAGE followed by Coomassie Blue staining.
The BAR domain of Bin1 is sufficient for F‐actin binding
In order to identify the region of Bin1 driving the interaction with F‐actin, we performed crosslinking experiments between hBin1 and actin using the zero‐length crosslinker 1‐ethyl‐3‐(3‐dimethylaminopropyl) carbodiimide (EDC). hBin1 (isoform 1) and F‐actin were incubated at different molar ratios and crosslinked with EDC. On a SDS–PAGE, a band running at 135 kDa could be readily detected and increased with higher concentrations of F‐actin (Fig 2A). This band was absent in F‐actin and hBin1 control samples (Fig 2A), indicating the formation of specific crosslinks between the two proteins. Mass‐spectrometric analysis of this band confirmed the formation of a specific dimer between hBin1 and actin and revealed a selected number of crosslinked peptide sequences between the BAR domain as well as the N‐terminal amphiphatic helix of hBin1 to actin (Fig EV1C and Dataset EV1). This finding confirms a specific binding between hBin1 and F‐actin and indicates the BAR domain as interaction interface, in line with previous results, where the BAR domains of pacsin2 and PICK1 have been shown to directly interact with actin 5, 6.
Figure 2. The BAR domain of Bin1 is sufficient to bind F‐actin.
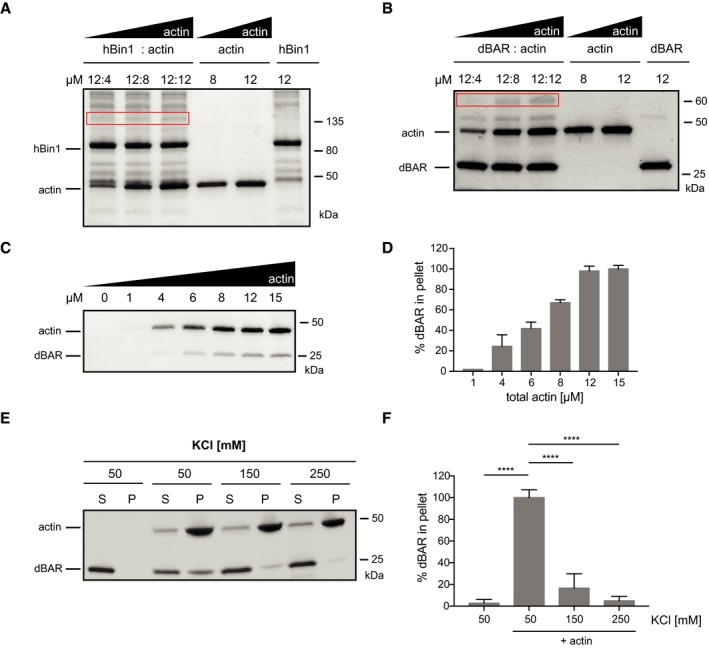
- Crosslinking of F‐actin with hBin1 (isoform 1). F‐actin and hBin1 were mixed at the indicated concentrations and crosslinked by addition of the EDC crosslinker. SDS–PAGE of crosslinked samples stained with Coomassie Blue is shown. The red box highlights the specific band, corresponding to ˜135 kDa, which appeared after crosslinking in a dose‐dependent manner.
- Crosslinking of F‐actin with dBAR. F‐actin and dBAR were mixed at the indicated concentrations and crosslinked by addition of the EDC crosslinker. SDS–PAGE of crosslinked samples stained with Coomassie Blue is shown. The red box highlights the specific band, around 65 kDa, which appeared after crosslinking in a dose‐dependent manner.
- Effect of increasing amounts of F‐actin on co‐sedimentation of dBAR. 1 μM of dBAR was mixed with F‐actin at indicated concentrations before subjecting the sample to high‐speed sedimentation. SDS–PAGE of pellet fraction visualized by Coomassie Blue staining is shown.
- Densitometric quantitation of dBAR found in the pellet fractions from (C). The error bars represent mean ± SD (n = 3).
- Effect of increasing KCl concentration on co‐sedimentation of 2 μM dBAR with 4 μM F‐actin. SDS–PAGE of supernatant (S) and pellet (P) fractions stained with Coomassie Blue.
- Densitometric quantitation of dBAR found in the pellet fractions from (E). Graph shows percentage of binding compared to the maximum binding; statistically significant differences are indicated (one‐way ANOVA, ****P < 0.0001, n = 3). The error bars represent mean ± SD.
The BAR domain is pivotal for the physiological function of hBin1 as a membrane‐interacting protein and its ability to induce membrane curvature 33, and is a highly conserved feature in the amphiphysin protein superfamily 14, 34. To test whether the actin‐binding property of hBin1 is conserved in other Bin1 orthologues, we turned to the invertebrate Bin1 orthologue from Drosophila. dBin1 is similar to hBin1 (isoform 1) in the overall structure of the BAR domain and the SH3 domain, while the middle domain of dBin1 lacks the defined CLAP and MBD domains (Fig EV1D). dBin1 displays 34% sequence identity with hBin1 (isoform 1; Fig EV1E), and its BAR domain (dBAR) has even 41% sequence identity to the BAR domain of hBin1 (Fig EV1F). Therefore, hBin1 is closer in identity to its evolutionary ancestor than to other human N‐BAR domains such as endophilin A1 (Endo A1; Fig EV1E). We first confirmed dBin1 binding to F‐actin by its localization in the F‐actin pellet fraction in our co‐sedimentation assay (Fig EV1G). Not only full‐length dBin1 but also only the dBAR domain was sufficient to directly bind actin filaments (Fig EV1G). Furthermore, our EDC crosslinking experiments confirm the direct interaction between dBAR domains with actin, as we observed a heterodimer band at 65 kDa (Fig 2B). To further investigate the specificity of this dBAR‐domain‐mediated binding, we tested co‐sedimentation assays with increasing actin or increasing salt concentrations (Fig 2C–E). Increasing the actin concentration also elevated the amount of dBAR in the pellet fraction (Fig 2C and D), indicating a specific interaction. Moreover, raising the KCl concentration from 50 to 250 mM decreased the amount of dBAR in the pellet to below 10% (Fig 2E and F), which suggests that the binding between actin and the dBAR domain is mainly driven by electrostatic interactions.
Although all these experiments suggest that the BAR domain of Bin1 harbors the conserved actin‐binding capacity, the SH3 domain was recently also suggested to bind F‐actin 35. We therefore compared the F‐actin‐binding abilities of human BAR domain, SH3 domain, and full‐length hBin1 to each other. In our experimental setup, the hSH3 domain did not co‐pellet with F‐actin, whereas hBAR binds F‐actin with a comparable K d (1.5 μM) to the full‐length protein (Fig EV2A–D). Interestingly the hBAR binding curve exhibits clear kinetic cooperativity, likely reflecting the stronger requirement for multimerization in the binding of the BAR domain compared to full‐length hBin1 (Fig EV2D).
Figure EV2. Comparison of binding abilities of hBin1, hBAR, and hSH3.
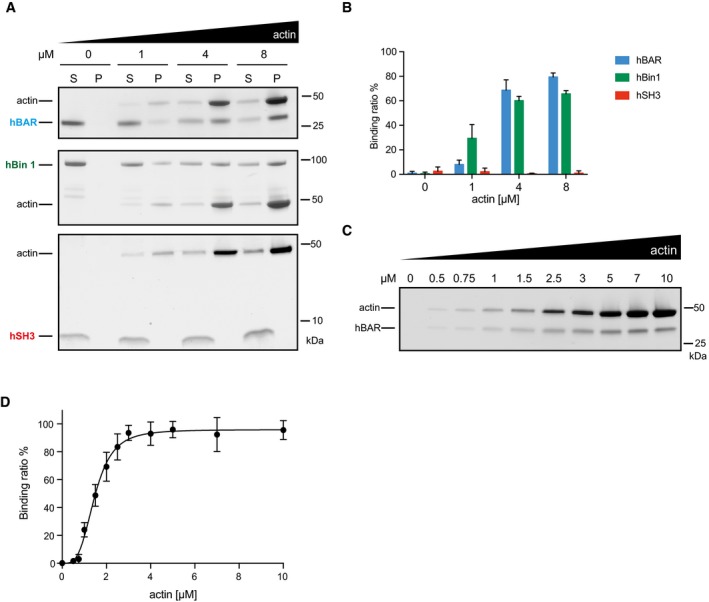
- Effect of increasing amounts of F‐actin on co‐sedimentation of hBAR, hBin1, and hSH3 (all 2 μM). Different Bin1 proteins were mixed with F‐actin at indicated concentrations before subjecting the samples to high‐speed sedimentation. SDS–PAGE of supernatant and pellet fraction visualized by Coomassie Blue staining is shown.
- Densitometric quantitation of hBAR, hBin1, and hSH3 found in the pellet fractions from (A). The error bars represent mean ± SD (n = 3).
- Effects of increasing amounts of F‐actin on co‐sedimentation of hBAR. F‐actin at indicated concentrations was incubated with 1 μM hBAR and then subjected to a co‐sedimentation assay. SDS–PAGE followed by a Coomassie Blue staining of the proteins found in the pellet fraction is shown.
- Binding of hBAR to F‐actin. Data represent mean values of three independent experiments similar to (C). Calculated K d of binding: 1.47 ± 0.05 μM. Error bars represent ± SD.
hBin1 stabilizes F‐actin
We next hypothesized that the binding of hBin1 to F‐actin could influence actin dynamics. To address this question biochemically, the polymerization of pyrene‐labeled actin was monitored via its fluorescence signal, which is drastically enhanced during its polymerization into filaments 36. Analysis of actin polymerization kinetics revealed no difference in polymerization rate between actin alone or actin incubated with increasing amounts of hBin1 (Fig 3A). In a second experimental setup, we tested whether hBin1 has an influence on cofilin‐induced actin severing. In a polymerization assay, cofilin enhances the rate of polymerization by severing the filaments and therefore producing more barbed ends which can be used for subsequent filament elongation 37. Our data confirm this effect by showing that cofilin could increase the rate of polymerization dramatically compared to actin alone (Fig 3B). Surprisingly, hBin1 addition was able to further promote this cofilin‐induced effect in a dose‐dependent manner (Fig 3B). This effect could result from enhanced filament severing and barbed‐end production upon hBin1 binding, as proposed for other actin‐binding proteins such as fascin and the Switch‐associated protein 70 (Swap‐70) 38, 39.
Figure 3. hBin1 stabilizes actin filaments.
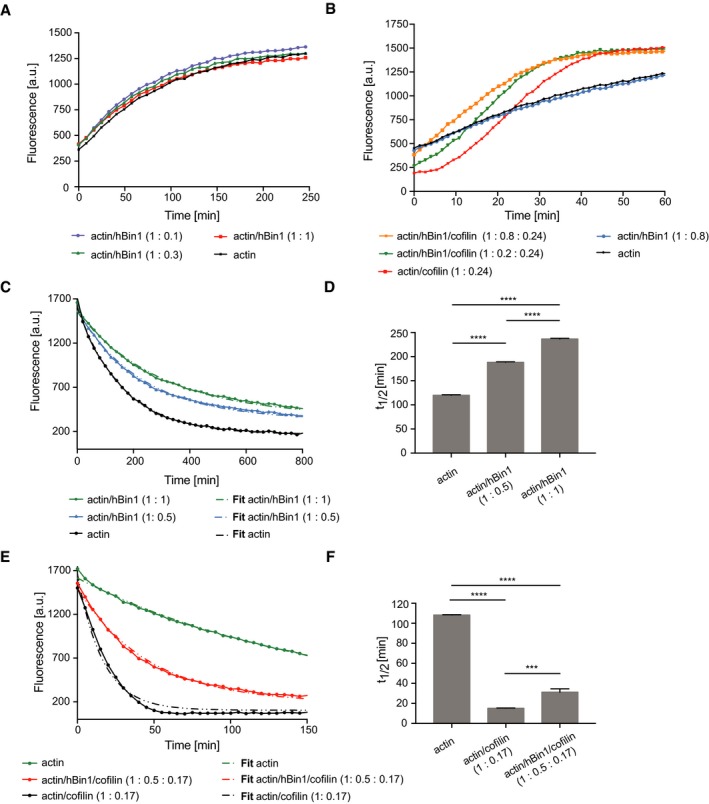
- Effect of hBin1 on the spontaneous polymerization of actin. In vitro polymerization of pyrene‐labeled actin was monitored in the presence or absence of hBin1 (isoform 1) at indicated molar ratios using time‐based fluorimetry.
- Effect of hBin1 on actin polymerization, increased by cofilin. Pyrene‐labeled actin was polymerized in the presence or absence of hBin1 (isoform 1) and/or cofilin at indicated molar ratios resulting in a Bin1‐dependent acceleration of the actin polymerization.
- Effect of hBin1 on the rate of actin depolymerization. Dilution‐induced depolymerization of pyrene‐labeled actin in the presence or absence of hBin1 (isoform 1) at indicated molar ratios was measured following pyrene fluorescence intensity over time. Dotted lines indicate the one‐phase exponential decay fitting.
- Calculated half‐time of actin depolymerization from (C) after one‐phase exponential decay fitting. Statistically significant differences are indicated (one‐way ANOVA, ****P < 0.0001, n = 3). The error bars represent mean ± SD.
- Effects of cofilin and Bin1 (isoform 1) on dilution‐induced depolymerization of F‐actin. Spontaneous disassembly of pyrene‐labeled F‐actin in the presence or absence of hBin1 (isoform 1) and cofilin was followed, as measured by pyrene fluorescence. Dotted lines indicate the one‐phase exponential decay fitting.
- Calculated half‐time of actin depolymerization from (E) after one‐phase exponential decay fitting. Statistically significant differences are indicated (one‐way ANOVA, ***P < 0.001, ****P < 0.0001, n = 3). The error bars represent mean ± SD.
To further determine the mechanism underlying the ability of hBin1 to increase cofilin‐induced actin severing, we followed actin polymerization in the presence of GFP‐tagged cofilin and hBin1 via TIRFM. In line with previous studies, severing events occurred at boundaries between GFP–cofilin‐decorated and non‐decorated filament segments 40. Intriguingly, we frequently observed that addition of hBin1 induced filament crosslinking and cofilin‐induced severing occurred often during incorporation of filaments into Bin1‐induced bundles (Movie EV1). Therefore, the promotion in severing is probably due to increased shear stress when filaments attach to adjacent filaments, in agreement with an overall stabilization function of hBin1 on F‐actin.
To evaluate the effect of hBin1 on filament disassembly, we performed dilution‐induced depolymerization experiments with actin filaments in the presence of different hBin1 concentrations. Interestingly, in this assay, hBin1 could decrease the depolymerization rate in a dose‐dependent manner, thus indicating a stabilization of actin filaments upon its binding (Fig 3C). By mixing equimolar concentrations of hBin1 and actin filaments, the half‐time of the depolymerization could be doubled (Fig 3D). This stabilization could be either due to an impairment of the hBin1‐actin complex to release actin monomers from the filament ends or due to the inhibition of actin filament breakage. To explore these possibilities, we monitored the spontaneous disassembly of actin filaments by cofilin by measuring the decrease in pyrene fluorescence upon dilution. As expected, the depolymerization rate was enhanced by cofilin, in line with previous studies 41, 42 (Fig 3E). The half‐time of the actin depolymerization could be reduced by fivefold by adding cofilin (Fig 3E and F). Interestingly, the stabilization of actin filaments by hBin1 binding resulted in an increased half‐life of the hBin1‐actin complex during its cofilin‐mediated disassembly. However, the half‐life was only partially rescued to the level of actin depolymerization without cofilin (Fig 3F). This suggests that cofilin partially antagonizes the stabilizing effect of hBin1 on actin disassembly by creating new filament ends.
Since the BAR domain seems to be responsible for binding actin, we tested whether hBAR exhibits also the actin–stabilization effect observed in the depolymerization assays using the full‐length hBin1. If hBAR was the main driver for the F‐actin interaction, it should be able to functionally replace full‐length hBin1 in the depolymerization assays. Importantly, the BAR domain itself was able to stabilize actin filaments during dilution‐induced depolymerization (Fig EV3A and B) and also during cofilin‐mediated disassembly (Fig EV3C and D). Overall, these findings implicate that hBin1 modulates actin dynamics by stabilizing existing actin filaments via interactions through its BAR domain.
Figure EV3. hBAR stabilizes F‐actin.
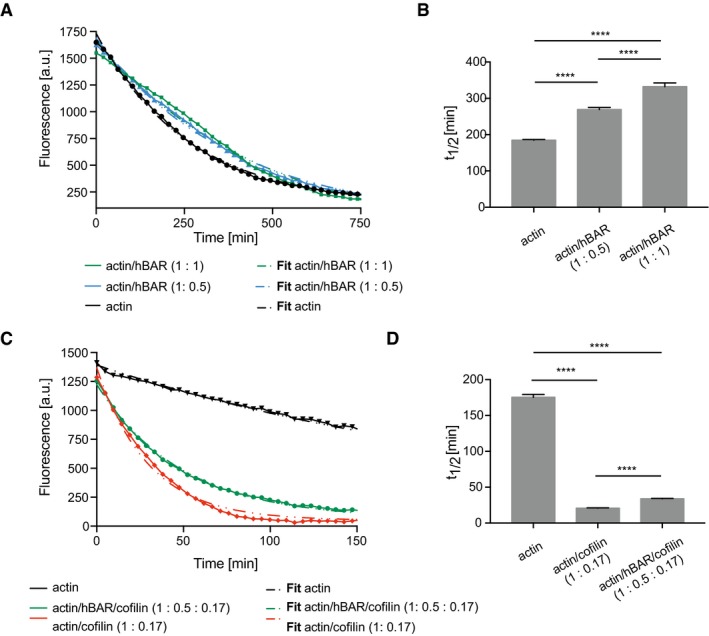
- Effect of hBAR on the rate of actin depolymerization. Dilution‐induced depolymerization of pyrene‐labeled actin in the presence or absence of hBAR at indicated molar ratios was measured following pyrene fluorescence intensity over time. Dotted lines indicate the one‐phase exponential decay fitting.
- Calculated half‐time of actin depolymerization from (A) after one‐phase exponential decay fitting. Statistically significant differences are indicated (one‐way ANOVA, ****P < 0.0001, n = 3). The error bars represent mean ± SD.
- Effects of cofilin and hBAR on dilution‐induced depolymerization of F‐actin. Spontaneous disassembly of pyrene‐labeled F‐actin in the presence or absence of hBin1 (isoform 1) and cofilin was followed, as measured by pyrene fluorescence. Dotted lines indicate the one‐phase exponential decay fitting.
- Calculated half‐time of actin depolymerization from (C) after one‐phase exponential decay fitting. Statistically significant differences are indicated (one‐way ANOVA, ****P < 0.0001, n = 3). The error bars represent mean ± SD.
hBin1 exhibits an actin bundling activity
Similar effects to hBin1 in the described actin polymerization and depolymerization assays were monitored for the actin crosslinking proteins fascin and SWAP‐70 38, 39. Additionally, we already observed filament crosslinking when hBin1 was added into the TIRF assay to study cofilin‐induced actin severing (Movie EV1). Therefore, we systematically explored hBin1's F‐actin bundling activity. Here, TIRFM allowed the visualization of freely diffusing, growing filaments in the presence or absence of hBin1 to observe possible F‐actin bundling events by real‐time imaging. While low concentrations of hBin1 did not exhibit any bundling activity (Fig 4A, top panels), at 250 nM hBin1 single actin filaments frequently fused with other filaments to form bundles with mixed polarity (Fig EV4A). This effect was enhanced further by higher concentrations of hBin1. Twenty minutes after initiation of actin polymerization, higher concentrations of Bin1 resulted in a network of actin filament bundles (Fig 4A, lower panels). Previous studies demonstrated that the fluorescence intensity of the actin bundles directly correlates with the number of filaments per bundle 38. We exploited this to determine the number of filaments in each hBin1‐induced bundle. The number of filaments per bundle increased to approximately three (250 nM hBin1) and four (500 nM hBin1), but could not be increased further when adding 1,000 nM hBin1 to actin (Fig 4B). These data show that hBin1 induces actin bundling in a concentration‐dependent manner.
Figure 4. hBin1 can bundle actin filaments.
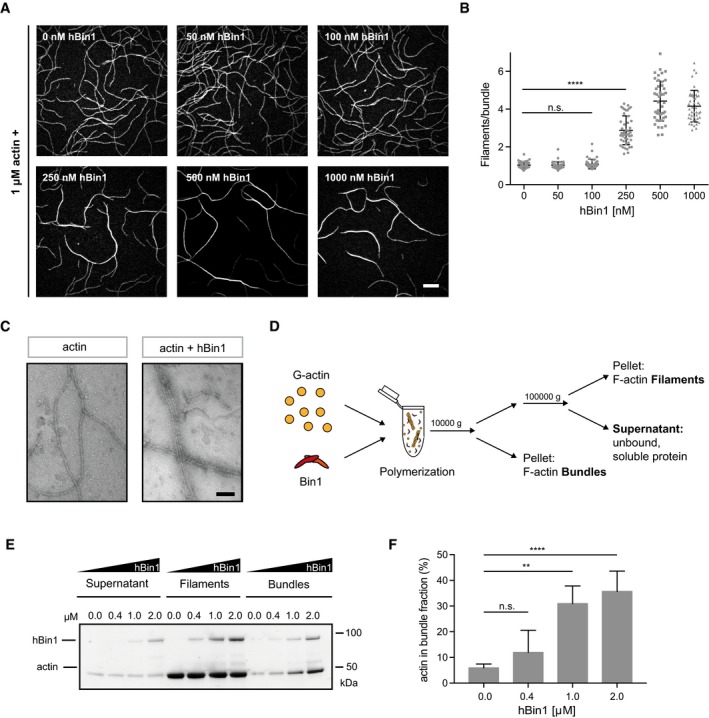
- Concentration‐dependent actin bundling induced by hBin1 (isoform 1). 1 μM actin (10% DY‐549 labeled) was polymerized for 20 min in the presence or absence of different concentrations of hBin1 (isoform 1) in TIRF buffer. Scale bar = 10 μm.
- Number of filaments/bundle formed in the presence of different hBin1 (isoform 1) concentrations. For each condition, 45 bundles were analyzed using fluorescence intensity measurements. Statistically significant differences are indicated (one‐way ANOVA, ****P < 0.0001, n.s. = not significant). The error bars represent mean ± SD. For reasons of clarity, not all significances are indicated.
- Negatively stained electron micrographs of hBin1 induced actin bundling. 4 μM G‐actin was polymerized alone or together with hBin1 (isoform 1; 2 μM) before diluting the sample 1:10. Scale bar = 50 nm.
- Experimental setup to monitor a possible bundling activity of Bin1 via a low‐ and high‐speed centrifugation protocol. G‐actin was polymerized in the presence of hBin1 (isoform 1) for 20 min. In order to separate bundles from filaments, the sample was centrifuged at 10,000 g for 15 min. The supernatant containing filaments and monomeric protein was further separated by high‐speed centrifugation 100,000 g for 1.5 h to spin down filamentous actin.
- hBin1 induces actin bundling as monitored by co‐sedimentation assays. 4 μM G‐actin was polymerized in the presence of different concentrations of hBin1 (isoform 1) for 20 min. Actin fractions were separated as illustrated in (D). SDS–PAGE of the fractions after Coomassie Blue staining is shown.
- Densitometric quantitation of the percentage of actin found in the bundle fraction of (E) at the indicated hBin1 concentrations. Statistically significant differences are indicated (one‐way ANOVA, **P < 0.01, ****P < 0.0001, n = 3). The error bars represent mean ± SD. For reasons of clarity, not all significances are indicated.
Figure EV4. hBin1, hBin1‐SNAP, and hBAR induce actin bundling.
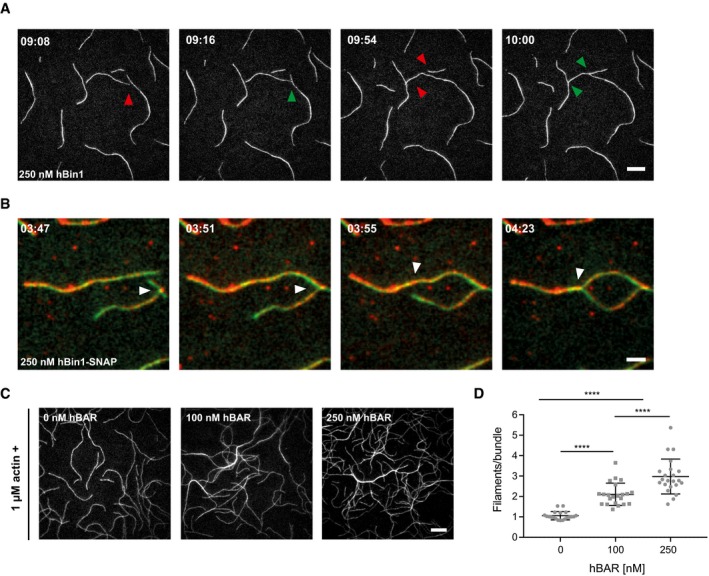
- Time course analysis visualizes hBin1 (isoform 1) induced actin bundling events visualized by TIRFM. Time is in minutes and seconds. Conditions are the same as in Fig 4A, hBin1 (isoform 1) = 250 nM. Red arrowheads indicate two filaments just before fusion and green arrowheads the filaments after the bundling event. Scale bar = 10 μm.
- Time course analysis visualizes hBin1‐SNAP594 (red) associating with actin filaments (green) and inducing actin bundling events visualized by dual‐color TIRFM. Time is in minutes and seconds. Conditions are the same as in Fig 4A, hBin1 (isoform 1) = 250 nM. White arrowheads indicate two filaments just before and after fusion. Scale bar = 4 μm.
- Concentration‐dependent actin bundling induced by hBAR. 1 μM actin (10% ATTO‐488 labeled) was polymerized for 20 min in the presence or absence of different concentrations of hBAR. Scale bar = 10 μm.
- Number of filaments/bundle formed in the presence of different hBAR concentrations. For each condition, 25 bundles were analyzed using fluorescence intensity measurements. Statistically significant differences are indicated (one‐way ANOVA, ****P < 0.0001). The error bars represent mean ± SD.
To better understand the role of hBin1 in actin bundling, we employed two‐color TIRF microscopy to directly visualize fluorescently labeled hBin1 molecules interacting with actin filaments. hBin1‐SNAP readily associated with actin filaments and remarkably adjacent filaments often fused at places where hBin1 was concentrated (Fig EV4B). We repeated the TIRF assay with hBAR in order to test whether the BAR domain is sufficient to induce actin bundling. Interestingly, the hBAR domain was not only able to bundle F‐actin, but this was also achieved at lower concentrations than needed for the full‐length hBin1 (Fig EV4C and D). To obtain an insight in the morphology of hBin1‐induced bundles, negative staining EM was employed. By using the same experimental conditions as in our TIRF assay, we detected hBin1 induced bundles, which were not observed in the absence of hBin1 (Fig 4C). These bundles consisted mostly of 2–5 filaments, which is comparable to our quantification of bundles as determined using TIRF. To confirm the bundling activity of hBin1 in an additional experimental setup, we used low‐ and high‐speed sedimentation protocols to first pellet bundles and subsequently pellet filaments (Fig 4D). Increasing the amount of hBin1 resulted in a concentration‐dependent increase in actin found in the bundle fraction collected at low centrifugation speeds (Fig 4E and F), in line with the results of our TIRFM study.
hBin1 stabilizes tau‐induced actin bundling
Tau is a microtubule‐binding protein, which has been proposed to also bind actin in order to link both the microtubule‐ and the actin network 43. Interestingly, tau has also been suggested to have actin bundling activity 44, 45, and in AD patients, the abundance of actin–cofilin‐rich rods correlates well with the extent of tau‐toxicity 23. We therefore asked whether the extent of tau‐induced actin bundle formation could be altered by changes in hBin1 levels. To answer this question, we purified recombinant human tau (0N4R) from Escherichia coli and tested its actin bundling activity as described above via TIRFM. Interestingly, we observed tau‐dependent bundling of actin already at 100 nM tau (Fig 5A) and even thicker bundles could be observed when actin was co‐polymerized with 300 nM tau (Fig 5A). At a concentration of 100 nM tau, actin bundles consisted of 2–6 filaments, whereas 300 nM tau resulted in bundles of up to 13 filaments (Fig 5B). Low‐speed co‐sedimentation assays could again confirm this dose‐dependent bundling of actin filaments (Fig 5C). Specifically, up to 80% of total actin could be found in the bundle fraction when co‐polymerized with 1 μM tau (Fig 5D). The bundling activity of tau in our experimental setup is in good agreement with results from previous work 43. We next investigated which effect a mixture of hBin1 and tau would have on actin bundling. Here, low‐ and high‐speed co‐sedimentation assays were used again to separate the actin fraction after co‐polymerizing it with hBin1, tau or hBin1, and tau. We could not observe a significant difference between the amount of actin in the pellet fraction when it was bundled by tau compared to a bundling by tau and hBin1 together (Fig 5E and F). Therefore, it seems that these two proteins are neither inhibiting each other nor working in a synergistic fashion. However, we reasoned that additional stabilization of tau‐induced actin bundles due to the addition of hBin1 might change the stability of the tau‐induced bundles and subjected the bundles to cofilin‐induced disassembly (Fig 5G). Pyrene‐labeled actin was co‐polymerized with tau to form bundles. These were pre‐incubated with different concentration of hBin1, before exposing them to cofilin. Tau‐mediated actin bundling by itself already stabilized F‐actin to some extend against cofilin‐induced disassembly. Most interestingly, hBin1 was able to further stabilize the tau‐induced actin bundles in a concentration‐dependent manner (Fig 5G). The half‐time of disassembly could be almost doubled when adding Bin1 to the actin bundles (Figs 5H and EV5A). Therefore, hBin1 is not only able to stabilize filaments but also bundles formed by the pathological action of tau. This discovery therefore provides a new possible mechanistic link between tau pathology and hBin1 as an AD risk factor.
Figure 5. hBin1 stabilizes tau‐induced actin bundles.
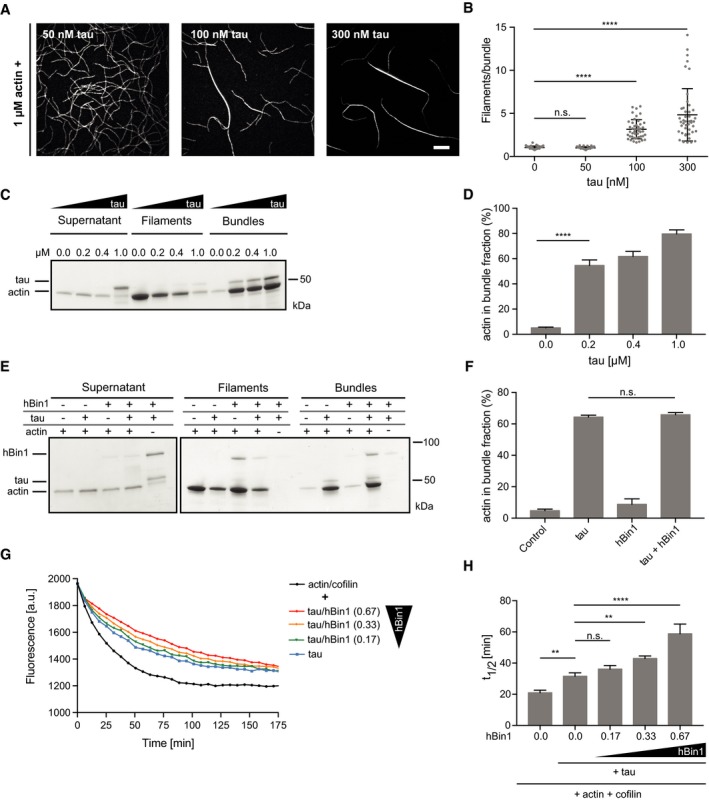
- Concentration‐dependent actin bundling induced by tau. 1 μM actin (10% DY‐549 labeled) was polymerized for 20 min in the presence of tau at the indicated concentrations. Scale bar = 10 μm.
- Number of filaments/bundle formed in the presence of different tau concentrations. For each condition, 45 bundles were analyzed using fluorescence intensity measurements. Statistically significant differences are indicated (one‐way ANOVA, n.s. = not significant, ****P < 0.0001). The error bars represent mean ± SD. For reasons of clarity, not all significances are indicated.
- Tau‐induced actin bundling monitored by co‐sedimentation assays. 4 μM actin was polymerized in the presence of different concentrations of tau for 20 min, and different fractions were separated as described in Fig 4D. SDS–PAGE of the fractions stained with Coomassie Blue is shown.
- Densitometric quantification of the percentage of actin found in the bundle fraction of (C) at the indicated tau concentrations. Statistically significant differences are indicated (one‐way ANOVA, ****P < 0.0001, n = 3). The error bars represent mean ± SD. For reasons of clarity, not all significances are indicated.
- hBin1 does not change the amount of tau‐induced actin bundles. Results of co‐sedimentation assay separating different actin fractions after polymerization 4 μM actin in the presence or absence of 0.2 μM tau and 1 μM hBin1 (isoform 1).
- Densitometric quantification of the percentage of actin found in the bundle fraction of (E) (one‐way ANOVA, n.s. = not significant, n = 3). The error bars represent mean ± SD.
- Effect of hBin1 on the disassembly rate of tau‐induced actin bundles. Pyrene‐labeled actin was polymerized in the presence of tau before subjecting it to depolymerization in the presence or absence of cofilin and hBin1 (isoform 1) at indicated molar ratios. Molar ratios of actin/cofilin/tau: 1:0.23:0.07. Disassembly was monitored using time‐based fluorimetry.
- Calculated half‐time of actin disassembly from (G) after one‐phase exponential decay fitting (Fig EV3). Statistically significant differences are indicated (one‐way ANOVA, n.s. = not significant, **P < 0.01, ****P < 0.0001, n = 4). The error bars represent mean ± SD. For reasons of clarity, not all significances are indicated.
Figure EV5. Biochemical validation of dBin1 downregulation in fly brains.
- Phalloidin staining of whole‐mount brain (22 days old) dissected from tauR406W transgenic flies and control flies. The zoom‐in images represent one single section in the middle of the central body of the fly brain, and the white arrows highlight actin rods. Scale bar of brain overview: 100 μm and zoom‐in: 25 μm.
- Knockdown efficiency of different dBin1 RNAi lines. The dBin1 lines were expressed pan‐neuronally under the Elav promoter, and fly heads were subjected to protein extraction 10 days post‐eclosion. Samples were resolved by SDS–PAGE and immunoblotted.
Loss of dBin1 decreases actin rods in tauR406W flies
To explore the effect of Bin1 on tau‐induced actin bundles in vivo, we turned to a well‐established Drosophila model of tau‐induced neurodegeneration 46. Specifically, expression of a disease‐associated mutant tau variant (R406W) in Drosophila neurons has been shown to induce the accumulation of F‐actin and the formation of actin‐rich rods in the fly brain 47. These rod‐like inclusions are phalloidin‐positive in whole‐mount fly brains (Fig EV5B). As we have shown the conserved function of the BAR domain, we reasoned that changes in dBin levels may alter the accumulation of actin in this in vivo model. To determine whether changes in dBin1 levels have an impact on these tau‐induced actin rods, we assessed the number of actin rods in a defined area in the midbrains of flies with different levels of dBin1 protein. Here, we used two dBin1 dsRNA constructs to modulate dBin1 levels, whereby the expression of dBin1 RNAi #1 resulted in a more efficient knockdown of dBin1 compared to RNAi line #2 (Fig EV5C). In control fly brains without human tau overexpression, very few actin rods were detected (Fig 6A). However, in agreement with previous reports, pan‐neuronal expression of tauR406W led to the development of significant amounts of actin rods in the midbrain 47 (Fig 6A). Following our biochemical data, lowering the levels of dBin1 might reduce the stability of these tau‐induced actin bundles and therefore foster their clearance. Indeed, expression of the strong RNAi line #1 significantly decreased the number of actin‐rich rods typically found in the midbrain of tau transgenic flies (Fig 6A and B). Intriguingly, the number of tau‐induced actin inclusions decreased by 50% when dBin1 was downregulated. We also observed a small decrease of actin rods in flies expressing the second, weaker RNAi line #2; however, this reduction did not reach significance. These results therefore directly link changes in dBin1 protein levels with tau‐induced pathology, suggesting that downregulation of Bin1 also modulates tau‐mediated pathobiological actin dynamics in vivo.
Figure 6. dBin1 modulates actin dynamics in vivo .
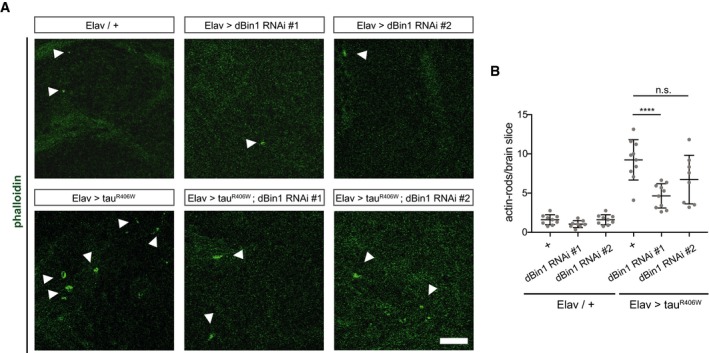
- TauR406W expressing Drosophila brains with endogenous dBin1 levels display more actin rods in the brain than in the brains of tauR406W transgenic flies in which dBin1 was downregulated. Phalloidin staining of whole‐mount brain (22 days old), dissected from tauR406W transgenic flies. Here, a zoom‐in is displayed (scale bar = 10 μm). White arrowheads indicate phalloidin‐positive actin inclusions.
- Quantification of actin rods per brain slice. Statistically significant differences are indicated (one‐way ANOVA, n.s. = not significant, ****P < 0.0001, n = 8–10). The error bars represent mean ± SD.
Discussion
The BAR domain protein superfamily has emerged, not only as determinants of membrane curvature, but also as linkers between membranes and the membrane‐associated cytoskeleton to coordinate membrane remodeling events 14. Directed actin polymerization beneath the plasma membrane is crucial for mechanical support and a driving force for movement 1. It can be triggered by Rho‐family GTPases and by activation of members of the WASP family 48, 49; both can be regulated by various BAR domain proteins. Previous observations indicated that the N‐BAR protein Bin1 may integrate membrane curvature and actin dynamics but mechanistic details have not been studied in detail. For example, the yeast Bin1 orthologous Rvs161/167 are involved in actin dynamics under environmental stress 50, 51 and Drosophila dBin1 colocalizes with actin in photoreceptor neurons 52. Moreover, hBin1 interacts with N‐WASP via its SH3‐domain which can lead to an induction of actin polymerization by the Arp2/3 complex 48 and the interaction with N‐WASP in myofibres is also crucial for nuclear positioning 35, 53.
Here, we show for the first time that hBin1 can directly bind to actin, an interaction that is mediated through its BAR domain. Remarkably, the neuronal isoform 1 of hBin1 as well as the conserved Drosophila orthologue of Bin1 was able to interact with F‐actin, indicating a conserved function. Our mass spectrometry experiments identified that most crosslinks connect the H0 helix and BAR domain of hBin1 to actin. This result is in line with previous reports identifying the BAR domain of pacsin2 and PICK1 as sufficient to interact with actin. Like PICK1, Bin1 is a member of the N‐BAR family, whereas pacsin2 belongs to the family of F‐BAR proteins. But since other N‐BAR members as endophilin have been shown not to bind to actin 5, the structural binding determinants need to be studied in further detail. We cannot exclude additional actin‐binding sites or cooperative binding events apart from the BAR domain, since the K d of hBAR was slightly lower than for hBin1. However, the SH3 domain alone does not contribute significantly to hBin1's actin‐binding ability under our experimental conditions as suggested previously 35. This discrepancy might be a result from different Bin1 isoforms tested and will be addressed in more detail in future studies. The direct binding of the BAR domain of Bin1 to actin also raises the question of how this interaction is regulated. It has already been demonstrated that there is a weak competition of liposomes and F‐actin for binding to the F‐BAR domain 5. In addition to lipid binding, post‐translational modifications of Bin1 and other protein interactions such as the binding to WASP could possibly modulate the Bin1‐actin interaction.
hBin1 and its BAR domain do not only bind to F‐actin, but also stabilize F‐actin in a depolymerization assays. This stabilization was also observed for pacsin2 at higher concentrations 5. In contrast to pacsin2, hBin1 binding to actin filaments could partly protect them from cofilin‐mediated disassembly. This difference indicates that the binding site and affinity might vary for each BAR domain protein. The stabilization of actin filaments has been observed for many actin‐binding proteins, in particular for actin bundling proteins 54. Notably, in vitro TIRFM and EM identified also an actin bundling activity of hBin1. Actin crosslinking/bundling requires at least two actin‐binding sites; dimerization of hBin1 could be a prerequisite for the induction of bundling. Although no actin bundling activity has been identified so far for pacsin2 and PICK1, filopodia‐inducing IRSp53 (insulin receptor phosphotyrosine 53 kDa substrate) can bind both membranes and bundles actin via its IMD domain, which has weak similarity with to a BAR domain 55, 56, 57. Whether the actin bundling activity of Bin1 is a physiological function or a pathological consequence to excess F‐actin is currently unclear.
hBin1 has been identified as the second most prevalent risk factor for late‐onset Alzheimer's disease (LOAD) 58. It has been hypothesized to be linked directly to two prominent hallmarks of Alzheimer's disease, both tau 21, 28 and Aβ 27, 26. hBin1 can directly bind tau via its SH3 domain and the Bin1‐tau complexes colocalize with the actin cytoskeleton in primary neurons 21, 59. dBin1 modulates tau‐induced neurotoxicity in Drosophila 21, 28, while in human cell culture studies, downregulation of hBin1 promotes tau propagation 60.
Here, we provide a mechanistic link between Bin1 and tau, by identifying actin dynamics as a possible causative connection. Tau overexpression has been shown to lead to the formation of actin rods in Drosophila models as well as in AD patient brains tau‐toxicity correlates with the extend of actin aggregates 32, 47. Moreover, mutant forms of tau were recently identified to increase F‐actin levels at the synapse and crosslink actin to synaptic vesicles, thus interfering with presynaptic functions 61. Tau itself has been demonstrated to bind and bundle actin filaments in vitro via its microtubule‐binding domain and proline‐rich domain 44, 45, 62. Therefore, it was suggested that tau might act as a linker between actin and microtubule cytoskeleton under physiological conditions 43. However, it is still unclear whether actin accumulations observed under pathological conditions in AD patients are only derived by tau‐induced actin bundling, since tau rarely colocalizes with these rods in post‐mortem human brain tissues 32, 63.
Given that Bin1 and tau interact directly and both also bind to F‐actin, we hypothesized that Bin1 might play a role in tau‐induced actin pathology. Although we could assign bundling activities to hBin1 as well as tau, no cooperative bundling activity of these two proteins could be observed, as reported in a study using fascin and α‐actinin 64. However, we obtained evidence that hBin1 can stabilize the tau‐induced actin bundles in vitro. Remarkably, dBin1 downregulation could be confirmed to have an effect on tau‐induced actin rods in vivo. Downregulation of dBin1 in Drosophila neurons decreased the amount of actin rods found in the brain. This in vivo phenotype is consistent with the proposed in vitro mechanism that Bin1 stabilizes actin bundles. Due to tau's involvement in the formation of the actin rods, its direct protein interactor hBin1 might be mislocalized toward tau‐induced actin bundles, thus further stabilizing them. However, we cannot exclude that Bin1 could modify actin dynamics in vivo via different mechanisms, for example by regulating the association with various actin regulators like N‐WASP. Future experiments will need to determine whether Bin1 alone is sufficient to stabilize actin rods in vivo and how this is mechanistically associated with tau pathology.
Several modifier screens and genomewide associations studies have been conducted in recent years in order to unravel novel pathways involved in AD 58, 65. Interestingly, many hits from these tau modifier screens also seem to have a role in axonal transport and actin dynamics, highlighting the actin cytoskeleton as an important pathway for further study 58, 66. For example, two independent screens identified the loss of Cheerio, an actin crosslinking protein and the Drosophila orthologue of the mammalian Filamin‐A, as an enhancer of tau‐toxicity 66, 67. Other identified modifiers from the actin field included Paxillin, CD2AP, and myosin 6 (MYO6) 65, 66, 68. Although MYO6 and Filamin‐A colocalize with tau tangles in post‐mortem brain stainings, their functional connection to AD needs to be further studied 69.
In conclusion, we have identified Bin1 as a novel and direct modulator of actin dynamics. This work provides important mechanistic information about the complex regulation of the actin cytoskeleton at the membrane and highlights Bin1 as a dual coordinator of membrane shaping and actin dynamics. Cytoskeleton impairments are emerging as a hallmark in AD and in neurodegeneration in general. Thus, a misbalance of functional membrane shaping versus actin bundle recruitment of Bin1 might contribute to AD pathology. It will be essential to study neuronal dysregulation of actin dynamics and the possible involvement of tau, Bin1 and other proteins, in order to decipher the molecular pathways leading to cytoskeleton impairments in AD.
Materials and Methods
Constructs and protein purification
hBin1 (isoform 1), hBAR, hSH3, hBin1‐SNAP, dBAR, dBin1 (isoform A), and human tau (0N4R) were all expressed with an Ulp1‐cleavable N‐terminal His6‐SUMO tag from a modified pCold vector (Clontech) in E. coli Rosetta expression host (gift from B. Bukau) by induction with 0.3 mM isopropyl‐1‐thio‐β‐D‐galactopyranoside at 16°C overnight (o/n). The proteins were purified using Ni‐IDA matrix (Protino; Macherey‐Nagel); eluted material containing 250 mM imidazole was supplemented with His6‐Ulp1 protease o/n at 4°C. Purified proteins were separated from Ulp1 by size exclusion using a Superdex200 16/60 column (GE Healthcare).
All Bin1 proteins were stored in LEW50 buffer (50 mM NaH2PO4, 50 mM NaCl, pH 8) at −80°C. Human tau (0N4R) was stored in HK buffer (25 mM HEPES‐KOH, 150 mM KCl, 2% glycerol, 2 mM DTT, pH 7.5) at −80°C. Before using proteins for any biochemical assay, they were subjected to a buffer exchange in the appropriate buffer to minimize buffer effects. Actin for TIRFM was purified from rabbit skeletal muscle according to Spudich and Watt 70 and was labeled on Cys374 with DY‐549 (Dyomics GmbH) or ATTO‐488 (Atto‐tec GmbH). GFP–cofilin was expressed in and purified from Rosetta 2 (Novagen) as previously described 71.
Actin filament co‐sedimentation assays
Actin co‐sedimentation assays were performed using the Actin Binding Protein Spin‐Down Assay Biochem Kit (Cytoskeleton) with slight modifications. Human‐platelet non‐muscle actin (APHL99, Cytoskeleton) was diluted in G‐Buffer (5 mM Tris–HCl pH 8.0, 0.2 mM CaCl2, 0.2 mM ATP) and allowed to depolymerize on ice for 1 h before centrifugation for 1 h at 100,000 g to remove residual non‐polymerized material. Actin was polymerized at either 8.4 or 20 μM for 1.5 h in polymerization buffer (50 mM KCl, 2 mM MgCl2 and 1 mM ATP in 10 mM Tris, pH 7.5). Actin was further mixed with the test proteins at the indicated concentrations, incubated for 30 min at room temperature (RT), and pelleted at 100,000 g for 1.5 h at 24°C. Equivalent volumes of supernatant and pellet fractions were analyzed by SDS–PAGE and Coomassie staining. SDS–PAGEs were quantified using the Image StudioLite 5.2.5 software (LI‐COR Biosciences). For co‐sedimentation experiments with increasing concentrations of KCl, dBAR in polymerization buffer supplemented with KCl of a final concentration of 50, 150, or 250 mM was added to polymerized actin and incubated for 30 min before centrifugation. For determination of the dissociation constant (K d), the amount of hBin1 (isoform 1) in the pellet fraction was fitted to a nonlinear regression and the amount of hBAR in the pellet fraction was fitted to a nonlinear regression with a Hill slope using Prism 7 (Graph Pad software).
G‐actin‐binding assay
G‐actin‐binding assays were performed using the G‐actin‐binding toolkit (Hypermol, 8020‐01) according to the manufacturer's instructions. Briefly, hBin1 and profilin (Cytoskeleton, PRO1‐A) were rebuffered into 1× MonoMix buffer, and 50 μl of each protein with a concentration of 1.25 μM was incubated with 50 μl of G‐actin beads and 400 μl of MonoMix buffer for 1 h at RT under constant agitation. Samples were centrifuged at 6,000 g for 4 min to pull down actin beads and proteins bound to it. Beads were washed five times with 1 ml of MonoMix buffer, and bound proteins were eluted by boiling the beads for 2 min at 95°C in 30 μl 1× Lämmli buffer. Equivalent volumes (30 μl) of input fraction, unbound fraction, and elution fraction were analyzed by SDS–PAGE and Coomassie staining.
Actin bundling assays
For low‐speed pelleting assays, 4 μM rabbit muscle actin was polymerized for 30 min in the presence of hBin1 (isoform 1) and tau at the indicated concentrations in polymerization buffer. Actin bundles were separated via centrifugation for 15 min at 10,000 g. The supernatant was subjected to a high‐speed centrifugation step (1.5 h at 100,000 g) to separate remaining actin filaments from soluble proteins. Equivalent volumes of supernatant and the two pellet fractions (filaments and bundles) were analyzed by SDS–PAGE and Coomassie staining. SDS–PAGEs were quantified using Image StudioLite software (LI‐COR Biosciences).
Crosslinking studies
The crosslinking was carried out as described in 5. Briefly, F‐actin and hBin1 (isoform 1) or dBAR were mixed at the indicated concentrations in 10 mM PIPES, 50 mM KCl, 2 mM MgCl2, pH 6.8 and incubated for 30 min, before adding the EDC crosslinker (1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide hydrochloride; Thermo Fisher Scientific) at a final concentration of 1 mM. After 30 min of crosslinking, the reaction was stopped by adding SDS‐loading buffer [60 mM Trizma base (pH 6.8), 2% (w/v) SDS, 5% (v/v) mercaptoethanol, 0.01% (w/v) Brilliant blue], and samples were analyzed via SDS–PAGE and Coomassie Blue staining.
Mass‐spectrometric analysis
Coomassie‐stained bands representing the crosslinked products were cut; then, the gel slices were transferred to a 96‐well plate and automatically reduced, alkylated, and digested with trypsin as described in 72. Peptides were extracted from the gel pieces with 50% acetonitrile/0.1% trifluoroacetic acid (TFA), concentrated nearly to dryness in a SpeedVac vacuum centrifuge and diluted to a total volume of 30 μl with 0.1% TFA. 10 μl of the sample was analyzed by a Dionex UltiMate 3000RSLCnano HPLC system (Thermo Scientific) coupled to a LTQ Orbitrap Elite mass spectrometer (Thermo Scientific) equipped with a Sonation column oven (set to 50°C). Samples were directly loaded onto a self‐pulled and self‐packed analytical emitter column (Reprosil Pur‐AQ C18, 3 μm, 100 μm i.d. × 400 mm) at a flow rate of 550 nl/min. Afterward, peptides were eluted into the mass spectrometer at a flow rate of 300 nl/min using a gradient. One Orbitrap survey scan was followed by 10 information‐dependent product ion scans in the Orbitrap. 3+, 4+ and higher charged ions were selected for data dependent HCD fragmentation with dynamic exclusion enabled. For the identification of crosslinked peptides, the uninterpreted MS/MS spectra were compared against a small database containing the actin and Bin1 (isoform 1) sequences using StavroX 3.6.0 73. StavroX was set to use trypsin as proteolytic enzyme (mis. cleavage: 4; blocked as XL‐site: K); EDC (comp: ‐H2O, Site 1: K, N‐term, Site 2: E, D, N‐term, mass: −18.0106 Da) was defined as crosslinker; methionine oxidation (147.0354 Da) was set as variable modification and carbamidomethylation of cysteine (57.0215 Da) as fixed modification. Mass tolerance was set to 10 and 20 ppm for MS and MS/MS, respectively. The search range was restricted from 400 to 6,000 Da, a S/N ratio of 2 was set and deisotoping enabled. No neutral losses were considered. Peptide score thresholds were automatically calculated using the target‐decoy approach with a FDR of 0.05. Visualization of the identified crosslinks was done using the xVis Crosslink Analysis Webserver 74.
Negative stain electron microscopy
4 μM F‐actin was incubated in the absence or presence of 4 μM hBin1 (isoform 1) for 30 min at RT. The samples were diluted 10‐fold, and 3 μl of the dilution was immediately pipetted onto carbon‐coated grids, where they were allowed to adsorb for 30 s before excess protein was blotted off. For identifying Bin1‐induced actin bundles, 4 μM G‐actin was polymerized in the presence of 4 μM hBin1 for 2 h, before diluting the samples 1:10 and transferring them on the grids. These were then stained with 3% aqueous uranyl acetate solution for 1 min and washed three times with distilled water, air‐dried, and imaged on an EM900 transmission electron microscope (Zeiss) at an accelerating voltage of 80 kV. Images were acquired at 85,000 magnification, resulting in a pixel (px) size of 0.6 nm/px.
Pyrene–actin assays
The pyrene actin polymerization and depolymerization assays were conducted using the Actin Polymerization Biochem Kit protocols (Cytoskeleton) with slight modifications. For polymerization assays, pyrene‐labeled actin (rabbit skeletal muscle, AP05, Cytoskeleton) was diluted in G‐buffer and allowed to depolymerize on ice for 1 h before centrifugation for 30 min at 21,000 g to remove remaining filaments. Then, 2.5–3 μM pyrene actin was polymerized in the presence or absence of hBin1 (isoform 1) and cofilin (CF01, Cytoskeleton) at the indicated molar ratios in 0.25× polymerization buffer. For dilution‐induced actin disassembly, 12 μM pyrene actin was polymerized for 1 h in the dark by adding 0.25× polymerization buffer. The F‐actin was then diluted four times in G‐buffer in the presence or absence of Bin1/hBAR and cofilin at the indicated molar ratios. For disassembly experiments using tau‐induced bundles, 12 μM pyrene actin was polymerized in the presence or absence of tau at indicated molar ratios for 1.5 h by adding 1× polymerization buffer before dilution. hBin1 (isoform 1) and cofilin were added at indicated molar ratios. Pyrene fluorescence was measured using a FLUORstar Omega plate reader (BMG LABTECH) with excitation of 355 nm and emission of 405 nm at RT. Background fluorescence of the buffer was subtracted from the pyrene actin fluorescence. For the halftime determination of the depolymerization experiments, a one‐phase exponential decay was fit using Prism 7 (Graph Pad software).
In vitro TIRF microscopy
Total internal reflection fluorescence microscopy (TIRFM) was essentially performed as described previously 75 with the following modifications: The experiments were conducted on a Nikon Eclipse TI‐E inverted microscope equipped with a TIRF Apo 100× objective and a motorized stage and were recorded by an Ixon3 897 EMCCD camera (Andor). hBin1‐SNAP was labeled with a 2.5‐fold excess of SNAP‐surface 549 (New England Biolabs) at 4°C in LEW 50 buffer supplemented with 1 mM DTT under slight agitation ON before it was rebuffered in polymerization buffer. hBin1, hBAR, hBin1‐SNAP594, tau, and GFP–cofilin were pre‐diluted in 1× TIRF buffer (20 mM imidazole pH 7.4, 50 mM KCl, 1 mM MgCl2, 1 mM EGTA, 25 mM DTT, 0.5 mM ATP, 15 mM glucose, 2.5 mg/ml methylcellulose (4,000 cP), 20 μg/ml catalase, 100 μg/ml glucose‐oxidase) prior to initiation of the assay. The assays were started by adding G‐actin (1 μM final concentration, 10% DY‐549‐or or ATTO‐488 labeled at Cys374) and flushing the mixtures into the pre‐coated flow chambers as described in 76. Images were captured with exposure times of 50 ms every 2 s at two positions of the coverslip for at least 10 min. The pixel size corresponded to 0.159 μm for the 100× objective. Images were background subtracted using a 50‐pixel rolling ball radius and a slight Gaussian blur filter of 0.7 pixel in Fiji software 77.
The fluorescence intensity of actin bundles was measured using the Plot Profile tool in Fiji 77 software. Bundles were normalized to a single actin filament from the same experiment to analyze the number of filaments per bundle.
Drosophila transgenic lines and husbandry
Flies were raised on standard cornmeal and molasses medium. Crosses were kept for 3 days at 25°C and then shifted to 29°C (60% rH). The progeny were collected in a 24 h window, and mated females were kept at 29°C. All flies were age matched when used for experiments. Pan‐neuronal expression of UAS‐tauR406W 46 and RNAi was achieved using the X‐Elav‐Gal4 promoter. Bin1 RNAi #1 was obtained from the Bloomington Drosophila stock center (stock number 39015), and Bin1 RNAi #2 was obtained from the Vienna Drosophila RNAi Center (VDRC ID 9264).
Fly head protein extraction and immunoblotting
Frozen Drosophila heads were homogenized in extraction buffer [50 mM Tris (pH 7.5), 2 mM sodium orthovanadate, 50 mM sodium fluoride, 50 mM β‐glycerophosphate disodium salt hydrate, 1× phosphatase inhibitor (Roche), 1× protease inhibitor (Santa Cruz Biotechnology Inc.), 150 mM sodium chloride, 2 mM magnesium chloride, 1% (v/v) Triton X‐100, and 1% (w/v) SDS]. After sonication for 15 min and further incubation on ice for 15 min, samples were centrifuged for 15 min at 21,000 g at 4°C. Overall protein concentrations were determined using Lowry quantification (DC™ Protein Assay, Bio‐Rad) and adjusted accordingly. Samples were boiled for 5 min in 1× Laemmli sample buffer before analyses by 4–20% gradient SDS–PAGE. After transfer, nitrocellulose membranes were subsequently blocked in 5% (w/v) milk powder in phosphate‐buffered saline (PBS, AppliChem) containing 0.1% (v/v) Tween‐20 for 1 h at room temperature (RT). Membranes were incubated with primary antibodies o/n at 4°C against anti dBin1 (1:350, gift from G. Boulianne) and α‐tubulin (1:300, AA4.3, DSHB). After washing, membranes were incubated with HRP‐conjugated secondary antibodies for 2 h at RT. Blots were developed with an enhanced chemiluminescent substrate (Thermo Scientific).
Immunohistochemistry and actin‐rod analysis
Adult flies were fixed in 4% paraformaldehyde (PFA) for 2 h at RT before brain dissection in PBS. Dissected brains were blocked in 5% (v/v) fetal bovine serum (FBS, biochrom) in PBST (PBS + 0.5% (v/v) Triton‐X100) for 2 h at RT before incubating them in the same buffer containing CF488‐A phalloidin (Biotium; 1:250 dilution) for 24 h at 4°C. The brains were then washed 3× in PBST, mounted in Vectashield (Biozol), and imaged using a Zeiss LSM 710 laser scanning confocal microscope and a 63× objective. The number of actin‐rich rods was determined by counting all rod‐shaped and round structures that positively stained for phalloidin in every single slice of the z‐stack from the Drosophila midbrain. Images were analyzed with Fiji. All images from Fig 6 are a single plane.
Author contributions
NMD conceived the project, performed the biochemical assays and the fly stainings, and analyzed the data. EN cloned and purified tau and Bin1 constructs. MW and SBr purified and labeled actin and performed the TIRFM experiments; PS performed the negative stain electron microscopy experiments. TK provided the recombinant tau. AAT, JF, and SBo helped with the experimental design and data interpretation. TRJ supervised the project. NMD and TRJ wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Movie EV1
Review Process File
Acknowledgements
We thank M. Feany, A. Mogk, B. Bukau, and G. Boulianne for reagents. Special thanks to R. Zahn for help with the protein purification. We thank the Core Facility for Mass Spectrometry & Proteomics (ZMBH Heidelberg) and especially R. Hardt for the mass‐spectrometrical analysis. This work was supported by the Chica and Heinz Schaller Foundation, a PhD Fellowship (by the Helmholtz International Graduate School for Cancer Research, DKFZ) to NMD and by the Deutsche Forschungsgemeinschaft (DFG) to JF (FA33010/1).
EMBO Reports (2017) 18: 2051–2066
References
- 1. Pollard TD, Cooper JA (2009) Actin, a central player in cell shape and movement. Science 326: 1208–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dawson JC, Legg JA, Machesky LM (2006) Bar domain proteins: a role in tubulation, scission and actin assembly in clathrin‐mediated endocytosis. Trends Cell Biol 16: 493–498 [DOI] [PubMed] [Google Scholar]
- 3. Frost A, Perera R, Roux A, Spasov K, Destaing O, Egelman EH, De Camilli P, Unger VM (2008) Structural basis of membrane invagination by F‐BAR domains. Cell 132: 807–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fricke R, Gohl C, Bogdan S (2010) The F‐BAR protein family Actin' on the membrane. Commun Integr Biol 3: 89–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kostan J, Salzer U, Orlova A, Toro I, Hodnik V, Senju Y, Zou J, Schreiner C, Steiner J, Merilainen J et al (2014) Direct interaction of actin filaments with F‐BAR protein pacsin2. EMBO Rep 15: 1154–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rocca DL, Martin S, Jenkins EL, Hanley JG (2008) Inhibition of Arp2/3‐mediated actin polymerization by PICK1 regulates neuronal morphology and AMPA receptor endocytosis. Nat Cell Biol 10: 259–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. David C, Mcpherson PS, Mundigl O, De Camillit P (1996) A role of amphiphysin in synaptic vesicle endocytosis suggested by its binding to dynamin in nerve terminals. Cell Biol 93: 331–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wigge P, Köhler K, Vallis Y, Doyle CA, Owen D, Hunt SP, McMahon HT (1997) Amphiphysin heterodimers: potential role in clathrin‐mediated endocytosis. Mol Biol Cell 8: 2003–2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pant S, Sharma M, Patel K, Caplan S, Carr CM, Grant BD (2009) AMPH‐1/Amphiphysin/Bin1 functions with RME‐1/Ehd1 in endocytic recycling. Nat Cell Biol 11: 1399–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Butler MH, David C, Ochoa G‐C, Freyberg Z, Daniell L, Grabs D, Cremona O, De Camilli P (1997) Amphiphysin II (SH3P9; BIN1), a member of the amphiphysin/Rvs family, is concentrated in the cortical cytomatrix of axon initial segments and nodes of ranvier in brain and around T tubules in skeletal muscle. J Cell Biol 137: 1355–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Razzaq A, Robinson IM, McMahon HT, Skepper JN, Su Y, Zelhof AC, Jackson AP, Gay NJ, O'Kane CJ (2001) Amphiphysin is necessary for organization of the excitation‐contraction coupling machinery of muscles, but not for synaptic vesicle endocytosis in Drosophila . Genes Dev 15: 2967–2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Prokic I, Cowling BS, Laporte J (2014) Amphiphysin 2 (BIN1) in physiology and diseases. J Mol Med 92: 453–463 [DOI] [PubMed] [Google Scholar]
- 13. Hong T, Yang H, Zhang S‐S, Cho HC, Kalashnikova M, Sun B, Zhang H, Bhargava A, Grabe M, Olgin J et al (2014) Cardiac BIN1 folds T‐tubule membrane, controlling ion flux and limiting arrhythmia. Nat Med 20: 624–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Frost A, Unger VM, De Camilli P (2009) The BAR domain superfamily: membrane‐molding macromolecules. Cell 137: 191–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McMahon HT, Gallop JL (2005) Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 438: 590–596 [DOI] [PubMed] [Google Scholar]
- 16. Micheva KD, Ramjaun AR, Kay BK, McPherson PS (1997) SH3 domain‐dependent interactions of endophilin with amphiphysin. FEBS Lett 414: 308–312 [DOI] [PubMed] [Google Scholar]
- 17. Yamada H, Padilla‐Parra S, Park S‐J, Itoh T, Chaineau M, Monaldi I, Cremona O, Benfenati F, De Camilli P, Coppey‐Moisan M et al (2009) Dynamic interaction of amphiphysin with N‐WASP regulates actin assembly. J Biol Chem 284: 34244–34256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ramjaun AR, McPherson PS (2002) Multiple amphiphysin II splice variants display differential clathrin binding: identification of two distinct clathrin‐binding sites. J Neurochem 70: 2369–2376 [DOI] [PubMed] [Google Scholar]
- 19. Adam J, Basnet N, Mizuno N, Zimmerberg J, Kozlov MM, Suetsugu S, Kurisu S, Takenawa T, McMahon HT, Boucrot E et al (2015) Structural insights into the cooperative remodeling of membranes by amphiphysin/BIN1. Sci Rep 5: 15452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tan M‐S, Yu J‐T, Tan L (2013) Bridging integrator 1 (BIN1): form, function, and Alzheimer's disease. Trends Mol Med 19: 594–603 [DOI] [PubMed] [Google Scholar]
- 21. Chapuis J, Hansmannel F, Gistelinck M, Mounier A, Van Cauwenberghe C, Kolen KV, Geller F, Sottejeau Y, Harold D, Dourlen P et al (2013) Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry 18: 1225–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Glennon EBC, Whitehouse IJ, Miners JS, Kehoe PG, Love S, Kellett KAB, Hooper NM (2013) BIN1 is decreased in sporadic but not familial Alzheimer's disease or in aging. PLoS One 8: e78806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Holler CJ, Davis PR, Beckett TL, Platt TL, Webb RL, Head E, Murphy MP (2014) Bridging integrator 1 (BIN1) protein expression increases in the Alzheimer's disease brain and correlates with neurofibrillary tangle pathology. J Alzheimers Dis 42: 1221–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Karch CM, Jeng AT, Nowotny P, Cady J, Cruchaga C, Goate AM (2012) Expression of novel Alzheimer's disease risk genes in control and Alzheimer's disease brains. PLoS One 7: e50976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haass C, Kaether C, Thinakaran G, Sisodia S (2012) Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2: a006270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ubelmann F, Burrinha T, Salavessa L, Gomes R, Ferreira C, Moreno N, Guimas Almeida C (2016) Bin1 and CD2AP polarise the endocytic generation of beta‐amyloid. EMBO Rep 18: 102–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miyagawa T, Ebinuma I, Morohashi Y, Hori Y, Young Chang M, Hattori H, Maehara T, Yokoshima S, Fukuyama T, Tsuji S et al (2016) BIN1 regulates BACE1 intracellular trafficking and amyloid‐β production. Hum Mol Genet 25: 2948–2958 [DOI] [PubMed] [Google Scholar]
- 28. Merlo P, Frost B, Peng S, Yang YJ, Park PJ, Feany M (2014) p53 prevents neurodegeneration by regulating synaptic genes. Proc Natl Acad Sci USA 111: 18055–18060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frost B, Götz J, Feany MB (2015) Connecting the dots between tau dysfunction and neurodegeneration. Trends Cell Biol 25: 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Spillantini MG, Goedert M (2013) Tau pathology and neurodegeneration. Lancet Neurol 12: 609–622 [DOI] [PubMed] [Google Scholar]
- 31. Bamburg JR, Bernstein BW (2016) Actin dynamics and cofilin‐actin rods in Alzheimer disease. Cytoskeleton 73: 477–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rahman T, Davies DS, Tannenberg RK, Fok S, Shepherd C, Dodd PR, Cullen KM, Goldsbury C (2014) Cofilin rods and aggregates concur with tau pathology and the development of Alzheimer's disease. J Alzheimers Dis 42: 1443–1460 [DOI] [PubMed] [Google Scholar]
- 33. Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJG, Evans PR, McMahon HT (2004) BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science 303: 495–499 [DOI] [PubMed] [Google Scholar]
- 34. Bhatia VK, Madsen KL, Bolinger P‐Y, Kunding A, Hedegård P, Gether U, Stamou D, Bagatolli L, Bendix P, Pedersen M et al (2009) Amphipathic motifs in BAR domains are essential for membrane curvature sensing. EMBO J 28: 3303–3314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. D'Alessandro M, Hnia K, Gache V, Koch C, Gavriilidis C, Rodriguez D, Nicot A‐S, Romero NB, Schwab Y, Gomes E et al (2015) Amphiphysin 2 orchestrates nucleus positioning and shape by linking the nuclear envelope to the actin and microtubule cytoskeleton. Dev Cell 35: 186–198 [DOI] [PubMed] [Google Scholar]
- 36. Kouyama T, Mihashi K (2005) Fluorimetry study of N‐(1‐Pyrenyl)iodoacetamide‐Labelled F‐Actin. Eur J Biochem 114: 33–38 [PubMed] [Google Scholar]
- 37. Carlier MF, Laurent V, Santolini J, Melki R, Didry D, Xia GX, Hong Y, Chua NH, Pantaloni D (1997) Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin‐based motility. J Cell Biol 136: 1307–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Breitsprecher D, Koestler SA, Chizhov I, Nemethova M, Mueller J, Goode BL, Small JV, Rottner K, Faix J (2011) Cofilin cooperates with fascin to disassemble filopodial actin filaments. J Cell Sci 124: 3305–3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chacón‐Martínez CA, Kiessling N, Winterhoff M, Faix J, Müller‐Reichert T, Jessberger R (2013) The switch‐associated protein 70 (SWAP‐70) bundles actin filaments and contributes to the regulation of F‐actin dynamics. J Biol Chem 288: 28687–28703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Suarez C, Roland J, Boujemaa‐Paterski R, Kang H, McCullough BR, Reymann A‐C, Guérin C, Martiel J‐L, De La Cruz EM, Blanchoin L (2011) Cofilin tunes the nucleotide state of actin filaments and severs at bare and decorated segment boundaries. Curr Biol 21: 862–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Andrianantoandro E, Pollard TD, Berry SR, Rice SA, Ross J, Blanchoin L, Pollard TD, Blanchoin L, Pollard TD, Mullins RD et al (2006) Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol Cell 24: 13–23 [DOI] [PubMed] [Google Scholar]
- 42. Blanchoin L, Pollard TD (1999) Mechanism of interaction of Acanthamoeba actophorin (ADF/Cofilin) with actin filaments. J Biol Chem 274: 15538–15546 [DOI] [PubMed] [Google Scholar]
- 43. Elie A, Prezel E, Guérin C, Denarier E, Ramirez‐Rios S, Serre L, Andrieux A, Fourest‐Lieuvin A, Blanchoin L, Arnal I (2015) Tau co‐organizes dynamic microtubule and actin networks. Sci Rep 5: 9964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moraga DM, Nuñez P, Garrido J, Maccioni RB (1993) A tau fragment containing a repetitive sequence induces bundling of actin filaments. J Neurochem 61: 979–986 [DOI] [PubMed] [Google Scholar]
- 45. He HJ, Wang XS, Pan R, Wang DL, Liu MN, He RQ (2009) The proline‐rich domain of tau plays a role in interactions with actin. BMC Cell Biol 10: 81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB (2001) Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293: 711–714 [DOI] [PubMed] [Google Scholar]
- 47. Fulga TA, Elson‐Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB (2007) Abnormal bundling and accumulation of F‐actin mediates tau‐induced neuronal degeneration in vivo . Nat Cell Biol 9: 139–148 [DOI] [PubMed] [Google Scholar]
- 48. Takenawa T, Suetsugu S (2007) The WASP–WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol 8: 37–48 [DOI] [PubMed] [Google Scholar]
- 49. Sit S‐T, Manser E (2011) Rho GTPases and their role in organizing the actin cytoskeleton. J Cell Sci 124: 679–683 [DOI] [PubMed] [Google Scholar]
- 50. Colwill K, Field D, Moore L, Friesen J, Andrews B (1999) In vivo analysis of the domains of yeast Rvs167p suggests Rvs167p function is mediated through multiple protein interactions. Genetics 152: 881–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sivadon P, Bauer F, Aigle M, Crouzet M (1995) Actin cytoskeleton and budding pattern are altered in the yeast rvs161 mutant: the Rvs161 protein shares common domains with the brain protein amphiphysin. Mol Gen Genet 246: 485–495 [DOI] [PubMed] [Google Scholar]
- 52. Zelhof AC, Bao H, Hardy RW, Razzaq A, Zhang B, Doe CQ (2001) Drosophila Amphiphysin is implicated in protein localization and membrane morphogenesis but not in synaptic vesicle endocytosis. Development 128: 5005–5015 [DOI] [PubMed] [Google Scholar]
- 53. Falcone S, Roman W, Hnia K, Gache V, Didier N, Lainé J, Auradé F, Marty I, Nishino I, Charlet‐Berguerand N et al (2014) N‐WASP is required for Amphiphysin‐2/BIN1‐dependent nuclear positioning and triad organization in skeletal muscle and is involved in the pathophysiology of centronuclear myopathy. EMBO Mol Med 6: 1455–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schmoller KM, Semmrich C, Bausch AR (2011) Slow down of actin depolymerization by cross‐linking molecules. J Struct Biol 173: 350–357 [DOI] [PubMed] [Google Scholar]
- 55. Scita G, Confalonieri S, Lappalainen P, Suetsugu S (2008) IRSp53: crossing the road of membrane and actin dynamics in the formation of membrane protrusions. Trends Cell Biol 18: 52–60 [DOI] [PubMed] [Google Scholar]
- 56. Yamagishi A, Masuda M, Ohki T, Onishi H, Mochizuki N (2004) A novel actin bundling/filopodium‐forming domain conserved in insulin receptor tyrosine kinase substrate p53 and missing in metastasis protein. J Biol Chem 279: 14929–14936 [DOI] [PubMed] [Google Scholar]
- 57. Millard TH, Bompard G, Heung MY, Dafforn TR, Scott DJ, Machesky LM, Fütterer K (2005) Structural basis of filopodia formation induced by the IRSp53/MIM homology domain of human IRSp53. EMBO J 24: 240–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Naj AC, Schellenberg GD (2017) Genomic variants, genes, and pathways of Alzheimer's disease: an overview. Am J Med Genet B Neuropsychiatr Genet 174: 5–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sottejeau Y, Bretteville A, Cantrelle F‐X, Malmanche N, Demiaute F, Mendes T, Delay C, Alves Dos Alves H, Flaig A, Davies P et al (2015) Tau phosphorylation regulates the interaction between BIN1's SH3 domain and Tau's proline‐rich domain. Acta Neuropathol Commun 3: 58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Calafate S, Flavin W, Verstreken P, Moechars D (2016) Loss of Bin1 promotes the propagation of tau pathology. Cell Rep 17: 931–940 [DOI] [PubMed] [Google Scholar]
- 61. Zhou L, Mcinnes J, Wierda K, Holt M, Herrmann AG, Rosemary J, Wang Y, Swerts J, Beyens J, Miskiewicz K et al (2017) Tau association with synaptic vesicles causes presynaptic dysfunction. Nat Commun 8: 1–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Roger B, Al‐Bassam J, Dehmelt L, Milligan RA, Halpain S (2004) MAP2c, but not tau, binds and bundles F‐actin via its microtubule binding domain. Curr Biol 14: 363–371 [DOI] [PubMed] [Google Scholar]
- 63. Whiteman IT, Gervasio OL, Cullen KM, Guillemin GJ, Jeong E V., Witting PK, Antao ST, Minamide LS, Bamburg JR, Goldsbury C (2009) Activated actin‐depolymerizing factor/cofilin sequesters phosphorylated microtubule‐associated protein during the assembly of Alzheimer‐like neuritic cytoskeletal striations. J Neurosci 29: 2994–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tseng Y, Kole TP, Lee JSH, Fedorov E, Almo SC, Schafer BW, Wirtz D (2005) How actin crosslinking and bundling proteins cooperate to generate an enhanced cell mechanical response. Biochem Biophys Res Commun 334: 183–192 [DOI] [PubMed] [Google Scholar]
- 65. Hannan SB, Dräger NM, Rasse TM, Voigt A, Jahn TR (2016) Cellular and molecular modifier pathways in tauopathies: the big picture from screening invertebrate models. J Neurochem 137: 12–25 [DOI] [PubMed] [Google Scholar]
- 66. Blard O, Feuillette S, Bou J, Chaumette B, Frébourg T, Campion D, Lecourtois M (2007) Cytoskeleton proteins are modulators of mutant tau‐induced neurodegeneration in Drosophila . Hum Mol Genet 16: 555–566 [DOI] [PubMed] [Google Scholar]
- 67. Shulman JM, Feany MB (2003) Genetic modifiers of tauopathy in Drosophila . Genetics 165: 1233–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shulman JM, Imboywa S, Giagtzoglou N, Powers MP, Hu Y, Devenport D, Chipendo P, Chibnik LB, Diamond A, Perrimon N et al (2014) Functional screening in Drosophila identifies Alzheimer's disease susceptibility genes and implicates Tau‐mediated mechanisms. Hum Mol Genet 23: 870–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Feuillette S, Deramecourt V, Laquerriere A, Duyckaerts C, Delisle M‐B, Maurage C‐A, Blum D, Buée L, Frébourg T, Campion D et al (2010) Filamin‐A and Myosin VI colocalize with fibrillary Tau protein in Alzheimer's disease and FTDP‐17 brains. Brain Res 1345: 182–189 [DOI] [PubMed] [Google Scholar]
- 70. Spudich JA, Watt S (1971) The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin‐troponin complex with actin and the proteolytic fragments of myosin. J Biol Chem 246: 4866–4871 [PubMed] [Google Scholar]
- 71. Lai FP, Szczodrak M, Block J, Faix J, Breitsprecher D, Mannherz HG, Stradal TE, Dunn GA, Small JV, Rottner K (2008) Arp2/3 complex interactions and actin network turnover in lamellipodia. EMBO J 27: 982–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bärenz F, Inoue D, Yokoyama H, Tegha‐Dunghu J, Freiss S, Draeger S, Mayilo D, Cado I, Merker S, Klinger M et al (2013) The centriolar satellite protein SSX2IP promotes centrosome maturation. J Cell Biol 202: 81–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Götze M, Pettelkau J, Schaks S, Bosse K, Ihling CH, Krauth F, Fritzsche R, Kühn U, Sinz A (2012) StavroX–a software for analyzing crosslinked products in protein interaction studies. J Am Soc Mass Spectrom 23: 76–87 [DOI] [PubMed] [Google Scholar]
- 74. Grimm M, Zimniak T, Kahraman A, Herzog F (2015) xVis : a web server for the schematic visualization and interpretation of crosslink‐derived spatial restraints. Nucleic Acids Res 43: W362–W369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Winterhoff M, Junemann A, Nordholz B, Linkner J, Schleicher M, Faix J (2014) The Diaphanous‐related formin dDia1 is required for highly directional phototaxis and formation of properly sized fruiting bodies in Dictyostelium . Eur J Cell Biol 93: 212–224 [DOI] [PubMed] [Google Scholar]
- 76. Junemann A, Winterhoff M, Nordholz B, Rottner K, Eichinger L, Gräf R, Faix J (2013) ForC lacks canonical formin activity but bundles actin filaments and is required for multicellular development of Dictyostelium cells. Eur J Cell Biol 92: 201–212 [DOI] [PubMed] [Google Scholar]
- 77. Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Movie EV1
Review Process File