

Abstract
Toxoplasma gondii encodes three protein kinase A catalytic (PKAc1‐3) and one regulatory (PKAr) subunits to integrate cAMP‐dependent signals. Here, we show that inactive PKAc1 is maintained at the parasite pellicle by interacting with acylated PKAr. Either a conditional knockdown of PKAr or the overexpression of PKAc1 blocks parasite division. Conversely, down‐regulation of PKAc1 or stabilisation of a dominant‐negative PKAr isoform that does not bind cAMP triggers premature parasite egress from infected cells followed by serial invasion attempts leading to host cell lysis. This untimely egress depends on host cell acidification. A phosphoproteome analysis suggested the interplay between cAMP and cGMP signalling as PKAc1 inactivation changes the phosphorylation profile of a putative cGMP‐phosphodiesterase. Concordantly, inhibition of the cGMP‐dependent protein kinase G (PKG) blocks egress induced by PKAc1 inactivation or environmental acidification, while a cGMP‐phosphodiesterase inhibitor circumvents egress repression by PKAc1 or pH neutralisation. This indicates that pH and PKAc1 act as balancing regulators of cGMP metabolism to control egress. These results reveal a crosstalk between PKA and PKG pathways to govern egress in T. gondii.
Keywords: acylation, cAMP‐dependent protein kinase A, cGMP‐dependent protein kinase G, egress, Toxoplasma gondii
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Signal Transduction
Introduction
Members of the phylum of Apicomplexa are obligate intracellular parasites posing a threat and unresolved challenge for human and animal health. Toxoplasma gondii is the most ubiquitous protozoan parasite of this phylum. It infects virtually all warm‐blooded animals and has established chronic infection in at least one‐third of the human population. This parasite is the causative agent of toxoplasmosis (Montoya & Liesenfeld, 2004), a disease affecting immuno‐compromised individuals and threatening the health of the unborn child in case of primary infection during pregnancy (Torgerson & Mastroiacovo, 2013; Torrey & Yolken, 2013). The lytic cycle of this parasite involves active entry into host cells, replication inside a parasitophorous vacuole and egress from the infected cells prior to reinvasion (Blader et al, 2015). To egress from host cells, parasites rely on timely secretion of specialised secretory organelles termed the micronemes. Multiple external stimuli have been shown to trigger microneme release in in vitro cultures including a drop in external K+ (Moudy et al, 2001) or pH (Roiko et al, 2014), and serum albumin (Brown et al, 2016).
The precise signalling pathways coordinating timely regulation of egress and microneme exocytosis are not fully defined. The cGMP‐dependent protein kinase G (PKG) acts as a central regulator of microneme secretion by connecting phospholipase C (PLC) and calcium‐mediated signalling. The effectors that regulate Ca2+‐dependent microneme secretion include Ca2+‐dependent protein kinases (CDPKs) such as CDPK1 (Lourido et al, 2010) and in some conditions CDPK3 (Lourido et al, 2012; McCoy et al, 2012) but also proteins potentially involved in membrane fusion such as DOC2.1 (Farrell et al, 2012). Recently, phosphatidic acid (PA) produced through phosphorylation of diacylglycerol by DGK1 was also shown to critically contribute to microneme exocytosis (Bullen et al, 2016). A role for cyclic adenosine monophosphate (cAMP)‐dependent signalling to control egress and invasion in Apicomplexa has been suspected but remains largely uncharacterised (Baker, 2011).
Protein kinase A (PKA) is regulated by fluctuating levels of cAMP that binds to the PKA regulatory subunit (PKAr) leading to the release of the catalytic subunit (PKAc). In T. gondii, four putative adenylyl cyclases (ACs) (Mueller et al, 2016) and 18 cyclic nucleotide phosphodiesterases (PDEs) (Howard et al, 2015) have been identified but have not been functionally characterised yet. The role of PKA in Apicomplexa has been best studied in malaria parasites (Haidar et al, 2016). In Plasmodium falciparum asexual erythrocytic stages, PfPKAc was shown to phosphorylate the apical membrane antigen 1 (AMA1), a key protein required for merozoite invasion (Leykauf et al, 2010). Consistent with this, fusion of the PfPKAr first 20 amino acids with GFP targeted the fluorescent protein to the apical pole of merozoites (Cabrera et al, 2012). Pharmacological studies also proposed that cAMP regulates Ca2+ signals important for microneme secretion in invasive merozoite (Dawn et al, 2014). As PKG was also shown to play a central role in controlling the mobilisation of Ca2+ from intracellular stores and microneme secretion in merozoites (Brochet et al, 2014; Collins et al, 2013 #138), it is possible that PKA‐dependent signals interplay with cGMP‐mediated signalling to regulate egress or invasion through Ca2+ regulation (Brochet & Billker, 2016).
PKA holoenzyme usually exists as a tetramer composed of two PKAr and two PKAc subunits. While only one PKAr and one PKAc have been identified in Plasmodium species (Li & Cox, 2000; Merckx et al, 2008), T. gondii expresses a single PKAr and three distinct PKAcs (TgPKAc1‐3). The N‐terminal dimerisation domain usually found in eukaryotic PKAr is absent in Toxoplasma PKAr, and as a consequence, the parasite holoenzyme is likely to be composed of a one‐to‐one ratio of both subunits (Kurokawa et al, 2011; Haste et al, 2012). PKAr typically possesses two cAMP‐binding domains, and the association with cAMP provokes the release of active PKAc that can then phosphorylate downstream targets to propagate cAMP signalling events. In T. gondii, PKAc1 and PKAc2 localise to the parasite periphery whereas PKAc3 is cytosolic (Sugi et al, 2016) indicating that the spatial control of cAMP levels and PKAc isoforms is crucial to differentially regulate PKA activity. Both PKAc1 and PKAc3 are expressed in tachyzoites and bradyzoites, while PKAc2 is highly expressed during the sexual stages in felids. Use of H89, an ATP competitive inhibitor showing high affinity for a wide range of PKAc isoforms (Lochner & Moolman, 2006), previously suggested that PKAc activity is important to regulate the rate of T. gondii division (Kurokawa et al, 2011) while transient elevation of cAMP by forskolin induced bradyzoite differentiation (Kirkman et al, 2001). Functional analysis of PKAc3 recently identified this isoform as a key cAMP effector in the regulation of stage conversion between tachyzoites and bradyzoites (Sugi et al, 2016).
In this study, we first showed that PKAc1 is selectively targeted to the inner membrane complex (IMC) by interacting with acylated PKAr. We found that conditional destabilisation of PKAr dramatically blocked cell division. A similar phenotype was observed when overexpressing PKAc1, the closest homologue of PfPKAc, indicating that the balance of PKAr and PKAc1 is crucial to regulate PKA1 activity. Conversely, we noticed that down‐regulation of PKAc1 triggered premature egress. Similarly, conditional titration of endogenous PKAr with a mutant unable to respond to cAMP and hence leading to a depletion in active PKAc induced microneme exocytosis and premature egress. Importantly, this untimely egress was dependent on parasite density and host cell acidification and led to unproductive invasion attempts and destruction of the host cell monolayer. Phosphoproteome analysis of PKA genetic inhibition suggested a plausible interplay between cAMP and cGMP signalling. This was further confirmed, as specific chemical inhibition of PKG blocked the premature egress induced by PKAc genetic inhibition. Altogether this study highlights for the first time a crosstalk between cAMP and cGMP signalling in an apicomplexan parasite, with PKA acting as a repressor of PKG‐mediated signalling to ensure timely pH‐dependent egress.
Results
PKAr acylation targets PKA1 to the inner membrane complex
To characterise the subcellular distribution of the PKA holoenzyme, 3Ty epitope tags were inserted in the carboxy‐terminus of PKAc1 and PKAr at the endogenous locus via single homologous recombination. Tagging was performed in ΔKu80 parasites deficient in non‐homologous end joining to prevent random integration (Fig EV1A). Genomic PCR analysis (Fig EV1B) and detection of the tagged proteins at a slightly larger molecular weight than the expected size (43.8 and 47 kDa, respectively) by Western blot analysis confirmed integration in the correct loci (Fig 1A and B). Indirect immunofluorescence assay (IFA) by confocal microscopy detected PKAc1‐3Ty and PKAr‐3Ty primarily at the parasite periphery and both proteins co‐localised with IMC1 and GAP45 (Fig 1C and D). The staining of nascent daughter cells by PKAc1‐3Ty and PKAr‐3Ty suggests that these proteins are targeted to the inner membrane complex (IMC). The localisation of PKAr‐3Ty to the IMC was further confirmed by aerolysin treatment, which causes a separation between the IMC and the plasma membrane as shown by anti‐SAG1 staining (Fig 1E).
Figure EV1. Generation of transgenic lines expressing tagged alleles of PKAr or PKAc1.
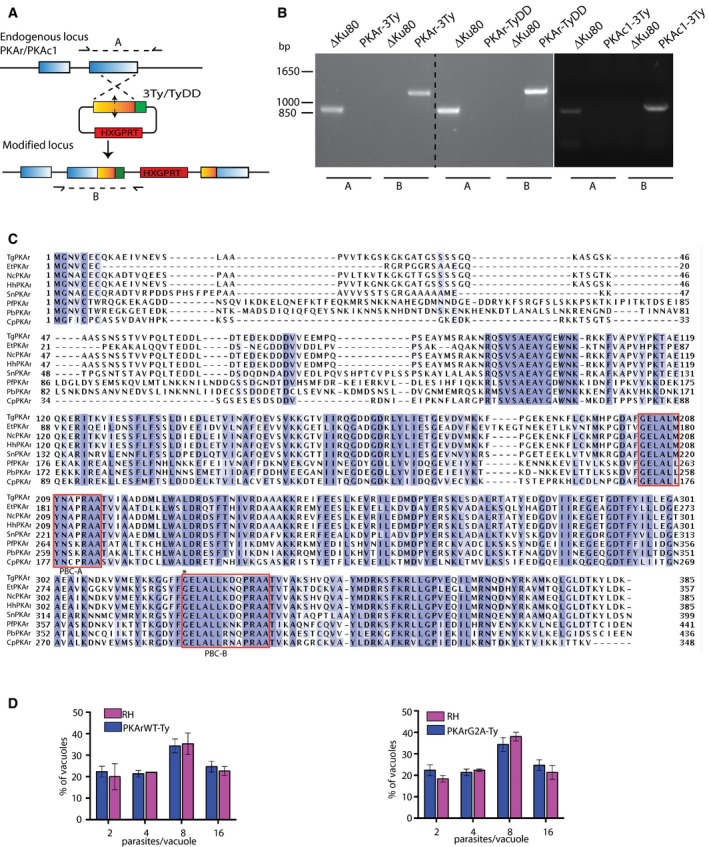
- Cloning strategy for the introduction of 3Ty or TyDD at the C‐terminus of PKAr or PKAc1 at their respective endogenous locus.
- PCR analysis showing correct insertion of the constructs coding for the epitope tags.
- Multiple sequence alignment of the PKAr genes in apicomplexan parasites. In all analysed apicomplexan orthologues (T. gondii TGGT1_242070, Neospora caninum NCLIV_017370, Eimeria tenella ETH_00011940, Hammondia hamondi HHA_242070, Sarcocystis neurona SN3_01200890, Plasmodium falciparum PF3D7_1223100, P. berghei PBANKA_143800 and Cryptosporidium cgd7_120), the N‐terminus of PKAr contains the putative myristoylation and palmitoylation sites and the C‐terminal domain contains two cAMP‐binding sites/red boxes). The star in the B site indicates glycine 321 that was mutated in this study.
- Expression of a second copy of PKArWT‐Ty or PKArG2A‐Ty does not affect parasite division. Error bars represent ±SD for 100 vacuoles counted in triplicate from three biological replicates.
Figure 1. PKA1 is targeted to the inner membrane complex (IMC) by dual acylation of PKAr.
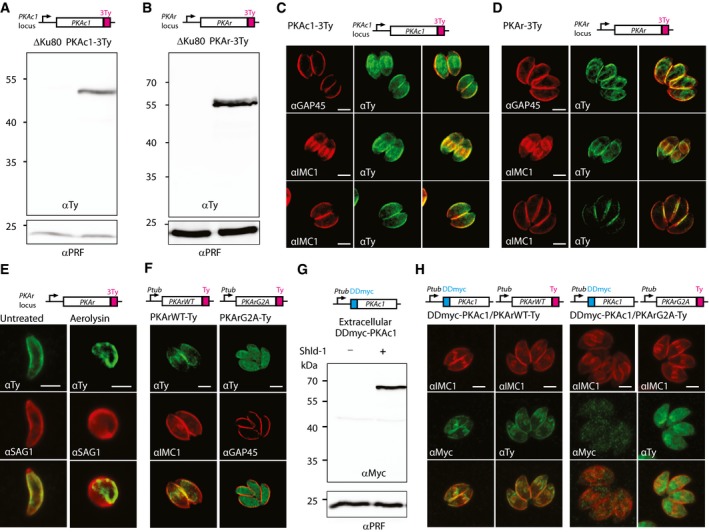
-
A, BWestern blot analysis of ΔKu80, PKAc1‐3Ty (A) and PKAr‐3Ty (B) parasite lysates probed with αTy antibodies. Profilin (αPRF) served as loading control.
-
C, DPeripheral localisation of PKAc1‐3Ty (C) and PKAr‐3Ty (D) shown by IFA using anti‐Ty antibodies. Antibodies against GAP45 and IMC1 were used as markers of plasma membrane and IMC, respectively.
-
EPlasma membrane stained with an αSAG1 antibody is separated from the IMC stained with anti‐Ty after aerolysin treatment, indicating that PKAr‐3Ty localises at the IMC.
-
FThe N‐terminus putative acylation site of PKAr is essential for its IMC targeting.
-
GA second copy of DDmyc‐PKAc1 is stabilised after 1 h in presence of Shld‐1.
-
HA second copy of DDmyc‐PKAc1 stabilised with Shld‐1 is targeted to the IMC in presence of a second copy of PKArWT‐Ty. When of a second copy of PKArG2A‐Ty is provided, stabilised DDmyc‐PKAc1 does not localise at the cell periphery.
Numerous proteins are targeted to the pellicle of T. gondii by dual acylation at a N‐terminal consensus motif (Frenal et al, 2014). In this motif, Gly2 is myristoylated, which then favours the recognition by palmitoyl acyltransferases that palmitoylate nearby cysteine residues (Yalovsky et al, 1999). PKAr contains such a motif with a glycine at position two and a pair of cysteine residues at positions 5 and 7 (Fig EV1C). Concordantly, PKAr was previously shown to be palmitoylated in T. gondii (Caballero et al, 2016). Attempts to introduce a Gly2 to Ala2 mutation in PKAr using CRISPR/Cas9 genome editing were unsuccessful, pointing to a critical role of PKAr acylation for T. gondii viability. To determine the importance of acylation for the association of PKAr to the IMC, vectors were constructed to stably express a second C‐terminally Ty‐tagged copy of PKArWT or PKArG2A under the control of the tubulin promoter. While PKArWT‐Ty exhibited the same localisation as the endogenous PKAr‐3Ty (Fig 1F), the mutation in the putative myristoylation site expectedly caused an essentially cytosolic distribution of PKArG2A‐Ty (Fig 1F) indicating that PKAr acylation is important for its IMC localisation. Intriguingly, overexpression of PKArWT‐Ty or PKArG2A‐Ty did not lead to any apparent fitness cost (Fig EV1D), in contrast to previous reports where PKAr controlled by strong GRA or HRP3 promoters was deleterious in T. gondii and P. falciparum, respectively (Merckx et al, 2008; Kurokawa et al, 2011).
PKAc1 has previously been shown to interact with PKAr in vitro (Kurokawa et al, 2011), and in the absence of any predictable acylation motif on PKAc1, we hypothesised that this kinase may be targeted to the IMC via its binding to PKAr. We thus conditionally overexpressed PKAc1 with the destabilising domain (DD) of FKBP fused at its N‐terminal end (Herm‐Gotz et al, 2007) in the PKArWT‐Ty and PKArG2A‐Ty strains. DDmyc‐PKAc1 was readily stabilised upon addition of Shield molecule (Shld‐1) in 2 h (Fig 1G). Upon stabilisation, DDmyc‐PKAc1 accumulated to the IMC in the PKArWT‐Ty strain whereas the protein remained cytosolic in the PKArG2A‐Ty strain (Fig 1H). Collectively, these results established that PKAc1 is anchored to the IMC via the acylation of PKAr.
Activation of PKA through either PKAr down‐regulation or PKAc1 overexpression dramatically blocks replication
To investigate the role of PKA, we first opted for the conditional destabilisation of PKAr. To do so, the endogenous locus was modified with the insertion of a C‐terminal TyDD (Fig EV1A and B). In presence of Shld‐1, PKAr‐TyDD expressing parasites were successfully selected and cloned. These parasites were not able to form plaques of lysis on human foreskin fibroblast (HFF) monolayers upon destabilisation of PKAr‐TyDD by removal of Shld‐1, as documented by plaque assays (Fig 2A). PKAr down‐regulation stopped parasite replication as suggested by the disappearance of the loading control protein, profilin (PRF), by Western blot analysis 12 h post‐Shld‐1 removal (Fig 2B). Severe anomalies in parasite division leading to a defect in apicoplast inheritance were also observed (Fig 2C) and intracellular growth scoring by counting the number of parasites per vacuole indicated a severe growth defect (Fig 2D). The block in parasite division was irreversible after 24 h of destabilisation as addition of Shld‐1 at this time point failed to revert the phenotype (Fig EV2A).
Figure 2. Down‐regulation of PKAr or overexpression of PKAc1 blocks parasite division.
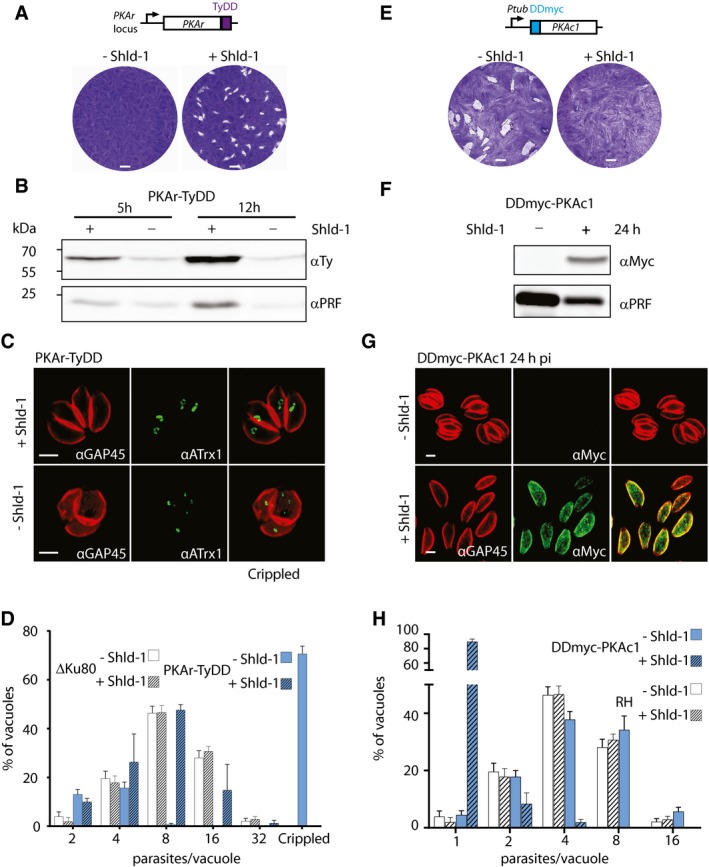
-
ALytic growth of intracellular wild‐type Toxoplasma gondii tachyzoite stages over a period of 8 days results in plaques within monolayers of host cells. Destabilisation of PKAr‐TyDD leads to the absence of plaque formation.
-
BA drop in PKAr‐TyDD expression is detected 5 h after Shld‐1 removal and a significant parasite growth defect is observed as early as 12 h later. Profilin (αPRF) served as loading control.
-
CIFA analysis showed that the down‐regulation of PKAr‐TyDD leads to the abnormal segregation of the apicoplast‐associated thioredoxin family protein (ATrx1) 24 h after Shld‐1 withdrawal.
-
DReplication assay of PKAr‐TyDD parasites grown for 24 h +/‐ Shld‐1 indicates that no more than four parasites are observed per vacuole upon PKAr destabilisation (100 parasites were counted in three independent replicates).
-
E, FStabilisation of a second copy of DDmyc‐PKAc1 leads to the complete block of cell growth indicated by plaque assay (E) and Western blot analysis (F).
-
G, HStabilisation of a second copy of DDmyc‐PKAc1 leads to the observation of mainly one parasite per vacuole in cells treated with Shld‐1 (100 parasites were counted in three independent replicates).
Figure EV2. Expression of a second copy of PKArWT or mistargeted PKArG321E rescues a conditional knockdown of PKAr‐TyDD.
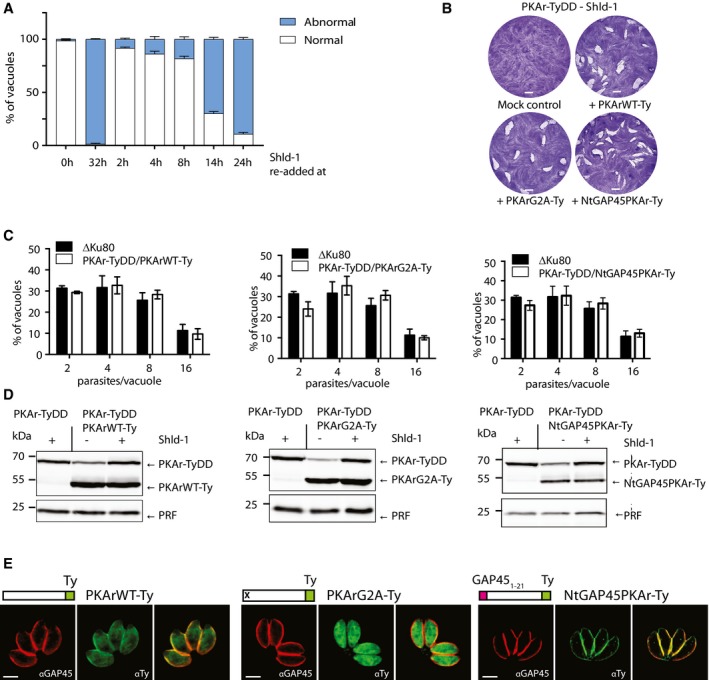
-
AConditional destabilisation of PKAr‐TyDD can be rescued by re‐addition of Shld‐1 up to 8 h after initial Shld‐1 removal (data are from three independent biological replicates).
-
B, CThe conditional partial destabilisation of PKAr‐TyDD is successfully rescued by introduction a second copy of PKArWT‐Ty, PKArG2A‐Ty or NtGAP45PKAr‐Ty as shown by plaque assays (B) and cell division assays (C) in the absence of Shld‐1 (100 parasites were counted in three independent replicates).
-
DWestern blot analysis of PKAr‐TyDD in presence of Shld‐1 and its derivatives complemented with the above‐mentioned PKAr variants ± Shld‐1 for 24 h. In the presence of a second copy, a partial drop of endogenous PKAr‐TyDD expression was observed upon removal of Shld‐1 while the expression levels of the second copies were significantly higher than their endogenous counterpart. Profilin (PRF) served as a loading control.
-
EImmunofluorescence assay indicate that PKArWT‐Ty is targeted to the IMC, PKArG2A‐Ty to the cytosol and NtGAP45PKAr‐Ty to the plasma membrane.
Expression of a second copy of PKArWT‐Ty under the tubulin promoter when PKAr‐TyDD were grown in absence of Shld‐1 rescued the block in division (Fig EV2B–D), whereas mock‐transfected parasites did not grow as confirmed by plaque and intracellular growth assays. Overexpression of a second copy of mislocalised PKArG2A‐Ty also rescued conditionally destabilised endogenous PKAr‐TyDD. This is likely explained by the fact that the dramatic overexpression of PKArG2A‐Ty might be sufficient to target mutant isoforms to the IMC to complement the partial destabilisation of endogenous PKAr‐TyDD (Fig EV2C). A similar result was observed when the 20 first amino acids of PKAr‐Ty were replaced with the first 20 amino acids of GAP45 (NtGAP45‐PKAr‐Ty), targeting the protein to the parasite plasma membrane (Fig EV2B–D). Taken together, these results establish that overexpression of PKAr can rescue the PKAr knockdown phenotype and is well tolerated by the parasite even when mislocalised.
Since PKAr is predicted to act as a negative regulator of PKAc1, overexpression of the latter should lead to a comparable phenotype to that of PKAr down‐regulation. We thus documented the impact of PKAc1 overexpression upon stabilisation of a second copy of DDmyc‐PKAc1. In presence of Shld‐1, DDmyc‐PKAc1 expressing parasites were unable to form plaque of lysis on HFF monolayers (Fig 2E). After 24 h of Shld‐1 treatment, parasites stopped replicating as predicted by the drop in the level of PRF observed by Western blot (Fig 2F). This was confirmed by the absence of vacuoles containing more than two parasites (Fig 2G and H). These results implicate a tight regulation of PKAc1‐dependent phosphorylation to ensure proper cell division. Altogether these results reveal a considerable impact of PKAc1 on parasite division as well as a requirement for a fine‐tuned balance between the level of PKAr and PKAc1 at the IMC. An imbalance towards PKAc1 activity or mislocalisation leads to a complete block in cell division.
Conditional down‐regulation of PKAc1 causes premature egress and “restless invasion”
While overexpression of PKAc1 or down‐regulation of PKAr is detrimental to parasite replication, we aimed at investigating the consequence of knocking PKAc1 down. An inducible knockdown (PKAc1‐iKD) was generated by tet‐repressive promoter replacement (Fig EV3A–C). Depletion of PKAc1‐iKD was readily and tightly achieved in 24 h upon anhydrotetracycline (ATc) treatment (Fig 3A). Plaque assays revealed that conditional down‐regulation of PKAc1‐iKD is detrimental to parasite survival with very small plaques of lysis formed after 8 days (Fig 3B). PKAc1 depletion in presence of ATc showed no alteration of parasite replication 24 h post‐inoculation (Fig EV3D). Instead, in heavily infected cells, parasites prematurely egressed, while at the same time point non‐treated parasites were still intracellular (Fig 3C and D). Prematurely egressed parasites were able to invade new host cells (Fig EV3E) but failed to further develop intracellularly as compared to their non‐treated counterpart. Instead, we observed a considerable lysis of the whole HFF monolayer after 24 h that we interpret as a “restless” phenotype of parasites unable to settle in the invaded cell (Fig 3E). This shows that tight down‐regulation of PKAc1 activity is also required after invasion to initiate a new productive lytic cycle. Altogether this reveals that depletion of PKAc1 induces a premature egress that is followed by abortive invasion attempts.
Figure EV3. Generation, genotyping and phenotyping of PKAc1‐iKD parasites.
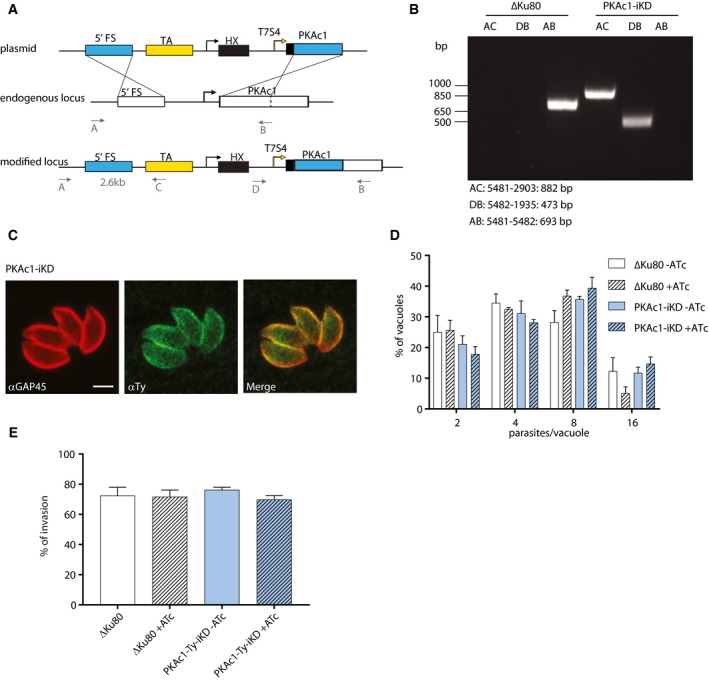
- Schematic representation of the strategy used to replace the endogenous promoter of PKAc1 by a tet‐repressive promoter to generate a PKAc1 inducible knockdown (PKAc1‐iKD).
- PCRs performed on gDNA extracted from a clone showing the correct integration of the construct.
- Peripheral localisation of PKAc1‐Ty‐iKD shown by IFA using an anti‐Ty antibody. An antibody against GAP45 is used as a marker of the plasma membrane. Scale bar = 2 μm.
- Replication assay of PKAc1‐iKD. Parasites grown for 24 h ± ATc did not show any defect in intracellular growth.
- The number of intracellular PKAc1‐Ty‐iKD parasites 30 min after invasion was not affected in presence or absence of ATc.
Figure 3. Down‐regulation of PKAc1 leads to premature egress and “restless invasion”.
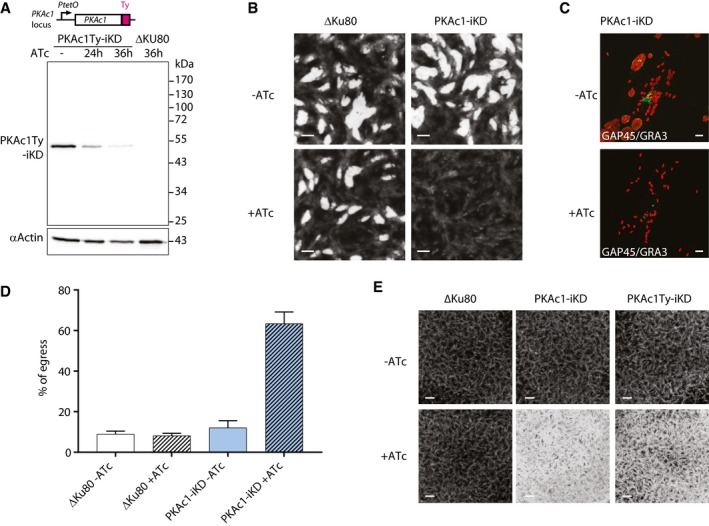
-
AA drop in PKAc1‐Ty‐iKD expression is detected as early as 24 h upon ATc treatment with almost no protein detectable after 36 h.
-
BConditional down‐regulation of PKAc1‐Ty‐iKD leads to the absence of plaque formation.
-
C, DDown‐regulation of PKAc1‐Ty‐iKD upon ATc treatment leads to increased parasite dispersion after 40 h and premature egress (data are from three independent biological replicates).
-
EPrematurely egressed PKAc1‐Ty‐iKD parasites after 24 h of ATc treatment invade and exit fully lysing the monolayer of HFF cells, while the non‐treated parasites invade and initiate a new lytic cycle.
Conditional overexpression of a non‐cAMP‐binding PKArG321E mutant causes premature egress
Like its counterpart in other organisms, T. gondii PKAr is predicted to possess two cAMP‐binding sites in the C‐terminal region (Fig EV1C). A single glycine to glutamic acid change in the site B of the cAMP‐binding domain is known to abolish this binding and to prevent activation of the holoenzyme in the presence of cAMP (Woodford et al, 1989) (Fig 4A). In PKAr, the conserved glycine residue is Gly321 and, expectedly, transfection of a vector expressing PKArG321E‐Ty failed to rescue PKAr‐TyDD strain in the absence of Shld‐1.
Figure 4. Overexpression of a non‐cAMP‐binding PKArG21E isoform prevents release of active PKAc1 causing premature egress and “restless invasion”.
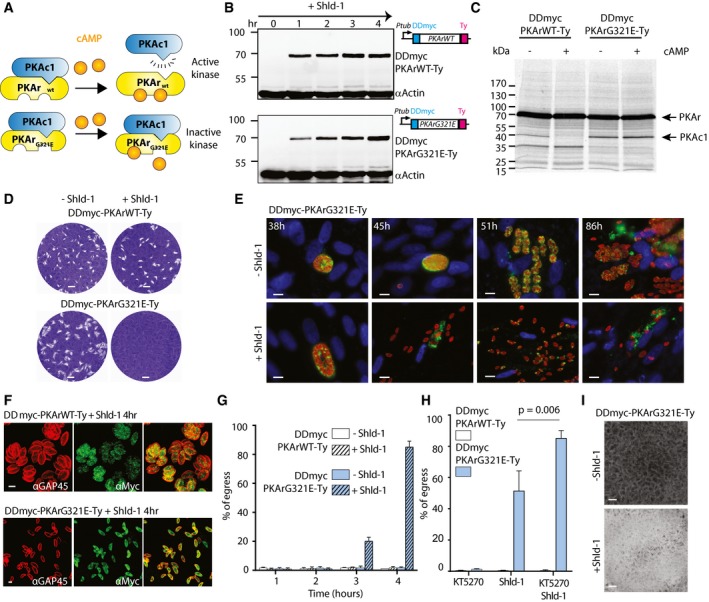
- PKAr is predicted to possess two cAMP‐binding sites. A single glycine to glutamic acid change in the site B of the cAMP‐binding domain is known to abolish cAMP binding and to prevent activation of the holoenzyme in the presence of cAMP. In PKAr, the conserved glycine residue is Gly321.
- Addition of Shld‐1 leads to the rapid stabilisation of DDmyc‐PKArWT‐Ty and DDmyc‐PKArG321E‐Ty.
- PKAc1 is co‐immunoprecipitated with DDmyc‐PKArWT‐Ty in absence of cAMP but is not recovered in presence of 20 μM cAMP as revealed with [35S]‐labelled methionine/cysteine metabolic labelling. Conversely, PKAc1 is co‐immunoprecipitated with DDmyc‐PKArG321E‐Ty in presence or absence of cAMP indicating the G321E substitution prevents the release of active PKAc1 in presence of cAMP. The autoradiogram shown is representative of two independent biological replicates.
- Conditional overexpression of a dominant‐negative DDmyc‐PKArG321E‐Ty that cannot bind cAMP leads a dramatic reduction in plaque formation while overexpression of DDmyc‐PKArWT‐Ty is well tolerated by the parasite.
- Overexpression of the dominant‐negative DDmyc‐PKArG321E‐Ty triggers premature egress from infected host cells and prevents initiation of a new lytic cycle as revealed by IFA. Images are representative of ˜80% of the parasite population. Blue = DAPI; red = GAP45; green = GRA3.
- In heavily infected cells, addition of Shld‐1 at 32 h post‐inoculation for 4 h leads to premature egress of DDmyc‐PKArG321E‐Ty parasites but not of the DDmyc‐PKAr‐Ty control line as assessed by IFA.
- Quantification of extracellular DDmyc‐PKArG321E‐Ty parasites released from heavily infected cells after addition of Shld‐1 at 32 h post‐inoculation for 4 h (100 parasites were counted in three independent replicates).
- A non‐selective PKA inhibitor, KT5270, significantly enhances the premature egress of DDmyc‐PKArWT‐Ty parasites in presence of Shld‐1 (data are from three independent biological replicates; statistical analysis was done by two‐tailed t‐test).
- Prematurely egressed DDmyc‐PKArG321E‐Ty parasites after 4 h of Shld‐1 treatment invade and exit host cells, fully lysing the monolayer of HFF cells while the non‐treated parasites invade and initiate a new lytic cycle.
To investigate the role of this mutation in the interaction between PKAr and PKAc1, chimeric genes controlled by a tubulin promoter encoding DDmyc‐PKArG321E‐Ty or DDmyc‐PKArWT‐Ty were stably introduced in the RH strain in the absence of Shld‐1. Both DDmyc‐PKArG321E‐Ty and DDmyc‐PKArWT‐Ty were readily stabilised after 1 h of Shld‐1 treatment and reached a saturation level of expression after 4 h (Fig 4B). Overexpression of either isoform did not impede intracellular growth (Fig EV4A). Immunoprecipitation of DDmyc‐PKArWT‐Ty in presence or absence of cAMP revealed the presence of a protein (single band) migrating just below 40 kDa in a cAMP‐dependent fashion (Fig 4C). This protein co‐immunoprecipitated more prominently with DDmyc‐PKArG321E‐Ty and persisted in presence of 20 μM cAMP. Analysis of this band by mass spectrometry only recovered peptides mapping on PKAc1 (Fig EV4B). In consequence, PKAc1 appears to be the only protein that is released from PKAr in presence of cAMP. This indicates that PKArG321E substitution acts as dominant mutant, as previously reported in various other organisms, by preventing cAMP binding and thus the release of active PKAc1.
Figure EV4. Phenotypic analysis of DDmyc‐PKArG321E‐Ty and DDmyc‐PKArWT‐Ty parasites.
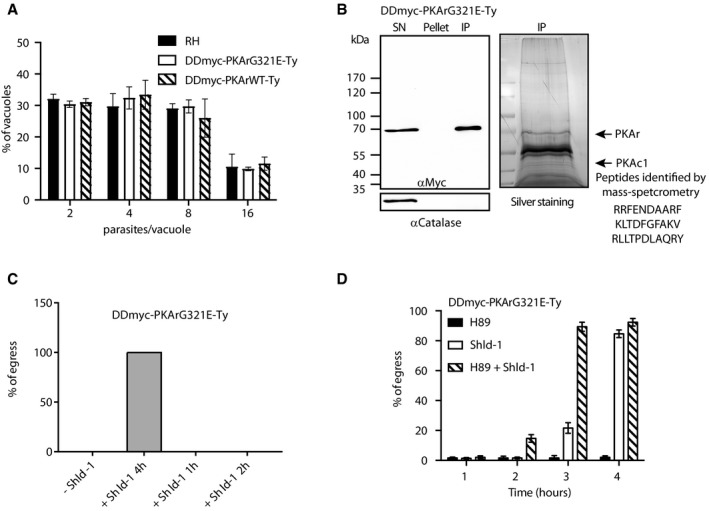
- Intracellular growth in presence of Shld‐1 for 24 h is not impaired in both DDmyc‐PKArG321E‐Ty and DDmyc‐PKArWT‐Ty parasites (100 parasites were counted in three independent replicates).
- Immunoprecipitation of DDmyc‐PKArG321E‐Ty parasites. Mass spectrometry‐based analysis of the band migrating at 40 kDa recovered three peptides from PKAc1 only.
- Conditional stabilisation of DDmyc‐PKArG321E‐Ty can be rescued by washing Shld‐1 off up to 2 h after initial Shld‐1 addition.
- A pharmacological PKA inhibitor, H89, significantly enhances the premature egress of DDmyc‐PKArG321E‐Ty parasites in presence of Shld‐1 (data are from three independent biological replicates).
We then tested the effect of conditional overexpression of both DDmyc‐PKArG321E‐Ty and DDmyc‐PKArWT‐Ty on parasite growth. Plaque assays revealed that stabilisation of DDmyc‐PKArG321E‐Ty is lethal with no visible plaque of lysis formed after 8 days whereas overexpression of DDmyc‐PKArWT‐Ty caused no apparent defect (Fig 4D). As observed for PKAc1 down‐regulation, stabilisation of DDmyc‐PKArG321E‐Ty led to premature egress from infected cells (Fig 4E) without alteration of parasite replication 24 h post‐inoculation (Fig EV4C). In heavily infected cells, stabilisation of DDmyc‐PKArG321E‐Ty led to premature egress in < 4 h after Shld‐1 induction, while at the same time point DDmyc‐PKArWT‐Ty parasites remained intracellular (Fig 4F and G). Similarly, DDmyc‐PKArWT‐Ty and DDmyc‐PKArG321E‐Ty in absence of Shld‐1 remained intracellular (Fig 4G). This phenotype was reversible when Shld‐1 was removed up to 2 h after stabilisation (Fig EV4C). We then investigated whether the addition of the PKA inhibitors KT5270 or H89 (Kurokawa et al, 2011) could speed up the process of the premature egress. Concordantly, 5 μM of KT5270 or 200 μM of H89 significantly accelerated the premature egress of the DDmyc‐PKArG321E‐Ty strain in presence of Shld‐1 (Figs 4H and EV4D). Finally, DDmyc‐PKArG321E‐Ty prematurely egressed parasites showed a restless invasion phenotype when added to a fresh monolayer of HFF (Fig 4I), as observed when PKAc1 was down‐regulated. Collectively, these results demonstrate that PKAc1 depletion or prevention of its release from the PKAr subunit is deleterious to the parasites by triggering premature egress followed by uncontrolled invasion.
Phosphoproteome analysis of PKAc inhibition points to an interplay between cGMP and cAMP in controlling parasite egress
The tightly controlled premature egress triggered by conditional stabilisation of DDmyc‐PKArG321E‐Ty offered a unique opportunity to get a first insight into the pathways regulated by PKA1. To do so, we quantitatively compared the phosphorylation events in DDmyc‐PKArWT‐Ty and DDmyc‐PKArG321E‐Ty parasites at 0, 20 and 60 min after Shld‐1 addition. In total, we identified 11,247 phosphorylated peptides mapping on 2,652 T. gondii proteins, 191 of which showed a > twofold change (up or down) in phosphorylation between the two lines at 20 or 60 min (Dataset EV1). Among the biological processes differentially phosphorylated upon PKA inhibition are MAP kinase activity and nucleic acid binding transcription factor activity (Fig 5A). In addition, PKA‐dependent phosphorylation of 83 proteins without functional annotation was also observed (Dataset EV1). Multiple kinases involved in cell cycle regulation including the MAPK1 pathway were differentially phosphorylated upon PKAc inhibition. This may be associated with the proposed role of PKAc1 in the regulation of cell cycle progression (Kurokawa et al, 2011). The role of PKAc3 in transition to bradyzoites may also rely on the regulation of seven AP2 domain transcription factors (Dataset EV1). However, the exact roles of these AP2 regulators and of MAPK1 pathway remain to be determined in T. gondii. Two protein phosphatases were also found to be differentially phosphorylated possibly explaining the broad spectrum of phosphorylation profiles observed.
Figure 5. Premature egress induced by PKA genetic inhibition is blocked by PKG chemical inhibition.
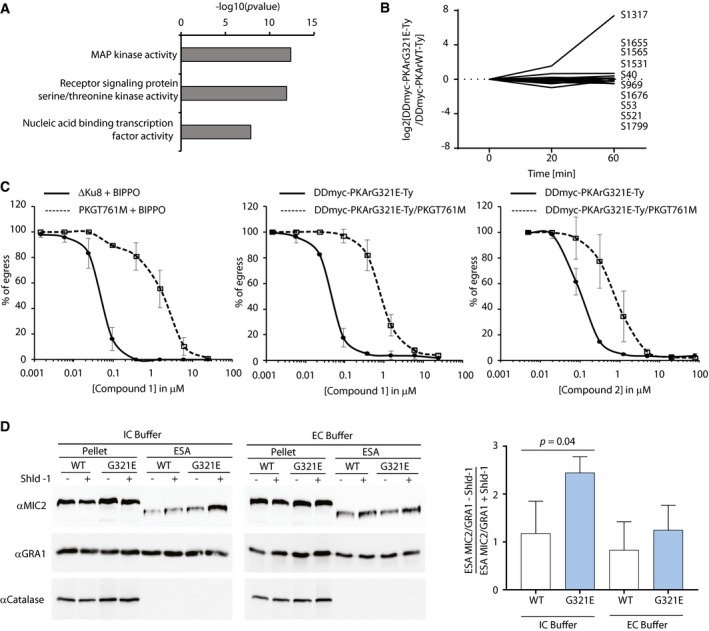
- GO term enrichment analysis of proteins differentially phosphorylated upon DDmyc‐PKArG321E‐Ty stabilisation compared with its wild‐type counterpart DDmyc‐PKArWT‐Ty. Bonferroni corrected P‐values are indicated.
- Upon Shld‐1 addition, a putative cGMP‐specific PDE, PDE2 was more phosphorylated at Serine 1317 in DDmyc‐PKArG321E‐Ty parasites compared with DDmyc‐PKArWT‐Ty parasites suggesting a crosstalk between cAMP and cGMP signalling.
- Chemical inhibition of the cGMP‐dependent PKG by Compound 1 and Compound 2 blocks egress induced by 5 μM BIPPO, a PDE inhibitor, or by stabilisation of DDmyc‐PKArG321E‐Ty in presence of Shld‐1. A PKGT761M substitution that renders PKG resistant to both compounds indicates that the block in egress in only mediated by PKG when 0.2 μM Compound 1 is used while the block in egress associated with C2 is not specific to PKG inhibition only (data are from three independent biological replicates).
- Genetic inhibition of PKA triggers MIC2 secretion in intracellular buffer. Anti‐GRA1 antibodies were used as a loading control and anti‐catalase antibodies served as a lysis control (data are from three independent biological replicates; statistical analysis was done by two‐tailed t‐test).
Interestingly, we found that serine 17 of PKAr becomes less phosphorylated upon PKAc inhibition suggesting a potential cyclic nucleotide feedback loop. This was further strengthened by higher phosphorylation levels of serine 1317 of PDE2 (TGGT1_293000), a 3′5′‐cyclic nucleotide phosphodiesterase domain‐containing protein (Fig 5B). Sequence analysis of this protein confirmed the presence of HDc and GAF domains that are commonly found in eukaryotic 3′,5′‐cGMP phosphodiesterases where cGMP regulates catalytic activity by binding to the GAF domain (Zoraghi et al, 2004). As elevation of cGMP levels was previously shown to induce egress of T. gondii tachyzoites by activation of PKG (Brown et al, 2016), we decided to further investigate whether an interplay between cAMP and cGMP signalling could operate to govern parasite egress.
Genetic inhibition of PKAc triggers egress through the activation of PKG‐dependent pathways
Attempts to knockout or knockdown the expression of PDE1 (TGGT1_202540) and PDE2 (TGGT1_293000), two PDEs recently predicted to be essential (Sidik et al, 2016), were unsuccessful. We were also unable to reliably quantify cGMP levels of intracellular parasites to ascertain cGMP accumulation upon genetic inhibition of PKAc. Alternatively, to investigate the possibility that cAMP and cGMP interact to control parasite egress, we focused on the requirement for PKG activity to control the PKAc1‐dependent premature egress. Two inhibitors, C1 and C2, were previously shown to block egress by targeting PKG (Donald et al, 2006). To determine whether genetic inhibition of PKAc1 indeed activates PKG‐dependent pathways, we tested the effect of C1 and C2 upon stabilisation of DDmyc‐PKArG321E‐Ty or DDmyc‐PKArWT‐Ty. Both C1 and C2 completely blocked egress induced by PKAc genetic inhibition with an EC50 of 0.3 and 0.01 μM, respectively (Fig 5C). To ascertain that C1 and C2 blocked PKA‐induced egress through PKG inhibition but not via off‐target effects, we introduced a mutation at Thr761 to Met that renders PKG resistant to both compounds (Donald et al, 2002a) in DDmyc‐PKArG321E‐Ty and DDmyc‐PKArWT‐Ty parasites with the assistance of CRISPR/Cas9 genome editing (Appendix Fig S1). The PKGT761M substitution conferred a 10‐fold and a fivefold increase in resistance to C1 and C2, respectively (Fig 5C). At 0.2 μM, C1 completely blocked PKA‐dependent egress with no obvious off‐target effect since at the same concentration, no inhibition was observed in the resistant DDmyc‐PKArG321E‐Ty/PKGT761M line. Conversely, C2 appeared to be less specific as no C2 concentrations were found to fully block egress in the DDmyc‐PKArG321E‐Ty with no effect on the counterpart DDmyc‐PKArG321E‐Ty/PKGT761M‐resistant line. Importantly, both compounds were previously shown to also target CDPK1 (Donald et al, 2002a, 2006), a critical regulator of parasite egress downstream of a PKG‐mediated calcium signal (Lourido et al, 2010, 2012). Dual inhibition of PKG and CDPK1 by C2 likely explains the lack of specificity for the observed phenotype.
As activation of both PKG and CDPK1 has been shown to stimulate apical microneme secretion (Wiersma et al, 2004; Lourido et al, 2010), we reasoned that genetic inhibition of PKA would trigger microneme release. To test this hypothesis, DDmyc‐PKArWT‐Ty and DDmyc‐PKArG321E‐Ty parasites were resuspended in intracellular buffer or extracellular buffer. After 30 min of incubation at 37°C, cell‐free excreted secreted antigens (ESA) samples were collected and microneme secretion was assessed by measuring the level of the post‐exocytosis processed form of MIC2 in ESA fractions. Enhanced MIC2 secretion was only detected in Shld‐1‐treated DDmyc‐PKArG321E‐Ty parasites in intracellular conditions (Fig 5D) indicating that genetic inhibition of PKA triggers microneme exocytosis in intracellular parasites. Collectively, these results confirm that PKA‐dependent control of parasite egress is targeting PKG‐mediated signalling and the downstream regulation of microneme secretion by CDPK1.
PKAc prevents PKG‐dependent premature egress induced by host cell acidification
We noticed that a high multiplicity of infection (number of parasitophorous vacuoles per host cell—MOI) exacerbated premature egress observed upon stabilisation of DDmyc‐PKArG321E‐Ty (Fig 6A). This strongly suggested that an intrinsic solute able to diffuse between vacuoles serves as trigger for egress. Consistent with this, transferring supernatants of Shld‐1‐treated cultures from heavily infected cells to cells infected with a low MOI significantly triggered premature egress of these latter (Fig 6B).
Figure 6. PKA represses PKG‐dependent egress triggered by environmental acidification.
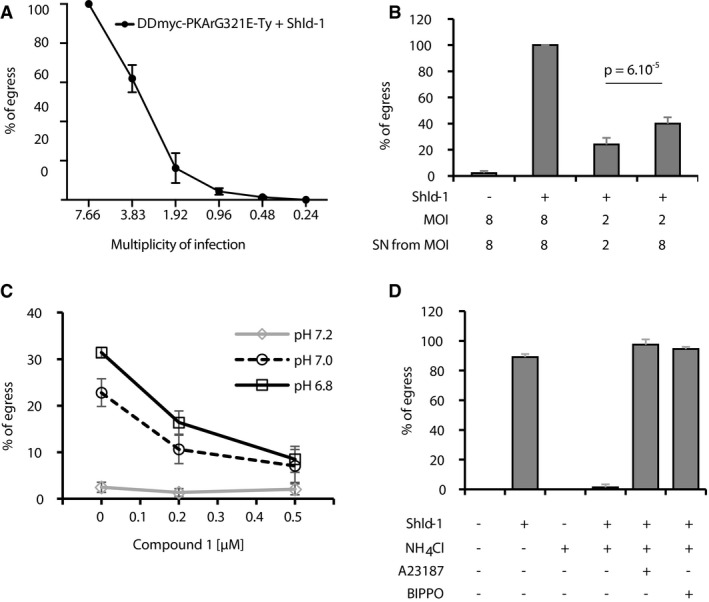
- The premature egress induced by PKA genetic inhibition is exacerbated when host cells are heavily infected (data are from three independent biological replicates).
- Transferring supernatants of Shld‐1‐treated cultures from cells infected with DDmyc‐PKArG321E‐Ty parasites at a MOI of 8 induces egress of parasites from cells infected at a MOI of 2 within 3 h (data are from three independent biological replicates; statistical analysis was done by two‐tailed t‐test).
- Acidification of the culture medium induces parasite egress in < 10 min. The effect is inhibited in presence of Compound 1 (data are from two independent biological replicates).
- Neutralisation of cultures with the weak base NH4Cl blocks premature egress induced by DDmyc‐PKArG321E‐Ty stabilisation. This block is circumvented by addition of the Ca2+ ionophore A23187 or the PDE inhibitor BIPPO (data are from three independent biological replicates).
A pH decrease in the parasitophorous vacuole towards the end of the replication cycle was previously shown to stimulate motility and egress by triggering Ca2+‐dependent secretion of micronemes (Roiko et al, 2014). This effect was prevented by the addition of NH4Cl, a weak base that accumulates in acidic compartments. Acidification‐dependent egress was also blocked by C1, indicating that the signalling pathways sensing a drop in pH lead to PKG activation (Fig 6C).
To determine whether a rise in H+ concentration in heavily infected cells serves as a key parameter for premature egress, cells infected with DDmyc‐PKArG321E‐Ty were neutralised in presence of 20 μM NH4Cl 1 h after the addition of Shld‐1 and further incubated for 2 h. As expected, addition of NH4Cl completely blocked the PKA‐dependent premature egress of DDmyc‐PKArG321E‐Ty parasites (Fig 6D). This revealed that a decrease in pH is necessary for premature egress when PKA is genetically inhibited. This indicates that in WT parasites, PKA activity prevents egress induced by parasite‐dependent acidification. Inhibition of PKA‐dependent egress by NH4Cl could be due to the inhibition of proximal or distal effectors downstream of PKA. Addition of a Ca2+ ionophore on neutralised and Shld‐1‐treated DDmyc‐PKArG321E‐Ty parasites restored egress indicating that Ca2+‐dependent pathways are blocked in the presence of NH4Cl, as previously described (Roiko et al, 2014). We then tested the effect of BIPPO, a potent phosphodiesterase inhibitor that was previously shown to stimulate egress and microneme secretion by activating PKG upstream of Ca2+ mobilisation (Howard et al, 2015). Addition of BIPPO also circumvented the neutral pH block of DDmyc‐PKArG321E‐Ty and induced premature egress (Fig 6D) indicating that under the tested conditions, (i) BIPPO is triggering egress through the inhibition of a cGMP‐specific PDE, and (ii) cGMP degradation is a limiting factor to stimulate PKG‐dependent egress. Collectively, these results demonstrate that PKA‐dependent regulation of cGMP metabolism acts as a focal regulatory point upstream of PKG to control pH‐dependent egress.
The apical pole and the pellicle represent critical cellular compartments to integrate cAMP and cGMP signals
The spatial properties of intracellular signals are often determined by a precise organisation of signalling components. We thus set out to investigate the localisation of the crucial cAMP and cGMP components possibly involved in regulating T. gondii tachyzoites egress. We first localised the two PDEs predicted to be essential for the parasite lytic cycle by inserting 3Ty epitope tags at the carboxy‐terminus via single homologous recombination in ΔKu80 parasites (Fig EV5A). PDE1‐3Ty which encodes four transmembrane domains showed a punctate pattern with an enrichment at the parasite periphery (Fig 7A) while PDE2‐3Ty showed a cytoplasmic punctate pattern with no signal in the nucleus (Fig 7B). We then characterised the subcellular distribution of the only putative guanylyl cyclase (an orthologue of Plasmodium GCα, TGGT1_254370), using the same epitope tagging strategy (Fig EV5A). GCα is a large polytopic protein that is predicted to code for a putative phospholipid P‐type ATPase translocating domain at its N‐terminus and two guanylate cyclase domains at its C‐terminal end. GCα‐3Ty distributes between the plasma membrane at the apical pole and within the residual body (Fig 7C) and by Western blot analysis revealed multiple isoforms (Fig 7E). PKG was also tagged by inserting 3Ty epitope tags at the carboxy‐terminus (Fig EV5A). IFA revealed a relatively broad distribution of PKG‐3Ty with enrichment at the cell periphery, the tip of the apical pole, and the residual body suggesting that PKG is required at multiple subcellular localisations (Fig 7D). Western blot analysis of PKG‐3Ty confirmed the expression of at least two isoforms as previously described (Brown et al, 2017), with an acylated large isoform (~130 kDa) and a non‐acylated shorter isoform (~100 kDa) (Fig 7F). Altogether, this shows that the apical pole and the pellicle represent crucial subcellular compartments integrating both cAMP and cGMP signals.
Figure EV5. Generation and characterisation of PDE1‐3Ty, PDE2‐3Ty, GCα‐3Ty, PKG‐3Ty, ACα‐KO, Myc‐ACβ‐iKD and ACα‐KO/Myc‐ACβ‐iKD parasites.
- PCR analysis showing correct insertion of the constructs coding for the epitope tags.
- Schematic representation of the strategy used to replace the endogenous promoter of ACβ by a Tet‐inducible promoter.
- PCRs performed on gDNA extracted from a clone showing the correct integration of the construct.
- Down‐regulation of Myc‐ACβ‐iKD after 8 days in presence of ATc leads to the formation of normal plaques.
- Cloning strategy for the deletion of ACα in the Myc‐ACβ‐iKD background.
- PCR analysis showing correct deletion of ACα in the Myc‐ACβ‐iKD background.
- Down‐regulation of Myc‐ACβ‐iKD in the absence of ACα after 8 days in presence of ATc leads to the formation of smaller plaques compared with non‐treated parasites or ACα‐KO parasites.
Figure 7. Cellular localisation of cAMP and cGMP signalling components possibly involved in egress.
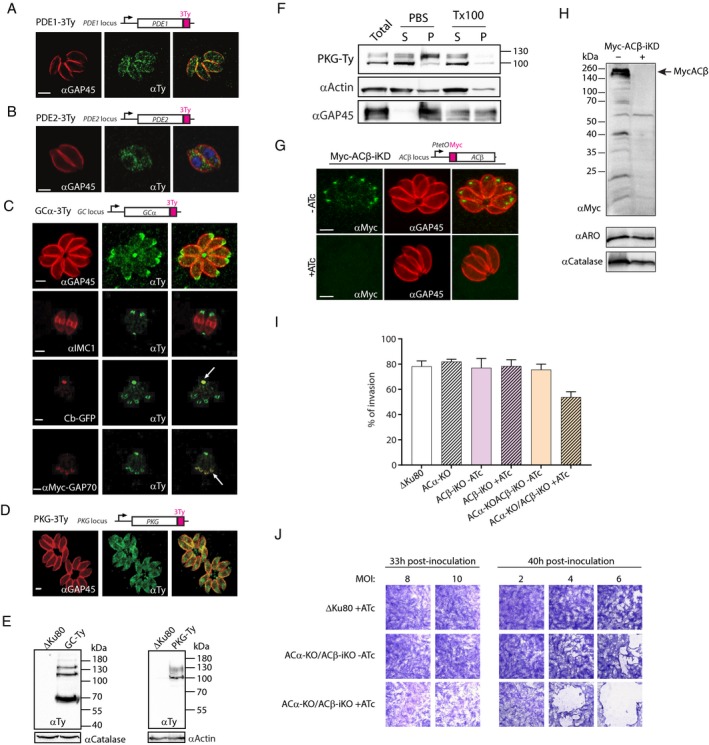
-
A–DLocalisation of PDE1‐3Ty (A), PDE2‐3Ty (B), GCα‐3Ty (C) and PKG‐3Ty (D) shown by IFA using an anti‐Ty antibody. Antibodies against GAP45, IMC1, cb‐GFP and myc‐GAP70 were used as markers of the plasma membrane, IMC, residual body and apical pole, respectively.
-
EWestern blot analysis indicates that GCα‐3Ty is present in three distinct isoforms. A PKG‐3Ty Western blot also shows three immune reactive isoforms. Catalase or actin was used as loading controls and ΔKu80 as the parental strain.
-
FPKG‐3Ty parasites were solubilised in either PBS or 1% Triton X‐100 (TX100) and split into soluble (S) and pellet (P) fractions. Total lysate is also shown.
-
G, HThe signal of Myc‐ACβ‐iKD followed by IFA (G) or Western blot (H) after 48 h ± ATc.
-
IInvasion of ACα‐KO tachyzoites or Myc‐Acβ‐iKD after 48 h ± ATc is not affected. However, addition of ATc for 48 h reduces the invasion efficiency of ACα‐KO/Myc‐Acβ‐iKD (data are from three independent biological replicates). Error bars represent ±SD for 100 vacuoles counted in triplicate from three biological replicates.
-
JAs for destabilisation of PKAc1‐iKD or stabilisation of DDmyc‐PKArG321E‐Ty, ACα‐KO/Myc‐Acβ‐iKD tachyzoites treated with ATc for 33 or 40 h, invade and exit the monolayer of HFF cells leading to lysis, while the non‐treated parasites invade and initiate a new lytic cycle.
We then explored the role of ACα and ACβ in Toxoplasma egress. A Myc‐ACβ‐iKD strain was generated by tet‐repressive promoter replacement at the endogenous locus and confirmed by genomic PCR (Fig EV5B and C). Myc‐ACβ was efficiently depleted upon 48 h treatment with ATc as observed by IFA (Fig 7G) and Western blot (Fig 7H). Since the absence of ACβ led to no significant loss of fitness based on plaque assay (Fig EV5D), we generated single ACα and double ACα‐KO/Myc‐ACβ‐iKD mutants to test whether both enzymes could be at play upstream of PKAc1. Disruption of ACα was generated by homologous replacement of the coding sequence assisted by CRISPR/Cas9 editing and confirmed by genomic PCR (Fig EV5E and F). When comparing the various mutant strains, only ACα‐KO/ACβ‐iKD exhibited a slight defect in invasion (Fig 7I). Remarkably, although less pronounced than in the cases of PKAc1‐iKD or PKArG231E‐Ty, the dual absence of ACα and depletion of ACβ exhibited a combined premature egress and “restless invasion” phenotype observed by the partial lysis of the HFF monolayer cells at 33 and 40 h post‐inoculation (Fig 7J). Altogether ACα and ACβ participate in cAMP synthesis upstream of PKAc1 to control timely egress. This modest defect is recapitulated by smaller plaques of lysis (Fig EV5G).
Discussion
Little is known about PKAc1 and its substrates in T. gondii. In contrast, more is known about the contribution of PfPKA in P. falciparum physiology (Haidar et al, 2016). Notably, PfPKAc was reported to regulate host erythrocyte invasion and the proliferation of merozoites. Concordantly, the glideosome‐associated protein GAP45 and a key component of the moving junction, PfAMA1, were reported to be substrates of PfPKAc in vitro (Leykauf et al, 2010; Lasonder et al, 2012). A phosphoproteomic analysis of P. falciparum schizonts suggested that PKA signalling is involved in a wide variety of processes including signalling mediated by PfCDPK1, the closest homologue of TgCDPK3, as well as other proteins of the glideosome (Lasonder et al, 2012). Recently, PfPKAr was confirmed to hold PfPKAc in an inactive state by exerting an allosteric inhibitory effect that is freed by the binding of cAMP (Littler et al, 2016). PKA is regulated by cAMP, which is produced by ACs. T. gondii tachyzoites express two ACs, ACα and ACβ, the latter being anchored at the surface of the rhoptries as part of a complex with ARO and AIP (Mueller et al, 2016). Both appear to be dispensable during the lytic cycle of this parasite (Sidik et al, 2016). Bicarbonate produced by carbonic anhydrase is known to activate some ACs. Toxoplasma gondii possesses a carbonic anhydrase‐related protein (TgCA), which has been identified at the surface of the rhoptries (Mueller et al, 2016). A recent study reports that TgCA is GPI‐anchored and localises to rhoptry bulbs in mature parasites and to the outer membrane of nascent rhoptries. Parasites lacking TgCA display a growth and invasion phenotype (Chasen et al, 2017). Degradation of cAMP by PDEs is much less characterised with at least 18 putative PDEs encoded in the genome of T. gondii. Only two of them appear to be non‐redundant in the lytic stages but their specificities for cAMP or cGMP remain unknown (Sidik et al, 2016).
In this study, we demonstrate that uncontrolled PKAc1 activity, either by down‐regulating PKAr or overexpressing PKAc1, is highly deleterious for parasite replication. In sharp contrast to PKAc1 activation, PKAc1 down‐regulation or indirect depletion of PKA activity by conditional stabilisation of DDmyc‐PKArG321E‐Ty, a mutant unable to bind cAMP and to release active PKAc, revealed a fundamentally distinct phenotype. While intracellular parasite replication was unaffected, the depletion of PKAc1‐iKD or transient stabilisation of DDmyc‐PKArG321E‐Ty caused premature egress of parasites from infected host cells. Early egress upon PKA genetic inhibition critically depended on the multiplicity of infection. This implied that a diffusible solute was accumulating in densely infected host cells and triggered untimely egress in the absence of functional PKAc. Environmental acidification was previously shown to participate in parasite motility and egress (Roiko et al, 2014) and we showed here that the premature egress induced by PKA genetic inhibition could be blocked by the neutralising action of the weak base NH4Cl. This suggests that PKA activity is required to prevent egress induced by the acidification of the parasite environment.
Parasite egress can be artificially triggered by cGMP‐PDE inhibitors such as zaprinast or BIPPO that elevate cGMP levels and lead to PKG activation and microneme secretion (Lourido et al, 2012; Stewart et al, 2016). This suggests that, similarly, PKA could repress egress by activating cGMP‐PDEs. This hypothesis is consistent with the fact that some PDEs in other organisms are known to be activated via phosphorylation by PKA (Rybalkin et al, 2002; Omori & Kotera, 2007). This mechanism constitutes an important feedback loop that results in the restoration of basal cAMP and cGMP levels. In consequence, it is plausible that microneme secretion in intracellular parasites is controlled by a fine‐tuned balance of the levels of cAMP and cGMP with PKA playing a central role in this process. Concordantly, our comparative phosphoproteome analysis of DDmyc‐PKArG321E‐Ty and DDmyc‐PKArWT‐Ty parasites identified a potential feedback loop between cAMP and cGMP signalling via differential phosphorylation of PKAr and PDE2. Consistent with a key role of cGMP level fluctuations in the integration of PKA signals, premature egress caused by genetic inhibition of PKA could be blocked by inhibiting PKG. Importantly, in all tested conditions, BIPPO was able to induce egress indicating that GCα shows a basal level of activity and that cGMP‐PDE regulation is also a critical regulatory point to control PKG activity. However, the exact contribution of GCα and the cGMP‐PDEs in controlling the delicate balance of cGMP levels in response to changing pH and other stimuli remains to be characterised.
This interplay between cGMP and cAMP signalling relies on a precise spatial organisation of its molecular components (Fig 8). The cAMP synthesising ACβ was previously shown to be present at the surface of the rhoptry organelle facing the parasite cytosol (Mueller et al, 2016). Here, we show that the cAMP effector PKAc1 is targeted to the IMC by its interaction with acylated PKAr. Upon cAMP binding, active PKAc1 is released to phosphorylate its cellular targets including some PKG‐dependent pathways controlling microneme secretion. Consistent with such a role, we found that a subpopulation of the predicted cGMP synthesising GCα and of PKG localises at the apical pole of the parasites. Remarkably, both PKG and GCα were also observed in the residual body suggesting that cGMP signalling may be involved to coordinate microneme secretion at the apical pole and the sealing of the existing connection between parasites via the residual body at the posterior pole (Frenal et al, 2017). When expressed as a second copy, PKG was previously shown to be targeted to the pellicle (Donald & Liberator, 2002b). The localisation of both kinases likely represents a strategic position to react fast on the need to coordinate glideosome activation and microneme secretion to power motility. The localisation of two essential PDEs was less marked, and their potential implication in an interplay between cAMP and cGMP remains to be established.
Figure 8. Geography of cyclic nucleotide signalling and functional relationship between cAMP and cGMP signalling to control pH‐dependent egress.
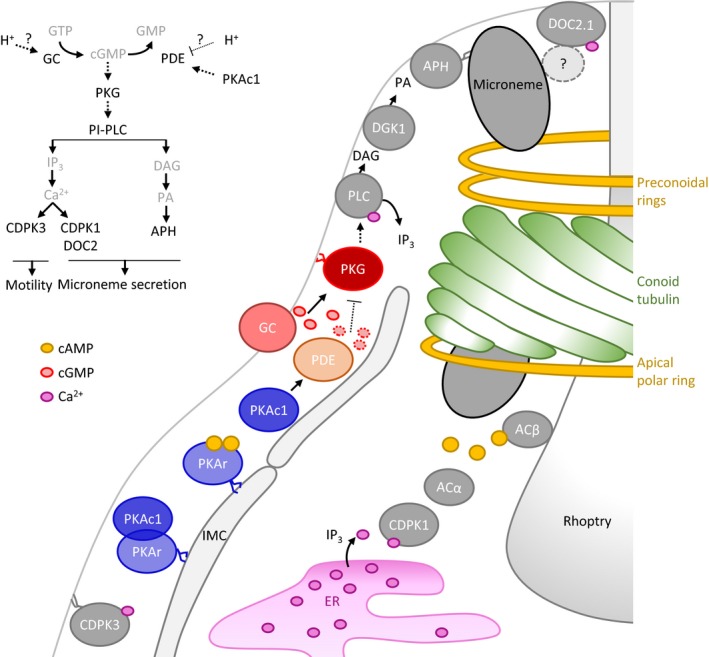
In presence of cAMP produced by ACα and ACβ, active PKAc1 is released from its regulatory subunit PKAr anchored at the inner membrane complex by dual acylation. At low pH, PKAc1 activates an unidentified PDE leading to the degradation of cGMP and down‐regulation of PKG preventing parasite egress. Upon unidentified stimuli, PKAc1 activity is repressed and/or GCα activity stimulated to increase cGMP levels and activate PKG. PKG stimulates Ca2+ mobilisation from intracellular store and PLC activity to produce IP3 and DAG leading to microneme secretion and gliding motility necessary for parasite egress and invasion.
PKG has recently emerged as a central regulator of egress in Apicomplexa by controlling Ca2+ signals upstream of proteins required for egress (Brochet & Billker, 2016; Brown et al, 2016; Stewart et al, 2016). However, how PKG activity is regulated remained a mystery. Here, we have shown that the kinase acts at a central regulatory point that integrates balancing signals to control the timely egress of T. gondii. First, PKG is important to relay intracellular signals induced by environmental acidification to trigger egress. Second, active PKA down‐regulates PKG‐dependent signalling pathways to prevent premature egress induced by environmental acidification in the absence of complementary or more alarming signals (Fig 8).
Materials and Methods
Antibodies and reagents
Primary antibodies used in this study include mouse anti‐Ty tag BB2, mouse anti‐Myc 9E10, mouse anti‐MIC2 (gift from JF Dubremetz), mouse anti‐SAG1 (T4‐1E5), mouse anti‐actin (Herm‐Gotz et al, 2002), mouse anti‐GRA1 (gift from JF Dubremetz), rabbit anti‐IMC1 (Mann & Beckers, 2001), rabbit anti‐GAP45 (Plattner et al, 2008), rabbit anti‐profilin (Plattner et al, 2008), rabbit anti‐ATrx1 (DeRocher et al, 2008) and anti‐catalase (Ding et al, 2000). Secondary antibodies Alexa Fluor 488 and Alexa Fluor 594‐conjugated goat anti‐mouse/rabbit antibodies (Molecular Probes) were used for immunofluorescence analysis and horseradish peroxidase (HRP)‐conjugated goat anti‐mouse/rabbit antibodies (Molecular Probes) for Western blot analysis. PKA inhibitors (H89 and KT‐5270) and NH4Cl were purchased from Sigma. Compound 1 (tri‐substituted pyrrole 4‐[2‐(4‐fluorophenyl)‐5‐(1‐methylpiperidine‐4‐yl)‐1H‐pyrrol‐3‐yl]pyridine) and Compound 2 (4‐[7‐[(dimethylamino) methyl]‐2‐(4‐fluorophenyl) imidazo[1,2‐a] pyridin‐3‐yl] pyrimidin‐2‐amine) were kind gift from David Baker and Oliver Billker, respectively.
Parasite culture and transfection
Tachyzoites of the T. gondii RH strains lacking HXGPRT or KU80 (RH and ΔKu80) were grown in confluent HFF monolayers maintained in Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% foetal calf serum (FCS), 2 mM glutamine and 25 mg/ml gentamicin. Parasites were transfected and selected as described previously (Soldati & Boothroyd, 1993).
Cloning of DNA constructs
All primers used in this study are listed in Appendix Table S1. Genomic DNA was isolated with the Wizard SV genomic DNA purification system (Promega). RNA was purified using Trizol (Invitrogen) extraction. Total cDNA was generated by RT–PCR using the Superscript II reverse transcriptase (Invitrogen) according to the manufacturer's protocol.
To generate C‐terminal triple Ty‐tagged PKAc1, PKAr, PDE1, PDE2, PKG and GCα at the endogenous locus, genomic fragments were amplified with Phusion DNA polymerase (NEB) using primer pairs 6046–6047, 4085–4086, 5284–5285, 5301–5302, 5819–6232 and 6637–6638, respectively. PCR products were cloned into KpnI/NsiI (SbfI) sites of the pT8‐TgMIC13‐3Ty‐HXGPRT (Friedrich et al, 2010) or ApaI/Nsi (SbfI) sites of the pTgASP5‐3Ty‐DHFR and linearised with PstI, EcoRI, EcoRV, BglII, MfeI and BstBI respectively, before transfection.
To generate C‐terminal TyDD‐tagged PKAr (PKAr‐TyDD) at the endogenous locus, Ty‐tagged PKAr coding sequence was amplified from pPKAr‐3Ty‐ki using primer pair 4086–4253 and cloned into KpnI/SbfI (PstI) sites of pT8‐MycGFPDD‐HXGPRT (Herm‐Gotz et al, 2007). pPKAr‐TyDD‐ki were linearised with EcoRI prior to transfection. For the PKAr‐TyDD, transfected parasites were maintained in presence of 0.5 μM Shld‐1 with mycophenolic acid (MPA) and xanthine selection. Integration at both sites of the endogenous PKAr locus was confirmed by PCR of genomic DNA (Fig EV1B).
The vector pT8‐PKAr‐Ty‐HXGPRT was obtained by cloning the PCR product of primer pair 4084–4085 amplified from PKAr cDNA into the EcoRI/NsiI (SbfI) sites in pT8Myc‐GFP‐PfmyoATail‐Ty‐HXGPRT (Herm‐Gotz et al, 2007). Mutation of the putative myristoylation site (G2A) was introduced using primer pair 4462‐p30a, and the PCR product was cloned into EcoRI/PacI sites of pT8Myc‐GFP‐PfmyoATail‐Ty‐HXGPRT. To swap the N‐terminal PKAr with the N‐terminal of GAP45, the coding region of PKAr lacking the first 20 codons was amplified using primer pair 4466‐p30a and the corresponding PCR product was introduced into the NsiI (SbfI)/PacI site of pT8‐GAP45N21‐GFP‐HXGPRT (Frenal et al, 2010). For pT8‐DDmyc‐PKAc1‐HXGPRT and pT8‐DDmyc‐PKArWT‐Ty‐HXGPRT, full‐length PKAc1 and full‐length PKAr were amplified by PCR using primer pairs 5247–5248 and 5084‐p30a from respective cDNA and pT8‐PKArWT‐Ty and cloned into the NsiI(PstI)/PacI sites of pT8‐DDmyc‐GFP‐HXGPRT (Herm‐Gotz et al, 2007). PKArG321E point mutations in the pT8‐PKArG321E‐Ty and pT8‐DDmyc‐PKArG321E‐Ty were introduced with templates pT8‐PKArWT‐Ty and pT8‐DDmyc‐PKArWT‐Ty, respectively, using the QuickChange II Site‐Directed Mutagenesis kit (Stratagene) with primer pair 4471–4472 according to the manufacturer's instructions.
To generate the construct for the direct knockout of endogenous PKAc1 in the DDmycPKAc1/PKArWT‐Ty strain, 5′ UTR and C‐terminal coding region of PKAc1 (1.134 kb and 2.628 kb, respectively) were PCR amplified using primer pairs 5587–5588 for the 5′ homologous region and 5589–5590 for 3′ homologous region and subsequently cloned into of the pT5DHFRSag1 plasmid using HindIII/NheI and NotI/SpeI sites, respectively. The final construct 5′PKAc1‐pT5DHFRSag1‐3′PKAc1 was linearised with NotI/HindIII prior to transfection. Transfected parasites were maintained in presence of 0.5 μM Shld‐1 with pyrimethamine (1 μg/ml).
The gRNA/Cas9 vectors used for endogenous PKAr, PKAc1, PDE1, PDE2 and PKG genome editing were generated using the Q5 site‐directed mutagenesis kit (New England Biolabs) with pSAG1::CAS9‐GFP‐U6::sgUPRT (Shen et al, 2014) as template using primer pairs 5198–4483, 5199–4483, 5303–4483, 5730–4483 and 6319–4483, respectively. For the CRISPR/Cas9‐mediated N‐terminus DDmyc‐tagging of endogenous PKAc1, and PDE1, 20 μg of the specific gRNA/Cas9 vectors and purified specific PCR products using primer pairs 5352–5353 and 5304–5305 amplified from pT5DHFRSag1 plasmid was transfected. To generate the conditional knockdown of PKAc1, PDE1 and PDE2, 20 μg of the respective gRNA/Cas9 vectors and purified PCR products using primer pairs 5584–5585, 5592–5593 and 5731–5733 amplified from 5′COR‐pT8TATi‐HX‐TetO7S1mycNtCOR‐Ty plasmid was transfected. GFP‐positive parasites were FACS sorted 24 h post‐transfection and cloned into 96‐well plates.
To generate endogenous the PKArG2A line, 20 μg of pSAG1::CAS9‐U6::sgPKAr and annealed oligonucleotides 5200–5201 was transfected into the ΔKu80 strain. To generate DDmyc‐PKArWT‐Ty/PKGT761M and DDmyc‐PKArG321E‐Ty/PKGT761M lines, 20 μg of pSAG1::CAS9‐U6::sgPKG and annealed oligonucleotides 6320–6321 was transfected in the DDmyc‐PKArWT‐Ty and DDmyc‐PKArG321E‐Ty strains. Both mutants were confirmed by sequencing.
To generate the PKAc1‐iKD vector, two KOD (Merck) PCRs using primers 5584‐5585 were performed using 5′COR‐pT8TATi1‐HX‐tetO7S1mycNtCOR (Salamun et al, 2014) as template. A specific gRNA/Cas9 vector used for PKAc1‐iKD was generated using the Q5 site‐directed mutagenesis kit (New England Biolabs) with pSAG1::CAS9‐GFP‐U6::sgUPRT as template (Shen et al, 2014) using primers 5199–4483. To endogenously tag PKAc1‐iKD at the C‐terminus, the C‐terminal part was amplified from genomic DNA using primers 7244–6049 digested with KpnI/NsiI and cloned in pG152‐3Ty‐LoxP‐3′UTR‐Sag1‐HXGPRT‐LoxP‐U1 (Pieperhoff et al, 2015). The HXGPRT cassette was exchanged with DHFR using the restriction sites SacII with p2854‐DHFR as donor plasmid.
To generate the ACβ‐iKD vector, parasite gDNA was used as template to amplify ACβ 5′ UTR with primers 4534–4535 and ACβ N‐terminus with primers 4335–4337. ACβ N‐terminus was digested with BglII and NotI and ligated into the BglII/NotI digested 5′COR‐pT8TATi1‐HX‐tetO7S1mycNtCOR (Salamun et al, 2014). ACβ 5′UTR was then digested with SnaBI and BamHI and ligated into 5′COR‐T8TATi1‐HXtetS1mycNtTgACβ that had been digested with the same two enzymes.
To generate the ACα‐KO vector, two KOD (Merck) PCRs using primers 5218–5219 with p2854‐DHFR as template were performed. Specific gRNA was generated as described above with primers 4483–5115.
The vector pT8‐Cb‐GFP‐HXGPRT was obtained by cloning the PCR product of the actin chromobody (Cb) using the primer pair 7110–7111 amplified from pT8‐Cb‐Halo‐HXGPRT (Periz et al, 2017) into a pT8‐Cb‐GFP‐Ty‐HXGPRT (EcoRI/NsiI) and subsequently amplified by PCR using the primers 7110–7212 and subconed in the in pT8‐Cb‐GFP‐Ty‐HXGPRT (EcoRI/PacI) in order to remove the Ty epitope.
Indirect immunofluorescence analysis (IFA)
HFF monolayers grown on 24‐well coverslips were infected with tachyzoites and grown ± 0.5 μM Shld‐1 for 24 h. Immunofluorescence analysis was performed as previously described (Daher et al, 2010). Nuclei were stained with DAPI, and coverslips were mounted in Fluoromount G (Southern Biotech). Images were acquired with a LSM700 confocal scanning microscope (Zeiss).
Aerolysin treatment
Freshly lysed parasites were harvested and washed once in PBS buffer. Plasma membrane and IMC separation were achieved after aerolysin treatment, as previously described (Frenal et al, 2010).
Western blotting
Freshly lysed extracellular parasites were added to HFF monolayers in presence or absence of 0.5 μM Shld‐1. Freshly lysed parasites were harvested, washed once in PBS and analysed with indicated antibodies as described (Frenal et al, 2010).
Plaque assays
Freshly egressed parasites were inoculated on HFF monolayers in presence or absence of 0.5 μM Shld‐1. Parasites were grown for 8 days prior to fixation with 4% paraformaldehyde/0.005% glutaraldehyde in PBS and stained with 0.1% crystal violet.
Intracellular growth assay
HFF monolayers were inoculated with freshly egressed parasites and incubated for 24 h ± 0.5 μM Shld‐1 before fixation with 4% paraformaldehyde/0.005% glutaraldehyde in PBS. Cells were stained using α‐GAP45 (1/5,000) to detect individual parasites. Results are shown as the mean and the standard deviation from 100 parasites counted in triplicate from three independent biological replicates.
Metabolic labelling and co‐immunoprecipitation
Freshly released parasites were harvested and incubated for 15 min in methionine/cysteine‐free DMEM (Sigma). Parasites were harvested and resuspended in DMEM containing 10 μCi [35S]‐labelled methionine/cysteine (Hartmann analytic GmbH) per ml for 4 h. Parasites were lysed in CoIP buffer (1% (v/v) Triton X‐100, 50 mM Tris–HCl, pH 8, 150 mM NaCl) in presence of a protease inhibitor cocktail (Roche). Cells were incubated for 10 min on ice, frozen and thawed, sonicated and centrifuged at 14,000 g for 30 min. Supernatants were incubated with the corresponding antibodies as previously described (Gaskins et al, 2004). All samples were boiled prior to SDS–PAGE.
Egress assay
Freshly egressed parasites were inoculated on HFF monolayers and allowed to grow for 30 h. Infected host cells were incubated ± 0.5 μM Shld‐1 or combined with H89 and sampled and fixed every hour prior to fixation. Cells were labelled with anti‐GAP45 antibodies, and the proportion of egressed versus non‐egressed vacuoles was calculated by counting 100 vacuoles in triplicate from three independent biological replicates. To investigate the effect of PKA genetic inhibition, freshly egressed DDmyc‐PKArG321E‐Ty parasites were inoculated on HFF monolayers and allowed to grow, ± 0.5 μM Shld‐1. Parasites were fixed at different time points (38, 45, 51 and 86 h), and IFA was performed using α‐GRA3 and α‐GAP45 antibodies. To test the effect of the supernatant of heavily infected cell (MOI 8) on less heavily infected cells (MOI 2), the cells were fixed after 3 h of incubation with 4% paraformaldehyde/0.005% glutaraldehyde in PBS and stained with α‐GRA3 and α‐GAP45 antibodies. To test the effect of host cell pH, parasites were first pre‐treated with C1 or its vehicle for 10 min and subsequently incubated in intracellular buffer at pH 7.2, 7.0 and 6.8 for 10 min. Cells were then fixed with 4% paraformaldehyde/0.005% glutaraldehyde in PBS. In the case where parasites were treated with NH4Cl, the cells were pre‐treated with Shld‐1 for 1 h and fixed after 2‐h incubation with NH4Cl. Treatments with A23187 or BIPPO were performed 10 min prior to the end of this 2‐h incubation.
Density assay
Freshly egressed DDmyc‐PKArG321E‐Ty parasites were inoculated on HFF cells with a multiplicity of infection (MOI) of 7.66, 3.83, 1.92, 0.96, 0.48 and 0.24. Parasites were grown for 26 h before adding Shld‐1 for 4 h. Parasites were then fixed with 4% paraformaldehyde/0.005% glutaraldehyde and stained with α‐GRA3 and α‐GAP45 antibodies. The average number of ruptured vacuoles was determined by counting 300 vacuoles for each condition and for three independent experiments.
Phosphoproteome analysis
Confluent HFFs were heavily infected with the DDmyc‐PKArWT‐Ty or DDmyc‐PKArG321E‐Ty parasites. Parasites were grown for 26 h before adding ± Shld‐1 for 20 min or 1 h. Parasites were washed twice with pre‐warmed PBS to deplete residual serum‐containing media. Parasites were lysed for 30 min on ice in 5 ml lysis buffer (8 M urea, 25 mM Tris pH 8, 100 mM sodium chloride, containing Halt™Protease & Phosphatase Inhibitor Cocktail from Life Technology). Samples were sonicated three times for 30 s on ice with 30% output. Samples were spin down at 15,000 ×g for 10 min to remove cellular debris. The protein concentration was estimated using the Pierce™ BCA Protein Assay Kit. 100 μg of proteins in 150 μl was reduced with 20 mM TCEP (Sigma) at 56°C for 20 min and then was alkylated for 2 h at room temperature in the dark by adding iodoacetamide (Sigma) to a final concentration of 30 mM. Proteins were precipitated with 20% trichloroacetic acid on ice to remove the detergent and the excess reagent. Protein pellets were subsequently digested with 4 μg trypsin at 37°C in 100 μl of 100 mM TEAB (triethylammonium bicarbonate) for 12 h. The resulting tryptic peptides were labelled with TMT6plex (Life Technologies), and then, samples were pooled together and dried in SpeedVac. The mixed peptides were fractionated on a 2.1 mm i.d. × 150 mm XBridge BEH C18 column (130 Å, 3.5 μm, Waters) at pH 10 over a linear gradient of 5–35% ACN/0.1% NH3 in 30 min/60 min cycle time. Fractions were collected every minute for the first 41 min, while fractions from the first 6 min and last 8 min were pooled at 2‐ or 3‐min intervals.
Enrichment for phosphopeptides was performed with IMAC with PHOS‐Select Iron Affinity Gel (Sigma) and then TiO2 tips (Thermo Fisher). All procedures followed s manufacturer's instruction with some modifications. 100 μl of 50% suspension of PHOS‐Select Iron Affinity Gel was used for each fraction. The peptides were redissolved in 50% CH3CN/250 mM acetic acid/0.1% TFA (trifluoroacetic acid) (binding solution) and then added to the prewashed beads. After binding at room temperature with end‐to‐end rotation for 30 min, the beads were washed three times with the binding solution and once with H2O. Phosphopeptides were eluted twice with 100 μl of 1.5% NH3/25% ACN then dried in SpeedVac. The flow through and the first wash of IMAC beads were collected and dried in SpeedVac, and then, phosphopeptides were enriched with TiO2 tips. Phosphopeptides were eluted from the tip with 1.5% NH4OH and then 5% pyrrolidine. Both eluates were pooled, acidified and desalted on Graphite Spin Columns (Thermo) as instructed by the manufacturer's protocol.
IMAC‐ and TiO2‐enriched phosphopeptides were redissolved in 0.5% FA before LC‐MS/MS analysis on an Orbitrap Fusion Tribrid mass spectrometer coupled with an Ultimate 3000 RSLCnano system. Peptides from same fraction were loaded on a trap column (Acclaim PepMap C18, 100 μm i.d. × 20 mm) and then separated on a 75 μm i.d. × 500 mm column (Acclaim PepMap C18) with a linear gradient of 4‐36% CH3CN/0.1% FA for 100 min with a total of 130 min per cycle. The Orbitrap Fusion was operated at top 10 methods. The MS full scan was in Orbitrap (m/z 380–1580) with a lock mass at 445.120025, a resolution at 120,000 at m/z 200 and an AGC at 5 × 105 with a maximum injection time at 150 msec. The 10 most abundant multiply‐charged precursor ions (2+ to 7+), with a minimal signal above 1,000 counts, were dynamically selected for HCD fragmentation (MS/MS) and detected in Orbitrap with a resolution at 60,000 at m/z 200, and the AGC at 2 × 105 with the maximum injection time at 100 ms. The isolation width was 1.2 Da in quadrupole, and the collision energy was set at 40%. The dynamic exclusion duration time was set for 30 s with ± 10 ppm exclusion mass width.
Raw data were processed in Proteome Discoverer 1.4 with SequestHT search engine against a combined protein database of Toxoplasma gondii (ToxoDB‐25_TgondiiGT1_AnnotatedProteins.fasta from Toxodb.org) and mouse (UniprotKB). Carbamidomethyl (C) and TMT6plex were set as fixed modifications, and deamidated (NQ) and phospho (STY) were set as dynamic modifications. Precursor mass tolerance was set at 20 ppm and fragment ions at 0.2 Da. The search result was filtered with percolator where the q‐value was set at 0.01. Only unique peptides were used in quantification, but normalisation was not applied, and co‐isolation was set at 100%.
Invasion assays
Invasion assays were performed using the RH‐2YFP strain as internal standard as described in (Sawmynaden et al, 2008). Briefly, confluent HFF cells were heavily infected with a mixture of the DDmyc‐PKArG321E‐Ty and RH‐2YFP parasites and washed with DMEM medium after 2 h. Parasites were grown in the absence of Shld‐1 for 26 h, before adding ± 0.5 μM Shld‐1for 3 h. Extracellular parasites were then collected, and the ratio of non‐YFP to YFP parasites was determined. At the same time, these parasites were transferred on new host cells and allowed to invade for 30 min at 37°C ± Shld‐1 before washing with DMEM medium. Parasites were allowed to further develop for 24 h ± Shld‐1, and cells were fixed with 4% paraformaldehyde/0.05% glutaraldehyde. Parasites were stained with αGAP45, and the ratio between non‐YFP and YFP parasite vacuoles was calculated. The efficiency of invasion was determined by counting around 250 vacuoles for each condition and for three independent experiments.
To confirm the results obtained using strain RH‐2YFP as an internal standard, a red/green invasion assay was used to distinguish non‐invaded from invaded parasites. Freshly egressed parasites were inoculated on 24‐well plates containing coverslips seeded with HFF monolayers and were centrifuged at 250 g for 1 min at RT. Invasion was allowed to take place for 30 min at 37°C prior to fixation with 4% paraformaldehyde/0.005% glutaraldehyde. Standard non‐permeabilising IFA was performed by staining extracellular parasites with a monoclonal anti‐SAG1 antibody, followed by permeabilisation with PBS/Triton and subsequent staining of invaded parasites with a polyclonal anti‐GAP45 antibody. For each experiment, at least 50 parasites were counted, each time distinguishing non‐invaded (red) from invaded (green) parasites. Three independent biological replicates were completed.
Restless invasion assay
To monitor parasite‐induced destruction of the fibroblasts monolayer by abortive invasion, HFF was heavily infected with PKAc1‐iKD or control parasites and treated for 36 h ± ATc (up to the premature egress of the PKAc1‐iKD + ATc). All cultures were passed through a needle to release the remaining intracellular parasites. Parasites were let to invade new HFF for 10 h before fixation with PFA/GA for 10 min. Samples were finally stained with Crystal violet. The ACβ‐iKD/ACαKO strain was processed identically, but parasites were pre‐treated for 48 h ± ATc before starting the assay. The DDmyc‐PKArG321E‐Ty strain was pre‐treated ± Shld‐1 for 4 h at 32 h post‐infection and processed like PKAc1‐iKD.
Microneme secretion assay
Confluent HFFs were heavily infected with the DDmyc‐PKArWT‐Ty or DDmyc‐PKArG321E‐Ty strains. Parasites were grown for 26 h before adding ± Shld‐1 for 1 h. Dishes were rinsed twice with intracellular (IC) buffer (5 mM NaCl, 142 mM KCl, 1 mM MgCl2, 2 mM EGTA, 5.6 mM glucose, 25 mM HEPES, pH to 7.2 with KOH), scrapped in 2 ml of IC buffer, passed three times through a 30G needle and centrifuged at 1,000 g for 3 min at room temperature. The pellet was resuspended in IC buffer and incubated at 37°C for 70 min. Parasites were centrifuged at 1,000 g for 5 min at 4°C, and supernatants were transferred to new tubes and centrifuged again. Final supernatants and pellets were resuspended in sample buffer for further Western blotting analysis.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD006045.
Author contributions
YJ, J‐BM, HB, DJ and CM performed experiments. LY, MB and JC analysed the phosphoproteome data. DS‐F and MB supervised the project. DS‐F, MB and YJ wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7
Source Data for Expanded View
Acknowledgements
This work was supported by the Swiss National Foundation (FN310030B_166678 to D.S.‐F. and BSSGI0_155852 to M.B.). H.B.S is recipient of a Swiss Government Excellence Scholarship with Uruguay. D.S.‐F. is a HHMI senior international research scholar, and M.B. is an INSERM investigator. We thank Dr. C. Tonkin for fruitful discussion and sharing unpublished data and Dr. M. Meissner for providing an actin chromobody construct.
The EMBO Journal (2017) 36: 3250–3267
Contributor Information
Mathieu Brochet, Email: mathieu.brochet@unige.ch.
Dominique Soldati‐Favre, Email: dominique.soldati-favre@unige.ch.
References
- Baker DA (2011) Cyclic nucleotide signalling in malaria parasites. Cell Microbiol 13: 331–339 [DOI] [PubMed] [Google Scholar]
- Blader IJ, Coleman BI, Chen CT, Gubbels MJ (2015) Lytic cycle of Toxoplasma gondii: 15 years later. Annu Rev Microbiol 69: 463–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochet M, Collins MO, Smith TK, Thompson E, Sebastian S, Volkmann K, Schwach F, Chappell L, Gomes AR, Berriman M, Rayner JC, Baker DA, Choudhary J, Billker O (2014) Phosphoinositide metabolism links cGMP‐dependent protein kinase G to essential Ca2+ signals at key decision points in the life cycle of malaria parasites. PLoS Biol 12: e1001806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochet M, Billker O (2016) Calcium signalling in malaria parasites. Mol Microbiol 100: 397–408 [DOI] [PubMed] [Google Scholar]
- Brown KM, Lourido S, Sibley LD (2016) Serum albumin stimulates protein kinase G‐dependent microneme secretion in Toxoplasma gondii . J Biol Chem 291: 9554–9565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KM, Long S, Sibley LD (2017) Plasma membrane association by N‐acylation governs PKG function in Toxoplasma gondii . MBio 8: e00375‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullen HE, Jia Y, Yamaryo‐Botte Y, Bisio H, Zhang O, Jemelin NK, Marq JB, Carruthers V, Botte CY, Soldati‐Favre D (2016) Phosphatidic acid‐mediated signaling regulates microneme secretion in Toxoplasma . Cell Host Microbe 19: 349–360 [DOI] [PubMed] [Google Scholar]
- Caballero MC, Alonso AM, Deng B, Attias M, de Souza W, Corvi MM (2016) Identification of new palmitoylated proteins in Toxoplasma gondii . Biochim Biophys Acta 1864: 400–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera A, Herrmann S, Warszta D, Santos JM, John Peter AT, Kono M, Debrouver S, Jacobs T, Spielmann T, Ungermann C, Soldati‐Favre D, Gilberger TW (2012) Dissection of minimal sequence requirements for rhoptry membrane targeting in the malaria parasite. Traffic 13: 1335–1350 [DOI] [PubMed] [Google Scholar]
- Chasen NM, Asady B, Lemgruber L, Vommaro RC, Kissinger JC, Coppens I, Moreno SNJ (2017) A glycosylphosphatidylinositol‐anchored carbonic anhydrase‐related protein of Toxoplasma gondii is important for rhoptry biogenesis and virulence. mSphere 2: e00027‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CR, Hackett F, Strath M, Penzo M, Withers‐Martinez C, Baker DA, Blackman MJ (2013) Malaria parasite cGMP‐dependent protein kinase regulates blood stage merozoite secretory organelle discharge and egress. PLoS Pathog 9: e1003344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daher W, Plattner F, Carlier MF, Soldati‐Favre D (2010) Concerted action of two formins in gliding motility and host cell invasion by Toxoplasma gondii . PLoS Pathog 6: e1001132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawn A, Singh S, More KR, Siddiqui FA, Pachikara N, Ramdani G, Langsley G, Chitnis CE (2014) The central role of cAMP in regulating Plasmodium falciparum merozoite invasion of human erythrocytes. PLoS Pathog 10: e1004520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRocher AE, Coppens I, Karnataki A, Gilbert LA, Rome ME, Feagin JE, Bradley PJ, Parsons M (2008) A thioredoxin family protein of the apicoplast periphery identifies abundant candidate transport vesicles in Toxoplasma gondii . Eukaryot Cell 7: 1518–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding M, Clayton C, Soldati D (2000) Toxoplasma gondii catalase: are there peroxisomes in Toxoplasma? J Cell Sci 113: 2409–2419 [DOI] [PubMed] [Google Scholar]
- Donald RG, Allocco J, Singh SB, Nare B, Salowe SP, Wiltsie J, Liberator PA (2002a) Toxoplasma gondii cyclic GMP‐dependent kinase: chemotherapeutic targeting of an essential parasite protein kinase. Eukaryot Cell 1: 317–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donald RG, Liberator PA (2002b) Molecular characterization of a coccidian parasite cGMP dependent protein kinase. Mol Biochem Parasitol 120: 165–175 [DOI] [PubMed] [Google Scholar]
- Donald RG, Zhong T, Wiersma H, Nare B, Yao D, Lee A, Allocco J, Liberator PA (2006) Anticoccidial kinase inhibitors: identification of protein kinase targets secondary to cGMP‐dependent protein kinase. Mol Biochem Parasitol 149: 86–98 [DOI] [PubMed] [Google Scholar]
- Farrell A, Thirugnanam S, Lorestani A, Dvorin JD, Eidell KP, Ferguson DJ, Anderson‐White BR, Duraisingh MT, Marth GT, Gubbels MJ (2012) A DOC2 protein identified by mutational profiling is essential for apicomplexan parasite exocytosis. Science 335: 218–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenal K, Polonais V, Marq JB, Stratmann R, Limenitakis J, Soldati‐Favre D (2010) Functional dissection of the apicomplexan glideosome molecular architecture. Cell Host Microbe 8: 343–357 [DOI] [PubMed] [Google Scholar]
- Frenal K, Kemp LE, Soldati‐Favre D (2014) Emerging roles for protein S‐palmitoylation in Toxoplasma biology. Int J Parasitol 44: 121–131 [DOI] [PubMed] [Google Scholar]
- Frenal K, Jacot D, Hammoudi PM, Graindorge A, Maco B, Soldati‐Favre D (2017) Myosin‐dependent cell‐cell communication controls synchronicity of division in acute and chronic stages of Toxoplasma gondii . Nat Commun 8: 15710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich N, Santos JM, Liu Y, Palma AS, Leon E, Saouros S, Kiso M, Blackman MJ, Matthews S, Feizi T, Soldati‐Favre D (2010) Members of a novel protein family containing microneme adhesive repeat domains act as sialic acid‐binding lectins during host cell invasion by apicomplexan parasites. J Biol Chem 285: 2064–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaskins E, Gilk S, DeVore N, Mann T, Ward G, Beckers C (2004) Identification of the membrane receptor of a class XIV myosin in Toxoplasma gondii . J Cell Biol 165: 383–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haidar M, Ramdani G, Kennedy EJ, Langsley G (2016) PKA and apicomplexan parasite diseases. Horm Metab Res 22: 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haste NM, Talabani H, Doo A, Merckx A, Langsley G, Taylor SS (2012) Exploring the Plasmodium falciparum cyclic‐adenosine monophosphate (cAMP)‐dependent protein kinase (PfPKA) as a therapeutic target. Microbes Infect 14: 838–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herm‐Gotz A, Weiss S, Stratmann R, Fujita‐Becker S, Ruff C, Meyhofer E, Soldati T, Manstein DJ, Geeves MA, Soldati D (2002) Toxoplasma gondii myosin A and its light chain: a fast, single‐headed, plus‐end‐directed motor. EMBO J 21: 2149–2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herm‐Gotz A, Agop‐Nersesian C, Munter S, Grimley JS, Wandless TJ, Frischknecht F, Meissner M (2007) Rapid control of protein level in the apicomplexan Toxoplasma gondii . Nat Methods 4: 1003–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard BL, Harvey KL, Stewart RJ, Azevedo MF, Crabb BS, Jennings IG, Sanders PR, Manallack DT, Thompson PE, Tonkin CJ, Gilson PR (2015) Identification of potent phosphodiesterase inhibitors that demonstrate cyclic nucleotide‐dependent functions in apicomplexan parasites. ACS Chem Biol 10: 1145–1154 [DOI] [PubMed] [Google Scholar]
- Kirkman LA, Weiss LM, Kim K (2001) Cyclic nucleotide signaling in Toxoplasma gondii bradyzoite differentiation. Infect Immun 69: 148–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa H, Kato K, Iwanaga T, Sugi T, Sudo A, Kobayashi K, Gong H, Takemae H, Recuenco FC, Horimoto T, Akashi H (2011) Identification of Toxoplasma gondii cAMP dependent protein kinase and its role in the tachyzoite growth. PLoS ONE 6: e22492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasonder E, Green JL, Camarda G, Talabani H, Holder AA, Langsley G, Alano P (2012) The Plasmodium falciparum schizont phosphoproteome reveals extensive phosphatidylinositol and cAMP‐protein kinase A signaling. J Proteome Res 11: 5323–5337 [DOI] [PubMed] [Google Scholar]
- Leykauf K, Treeck M, Gilson PR, Nebl T, Braulke T, Cowman AF, Gilberger TW, Crabb BS (2010) Protein kinase a dependent phosphorylation of apical membrane antigen 1 plays an important role in erythrocyte invasion by the malaria parasite. PLoS Pathog 6: e1000941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Cox LS (2000) Isolation and characterisation of a cAMP‐dependent protein kinase catalytic subunit gene from Plasmodium falciparum . Mol Biochem Parasitol 109: 157–163 [DOI] [PubMed] [Google Scholar]
- Littler DR, Bullen HE, Harvey KL, Beddoe T, Crabb BS, Rossjohn J, Gilson PR (2016) Disrupting the allosteric interaction between the Plasmodium falciparum cAMP‐dependent kinase and its regulatory subunit. J Biol Chem 291: 25375–25386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochner A, Moolman JA (2006) The many faces of H89: a review. Cardiovasc Drug Rev 24: 261–274 [DOI] [PubMed] [Google Scholar]
- Lourido S, Shuman J, Zhang C, Shokat KM, Hui R, Sibley LD (2010) Calcium‐dependent protein kinase 1 is an essential regulator of exocytosis in Toxoplasma . Nature 465: 359–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourido S, Tang K, Sibley LD (2012) Distinct signalling pathways control Toxoplasma egress and host‐cell invasion. EMBO J 31: 4524–4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann T, Beckers C (2001) Characterization of the subpellicular network, a filamentous membrane skeletal component in the parasite Toxoplasma gondii . Mol Biochem Parasitol 115: 257–268 [DOI] [PubMed] [Google Scholar]
- McCoy JM, Whitehead L, van Dooren GG, Tonkin CJ (2012) TgCDPK3 regulates calcium‐dependent egress of Toxoplasma gondii from host cells. PLoS Pathog 8: e1003066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merckx A, Nivez MP, Bouyer G, Alano P, Langsley G, Deitsch K, Thomas S, Doerig C, Egee S (2008) Plasmodium falciparum regulatory subunit of cAMP‐dependent PKA and anion channel conductance. PLoS Pathog 4: e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya JG, Liesenfeld O (2004) Toxoplasmosis. Lancet 363: 1965–1976 [DOI] [PubMed] [Google Scholar]
- Moudy R, Manning TJ, Beckers CJ (2001) The loss of cytoplasmic potassium upon host cell breakdown triggers egress of Toxoplasma gondii . J Biol Chem 276: 41492–41501 [DOI] [PubMed] [Google Scholar]
- Mueller C, Samoo A, Hammoudi PM, Klages N, Kallio JP, Kursula I, Soldati‐Favre D (2016) Structural and functional dissection of Toxoplasma gondii armadillo repeats only protein. J Cell Sci 129: 1031–1045 [DOI] [PubMed] [Google Scholar]
- Omori K, Kotera J (2007) Overview of PDEs and their regulation. Circ Res 100: 309–327 [DOI] [PubMed] [Google Scholar]
- Periz J, Whitelaw J, Harding C, Gras S, Del Rosario Minina MI, Latorre‐Barragan F, Lemgruber L, Reimer MA, Insall R, Heaslip A, Meissner M (2017) Toxoplasma gondii F‐actin forms an extensive filamentous network required for material exchange and parasite maturation. Elife 6: e24119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieperhoff MS, Pall GS, Jimenez‐Ruiz E, Das S, Melatti C, Gow M, Wong EH, Heng J, Muller S, Blackman MJ, Meissner M (2015) Conditional U1 gene silencing in Toxoplasma gondii . PLoS ONE 10: e0130356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner F, Yarovinsky F, Romero S, Didry D, Carlier MF, Sher A, Soldati‐Favre D (2008) Toxoplasma profilin is essential for host cell invasion and TLR11‐dependent induction of an interleukin‐12 response. Cell Host Microbe 3: 77–87 [DOI] [PubMed] [Google Scholar]
- Roiko MS, Svezhova N, Carruthers VB (2014) Acidification activates Toxoplasma gondii motility and egress by enhancing protein secretion and cytolytic activity. PLoS Pathog 10: e1004488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybalkin SD, Rybalkina IG, Feil R, Hofmann F, Beavo JA (2002) Regulation of cGMP‐specific phosphodiesterase (PDE5) phosphorylation in smooth muscle cells. J Biol Chem 277: 3310–3317 [DOI] [PubMed] [Google Scholar]
- Salamun J, Kallio JP, Daher W, Soldati‐Favre D, Kursula I (2014) Structure of Toxoplasma gondii coronin, an actin‐binding protein that relocalizes to the posterior pole of invasive parasites and contributes to invasion and egress. FASEB J 28: 4729–4747 [DOI] [PubMed] [Google Scholar]
- Sawmynaden K, Saouros S, Friedrich N, Marchant J, Simpson P, Bleijlevens B, Blackman MJ, Soldati‐Favre D, Matthews S (2008) Structural insights into microneme protein assembly reveal a new mode of EGF domain recognition. EMBO Rep 9: 1149–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Brown KM, Lee TD, Sibley LD (2014) Efficient gene disruption in diverse strains of Toxoplasma gondii using CRISPR/CAS9. MBio 5: e01114‐14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidik SM, Huet D, Ganesan SM, Huynh MH, Wang T, Nasamu AS, Thiru P, Saeij JP, Carruthers VB, Niles JC, Lourido S (2016) A genome‐wide CRISPR screen in Toxoplasma identifies essential apicomplexan genes. Cell 166: 1423–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldati D, Boothroyd JC (1993) Transient transfection and expression in the obligate intracellular parasite Toxoplasma gondii . Science 260: 349–352 [DOI] [PubMed] [Google Scholar]
- Stewart RJ, Whitehead L, Nijagal B, Sleebs BE, Lessene G, McConville MJ, Rogers KL, Tonkin CJ (2016) Analysis of calcium mediated signaling regulating Toxoplasma infectivity reveals complex relationships between key molecules. Cell Microbiol 26: e12685 [DOI] [PubMed] [Google Scholar]
- Sugi T, Ma YF, Tomita T, Murakoshi F, Eaton MS, Yakubu R, Han B, Tu V, Kato K, Kawazu S, Gupta N, Suvorova ES, White MW, Kim K, Weiss LM (2016) Toxoplasma gondii cyclic AMP‐dependent protein kinase subunit 3 is involved in the switch from tachyzoite to bradyzoite development. MBio 7: e00755‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torgerson PR, Mastroiacovo P (2013) The global burden of congenital toxoplasmosis: a systematic review. Bull World Health Organ 91: 501–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrey EF, Yolken RH (2013) Toxoplasma oocysts as a public health problem. Trends Parasitol 29: 380–384 [DOI] [PubMed] [Google Scholar]
- Wiersma HI, Galuska SE, Tomley FM, Sibley LD, Liberator PA, Donald RG (2004) A role for coccidian cGMP‐dependent protein kinase in motility and invasion. Int J Parasitol 34: 369–380 [DOI] [PubMed] [Google Scholar]
- Woodford TA, Correll LA, McKnight GS, Corbin JD (1989) Expression and characterization of mutant forms of the type I regulatory subunit of cAMP‐dependent protein kinase. The effect of defective cAMP binding on holoenzyme activation. J Biol Chem 264: 13321–13328 [PubMed] [Google Scholar]
- Yalovsky S, Rodr Guez‐Concepcion M, Gruissem W (1999) Lipid modifications of proteins ‐ slipping in and out of membranes. Trends Plant Sci 4: 439–445 [DOI] [PubMed] [Google Scholar]
- Zoraghi R, Corbin JD, Francis SH (2004) Properties and functions of GAF domains in cyclic nucleotide phosphodiesterases and other proteins. Mol Pharmacol 65: 267–278 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7
Source Data for Expanded View
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD006045.