

In this study, Singh et al. investigated Bim structure and activity and show that Bim is regulated by the formation of large protein complexes containing dynein light chain 1 (DLC1). Their findings demonstrate that control of apoptosis at mitochondria extends beyond the interaction of monomers of proapoptotic and anti-apoptotic Bcl-2 family members and involves more complex structures of proteins at the mitochondrial outer membrane.
Keywords: Bim; Bcl-2; BH3-only; DLC1, DYNLL1; apoptosis; complexes
Abstract
The Bcl-2 family protein Bim triggers mitochondrial apoptosis. Bim is expressed in nonapoptotic cells at the mitochondrial outer membrane, where it is activated by largely unknown mechanisms. We found that Bim is regulated by formation of large protein complexes containing dynein light chain 1 (DLC1). Bim rapidly inserted into cardiolipin-containing membranes in vitro and recruited DLC1 to the membrane. Bim binding to DLC1 induced the formation of large Bim complexes on lipid vesicles, on isolated mitochondria, and in intact cells. Native gel electrophoresis and gel filtration showed Bim-containing mitochondrial complexes of several hundred kilodaltons in all cells tested. Bim unable to form complexes was consistently more active than complexed Bim, which correlated with its substantially reduced binding to anti-apoptotic Bcl-2 proteins. At endogenous levels, Bim surprisingly bound only anti-apoptotic Mcl-1 but not Bcl-2 or Bcl-XL, recruiting only Mcl-1 into large complexes. Targeting of DLC1 by RNAi in human cell lines induced disassembly of Bim–Mcl-1 complexes and the proteasomal degradation of Mcl-1 and sensitized the cells to the Bcl-2/Bcl-XL inhibitor ABT-737. Regulation of apoptosis at mitochondria thus extends beyond the interaction of monomers of proapoptotic and anti-apoptotic Bcl-2 family members but involves more complex structures of proteins at the mitochondrial outer membrane, and targeting complexes may be a novel therapeutic strategy.
Apoptosis is a common form of programmed cell death and plays important roles in development, homeostasis, and defense (Vaux et al. 1994). The mitochondrial apoptosis pathway contributes to most forms of apoptosis. During mitochondrial apoptosis, the release of cytochrome c from mitochondria causes the activation of caspase proteases in the cytosol and eventually the death of the cell (Youle and Strasser 2008).
The Bcl-2 protein family regulates apoptosis by initiating or inhibiting the release of cytochrome c from mitochondria (Youle and Strasser 2008). Within the Bcl-2 protein family, the BH3-only protein group activates the proapoptotic effector proteins Bax and Bak, which then oligomerize in the mitochondrial outer membrane and release cytochrome c (Dewson 2016). The anti-apoptotic Bcl-2 proteins (such as Bcl-2, Bcl-XL, and Mcl-1) inhibit mitochondrial apoptosis by binding to either Bax/Bak or BH3-only proteins (Llambi et al. 2011).
Bim is one of the most prominent BH3-only proteins, with far-reaching roles in biology. Bim is expressed in all tissues investigated (O'Reilly et al. 2000), and the deletion of Bim perturbs homeostasis in the immune system (Bouillet et al. 1999) as well as the apoptotic response to many stimuli (Bouillet et al. 1999; Tan et al. 2005; Kuroda et al. 2006). Loss of Bim expression is associated with a number of human tumors such as B-cell lymphoma (Mestre-Escorihuela et al. 2007) and renal cell carcinoma (Zantl et al. 2007). A role of Bim as a tumor suppressor has been confirmed in epithelial (Tan et al. 2005) and haematopoietic (Egle et al. 2004) cells, and Bim functions to determine the response of tumor cells to chemotherapy (Tan et al. 2005).
Bim can directly activate Bax and Bak, initiating cytochrome c release as well as inhibiting the anti-apoptotic Bcl-2 proteins (Bhola and Letai 2016). How the activation of BH3-only proteins, including Bim itself, is regulated is less clear. The best-understood BH3-only protein is Bid, which is proteolytically activated to truncated Bid (tBid). tBid rapidly inserts into membranes, where it can activate recombinant Bax to permeabilize the membrane, but Bid is considered an unusual BH3-only protein with peculiar characteristics (Billen et al. 2008; Lovell et al. 2008).
No molecular data are available for Bim protein beyond its principal ability to initiate the release of cytochrome c and depolarize mitochondria (Sarosiek et al. 2013). Regulation of Bim may be achieved through adjustment of its protein levels. A prominent pathway is ERK-dependent phosphorylation (Ley et al. 2003) and ubiquitination/deubiquitination, regulating the turnover and thereby the levels of Bim (Dehan et al. 2009; Weber et al. 2016). Bim may be further regulated at mRNA levels; for instance, by the transcription factor FOXO3a. However, this transcriptional regulation plays only a minor role at least in hematopoietic cells that die in a Bim-dependent fashion (Herold et al. 2013). In T cells, it was found that although Bim levels increased with the initiation of Bim-dependent apoptosis, this increase was only marginal over the already expressed Bim protein (Parish et al. 2009), and examples have even been described in myeloid and lymphoid cells, where Bim levels were inversely correlated with the induction of Bim-dependent apoptosis (Bauer et al. 2007; Shenoy et al. 2014). The data suggest that additional Bim-regulating mechanisms exist.
Apart from regulation through abundance, the only proposed mechanism of regulating Bim activity is through a site that confers binding to dynein light chain 1 (DLC1, also known as DYNLL1 and LC8) (Puthalakath et al. 1999). It was initially suggested that Bim–DLC1 binding sequesters Bim to the microtubule cytoskeleton (since DLC1 is also found in the dynein motor complex), from where it may be released by an unknown mechanism and translocate to mitochondria when apoptosis is initiated (Puthalakath et al. 1999). However, more recent data have shown that Bim in nonapoptotic cells is already found on mitochondria, where it is C-terminally inserted in the outer membrane (Gomez-Bougie et al. 2005; Huang and Sinicrope 2008; Aranovich et al. 2012; Wilfling et al. 2012).
Here we examined the molecular mechanism of Bim regulation. We combined the analysis of Bim function in a synthetic system of recombinant full-length proteins and lipid membranes with models of cellular expression and the analysis of endogenous proteins. Using biophysical methods, we found that Bim inserts specifically into cardiolipin (CL)-containing membranes, which explains its mitochondrial localization inside cells. We confirmed Bim binding to DLC1 with recombinant proteins and report that membrane-inserted Bim can recruit DLC1. This interaction caused the dimerization of Bim, which initiated the formation of large Bim–DLC1 complexes on liposomes as well as on mitochondria, both isolated and in intact cells. Although complex formation appeared not to be required for its Bax-activating activity, Bim complexes were more active at binding anti-apoptotic Bcl-2 proteins than noncomplexed Bim. In intact cells and at endogenous levels, we found that, of the anti-apoptotic proteins, Bim–DLC1 complexes specifically recruited Mcl-1. The disruption of the Bim complexes caused the dissociation of Mcl-1 and exposed Mcl-1 to down-regulation through proteasomal degradation, sensitizing the cells to apoptosis induction by the Bcl-2/Bcl-XL inhibitor ABT-737.
Results
Bim inserts into synthetic membranes, and Bim–Bim complexes are formed through binding of DLC1
In order to avoid uncontrollable signals and binding partners inside cells we first used a synthetic system of purified components. We produced the recombinant full-length splice form BimL in Escherichia coli (Supplemental Fig. S1A,B). Three forms of Bim can be detected in mouse and human cells, originating from differential splicing. BimEL is the major form, with less BimL and even less BimS produced in most cells (O'Connor et al. 1998; Bouillet et al. 2001). We were unable to achieve sufficient expression of BimEL in E. coli and therefore chose BimL for analysis.
Recombinant BimL was capable of activating Bax in a way similar to and at concentrations similar to recombinant tBid (Supplemental Fig. S2). Fluorescent-labeled BimL inserted efficiently into giant unilamellar vesicles (GUVs), as observed by confocal microscopy (Fig. 1A,B). We used a number of different lipid compositions for this experiment (phosphatidylcholine [PC] alone or a mixture of 80% PC with 20% different negatively charged phospholipids or “mitomix”). The lipid composition of GUV membranes proved to be important to the membrane binding of Bim; the most efficient binding by far was detected with GUVs containing 20% CL followed by GUVs mimicking the lipid composition of mitochondrial outer membrane (mitomix with 8% CL) (see the Materials and Methods). A similar preference for CL has been reported for tBid (Lutter et al. 2000). This preference may be relevant to the mitochondrial targeting of Bim in intact cells (Wilfling et al. 2012). We therefore decided to use GUVs composed of PC:CL (80:20 [mol/mol]) for further studies.
Figure 1.
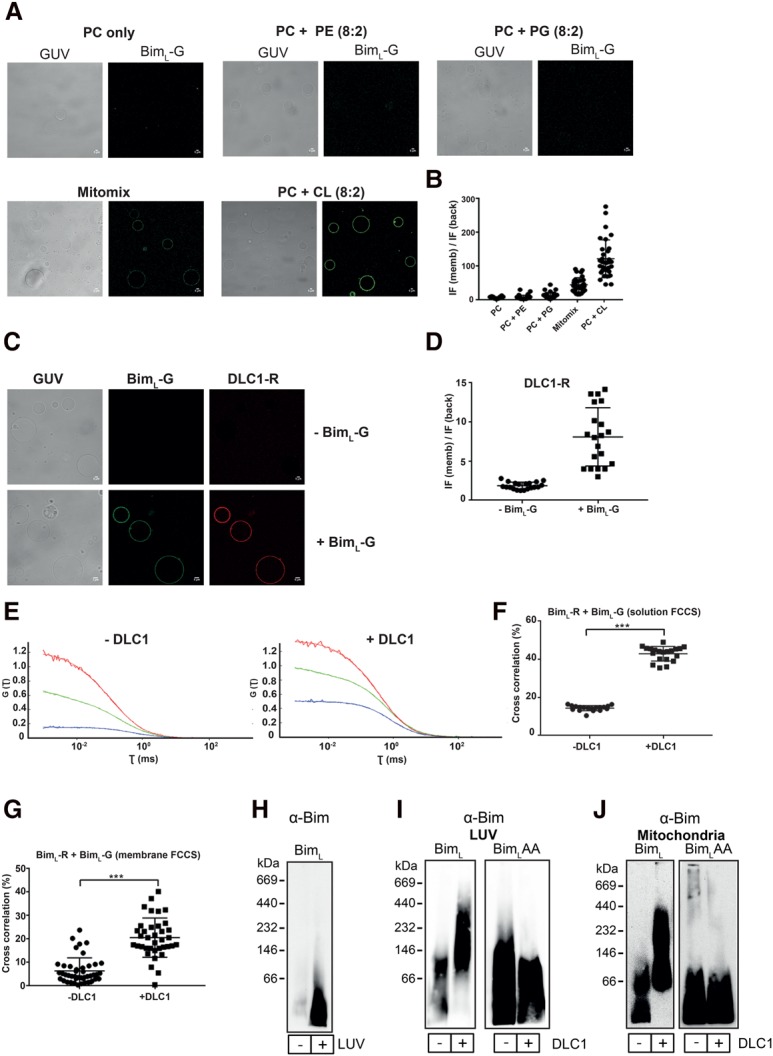
Characterization of recombinant BimL using biophysical methods. (A) Bim binding to GUVs requires CL. Confocal images showing the binding of BimL-Al488 (BimL-green [BimL-G]) to GUVs of different lipid compositions (PC alone or a mixture of 80% PC with 20% different phospholipids [phosphatidylethanolamine {PE} or phosphatidylglycerol {PG} or CL and mitomix]). Mitomix is a complex mixture of lipids mimicking the mitochondrial outer membrane with 8% CL. The GUVs were incubated with 20 nM BimL-G for 30 min at room temperature prior to image acquisition. Strong binding of BimL-G was observed only in CL-containing GUVs (mitomix and PC:CL). Images are representative of three independent experiments. Bars, 5 µm. (B) Quantification of BimL-G binding to GUVs of different lipid compositions. The dot plot shows the BimL-G fluorescence intensity at the rim (memb) of GUVs normalized against the background (back) intensity. For quantification, images of individual GUVs were acquired after zooming in, and each dot represents the fluorescence intensity of BimL-G on an individual GUV. BimL-G binding to GUVs was directly related to their CL content (mitomix has 8%, and PC:CL has 20% CL). Data are shown as mean ± SD of three independent experiments. (C) Recruitment of DLC1 to GUVs requires BimL. Confocal images were acquired after GUVs (PC:CL; 80:20 [mol/mol]) were incubated with 40 nM DLC1-Al647 (DLC1-red [DLC1-R]) alone or 20 nM BimL-G for 30 min. No DLC1 binding to GUVs was observed in the absence of Bim. Images are representative of three independent experiments. Bars, 5 µm. (D) Quantification of DLC1 binding to GUVs in the presence or absence of BimL. The dot plot depicts the DLC1-R fluorescence intensity at the rim (memb) of GUVs normalized against the background (back) intensity. Each dot represents the fluorescence intensity of DLC1-R bound to an individual GUV. Data are shown as mean ± SD of three independent experiments. (E) DLC1 facilitates Bim–Bim interaction in solution and on GUVs. Solution fluorescence cross-correlation spectroscopy (FCCS) analysis showing the autocorrelation curves (defined as the correlation between the fluctuations in particle number due to diffusion through the detection volume and the resulting fluctuations in the fluorescence) of BimL-Al647 (BimL-red [BimL-R]; red line), BimL-G (green line), and the cross-correlation (blue line; indicates the interaction between the two fluorescently labeled proteins diffusing together as a complex) between them in the presence or absence of DLC1. The zigzag and the straight lines represent the raw data and the fitted autocorrelation curves, respectively. The Y-axis represents the inverse of the number of particles [G(τ) = 1/N] diffusing through the detection volume at any given time. The intersection on the X-axis from a point of the autocorrelation curve corresponds to the diffusion time (τ, milliseconds) of those particles. Note that in the presence of DLC1, the diffusion time of the particles as well as the G(τ) value increased (diffusion of lower number of particles), indicating an increase in molecular weight (MW) due to formation of Bim–Bim complexes. (F) Quantification of the percentage cross-correlation (percentage complex formation) between the BimL-R and BimL-G in solution. Cross-correlation (percentage) was calculated from three independent experiments (five runs per experiment). The data are shown as mean ± SD. (***) P < 0.001, two tailed paired t-test. (G) Quantification of Bim–Bim interaction on GUVs using scanning FCCS. GUVs composed of PC:CL (80:20, mol/mol) were incubated with BimL-R and BimL-G in the presence or absence of DLC1, and scanning FCCS was performed to detect the complex formation. Note that in the presence of DLC1, the percentage of cross-correlation (percentage complex formation) between the two differently labeled Bim molecules increased. Data (mean ± SD) are representative of three independent experiments (at least 10 GUVs were scanned per sample per experiment). (***) P < 0.001, two-tailed paired t-test. (H) Recombinant BimL inserts spontaneously into large unilamellar vesicles (LUVs). BimL (50 nM) was incubated for 30 min with or without LUVs. After centrifugation, the pellet was solubilized in buffer containing 1% digitonin. The soluble fraction was then loaded onto a 6%–16.5% gradient gel, separated by blue native (BN)-PAGE, and subjected to Western blotting for Bim. Data are representative of three independent experiments. (I,J) Recombinant DLC1 induces the formation of high-MW BimL complexes. LUVs or isolated mitochondria from Bax−/−Bak−/− mouse embryonic fibroblasts (MEFs) were incubated for 30 min with 50 nM recombinant BimL or BimLT54A Q55A (BimLAA) in the absence or presence of 100 nM DLC1. The pellet containing the LUVs or mitochondria was then solubilized in 1% digitonin buffer, loaded onto a 6%–16.5% gradient BN gel, and blotted for Bim. Data are representative of three independent experiments.
DLC1 is not known as a mitochondria-associated protein, and we asked whether the interaction of Bim and DLC1 indeed may occur at membranes. Fluorescence-labeled DLC1 (DLC1-Al647; referred to here as DLC1-red [DLC1-R]) on its own did not localize to GUVs but was efficiently recruited to membranes by coincubation with Bim, suggesting that Bim recruits and interacts with DLC1 on mitochondria (Fig. 1C,D).
A known function of DLC1 is the binding to and dimerization of a number of proteins of different biological functions (Rapali et al. 2011). We therefore tested whether DLC1 could also dimerize Bim. We labeled recombinant BimL protein with two different fluorescent dyes (Alexa 488 [BimL-green {BimL-G}]) and Alexa 647 [BimL-red {BimL-R}] and measured Bim–Bim interaction by solution fluorescence cross-correlation spectroscopy (FCCS). Under control conditions, we detected little cross-correlation, indicating that soluble Bim was largely a monomer. Addition of DLC1, however, gave a clear cross-correlation signal between the two different fluorescently labeled Bim molecules, indicating that DLC1 induced Bim assembly at least into dimers (Fig. 1E,F). Moreover, the diffusion time of labeled Bim increased in the presence of DLC1, in agreement with complex formation (Supplemental Fig. S3A,B).
These results indicate that DLC1 induces the association of Bim molecules in solution. We repeated these experiments on GUVs and obtained very similar results: DLC1 again induced Bim–Bim complex formation (Fig. 1G), indicating that assembly also occurred in membranes. We next assessed complex formation on large unilamellar vesicles (LUVs) using native gel electrophoresis. Membrane insertion of Bim was again seen by Western blotting (Fig. 1H). In the absence of DLC1, low-molecular-weight (MW) Bim complexes were observed by blue native (BN)-PAGE. Addition of DLC1 caused an increase in size, with Bim running in complexes of ∼100–400 kDa, while no such change was observed when a Bim mutant with two inactivating point mutations in the DLC1-binding site (BimLT54A Q55A [BimLAA]) was used (Fig. 1I; Puthalakath et al. 1999). Very similar complex formation was observed with heavy membrane fractions (we refer to these fractions here as mitochondria) isolated from Bax−/−Bak−/−-deficient mouse embryonic fibroblasts (MEFs). When recombinant Bim was added to these preparations, it inserted into the membranes and formed large complexes in the presence of DLC1; again, no complex formation was observed for BimLAA (Fig. 1J).
Thus, Bim preferentially and rapidly inserts into membranes containing CL and recruits DLC1 to the membrane. In addition, Bim binding to DLC1 is sufficient for the assembly of Bim molecules, leading to dimerization and multimerization of Bim.
In cells, Bim forms complexes on mitochondria through binding to DLC1
We expressed the main splice variant of Bim, BimEL, in MEFs from Bax−/− Bak−/− mice (Bax−/− Bak−/− MEFs; overexpression of Bim in the presence of Bax or Bak induces apoptosis). In the absence of proapoptotic stimuli, the endogenous Bim expression in MEFs is low (Supplemental Fig. S4; data not shown). Cells expressing Bim under an 800-kb fragment of the endogenous Bim promoter had only mitochondrial Bim, while MEFs expressing it from a strong viral promoter showed both mitochondrial and cytosolic Bim (Supplemental Fig. S4).
BimEL was found at sizes ∼600 kDa on mitochondria isolated from BimEL-overexpressing cells, while BimELAA (BimELT110A Q111A; the mutant lacking the DLC1-interacting domain) moved at much smaller sizes in a pattern very similar to the one observed with recombinant BimL (cf. Figs. 2A and 1I,J). By Western blotting, we also detected a strong signal for DLC1 at mitochondria from cells expressing wild-type Bim (and at similar size complexes) but only very little at mitochondria from cells expressing mutant BimELAA, suggesting that, as found with GUVs, binding to Bim caused the translocation of DLC1 to mitochondria (Fig. 2B). Dynein intermediate chain 1/2 (DIC1/2), a component of the dynein motor complex, was found at equal amounts in the preparations from both cells and at a different size, again demonstrating that DLC1 on mitochondria was not bound to the dynein motor complex (Fig. 2C).
Figure 2.
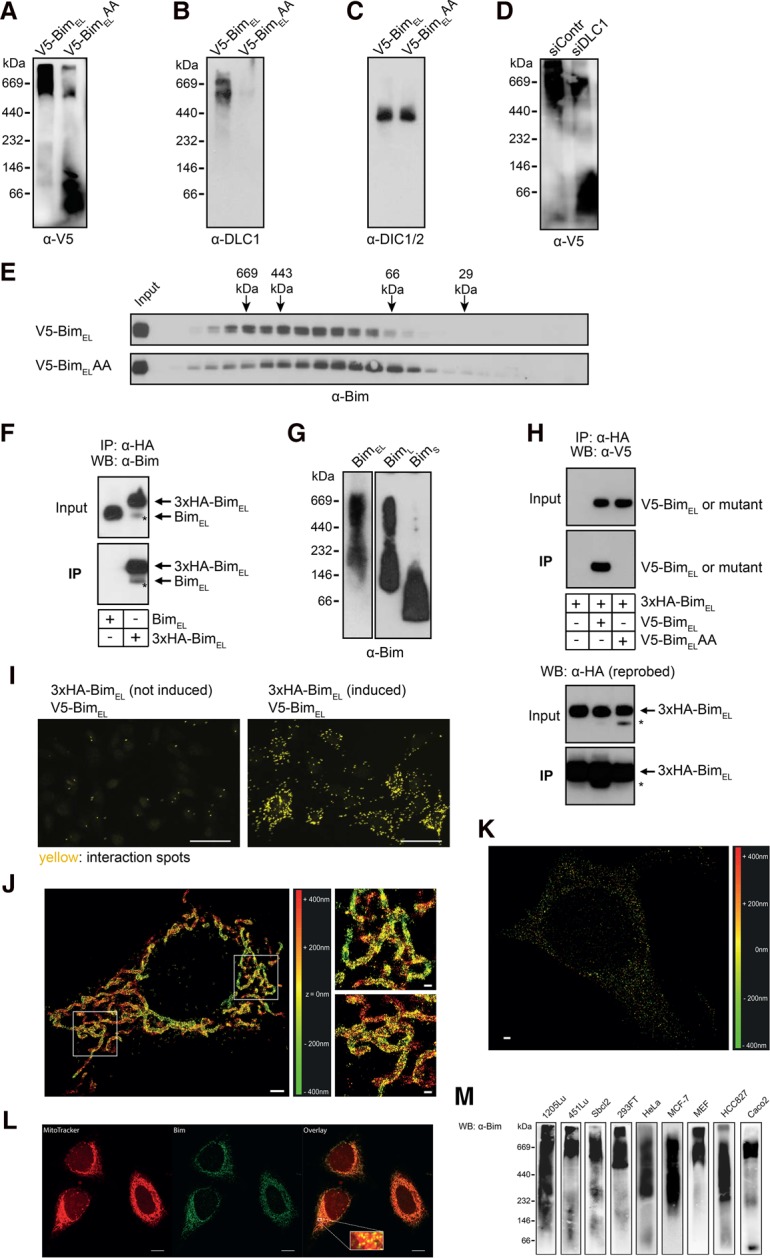
DLC1 promotes oligomerization and formation of high-MW Bim complexes. (A) Wild-type BimEL forms high-MW complexes, while BimELAA forms mainly low-MW complexes. One percent digitonin lysates of mitochondria from Bax−/−Bak−/− MEFs overexpressing either murine V5-tagged wild-type BimEL or the DLC1-binding mutant BimELAA were separated by BN-PAGE on a 6%–16.5% gradient gel and subjected to anti-V5 Western blotting. Data are representative of four independent experiments. (B,C) BN-PAGE was performed with the mitochondrial lysates of wild-type or BimELAA-overexpressing Bax−/− Bak−/− MEFs. After blotting on PVDF membrane, membranes were probed for DLC1 or DIC. Results are representative of three independent experiments. (D) RNAi against DLC1 reduces the amount of high-MW Bim complexes. Mitochondrial lysates from Bax−/−Bak−/− MEFs overexpressing murine V5-tagged BimEL were prepared 28 h after transfection of the cells with siRNA against DLC1 or control siRNA. Proteins were separated on a 6%–16.5% gradient gel, and Bim was detected as in A. Data are representative of three independent experiments. (E) Bim complexes as assessed by gel filtration. Mitochondrial lysates from Bax−/−Bak−/− MEFs overexpressing either murine V5-tagged BimEL or BimELAA were separated by gel filtration on a Superdex200 10/300 GL column. Individual fractions were collected and tested for the presence of BimEL by Western blotting. Sizes of marker proteins are indicated. Data are representative of two independent experiments. (F) HA-tagged BimEL copurifies with untagged BimEL. Anti-HA immunoprecipitation was performed with whole-cell lysates (1% Triton X-100) from Bax−/− Bak−/− MEFs overexpressing either untagged or 3xHA-tagged murine BimEL. Note the precipitation of endogenous BimEL with HA-BimEL. Data are representative of five independent experiments. An asterisk indicates endogenous BimEL signal. (G) BimEL and BimL but not BimS form high-MW complexes. Western blots of BN-PAGE (6%–16.5% gradient gel) from mitochondrial fractions (1% digitonin lysates) of Bax−/−Bak−/− MEFs stably overexpressing the indicated murine Bim splice variants. Data are representative of two independent experiments. (H) An inactivating mutation of the DLC1-binding site in BimEL abolishes its ability to form Bim–Bim complexes. Anti-HA immunoprecipitation was conducted with lysates of Bax−/−Bak−/− MEFs overexpressing murine V5-tagged wild-type BimEL or mutant BimEL (BimELAA) and carrying an inducible murine 3xHA-tagged BimEL construct. 3xHA-BimEL was induced for 48 h with 100 nM tamoxifen (4-hydroxytamoxifen [4HT]) and whole-cell lysates were prepared with 1% Triton X-100. Membranes were reprobed with anti-HA-antibodies. Data are representative of three independent experiments. The asterisk indicates anti-V5 signal from the first blot. (I) In situ identification of complexes containing at least two BimEL molecules by proximity ligation assay (PLA). Bax−/−Bak−/− MEFs expressing murine V5-tagged BimEL and carrying a 4HT-inducible murine 3xHA-tagged BimEL construct were fixed and permeabilized directly (left) or upon induction of 3xHA-tagged BimEL by treatment with 100 nM 4HT for 24 h (right). Images were acquired under identical conditions and exposure times and are representative of three independent experiments. Bars, 50 µm. (J) Bax−/−Bak−/− MEFs overexpressing 3xHA-BimEL were labeled with HA-specific primary and Alexa 647 secondary antibodies, and three-dimensional (3D) stochastic optical reconstruction microscopy (STORM) imaging was performed. The rectangular boxes indicate regions of the zoom-ins (shown in the right panel). The individual localizations are colored according to their z-positions (see the color bar). Bim was visualized by the HA labeling and exhibits structures that might suggest the localization of Bim in complexes. Bars: left images, 2 µm; right images, 500 nm. (K) Endogenous Bim in wild-type HeLa cells was detected with anti-Bim primary and Alexa 647 secondary antibodies, and 3D STORM imaging was performed similar to J. The individual localizations are colored as in J. Bar, 1 µm. (L) Confocal microscopy was performed with HeLa cells after staining the mitochondria with MitoTracker (red) and endogenous Bim with anti-Bim primary and Alexa 488 (green) secondary antibody. The overlay shows the mitochondrial localization of Bim in HeLa cells. The rectangular box depicts the region of a zoom-in (overlay). Bars, 10 µm. (M) Endogenous Bim forms high-MW complexes on mitochondria. BN-PAGE was performed with solubilized mitochondrial fractions (1% digitonin) of the indicated cell lines (except for MEFs, cell lines are human tumor lines). Proteins were transferred to PVDF membrane and were detected by Western blotting as indicated. In order to achieve good detection, the amounts of proteins loaded and exposure times were sometimes different for different cell lines. Data are representative of at least three independent experiments with each cell line.
In agreement with these observations, when DLC1 levels were reduced by RNAi, a shift of BimEL from large to small sizes was observed (Fig. 2D). In the analysis of mitochondrial complexes by gel filtration, BimEL also ran at much larger sizes than BimELAA (Fig. 2E). DLC1 binding to Bim at mitochondria therefore induces the formation of large protein complexes.
Coimmunoprecipitation (co-IP) experiments confirmed the presence of at least two Bim molecules in one complex, since overexpressed epitope-tagged BimEL coimmunoprecipitated endogenous BimEL (Fig. 2F). We then tested the three main splice variants of Bim (see Supplemental Fig. S5A for a schematic of the splice forms). Such interaction was observed (using tagged and untagged Bim molecules coexpressed in the same cell) for BimEL with BimEL as well as for BimL with BimL. BimEL could not coimmunoprecipitate BimS, and no interaction between tagged and untagged BimS was observed by this approach (Supplemental Fig. S5B). Since BimEL and BimL, but not BimS, contain the DLC1-binding site (Supplemental Fig. S5A), this is consistent with DLC1 binding being the relevant feature for the Bim–Bim interaction. As expected on the basis of these data, BimEL and BimL, but not BimS, formed large complexes when analyzed by native PAGE (Fig. 2G). Tests with truncation mutants further mapped the site required for Bim–Bim interaction to amino acids 99–113 (the DLC1-binding site is localized at amino acid residues 107–112 of mouse BimEL) (Supplemental Fig. S6A). The same truncation mutants that bound to BimEL (Supplemental Fig. S6A) also pulled down DLC1 (Supplemental Fig. S6B).
Co-IP experiments from MEF Bax−/− Bak−/− cells overexpressing 3xHA-BimEL with either V5-tagged wild type or BimELAA, showed that only wild-type V5-BimEL, but not the mutant, was coprecipitated with the 3xHA-BimEL, (Fig. 2H). In a complementary approach, we also assessed the proximity of two Bim molecules by microscopy using in situ proximity ligation assay (PLA). A strong PLA signal (higher number of interaction spots, indicating the proximity of two Bim molecules) was observed between the two differently tagged BimEL molecules (Fig. 2I), and the signal was much weaker when one of the two molecules lacked the DLC1 interaction site (Supplemental Fig. S7).
All of these results indicate that on mitochondria, binding of DLC1 induces complex formation between Bim molecules, leading to the formation of large Bim- and DLC1-containing complexes. By superresolution microscopy (stochastic optical reconstruction microscopy [STORM]), we further observed structures suggestive of localized Bim complexes on mitochondria of Bax−/−Bak−/− MEFs overexpressing Bim (Fig. 2J). Weaker but similarly localized expression was also detected for endogenous Bim by both STORM and high-resolution confocal microscopy (Fig. 2K,L).
Finally, we tested a number of cell lines for endogenous mitochondrial Bim complexes. In all cell lines investigated (eight human tumor cell lines and MEFs), endogenous Bim was exclusively or almost exclusively found in large complexes. Complex sizes ranged from ∼200 to >600 kDa (Fig. 2M). Complex formation of endogenous Bim on mitochondria is thus a constitutive feature of at least many cells.
Both monomeric and complexed Bim can activate Bax
Bim has two proapoptotic molecular functions; namely, the direct activation of Bax/Bak (Letai et al. 2002) and the inhibition of anti-apoptotic Bcl-2-like proteins (Chen et al. 2005). Both functions require the same secondary structure of Bim: the α-helical BH3 domain. When Bim was purified from E. coli, it was found to be intrinsically unstructured (Hinds et al. 2007), although the interpretation of that study may be limited by its use of a C-terminal truncation of Bim, excluding the membrane anchor.
We used the reconstitution system of purified components to test whether mutant BimLAA (monomeric in membranes) and wild-type Bim (complexed following DLC1 binding) could activate Bax and whether they differed in their activating capacity. LUV-inserted monomeric BimL appeared slightly more active than complexed Bim (wild-type BimL + DLC1) at activating recombinant Bax, measured as oligomerization and membrane insertion of Bax (Fig. 3A,B). A similar inhibitory effect of DLC1 was observed on the Bax-activating activity of BimL by cytochrome c release assay performed with isolated mitochondria from Bax−/− Bak−/− MEFs (Fig. 3C).
Figure 3.
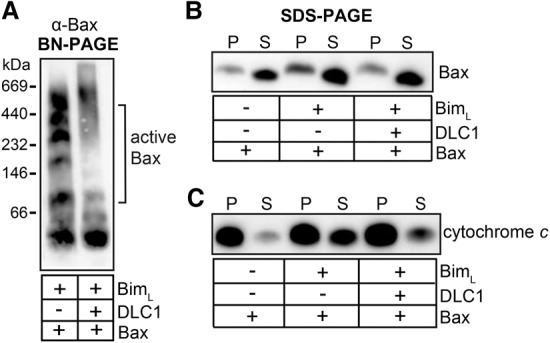
DLC1-mediated Bim complexes have reduced Bax-activating potency. (A,B) DLC1 binding reduces the Bax activation capacity of recombinant BimL. LUVs were incubated for 30 min at 30°C with 50 nM recombinant BimL, 100 nM DLC1, and 100 nM Bax as indicated. After centrifugation, the pellet was solubilized in 1% digitonin buffer and divided into two parts. (A) One part was loaded on a 6%–16.5% BN gradient gel and analyzed by Western blotting for Bax. Bax activation is suggested by the appearance of Bax multimers. (B) The other part and the supernatant (S) of the pelleted LUVs (P) was analyzed for Bax insertion into LUVs by SDS-PAGE and Western blotting. The inhibitory effect of DLC1 on BimL-mediated Bax activation is evidenced by reduced insertion of Bax into LUVs. Data are representative of three independent experiments. (C) Bim in complexes is less efficient at inducing Bax-mediated cytochrome c release from mitochondria. Mitochondria were isolated from Bax−/− Bak−/− MEFs and incubated with the recombinant proteins (10 nM BimL, 50 nM DLC1, and 100 nM Bax) for 30 min at 30°C as indicated. Mitochondria were then pelleted by centrifugation, and the presence of cytochrome c in the pellet (P) or supernatant (S) was determined by Western blot.
We used MEFs to test the proapoptotic activity of complexed versus monomeric Bim in intact cells. Apoptosis induced by thapsigargin in MEFs strongly depends on Bim (Fig. 4A; Puthalakath et al. 2007). RNAi against DLC1, which disrupts Bim complexes (see above; Fig. 2D), increased thapsigargin-induced apoptosis in wild-type MEFs by a small but statistically significant amount (Fig. 4A). RNAi against DLC1 also had a small effect on thapsigargin-induced apoptosis in the absence of Bim. One possibility is that Bmf here plays a role. Bmf can bind DLC1 in a way very similar to Bim (Day et al. 2004) and may induce the residual apoptosis upon thapsigargin treatment in the absence of Bim.
Figure 4.

Loss of BimEL–DLC1 binding enhances the proapoptotic activity of BimEL. (A) RNAi specific for DLC1 sensitizes MEFs for apoptosis induced by thapsigargin in a predominantly Bim-dependent manner. Cells were transfected with control siRNA or siRNA specific for DLC1 16 h prior to treatment with 200 nM thapsigargin for 24 h. QVD-OPh (10 µM) was added to control samples together with thapsigargin to test for caspase dependency. Data show means/SEM of three independent experiments. (*) P = 0.015 wild type; (*) P = 0.005 Bim−/−, two-tailed paired t-test. (B) BimELAA has higher proapoptotic activity than wild-type BimEL when expressed in MEFs. MEFs carrying a 4HT-inducible construct for the expression of either wild-type BimEL or BimELAA were treated with 100 nM 4HT for 24 h. Apoptosis was measured as the percentage of cells positive for active caspase-3. Data show means/SEM of four independent experiments. (**) P = 0.010, two-tailed paired t-test. Both BimEL and BimELAA were expressed at comparable levels (see Supplemental Fig. S13). (C,D) RNAi specific for DLC1 can sensitize MEFs to the induction of wild-type BimEL but not BimELAA. MEFs carrying a 4HT-inducible construct for the expression of either wild-type BimEL (C) or BimELAA (D) were transfected with control or DLC1-specific siRNA 16 h prior to Bim induction with 100 nM 4HT for 24 h. Cells with active caspase-3 were detected by flow cytometry as above. Data show means/SEM of four independent experiments. (*) P = 0.014 (C); (ns) nonsignificant (P = 0.679) (D), two-tailed paired t-test.
We next generated MEFs that carried either wild-type BimEL or BimELAA under a tamoxifen (4-hydroxytamoxifen [4HT])-inducible promoter. When the expression of BimEL or BimELAA was induced by 4HT, BimELAA was somewhat more active at inducing apoptosis than wild-type BimEL (Fig. 4B). When DLC1 was targeted by RNAi in the same cells prior to the induction of BimEL, DLC1 knockdown increased apoptosis induced by wild-type BimEL (Fig. 4C) to about the same level as apoptosis induced by BimELAA. RNAi against DLC1 had no apoptosis-increasing effect during induction of BimELAA, as expected (Fig. 4D). These results demonstrate that BimELAA has a higher proapoptotic activity than wild-type Bim. Apoptosis induced by the EGFR inhibitor gefitinib in a non-small cell lung cancer cell line, a known Bim-dependent apoptosis stimulus (Cragg et al. 2007), was also enhanced by RNAi against DLC1 (Supplemental Fig. S8).
DLC1-induced complex formation increases the ability of Bim to bind anti-apoptotic Bcl-2 proteins
The second molecular function of Bim requires its binding to anti-apoptotic Bcl-2 proteins. Through its BH3 domain, Bim can bind to all anti-apoptotic Bcl-2 family proteins (Chen et al. 2005). This binding inactivates Bim, but the association may also be important to inhibit anti-apoptotic proteins, and this inhibition can contribute to the induction of apoptosis (Chipuk et al. 2010). With the exception of activated Bax and Bak, Bcl-2 family members have so far been considered to be present as monomers or heterodimers at mitochondria. The finding that Bim is in a large complex at mitochondria suggests implications for the binding to anti-apoptotic Bcl-2 proteins.
We first tested whether complex formation of Bim affected its binding capacity for anti-apoptotic Bcl-2 proteins using MEFs overexpressing wild-type BimEL or BimELAA. Immunoprecipitation of wild-type Bim coprecipitated substantial amounts of Bcl-2, Bcl-XL, and Mcl-1, as expected. However, the amount coprecipitated of all three anti-apoptotic Bcl-2 proteins was substantially lower when BimELAA was expressed and precipitated (Fig. 5A). In a second approach, DLC1 was targeted by RNAi to disrupt the Bim complexes. Again, the amount of anti-apoptotic Bcl-2 proteins coprecipitated with noncomplexed Bim (siDLC1) was strongly reduced compared with complexed Bim (Fig. 5B). Analysis by native PAGE further showed large complexes of Bcl-2 and Bcl-XL when wild-type Bim but not BimELAA was overexpressed, and RNAi targeting DLC1 reduced the large complexes containing Bcl-XL in cells overexpressing wild-type BimEL (Fig. 5C,D).
Figure 5.
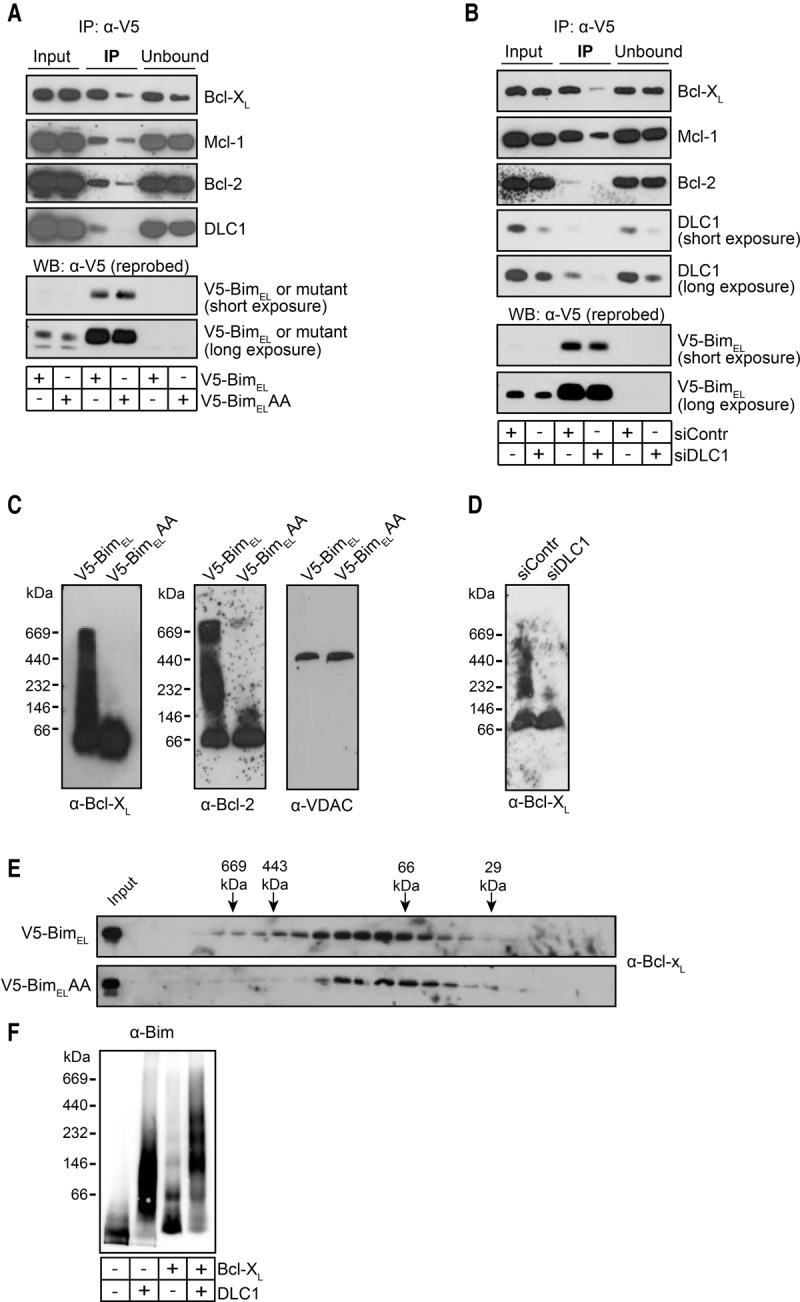
Bim–DLC1 binding is required for efficient binding of BimEL to anti-apoptotic Bcl-2 proteins. (A) Binding of wild-type BimEL versus BimELAA to anti-apoptotic Bcl-2 proteins. Whole-cell lysates (1% Triton X-100) of Bax−/−Bak−/− MEFs overexpressing either murine V5-tagged BimEL or the DLC1-binding mutant V5-BimELAA were subjected to anti-V5 immunoprecipitation. Precipitated proteins were identified by Western blotting with the antibodies indicated. Data are representative of three independent experiments. (B) BimEL binding to anti-apoptotic proteins is reduced by RNAi against DLC1. Bax−/−Bak−/− MEFs overexpressing murine V5-tagged BimEL were transfected with control siRNA or siRNA against DLC1. After 48 h, whole-cell lysates (1% Triton X-100) were prepared and subjected to anti-V5 immunoprecipitation followed by Western blotting for the proteins indicated. Data are representative of three independent experiments. (C) In the presence of overexpressed wild-type BimEL but not BimELAA, Bcl-2 and Bcl-XL run in higher-MW complexes in BN-PAGE. Mitochondrial lysates (1% digitonin) from Bax−/−Bak−/− MEFs overexpressing either murine V5-tagged BimEL wild type or BimELAA were run on a 6%–16.5% BN-PAGE gradient gel and subjected to Western blotting with the antibodies indicated. VDAC was probed to test whether it may be contained in the complexes. Data are representative of at least three independent experiments. (D) Bim-induced high-MW Bcl-XL complexes are reduced upon RNAi against DLC1. Mitochondria were prepared from Bax−/−Bak−/− MEFs overexpressing murine V5-tagged BimEL 28 h after transfection with control or DLC1-specific siRNA. Complexes were separated on a 6%–16.5% gradient BN-PAGE gel and subjected to Western blotting with antibodies specific for Bcl-XL. Data are representative of three independent experiments. (E) Overexpression of wild-type BimEL, but not BimELAA, induces Bcl-XL high-MW complexes as assessed by gel filtration. Lysates of mitochondria from Bax−/−Bak−/− MEFs overexpressing murine V5-tagged BimEL wild type or BimELAA were separated by gel filtration on a Superdex 200 10/300 GL column. The same collected fractions were used as in Figure 2E. (F) Binding of Bcl-XL to Bim preferentially occurs in DLC1-induced Bim complexes. Recombinant proteins (BimL, DLC1, and Bcl-XL) were incubated with LUVs as in Figure 1I. Recombinant Bcl-XL (100 nM) was added as indicated, and Bim-containing complexes were detected by BN-PAGE and anti-Bim Western blotting. Data are representative of three independent experiments (two experiments with Bim + Bcl-XL only).
Analysis of mitochondrial lysates by gel filtration also demonstrated that Bcl-XL ran at higher sizes in lysates from MEFs overexpressing wild-type BimEL compared with cells expressing BimELAA (Fig. 5E). Finally, on LUVs containing Bim, there was only a small shift in size when recombinant Bcl-XL was added; this shift was considerably greater when Bim had been allowed to form complexes through the addition of DLC1 (Fig. 5F). These results provide evidence that the binding of Bim to anti-apoptotic Bcl-2 proteins is substantially more efficient when Bim had formed complexes through binding to DLC1.
At endogenous levels, Bim specifically complexes with Mcl-1
As shown above, endogenous Bim is found in large complexes in all cell lines that we tested (see Fig. 2M), and overexpression of Bim recruited Bcl-2 and Bcl-XL into similar complexes in MEFs. However, when we tested endogenous proteins in the cell lines shown above, we were surprised to find that Bcl-XL (except for 293FT cells, where a small fraction was noticed at very high-MW complexes) and Bcl-2 did not run as large complexes; rather, at least most of the protein was found at monomeric or dimeric sizes (Fig. 6A,B). In some cell lines, Bcl-2 ran as apparently somewhat larger species; however, this apparent size was still considerably smaller than that of the complexes containing Bim in the same cells (cf. Fig. 2M). This suggested that Bcl-2 and Bcl-XL at endogenous expression levels were not bound to Bim. Mcl-1, on the other hand, ran at a size similar to Bim in all cells, suggesting that it may be constitutively bound to Bim at endogenous levels (Fig. 6C). Immunoprecipitation experiments in three of these cell lines (MCF-7, HeLa, and 293FT) showed near-complete precipitation of Mcl-1 with endogenous Bim. No detectable Bcl-2 was coimmunoprecipitated with Bim. A small amount of Bcl-XL was found bound to Bim in one cell line (293FT cells) (Fig. 6D); this likely corresponds to the small amount of Bcl-XL running at higher MW in BN-PAGE (Fig. 6A).
Figure 6.
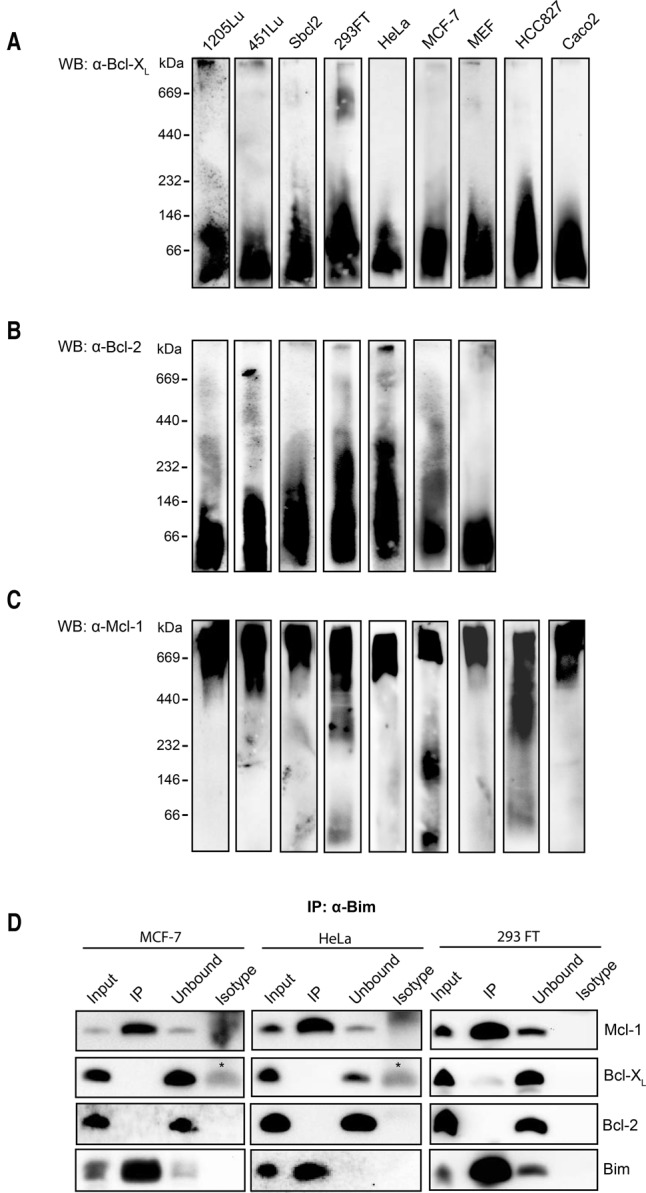
Endogenous Bim forms high-MW complexes together with Mcl-1 but not with Bcl-2 or Bcl-XL. (A–C) Complexes of endogenous Bcl-2 family proteins on mitochondria. BN-PAGE was performed as described earlier with solubilized mitochondrial fractions of the indicated cell lines. In order to achieve good detection, the amounts of proteins loaded and exposure times were sometimes different for different cell lines. Bcl-XL (except in 293FT, where a small amount of Bcl-XL ran at high-MW complexes) and Bcl-2 ran as low-MW proteins or small complexes, while Mcl-1 ran at high MW. Data are representative of at least three independent experiments with each cell line. We were unable to detect Bcl-2 in the HCC827 and Caco2 cell lines. (D) The majority of endogenous Bim is bound to MCL-1. Co-IP experiments were performed with whole-cell lysates from MCF-7, HeLa, and 293FT cells using anti-Bim antibody. The Bim-interacting partners were detected by Western blotting. (Isotype) Immunoprecipitation control with an isotype antibody of unrelated specificity; (*) antibody light chain. Data are representative of three independent experiments.
Thus, at endogenous protein levels, there appears to be a specific interaction between Bim complexed through DLC1-binding and Mcl-1. Bcl-2 and Bcl-XL are mostly not found in these complexes.
However, wild-type Bim did bind Bcl-2 and Bcl-XL when overexpressed (see Fig. 5C). When more Bim is available in a cell, as may be the case upon induction of Noxa (competing for Mcl-1-binding) or treatment with ABT-737 (blocking free Bcl-2/Bcl-XL, which may free other BH3-only proteins to compete for binding to Mcl-1), this may redistribute Bim from Mcl-1 to other Bcl-2 family members.
Bak has been reported earlier to be bound to Mcl-1. We also found Bak (but not Bax) in immunoprecipitation products with anti-Mcl-1-antibodies (Supplemental Fig. S9A) and (although at smaller amounts) when DLC1 was immunoprecipitated (Supplemental Fig. S9B).
DLC1–Bim–Mcl-1 complexes in the regulation of protein stability and apoptosis
The above data indicate that complex formation of Bim through DLC1 occurs in many or all cells and that the complex formation has a relatively minor effect on the Bax-activating capacity of Bim but a substantial effect on the binding of anti-apoptotic Bcl-2 proteins. This binding at endogenous levels is limited to Mcl-1, suggesting a specific mutual regulation of Bim and Mcl-1.
Mcl-1 is a critical anti-apoptotic Bcl-2 protein in many cells. In the hematopoietic system, most cells (all tested cells with the exception of macrophages) depend on Mcl-1 and die in its absence (Opferman et al. 2005; Steimer et al. 2009). An inhibitor of Mcl-1 function has recently proved potent in the induction of apoptosis in many tumor cells (Kotschy et al. 2016). The level of Mcl-1 is regulated by numerous upstream pathways, and its proteasomal degradation appears to be an important variable of a cell's apoptosis sensitivity (for review, see Mojsa et al. 2014). Notably, binding to Bim has been found to stabilize Mcl-1, possibly by protecting it from the degrading activity of its interaction with the BH3-only protein Noxa (Czabotar et al. 2007).
Bim, on the other hand, contains large intrinsically disordered stretches, which often make proteins unstable (Oldfield and Dunker 2014), and the regulation of Bim turnover by upstream kinase pathways has been found to be a substantial component of Bim activity (Ewings et al. 2007; Dehan et al. 2009). We therefore hypothesized that the interaction of Bim (in complexes) with Mcl-1 may be a mechanism regulating the stability of either protein or of both proteins. If this was indeed the case, then DLC1 would be expected to be an important regulator of Bim and/or Mcl-1 stability and activity.
To test this, we targeted DLC1 by RNAi in human cell lines. We first tested for sensitivity of cells to the Bcl-2/Bcl-XL inhibitor ABT-737; sensitivity to this inhibitor can be used as a surrogate of Mcl-1 function (van Delft et al. 2006). Indeed, RNAi against DLC1 sensitized HeLa epithelial cells as well as 1205Lu human melanoma cells to treatment with ABT-737 (Fig. 7A,B) accompanied by the near-complete loss of Mcl-1, while the levels of Bcl-2 and Bcl-XL were unaffected (Fig. 7C,D). The loss of Mcl-1 was reversed by inhibition of the proteasome (Fig. 7C,D). RNAi against DLC1 in combination with ABT-737 caused the activation of both Bax and Bak, as assessed by cross-linking studies and FACS analyses by staining with conformation-specific antibodies (Supplemental Fig. S10).
Figure 7.

Loss of DLC1 leads to proteasomal degradation of Bim and its binding partner, Mcl-1, and sensitizes cells to ABT-737-induced apoptosis. (A–D) HeLa (A) and 1205Lu (B) cells were transfected with control siRNA or siRNA specific to DLC1 24 h prior to 2 µM ABT-737 treatment as indicated. The MEK1/2 inhibitor UO126 (10 µM) was added at the time of siRNA transfection. Caspase-3-positive cells were identified by flow cytometry. Data (mean/SD) are representative of four independent experiments. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (ns) nonsignificant (P > 0.05), two-tailed paired t-test. For Western blotting experiments, cells were treated with either 10 µM UO126 or 20 µM MG132 together with the indicated siRNAs in presence of 10 µM QVD, and cell lysates were prepared in 1% Triton lysis buffer. Results are representative of three independent experiments. The asterisk indicates the previous signal, probably from the small Mcl-1 isoform.
The levels of Bim were also somewhat reduced in HeLa cells and strongly reduced in 1205Lu cells upon RNAi against DLC1 (Fig. 7C,D). Bim is subject to ERK-dependent ubiquitination and degradation, and 1205Lu cells harbor constitutively active BRAF (due to the oncogenic V600E mutation) and ERK. Inhibition of the Raf–MEK–ERK pathway with the MEK inhibitor UO126 stabilized Bim in 1205Lu cells (Fig. 7D). Treatment with UO126 further destabilized Mcl-1 in 1205Lu cells (Fig. 7D). This is consistent with the known Mcl-1-stabilizing activity of active ERK (Domina et al. 2004; Ding et al. 2008). One pathway of Mcl-1 degradation is triggered by its binding to Noxa. However, this pathway did not seem relevant here, since Noxa was not codegraded with Mcl-1 (Supplemental Fig. S12). Of note, RNAi against DLC1 had no significant effect on the cell cycle (Supplemental Fig. S11) or ERK phosphorylation (Supplemental Fig. S12).
Thus, the loss of DLC1 exposes both Bim and Mcl-1 to the turnover-regulating upstream signals, while the sequestration into Bim–Mcl-1 complexes stabilizes both proteins. By organizing the formation of Bim–Mcl-1 complexes, DLC1 has an important role in the regulation of apoptosis sensitivity of mammalian cells.
Discussion
The results of this study show that Bim associates with CL-containing membranes, recruiting DLC1 to mitochondria. DLC1 binding to Bim allows assembly of large complexes and strongly enhances Bim binding of anti-apoptotic Bcl-2 proteins. At endogenous levels and in non-apoptotic cells, this mechanism is relevant for the binding and stabilization of Mcl-1. Disruption of the complexes, such as by loss of DLC1, destabilizes Mcl-1 by exposing it to the regulation through upstream signaling.
The proapoptotic role of Bim has been abundantly clear for a long time. However, the known molecular aspects of its activation and activity have been limited almost exclusively to its BH3 domain, which can, on its own, bind to Bax and Bak as well as to anti-apoptotic Bcl-2-like proteins. Earlier data had already suggested that the binding of Bim to DLC1 does not have the once-proposed microtubule-sequestering function and that Bim is constitutively mitochondrial.
Bim has a C-terminal membrane anchor, typical of tail-anchored mitochondrial outer membrane proteins (Wilfling et al. 2012). How tail-anchored proteins are (1) specifically directed to the mitochondrial outer membrane and (2) inserted into the membrane is still not clear (Horvath et al. 2015). The requirement of the mitochondrial protein import machinery has been suggested for some (Thornton et al. 2010) but not other (Kemper et al. 2008) proteins; ergosterol content of membranes has also been proposed to determine insertion (Krumpe et al. 2012). We found an efficient insertion of Bim into pure lipid membranes that was greatly enhanced when CL was included. CL is the signature lipid of the inner mitochondrial membrane but is also known to be present in the outer mitochondrial membrane (Gebert et al. 2009). The observed preference of Bim for translocation to CL-containing membranes is a plausible way of directing Bim—and probably other BH3-only proteins—to mitochondria. Proteinaceous components appear to be dispensable for insertion.
The role of the interaction of Bim with DLC1 has been unclear. DLC1 is known to act as a dimerizing protein. In some instances, DLC1 has been reported to dimerize intrinsically disordered proteins, initiating secondary structure. At least three examples are known where DLC1 binding causes dimerization of its binding partner and where the formed dimers assemble into large complexes (for review, see Barbar and Nyarko 2015). This mode of assembly fits perfectly well for Bim. Bim is also an intrinsically disordered protein, and our data show that Bim–Bim interaction is mediated by DLC1 as well as the formation of large complexes. The BH3-only proteins Bad and Bmf were also found to be intrinsically disordered (Hinds et al. 2007), and Bmf can also bind DLC1/2 (Day et al. 2004). At least Bmf may therefore be regulated in a way similar to Bim.
For the proapoptotic activity of Bim, the α-helical BH3 domain is sufficient, and the area containing the BH3 domain appears to contain some structure in the otherwise disordered protein (Hinds et al. 2007); this is consistent with our finding that Bim–DLC1 binding was not required for its Bax-activating and proapoptotic activity. Non-DLC1-binding Bim was even more active at activating Bax and inducing apoptosis in the assays that we used. This may be linked to its inferior binding to anti-apoptotic proteins, a more localized expression of wild-type Bim due to the formation of large complexes, or a reduced availability of BH3 domains in the large complex structure.
At least one function of Bim–DLC1 complex formation appears to be the binding to Mcl-1. Most binding studies between BH3-only proteins and Bcl-2-like proteins have been conducted using only the BH3 domain, and the BH3 domain of Bim can bind well to all anti-apoptotic Bcl-2-like proteins (for example, see Chen et al. 2005). The interaction is far less well studied with intact proteins. Although Bim can be coimmunoprecipitated with Bcl-2-like proteins, this is, as far as we are aware, not typically done in a quantitative way. A recent study did use a semiquantitative approach and described an association of Bim specifically with Mcl-1 (but not Bcl-XL) in human tonsillar B cells (Peperzak et al. 2017). In the cell lines that we tested, Mcl-1 was almost exclusively bound to Bim.
Fitting with this notion of a specific physical interaction of Bim and Mcl-1, there are also reports about functional links of the two proteins. The survival of regulatory T cells was, for instance, found to be regulated through both Bim and Mcl-1 (in opposite directions) (Pierson et al. 2013). In another experimental model, the loss of one Mcl-1 allele prevented myc-driven lymphomagenesis, and the additional loss of Bim restored it (Grabow et al. 2016). A coregulation has further been reported: In granulocytes and lymphocytes, cytokines can enhance the levels of both Bim and Mcl-1 (Bauer et al. 2007; Shenoy et al. 2014). Intriguingly, in one of these studies, transcriptional up-regulation of Bim caused an increase of Mcl-1 protein by some unidentified post-translational mechanism (Shenoy et al. 2014). This may be the result of, for instance, a primary regulation of Bim, which may then stabilize and increase the levels of Mcl-1.
Previous results had suggested that DLC1 has a Bim-dependent anti-apoptotic role. DLC1 is a transcriptional target of the zinc finger protein ASCIZ, and ASCIZ-deficient mice exhibit B-cell lymphopenia, which is corrected by the concomitant loss of Bim (Jurado et al. 2012). Our results suggest that the mechanism of this inhibition is through the sequestration of Bim into large complexes. DLC1 was important for maintenance of the complexes, and its loss exposed Bim and Mcl-1 to regulation through upstream signaling pathways.
In summary, our findings reveal a new mechanism for the modulation of interactions between Bcl-2 proteins via the formation of high-MW DLC1–Bim–Mcl-1 complexes, with clear implications for the control of mitochondrial apoptosis and cell fate.
Materials and methods
Cell lines and cell culture
Bax−/− Bak−/− MEFs immortalized with SV40 large-T antigen (Dr. David Huang, Walter and Eliza Hall Institute [WEHI], Melbourne) were cultured in DMEM containing 10% FCS, antibiotics (100 U/mL penicillin G, 100 µg/mL streptomycin sulfate), and 50 µM 2-mercaptoethanol. 293FT cells (purchased from Invitrogen), MCF-7 breast cancer cells, HeLa cervical carcinoma cells, Caco2 colon carcinoma cells, HCC827 lung cancer cells, and wild-type or Bim-deficient MEFs (bim−/− immortalized with the 3T9 method) were cultured in DMEM containing 10% FCS and antibiotics as described above. Melanoma cell lines (451Lu, 1205Lu, and Sbcl2) were cultured as described previously (Weber et al. 2010). All cultures were incubated under standard culture conditions (37°C at 5% CO2).
Construction of Bim mutants
The genes coding for murine Bim and/or truncation mutants of Bim (N-terminal truncation mutants lacking amino acids 1–97 [Bim98–196], 1–113 [Bim114–196], or 1–139 [Bim140–196]) or murine BimEL (mBimEL) were amplified by PCR and subcloned into pENTR/SD/D-TOPO Gateway vector (Life Technologies). Transfer to expression vectors was done via Gateway LR recombinase reaction (Life Technologies). Point mutations of the non-DLC1-binding mutant (BimELAA) were generated with the QuickChange II site-directed mutagenesis kit (Stratagene). N-terminal tags (HA-tag, 3xHA-tag, or V5-tag) were added to the Bim constructs by overlap extension PCR followed by subcloning into pENTR/SD/D-TOPO. To inhibit splicing of BimEL to BimL and BimS, we used a mutant deficient for splicing (Shinjyo et al. 2001) for all BimEL-based constructs in this study.
Expression vectors and generation of cell lines
Retroviral constructs (pMIG-GW; vector carrying the gateway cassette [GW] for fast subcloning) of the indicated Bim versions were generated and transduced into Bax−/−Bak−/− MEF cells in order to generate cell lines stably overexpressing Bim constructs as described earlier (Wilfling et al. 2012). In some cells, we used a 4HT-regulated lentiviral system for the expression of additional Bim constructs in Bax−/−Bak−/− MEFs or for the analysis of cell death by Bim induction in wild-type MEFs. These cell lines were first transduced with pFU-G147EV16-PGK-Hygro and then with the lentiviral vector pF 5xUAS-SV40-Puro (Vince et al. 2008). Selection of cells was done using 800 mg/mL hygromycin (Invitrogen) and 5 µg/mL puromycin (InVivoGen) for 10 d. Bim constructs were induced with 100 nM 4HT (Sigma) for the indicated times.
For the generation of Bim constructs under the control of an 800-base-pair (bp) fragment of the Bim promoter, the 4HT-inducible promoter of the pF 5xUAS-GW-Puro construct was exchanged for the 800-bp fragment of the Bim promoter described in Bouillet et al. (2001). After virus transduction, MEF cells were selected with 5 µg/mL puromycin.
Mitochondrial isolation and subcellular fractionation
Cells were collected, washed once with PBS, and resuspended in MB-EDTA buffer prior to fractionation as described before (Wilfling et al. 2012). Hsp60 or tubulin (detected by Western blot) served as loading controls and marker proteins for cytosolic and mitochondrial fractions.
Gel electrophoresis and Western blotting
Whole cells or mitochondria-enriched fractions were extracted in buffer containing 1% detergent (Triton X-100 or digitonin), and protein concentrations were determined using the Bradford assay (Bio-Rad). Protein samples were separated by SDS-PAGE. BN-PAGE was performed essentially as described earlier (Vogtle et al. 2010). Mitochondrial fractions were prepared, and protein amounts were determined. For mitochondrial samples, equal protein amounts were solubilized with BN lysis buffer (20 mM Tris/HCl at pH 7.4, 50 mM NaCl, 0.1 mM EDTA, 1% digitonin, 1 mM PMSF). Samples were separated on a 6%–16.5% polyacrylamide gradient gel followed by wet transfer to a PVDF membrane and detection of proteins by Western blot.
Antibodies against Bim (Cell Signaling, no. C34C5), Bcl-XL (Cell Signaling, no. 54H6), Bcl-2 (Cell Signaling, no. 2870) Bax (Cell Signaling, no. 2772), HA (Cell Signaling, no. C29F4), VDAC (Cell Signaling, no. D73D12), Hsp60 (Cell Signaling, no. 4870), pERK (Cell Signaling, no. 4370), ERK (Cell Signaling, no. 4695), active Bax (6A7; Sigma, no. B8429), active Bak (NT; Millipore, no. 06536), tubulin (Sigma, no. T9026), DLC1 (Abcam, EP1660Y), Mcl-1 (Abcam, ab32087), Noxa (Abcam, ab13654), DIC1/2 (Santa Cruz Biotechnology, SC13524), V5 (Invitrogen, no. r960-25), cytochrome c (BD, no. 556432), and Bcl-2 (BD, no. 554218) as well as Mcl-1 (Rockland, no. 600401394) were used as suggested by the manufacturers. Signals were detected using horseradish peroxidase-conjugated secondary antibodies (anti-mouse, anti-hamster [both Dianova], or anti-rabbit [Sigma] IgG) and enhanced chemiluminescence (GE Healthcare or Thermo Scientific).
Immunoprecipitation
Four-hundred micrograms of whole-cell lysates (lysis buffer: 20 mM Tris/HCl at pH 7.4, 150 mM NaCl, 10% glycerol, 1% Triton X-100, protease inhibitor mix [Roche]) from Bax−/−Bak−/− double-knockout MEF or HEK293FT cells expressing the indicated Bim constructs were immunoprecipitated with antibodies to the triple-HA tag (anti-HA affinity matrix; Roche) or the V5 tag (anti-V5 agarose affinity matrix; Sigma). For testing the formation of Bim–Bim complexes in HEK293FT cells, we transiently cotransfected 3.5 × 106 HEK293FT cells per 10-cm dish (Invitrogen) with 4 µg of a pMIG-GW-3xHA-tagged Bim construct and 6 µg of an untagged Bim construct using FuGene HD (Promega). After preparation of whole-cell lysates, co-IP was performed as described above. Anti-Bim, anti-Mcl-1, or anti-DLC1 immunoprecipitation with 700–1000 µg of the whole-cell lysates of 293FT, HeLa, MCF-7, or 1205Lu cells was performed by incubating the protein G-agarose beads (Roche, no. 11719416001) with the respective antibodies (Bim [1:100; Cell Signaling, no. C34C5], Mcl-1 [1:100; Abcam, no. ab32087], and DLC1 [1:100; Abcam, no. EP1660Y]) for 1 h at 4°C. The coated beads were then incubated with the cell lysates for 4 h at 4°C. Anti-rabbit IgG (Cell Signaling) was used as an isotype control. The samples were analyzed by SDS-PAGE and Western blotting.
PLA
For PLA, the Duolink in situ kit (OLINK) was used. Cells were seeded on coverslips in 24-well cell culture plates. After induction of additional 3xHA-tagged mBimEL, cells were washed, fixed, permeabilized with 0.1% Triton X-100/PBS, and blocked with 5% BSA/PBS. Staining was done with α-V5 antibody (1:400; Invitrogen, no. r960-25) and α-HA antibody (1:1500; Cell Signaling, no. C29F4) in 5% BSA/0.5% saponin/PBS. Incubation with conjugated secondary antibodies and detection were performed according to the manufacturer's instructions. Cells were analyzed with the appropriate filters in a Keyence BZ-9000 fluorescence microscope.
RNAi against DLC1
MEF/HeLa/1205Lu cells were seeded in medium without antibiotics the day before transfection, and medium was again changed directly before RNAi. siRNA (20 nM final concentration) was mixed 1:0.83 with Lipofectamine RNAiMAX (Invitrogen) in serum-free medium (Optimem, Gibco), incubated for 20 min at room temperature, and added to the cells. siRNA specific for both human and mouse DLC1 (DYNLL1; Invitrogen, no. HSS189483) or scrambled siRNA (referred to here as siContr; negative control low GC [Invitrogen no. 12935-200] or siRNA against luciferase [GL2, 5′-CGUACGCGGAAUACUUCGA-3′]) (Elbashir et al. 2001) was used.
Cytochrome c release from mitochondria
Mitochondria were isolated as above and the cytochrome c release assay was performed as described earlier in Wilfling et al. (2012). The recombinant human Bax protein was purified according to the protocol by Suzuki et al. (2000) using pTYB1-Bax as an expression vector.
Production of recombinant proteins
For purification of recombinant murine BimL and BimLAA, we used a dual-tag purification strategy. The gene encoding for full-length murine BimL was cloned into the pTYB1 expression vector (New England Biolabs) to obtain a C-terminal fusion of an intein–chitin-binding domain (CBD) to BimL. Subsequently, the BimL-intein/CBD was subcloned into the pE-SUMOstar vector (Lifesensors), which yielded an N-terminal 6xHis-Sumo tag fused to the BimL-intein/CBD to obtain the pE-6xHis-Sumo-mBimL-intein/CBD. Full-length BimLAA was obtained using the pE-6xHis-Sumo-mBimL-intein/CBD as a template vector and the Q5 site-directed mutagenesis kit (New England Biolabs). The recombinant protein was purified from ArcticExpress E. coli strains (Agilent). Bacteria were lysed by mechanical disruption using a French press, and lysates were centrifuged at 100,000g for 1 h at 4°C. The supernatant was loaded on a chitin column (New England Biolabs). Cutting off the intein/CBD from BimL was induced by incubation with buffer containing 100 mM β-mercapthoethanol for 40 h at 4°C. The elution fraction was further purified using a Ni2+-NTA column (PerfectPro, Qiagen), and SUMO-BimL/BimLAA was eluted from the column with buffer containing 20 mM HEPES (pH 7.4), 100 mM NaCl, 20% glycerol, 0.3% CHAPS, and 300 mM imidazole. The purified protein was dialyzed against 20 mM HEPES, 300 mM NaCl, 20% glycerol, and 1% CHAPS. For some experiments, the 6xHis-SUMO tag was removed by cleavage overnight at 4°C with SUMO protease 1 (LifeSensors). After passing through a Ni2+-NTA column, BimL/BimLAA was collected in the flow-through, flash-frozen, and stored at −80°C.
The gene encoding full-length human DLC1 was cloned in the pETDuet-1 vector (Novagen). A DNA sequence was added to encode an N-terminal 6xHis tag. The recombinant protein was expressed in SOLUBL21 E. coli cells (Amsbio). Bacteria were treated as above, and the 6xHis-DLC1 was purified from the soluble fraction by a Ni2+-NTA column. 6xHis-DLC1 was eluted from the column with buffer containing 20 nM Tris-HCl (pH 8), 300 nM NaCl, and 300 nM imidazole. The purified protein was then flash-frozen and stored at −80°C. Recombinant full-length Bcl-XL protein was purified as before (Bleicken et al. 2013).
In vitro BimL complex formation assay
Mitochondria were isolated as above and treated for 20 min at 30°C with 250 nM SUMO-BimL in KCL buffer. Mitochondria were then washed and incubated in the same buffer with or without 500 nM recombinant DLC1. For BN-PAGE, mitochondria were solubilized in 1% digitonin buffer, and the complexes were analyzed by BN electrophoresis.
LUVs were centrifuged at 150.000g for 30 min. The pellet was resuspended in the LUV buffer (20 mM HEPES at pH 7.5, 138 mM NaCl). Recombinant proteins (50 nM BimL, 50 nM BimLAA, 100 nM DLC1, 100 nM Bcl-XL, and 100 nM Bax) were added, followed by incubation for 30 min at 30°C. The LUVs were then solubilized in 1% digitonin buffer, and the complexes were analyzed by BN electrophoresis.
Fluorescent labeling of proteins
Full-length recombinant BimL (Alexa 488 or Alexa 647 maleimide; referred to here as BimL-G and BimL-R) and DLC1 (Alexa 647-maleimide; DLC1-R) were labeled by incubating the proteins overnight with 10-fold molar excess of the respective reactive dyes at 4°C. The free dye and the salt were separated using a PD-10 desalting column packed with Sephadex G-25 resin (GE Healthcare).
Artificial membranes
The lipids used in the present study were purchased from Avanti Polar Lipids, Lipoid, or Sigma-Aldrich. GUVs of different lipid compositions (PC only or mixtures of 80% PC with 20% phosphatidylethanolamine [PE]/phosphatidylglycerol [PG]/or CL as indicated in the figures or mitomix, a complex mixture of lipid mimicking the mitochondrial outer membrane [PC:PE:PI:PS:CL, 46:25:11:10:8]) were prepared by the electroformation method as described (Bleicken et al. 2013). Briefly, 5 µg of the lipid mixtures dissolved in chloroform were applied to each platinum electrode of an electroformation chamber and air-dried before immersing in 300 µL of 300 mM sucrose solution. Next, electroformation was performed at 10 Hz for 2 h followed by 2 Hz for 30 min at room temperature. The indicated concentrations of the fluorescently labeled proteins were added in 220 µL of PBS in each well of BSA-blocked eight-well Lab-Tek chambers (Nunc). Eighty microliters of GUV suspension was added per well and incubated with the proteins for 30 min to attain equilibration before imaging and FCS acquisition.
Preparation of LUVs
LUVs containing egg phosphatidylcholine (Lipoid), bovine heart CL (Sigma-Aldrich), and rhodamine PE (Life Technologies) in a molar ratio of 79.8:20:0.2 were prepared with the thin film method. Lipids were dissolved in chloroform and mixed in a round-bottom flask. The solvent was removed using a rotary evaporator followed by further drying under vacuum to obtain a dry lipid film. The film was resuspended in a HEPES-buffered salt solution (20 mM HEPES, 134 mM NaCl at pH 7.5) to a total lipid concentration of 10 or 20 mM. After subsequent extrusion (51 times) through polycarbonate membranes with pores of 100 nm (Nuclepore Whatman), LUVs were obtained. Lipid content was determined using a phosphorous assay, and the hydrodynamic diameter (100–115 nm) was determined by dynamic light scattering (Zetasizer ZS, Malvern Instruments).
Confocal microscopy and FCCS
Images were acquired on a LSM710 confocal microscope with a C-Apo-chromat 40× N.A. 1.2 water immersion objective after excitation with Ar ion (488-nm) and/or HeNe (633-nm) lasers. Acquired images were processed, and the radial profile was analyzed using ImageJ.
For solution FCCS experiments, 20 nM BimL-G and BimL–R and 40 nM DLC1 were incubated in 200 µL of PBS (pH 7.4) for 30 min in a well of preblocked (1% BSA) Lab-Tek chambers (Nunc). Each acquisition lasted 20 sec (>10,000 times longer than the diffusion time of the labeled proteins) to ensure accumulation of sufficient data points for the generation of autocorrelation curves. The cross-correlation was calculated assuming three-dimensional (3D) diffusion of the proteins as described (Garcia-Saez et al. 2009).
The two focus linear scanning (membrane) FCCS experiments were performed, and the data were analyzed as described (Bleicken et al. 2013). Briefly, the proteins (concentration same as in the solution FCCS) were incubated in a sample containing 220 µL of PBS and 80 µL of the GUV suspension for 30 min. The data were acquired on a LSM710 confocal microscope at 22°C using a Confocor 3 module and Correlator Flex 02-01D/C for the recording of photon arrival times. The detection volumes (two perpendicular lines across the GUV equator) were consecutively repeatedly scanned over a period of 5 min. The photon stream was binned in 2 µsec and was arranged as a matrix so that each row corresponded to one line scan. The effect of membrane movements from scan to scan was corrected by aligning and summing up each intensity trace with respect to the position of maximum membrane contributions. A discrete intensity trace was obtained by fitting an average over all rows (only the elements of each row between −2.5 and +2.5 σ were added) with a Gaussian. Next, the irregular traces due to the distortions and undulations were removed, and autocorrelations, spectral cross-correlations, and spatial cross-correlations were calculated as in Garcia-Saez et al. (2009). The autocorrelation and cross-correlation functions obtained were fitted with a nonlinear least-square global fitting algorithm, and the percentage of cross-correlation was calculated using the formula in Supplemental Table 1.
Size exclusion chromatography
Mitochondria purified from cells expressing either wild-type V5-mBimEL or V5-mBimELAA were lysed in 20 mM Tris/HCl (pH 7.4), 50 mM NaCl, 0.1 mM EDTA, and 1% digitonin, and protein concentration was determined by the Bradford assay. Equal amounts were applied to a Superdex 200 10/300 GL column (AEKTA, Amersham Biosciences) equilibrated in 20 mM Tris/HCl (pH 7.4), 50 mM NaCl, 0.1 mM EDTA, and 0.1% digitonin. Fractions of 500 μL were collected, and 50 μL of each fraction was analyzed by SDS-PAGE and immunoblotting. A set of proteins for MW calibration (Sigma-Aldrich, no. MWGF1000) was run in parallel.
Apoptosis induction by Bim overexpression and thapsigargin, ABT-737, and gefitinib treatment
MEF cells carrying the 4HT-inducible wild-type BimEL or BimELAA were treated with 100 nM 4HT (Sigma) to induce Bim expression for 24 h. Cells were washed in PBS, fixed, and incubated in the presence of monoclonal anti-active caspase-3 antibody (1:500; BD Pharmingen, no. 559565) as described earlier (Weber et al. 2013). Flow cytometry was performed using a FACS Calibur (Becton Dickinson). Apoptosis was inhibited with 10 µM Q-VD-OPH (SM Biochemical) added together with stimulation with 4HT. Thapsigargin (200 nM; Sigma) was added after siRNA knockdown induction as described above. HeLa and 1205Lu cells were treated with 2 µM ABT-737 (Selleck Chemicals) after 24 h of siRNA knockdown and 10 µM MEK1/2 inhibitor UO126. Caspase-3 staining for FACS was performed as described above. The cells for Western blot were treated simultaneously with 10 µM pan-caspase inhibitor QVD-OPH (SM Biochemical) or 20 µM proteasome inhibitor MG132 (Enzo Life Sciences). HCC827 cells were treated with 10 µM gefitinib (Selleck Chemicals) upon DLC1-specific RNAi (24 h). The cells were harvested 72 h after gefitinib treatment, and caspase-3 activation (BD, no. 559565) was determined using flow cytometry.
Superresolution and confocal imaging of cells
Bax−/−Bak−/−double-knockout MEFs constitutively overexpressing 3xHA-tagged BimEL or wild-type HeLa were grown in glass-bottom Petri dishes (FluoroDish 35 mm, WPI), fixed in formaldehyde, and permeabilized. MEFs were labeled using an anti-HA primary antibody (1:100; Cell Signaling, no. 3724) and HeLa with the anti-Bim primary antibody, while, for both, an Alexa 647-labeled secondary antibody (1:100; Dianova, #711-605-152) was used. STORM was performed on a Nikon N-STORM system (Nikon Eclipse Ti-E inverted microscope, 100× objective, NA = 1.49, Andor iXon DU-897 EM-CCD camera, 300-mW 647-nm laser, astigmatism lens for 3D STORM). The imaging was performed in 3D STORM mode, similar to previously published studies (Huang et al. 2008). The imaging buffer was PBS with 50 mM mercaptoethlyamine (Sigma-Aldrich) for MEFs overexpressing 3xHA-tagged BimEL or PBS with 100 mM mercaptoethlyamine and GLOX-buffer (0.5 mg/mL glucose oxidase [Sigma-Aldrich], 40 µg/mL catalase [Sigma-Aldrich], 10% glucose) for wild-type HeLa cells labeled for endogenous Bim. The data were analyzed in Nikon's NIS-Elements imaging software. Gaussian fitting was applied to determine the single-molecule localizations (including Fit Overlapping Peaks function). The z-positions were calculated by comparing the fit parameters of the astigmatism-affected signals to a z calibration. In Figure 2, J and K, the relative position with respect to the focal plane is indicated using a color scale ranging from red (+400 nm) to green (−400 nm). For MEFs, a density filter was applied to the reconstructed image to reduce the number of localizations outside the mitochondria.
For confocal imaging, wild-type HeLa cells were treated for 20 min in culture conditions with 100 nM MitoTracker Deep Red FM (ThermoFischer Scientific, no. M22426). Afterward, they were fixed as described above on µ-slides (Ibidi, no. 80826) and stained using the anti-Bim primary and Alexa 488-labeled (Dianova, no. 711-545-152) secondary antibodies. Images were taken using a Zeiss LSM 880 with an Airyscan confocal microscope and analyzed with Zen (Zeiss) software.
Bax and Bak activation by cross-linking and flow cytometry
Mitochondria from HeLa or 1205Lu cells treated with siRNAs and ABT-737 were isolated as described above. The isolated mitochondria (30–60 µg) were incubated for 30 min in the dark at room temperature in 45 µL of cross-linking buffer (20 mM HEPES at pH 7.5, 250 mM sucrose, 1 mM EDTA, 50 mM KCl, 2.5 mM MgCl2) containing 0.5 mM BMH (1, 6–bismaleimidohexane; Thermo Scientific, no. 22331). After incubation, the reaction was stopped by adding 1 mM DTT for an additional 15 min at room temperature. Finally, samples were analyzed by SDS-PAGE, and proteins were detected by Western blotting. For flow cytometry experiments, treated cells were fixed in 4% formalin for 15 min and stained with conformation-specific primary antibodies (Bax 6A7 [Sigma, no. B8429] and Bak NT [Millipore, no. 06536]). Species-specific Alexa fluor 647 or Cy5-conjugated secondary antibodies were used to detect activated Bax and Bak by flow cytometry.
Cell cycle analysis
HeLa or 1205Lu cells were harvested 24 h after transfection with siRNA against DLC1 or control siRNA and fixed with 75% ethanol overnight at 4°C. The next day, samples were washed twice with wash buffer (0.5% BSA in PBS), and cells were resuspended in staining buffer (20 µM propidium iodide [Sigma, no. P4170], 25 µg/mL RNase A [Thermo Scientific, no. EN0531]) for 15 min at 37°C. The stained cells were then analyzed by flow cytometry.
Supplementary Material
Acknowledgments
This study was supported in part by the Excellence Initiative of the German Research Council DFG (DFG; GSC-4, Spemann Graduate School, and Excellence Cluster BIOSS Exc 294) and a grant from the DFG (FOR2036 to G.H. and A.G.-S.). W.R. also acknowledges support by the German Federal Ministry of Education and Research (BMBF) in the framework of the EU ERA SynBio project SynGlycTis and a starting grant from the European Research Council (Programme “Ideas,” ERC-2011-StG 282105).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.302497.117.
References
- Aranovich A, Liu Q, Collins T, Geng F, Dixit S, Leber B, Andrews DW. 2012. Differences in the mechanisms of proapoptotic BH3 proteins binding to Bcl-XL and Bcl-2 quantified in live MCF-7 cells. Mol Cell 45: 754–763. [DOI] [PubMed] [Google Scholar]
- Barbar E, Nyarko A. 2015. Polybivalency and disordered proteins in ordering macromolecular assemblies. Semin Cell Dev Biol 37: 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer A, Kirschnek S, Hacker G. 2007. Inhibition of apoptosis can be accompanied by increased Bim levels in T lymphocytes and neutrophil granulocytes. Cell Death Differ 14: 1714–1716. [DOI] [PubMed] [Google Scholar]
- Bhola PD, Letai A. 2016. Mitochondria-judges and executioners of cell death sentences. Mol Cell 61: 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billen LP, Shamas-Din A, Andrews DW. 2008. Bid: a Bax-like BH3 protein. Oncogene 27: S93–S104. [DOI] [PubMed] [Google Scholar]
- Bleicken S, Wagner C, Garcia-Saez AJ. 2013. Mechanistic differences in the membrane activity of Bax and Bcl-xL correlate with their opposing roles in apoptosis. Biophys J 104: 421–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, Adams JM, Strasser A. 1999. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 286: 1735–1738. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Zhang LC, Huang DC, Webb GC, Bottema CD, Shore P, Eyre HJ, Sutherland GR, Adams JM. 2001. Gene structure alternative splicing, and chromosomal localization of pro-apoptotic Bcl-2 relative Bim. Mamm Genome 12: 163–168. [DOI] [PubMed] [Google Scholar]
- Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. 2005. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 17: 393–403. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. 2010. The BCL-2 family reunion. Mol Cell 37: 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg MS, Kuroda J, Puthalakath H, Huang DC, Strasser A. 2007. Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS Med 4: 1681–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar PE, Lee EF, van Delft MF, Day CL, Smith BJ, Huang DC, Fairlie WD, Hinds MG, Colman PM. 2007. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc Natl Acad Sci 104: 6217–6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day CL, Puthalakath H, Skea G, Strasser A, Barsukov I, Lian LY, Huang DC, Hinds MG. 2004. Localization of dynein light chains 1 and 2 and their pro-apoptotic ligands. Biochem J 377: 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehan E, Bassermann F, Guardavaccaro D, Vasiliver-Shamis G, Cohen M, Lowes KN, Dustin M, Huang DC, Taunton J, Pagano M. 2009. βTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol Cell 33: 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewson G. 2016. Doughnuts, daisy chains and crescent moons: the quest for the elusive apoptotic pore. EMBO Jo 35: 371–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Huo L, Yang JY, Xia W, Wei Y, Liao Y, Chang CJ, Yang Y, Lai CC, Lee DF, et al. 2008. Down-regulation of myeloid cell leukemia-1 through inhibiting Erk/Pin 1 pathway by sorafenib facilitates chemosensitization in breast cancer. Cancer Res 68: 6109–6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domina AM, Vrana JA, Gregory MA, Hann SR, Craig RW. 2004. MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene 23: 5301–5315. [DOI] [PubMed] [Google Scholar]
- Egle A, Harris AW, Bouillet P, Cory S. 2004. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci 101: 6164–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Martinez J, Patkaniowska A, Lendeckel W, Tuschl T. 2001. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J 20: 6877–6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewings KE, Hadfield-Moorhouse K, Wiggins CM, Wickenden JA, Balmanno K, Gilley R, Degenhardt K, White E, Cook SJ. 2007. ERK1/2-dependent phosphorylation of BimEL promotes its rapid dissociation from Mcl-1 and Bcl-xL. EMBO J 26: 2856–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Saez AJ, Ries J, Orzaez M, Perez-Paya E, Schwille P. 2009. Membrane promotes tBID interaction with BCL(XL). Nat Struct Mol Biol 16: 1178–1185. [DOI] [PubMed] [Google Scholar]
- Gebert N, Joshi AS, Kutik S, Becker T, McKenzie M, Guan XL, Mooga VP, Stroud DA, Kulkarni G, Wenk MR, et al. 2009. Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: implications for Barth syndrome. Curr Biol 19: 2133–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Bougie P, Bataille R, Amiot M. 2005. Endogenous association of Bim BH3-only protein with Mcl-1, Bcl-xL and Bcl-2 on mitochondria in human B cells. Eur J Immunol 35: 971–976. [DOI] [PubMed] [Google Scholar]
- Grabow S, Delbridge AR, Aubrey BJ, Vandenberg CJ, Strasser A. 2016. Loss of a single Mcl-1 allele inhibits MYC-driven lymphomagenesis by sensitizing pro-B cells to apoptosis. Cell Rep 14: 2337–2347. [DOI] [PubMed] [Google Scholar]
- Herold MJ, Rohrbeck L, Lang MJ, Grumont R, Gerondakis S, Tai L, Bouillet P, Kaufmann T, Strasser A. 2013. Foxo-mediated Bim transcription is dispensable for the apoptosis of hematopoietic cells that is mediated by this BH3-only protein. EMBO Rep 14: 992–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds MG, Smits C, Fredericks-Short R, Risk JM, Bailey M, Huang DC, Day CL. 2007. Bim, Bad and Bmf: intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ 14: 128–136. [DOI] [PubMed] [Google Scholar]
- Horvath SE, Rampelt H, Oeljeklaus S, Warscheid B, van der Laan M, Pfanner N. 2015. Role of membrane contact sites in protein import into mitochondria. Protein Sci 24: 277–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Sinicrope FA. 2008. BH3 mimetic ABT-737 potentiates TRAIL-mediated apoptotic signaling by unsequestering Bim and Bak in human pancreatic cancer cells. Cancer Res 68: 2944–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Wang W, Bates M, Zhuang X. 2008. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 319: 810–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado S, Gleeson K, O'Donnell K, Izon DJ, Walkley CR, Strasser A, Tarlinton DM, Heierhorst J. 2012. The zinc-finger protein ASCIZ regulates B cell development via DYNLL1 and Bim. J Exp Med 209: 1629–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper C, Habib SJ, Engl G, Heckmeyer P, Dimmer KS, Rapaport D. 2008. Integration of tail-anchored proteins into the mitochondrial outer membrane does not require any known import components. J Cell Sci 121: 1990–1998. [DOI] [PubMed] [Google Scholar]
- Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, Chanrion M, Kelly GL, Gong JN, Moujalled DM, et al. 2016. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538: 477–482. [DOI] [PubMed] [Google Scholar]
- Krumpe K, Frumkin I, Herzig Y, Rimon N, Ozbalci C, Brugger B, Rapaport D, Schuldiner M. 2012. Ergosterol content specifies targeting of tail-anchored proteins to mitochondrial outer membranes. Mol Biol Cell 23: 3927–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda J, Puthalakath H, Cragg MS, Kelly PN, Bouillet P, Huang DC, Kimura S, Ottmann OG, Druker BJ, Villunger A, et al. 2006. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci 103: 14907–14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. 2002. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183–192. [DOI] [PubMed] [Google Scholar]
- Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. 2003. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem 278: 18811–18816. [DOI] [PubMed] [Google Scholar]
- Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, Dillon CP, Green DR. 2011. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell 44: 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, Andrews DW. 2008. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135: 1074–1084. [DOI] [PubMed] [Google Scholar]
- Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X. 2000. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nature Cell Biol 2: 754–761. [DOI] [PubMed] [Google Scholar]
- Mestre-Escorihuela C, Rubio-Moscardo F, Richter JA, Siebert R, Climent J, Fresquet V, Beltran E, Agirre X, Marugan I, Marin M, et al. 2007. Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood 109: 271–280. [DOI] [PubMed] [Google Scholar]
- Mojsa B, Lassot I, Desagher S. 2014. Mcl-1 ubiquitination: unique regulation of an essential survival protein. Cells 3: 418–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor L, Strasser A, O'Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. 1998. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J 17: 384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield CJ, Dunker AK. 2014. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu Rev Biochem 83: 553–584. [DOI] [PubMed] [Google Scholar]
- Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, Korsmeyer SJ. 2005. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 307: 1101–1104. [DOI] [PubMed] [Google Scholar]
- O'Reilly LA, Cullen L, Visvader J, Lindeman GJ, Print C, Bath ML, Huang DC, Strasser A. 2000. The proapoptotic BH3-only protein bim is expressed in hematopoietic, epithelial, neuronal, and germ cells. Am J Pathol 157: 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish IA, Rao S, Smyth GK, Juelich T, Denyer GS, Davey GM, Strasser A, Heath WR. 2009. The molecular signature of CD8+ T cells undergoing deletional tolerance. Blood 113: 4575–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peperzak V, Slinger E, Ter Burg J, Eldering E. 2017. Functional disparities among BCL-2 members in tonsillar and leukemic B-cell subsets assessed by BH3-mimetic profiling. Cell Death Differ 24: 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson W, Cauwe B, Policheni A, Schlenner SM, Franckaert D, Berges J, Humblet-Baron S, Schonefeldt S, Herold MJ, Hildeman D, et al. 2013. Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3+ regulatory T cells. Nat Immunol 14: 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthalakath H, Huang DC, O'Reilly LA, King SM, Strasser A. 1999. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell 3: 287–296. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, et al. 2007. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129: 1337–1349. [DOI] [PubMed] [Google Scholar]
- Rapali P, Szenes A, Radnai L, Bakos A, Pal G, Nyitray L. 2011. DYNLL/LC8: a light chain subunit of the dynein motor complex and beyond. FEBS J 278: 2980–2996. [DOI] [PubMed] [Google Scholar]
- Sarosiek KA, Chi X, Bachman JA, Sims JJ, Montero J, Patel L, Flanagan A, Andrews DW, Sorger P, Letai A. 2013. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell 51: 751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy AR, Kirschnek S, Hacker G. 2014. IL-15 regulates Bcl-2 family members Bim and Mcl-1 through JAK/STAT and PI3K/AKT pathways in T cells. Eur J Immunol 44: 2500–2507. [DOI] [PubMed] [Google Scholar]
- Shinjyo T, Kuribara R, Inukai T, Hosoi H, Kinoshita T, Miyajima A, Houghton PJ, Look AT, Ozawa K, Inaba T. 2001. Downregulation of Bim, a proapoptotic relative of Bcl-2, is a pivotal step in cytokine-initiated survival signaling in murine hematopoietic progenitors. Mol Cell Biol 21: 854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steimer DA, Boyd K, Takeuchi O, Fisher JK, Zambetti GP, Opferman JT. 2009. Selective roles for antiapoptotic MCL-1 during granulocyte development and macrophage effector function. Blood 113: 2805–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Youle RJ, Tjandra N. 2000. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 103: 645–654. [DOI] [PubMed] [Google Scholar]
- Tan TT, Degenhardt K, Nelson DA, Beaudoin B, Nieves-Neira W, Bouillet P, Villunger A, Adams JM, White E. 2005. Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell 7: 227–238. [DOI] [PubMed] [Google Scholar]
- Thornton N, Stroud DA, Milenkovic D, Guiard B, Pfanner N, Becker T. 2010. Two modular forms of the mitochondrial sorting and assembly machinery are involved in biogenesis of α-helical outer membrane proteins. J Mol Biol 396: 540–549. [DOI] [PubMed] [Google Scholar]
- van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, et al. 2006. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 10: 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux DL, Haecker G, Strasser A. 1994. An evolutionary perspective on apoptosis. Cell 76: 777–779. [DOI] [PubMed] [Google Scholar]
- Vince JE, Chau D, Callus B, Wong WW, Hawkins CJ, Schneider P, McKinlay M, Benetatos CA, Condon SM, Chunduru SK, et al. 2008. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1–TRAF2 complex to sensitize tumor cells to TNFα. J Cell Biol 182: 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogtle FN, Schmidt O, Chacinska A, Pfanner N, Meisinger C. 2010. Native techniques for analysis of mitochondrial protein import. Methods Mol Biol 619: 425–436. [DOI] [PubMed] [Google Scholar]
- Weber A, Kirejczyk Z, Besch R, Potthoff S, Leverkus M, Hacker G. 2010. Proapoptotic signalling through Toll-like receptor-3 involves TRIF-dependent activation of caspase-8 and is under the control of inhibitor of apoptosis proteins in melanoma cells. Cell Death Differ 17: 942–951. [DOI] [PubMed] [Google Scholar]
- Weber A, Auslander D, Hacker G. 2013. Mouse Noxa uses only the C-terminal BH3-domain to inactivate Mcl-1. Apoptosis 18: 1093–1105. [DOI] [PubMed] [Google Scholar]
- Weber A, Heinlein M, Dengjel J, Alber C, Singh PK, Hacker G. 2016. The deubiquitinase Usp27x stabilizes the BH3-only protein Bim and enhances apoptosis. EMBO Rep 17: 724–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfling F, Weber A, Potthoff S, Vogtle FN, Meisinger C, Paschen SA, Hacker G. 2012. BH3-only proteins are tail-anchored in the outer mitochondrial membrane and can initiate the activation of Bax. Cell Death Differ 19: 1328–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. 2008. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9: 47–59. [DOI] [PubMed] [Google Scholar]
- Zantl N, Weirich G, Zall H, Seiffert BM, Fischer SF, Kirschnek S, Hartmann C, Fritsch RM, Gillissen B, Daniel PT, et al. 2007. Frequent loss of expression of the pro-apoptotic protein Bim in renal cell carcinoma: evidence for contribution to apoptosis resistance. Oncogene 26: 7038–7048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.