

Abstract
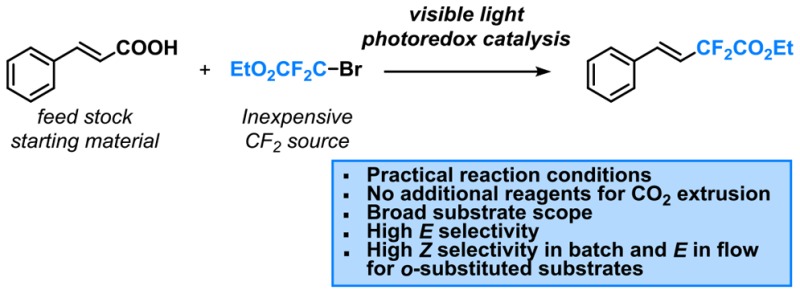
The development of synthetic methodologies which provide access to both stereoisomers of α,β-disubstituted olefins is a challenging undertaking. Herein, we describe the development of an operationally simple and stereoselective synthesis of difluoromethylated styrenes via a visible-light photocatalytic decarboxylation strategy using fac-Ir(ppy)3 as the photocatalyst. Meta- and para-substituted cinnamic acids provide the expected E-isomer. In contrast, ortho-substituted cinnamic acids yield selectively the less stable Z-product, whereas the E-isomer can be obtained via continuous-flow processing through accurate control of the reaction time. Furthermore, our protocol is amenable to the decarboxylative difluoromethylation of aryl propiolic acids.
Keywords: E/Z selectivity, flow chemistry, TTET, difluoromethylation, decarboxylation, photoredox catalysis
The introduction of fluorinated moieties into organic compounds has resulted in a dramatic enhancement of their physical, chemical, and biological properties, rendering medicinal and agrochemical compounds to be more potent.1 Consequently, in recent years, a tremendous amount of research effort has been devoted to develop new methods to enable the efficient incorporation of fluorinated moieties into parent molecules.2 Among these, the CF2 motif plays an increasingly important role, since the hydrogen bond donor properties of the difluoromethyl group increases acidity of its neighboring group, which enhances dipole moments and conformational changes in the molecules.3 In recent years, great progress has been made with regard to radical difluoroalkylation reactions, especially via visible-light photoredox catalysis.4 Visible light photoredox catalysis has become one of the most powerful tools in organic synthesis wherein both single electron transfer (SET) and triplet–triplet energy transfer (TTET) processes with organic substrates can be facilitated.5
With biomass feedstocks of vinyl carboxylic acids abundantly available, these inexpensive and stable compounds have attracted a great deal of attention as substrates for a wide variety of synthetic transformations.6 Perhaps the most widely used decarboxylative fluoroalkylation strategy involves transition-metal coordination in combination with high temperatures or strong oxidants to facilitate the CO2 extrusion process (Scheme 1A).7 It is evident that such harsh reaction conditions have repercussions on the substrate scope. Photocatalytic strategies have allowed the decarboxylative functionalization process to proceed at room temperature but still require stoichiometric amounts of strong oxidants or transition metals (Scheme 1B).8,9 In addition, all these methods give access to the thermodynamically more stable E-alkenes,10 while methods delivering selectively the Z-isomers are far less common.11
Scheme 1. (A) Classical Decarboxylative Cross-Coupling Strategies. (B) Recent Photocatalytic Approaches Still Require the Use of Metals or Hypervalent Iodine Reagents (HIR) to Enable the Decarboxylation Step. (C) Our Strategy for the Photocatalytic Radical Difluoromethylation of Cinnamic Acids.

The strategy we describe here involves a photocatalytic decarboxylation methodology to access difluoroalkenes, which is operationally simple, mild and requires no additional transition metals or oxidants to enable CO2 extrusion (Scheme 1C). Moreover, with ortho-substituted cinnamic acid substrates, Z-isomers could be obtained in high selectivity. The corresponding E-isomer could be accessed as well via continuous-flow processing through accurate control of the reaction time. To the best of our knowledge, having access to both stereoisomers simply by changing the reactor has never been reported before and constitutes a powerful approach to tune reaction selectivity for photoredox catalysis.
Building on our recent experience with the direct trifluoromethylation of styrenes,10a we commenced our investigations by using fac-Ir(ppy)3 as the photocatalyst (Table 1). The targeted product could be obtained in a 31% yield and with an E/Z ratio of 52:48 using 3 equiv of ethyl bromodifluoroacetate 2 and KOAc as a base (Table 1, entry 1). Interestingly, no metal cocatalyst or hypervalent iodine reagent (HIR) was required to facilitate the CO2 extrusion. The rather poor E/Z selectivity can be explained due to the high triplet energy level of the fac-Ir(ppy)3 photocatalyst (ET = 2.41 eV).12 Consequently, a triplet–triplet energy transfer occurs leading to an erosion of the stereoselectivity.11 Various solvents and bases were subsequently screened (Table 1, entries 1–6); the best results were obtained using 1,4 dioxane as a solvent and NaHCO3 as the base. Addition of water provided an improved yield but a decreased selectivity (Table 1, entry 7). Optimal results were obtained when the concentration was reduced to 0.1 M leading to a 68% yield and an excellent E/Z selectivity (94:6) (Table 1, entry 8). Lastly, control experiments confirmed the photocatalytic nature of our transformation, as no reaction was observed in the absence of photocatalyst and/or light (Table 1, entries 9 and 10).
Table 1. Reaction Discovery and Optimization Studies for the Photocatalytic Difluoromethylation of Cinnamic Acidsa.

| entry | base | solvent | yieldb (%) | E/Zb |
|---|---|---|---|---|
| 1d | KOAc | 0.2 M CH3CN | 31 | 52:48 |
| 2 | KOAc | 0.2 M EtOH | 44 | 57:43 |
| 3 | KOAc | 0.2 M 1,4-dioxane | 60 | 51:49 |
| 4 | Cs2CO3 | 0.2 M 1,4-dioxane | 46 | 71:29 |
| 5 | 2,6-lutidine | 0.2 M 1,4-dioxane | 70 | 46:54 |
| 6 | NaHCO3 | 0.2 M 1,4-dioxane | 75 | 75:25 |
| 7c,d | NaHCO3 | 0.2 M 1,4-dioxane | 83 | 50:50 |
| 8d | NaHCO3 | 0.1 M 1,4-dioxane | 68 | 94:6 |
Reaction conditions: fac-Ir(ppy)3 (1 mol %), cinnamic acid 1 (0.2 mmol), NaHCO3 (0.4 mmol), ethyl bromodifluoroacetate 2 (0.6 mmol), solvent (2 mL, 0.1 M), blue LEDs (3.12 W), room temperature, argon atmosphere, stirred for 24 h.
Yield and E/Z values are determined with 19F NMR using α,α,α-trifluorotoluene as internal standard.
10 equiv of H2O was added.
Reported yields are those of isolated compounds; E/Z values are determined by 1H NMR of isolated products.
Having identified the optimal reaction conditions for the photocatalytic difluoromethylation of cinnamic acid, we aimed to define the reaction scope (Table 2). Our protocol was found to readily accommodate a variety of para- and meta-substituted cinnamic acids, including electron-neutral (3a–d), electron-donating (3e–g), and electron-withdrawing substituents (3h–n). Overall, the E/Z ratio was good to excellent for all these examples. In addition, the presence of halogens was well tolerated, providing opportunities for further decoration of the molecule, e.g., via cross coupling (3k–n). In addition, heterocyclic substrates, such as pyridine (3o) and thiophene (3p), were found to be competent substrates. The pyridine analogue displayed an excellent E/Z selectivity (99:1), while the thiophene one was obtained with a lower stereoselectivity (61:39). Extended conjugation, e.g., for (2E,4E)-5-phenylpenta-2,4-dienoic acid, was tolerated as well, delivering the corresponding product (3q) in good yield and excellent selectivity (81%, 99:1). Also, β-substituted cinnamic acids, e.g., 1,1-diphenylethylene (3r), could be successfully subjected to our reaction conditions resulting in a good isolated yield (81% yield). The lower E/Z selectivity in some cases prompted us to evaluate the efficacy of fac-Ir(tBuppy)3. This photocatalyst was recently reported by Weaver et al. and was shown to lower the energy transfer rate due to the increased steric bulk.11a However, while an increase in yield was observed, the E/Z selectivity only marginally improved (Table 2, 3b and 3p).
Table 2. Decarboxylative Difluoromethylation: Scope of Meta- and Para-Substituted Cinnamic Acidsa,b.


Reaction conditions: cinnamic acid 1 (0.2 mmol, 1.0 equiv), ethyl bromodifluoroacetate 2 (0.6 mmol, 3.0 equiv), fac-Ir(ppy)3 (1 mol %), NaHCO3 (0.4 mmol, 2.0 equiv), 1,4-dioxane (2.0 mL), argon, blue LEDs (3.12 W), 24 h.
Reported yields are those of isolated compounds; E/Z values are determined by 1H NMR of isolated products.
fac-Ir(tBuppy)3 was used as the photocatalyst.
Reaction time, 30 h.
Due to the limited solubility of the substrate, the yield is lower for compound 3o. However, the yield could be increased by recycling the unreacted starting material.
However, when ortho-substituted cinnamic acids were evaluated, a selectivity switch was observed toward the Z product (Table 3, entry 1). Interestingly, the use of fac-Ir(tBuppy)3 completely altered the selectivity (Table 3, entry 2), confirming the observations of Weaver et al.11a We were delighted to find that an increase in concentration resulted in an improvement in both yield and selectivity (Table 3, entry 1 and 3). A further increase in catalyst loading and concentration contributed to an enhancement of the selectivity toward the Z isomer (Table 3, entries 4–6). Kinetic experiments revealed that the thermodynamically more stable E-isomer was formed first, after which the Z-isomer was obtained via a triplet–triplet energy transfer mechanism (TTET) (see the Supporting Information).11b Consequently, it should theoretically be possible to stop the reaction before E/Z isomerization occurs. In order to obtain high conversions in a short amount of time, we turned our attention to the use of continuous-flow microreactors which allow to accelerate photocatalytic reactions due to an improved irradiation profile and enhanced mass transfer characteristics (Table 3, entries 7–11).13,14 In flow, the reaction time could be reduced significantly resulting in a reversed E/Z selectivity (Table 3, entry 7). An increase in catalyst loading and concentration could further reduce the reaction time to 15 min resulting in an excellent E selectivity (62%, 92:8) (Table 3, entry 11).15 Longer residence times lead to an increase in yield but an erosion of the selectivity (Table 3, entries 8 and 9). However, it should be noted that it was possible to recover the starting material quantitatively, which can be subsequently reintroduced into the flow reactor obtaining higher overall conversions while maintaining a high stereoselectivity (see Table 4, 5g and 3p).
Table 3. Optimization Studies for the Photocatalytic Difluoromethylation of Ortho-Substituted Cinnamic Acids in Batcha or Continuous-Flowb.

| entry | conc (M) | fac-Ir(ppy)3 (mol %) | reaction time (h) | Yieldc (%) | E/Zc |
|---|---|---|---|---|---|
| Batch Conditionsa | |||||
| 1 | 0.1 | 1 | 24 | 67 | 21:79 |
| 2d | 0.1 | 1 | 24 | 60 | 79:21 |
| 3 | 0.2 | 1 | 24 | 86 | 15:85 |
| 4 | 0.2 | 3 | 24 | 88 | 10:90 |
| 5e | 0.5 | 3 | 24 | 77 | 6:94 |
| 6e | 1.0 | 2 | 24 | 57 | 5:95 |
| Continuous-Flow Conditionsb | |||||
| 7 | 0.05 | 0.5 | 2 | 51 | 75:25 |
| 8 | 0.1 | 0.5 | 2 | 68 | 68:32 |
| 9 | 0.1 | 1.0 | 2 | 46 | 26:74 |
| 10 | 0.1 | 1.5 | 0.5 | 55 | 78:22 |
| 11e | 0.15 | 1.0 | 0.25 | 62 | 92:8 |
Reaction conditions in batch: fac-Ir(ppy)3 (1 mol %), (E)-3-(o-tolyl)acrylic acid 4a (0.2 mmol), ethyl bromodifluoroacetate 2 (0.6 mmol), NaHCO3 (0.4 mmol), H2O (3.0 mmol), 1,4-dioxane (0.4 mL), blue LEDs (3.12 W), room temperature, argon atmosphere, stirred for 24 h.
Reaction conditions in continuous flow: fac-Ir(ppy)3 (1 mol %), (E)-3-(o-tolyl)acrylic acid 4a(1.0 mmol), ethyl bromodifluoroacetate 2 (3.0 mmol), 2,6-lutidine (2.0 mmol), 1,4-dioxane/EtOH (v/v 5:1, 6.7 mL, 0.15 M), blue LEDs (3.12 W), room temperature, argon atmosphere.
Yield and E/Z values are determined with 19F-NMR using α,α,α-trifluorotoluene as internal standard.
fac-Ir(tBuppy)3 was used as the photocatalyst.
Reported yields are those of isolated compounds; E/Z values are determined by 1H NMR of isolated products.
Table 4. Decarboxylative difluoromethylation: Scope of Ortho- and β-Substituted Cinnamic Acids in Batcha or Continuous-Flowb,c.

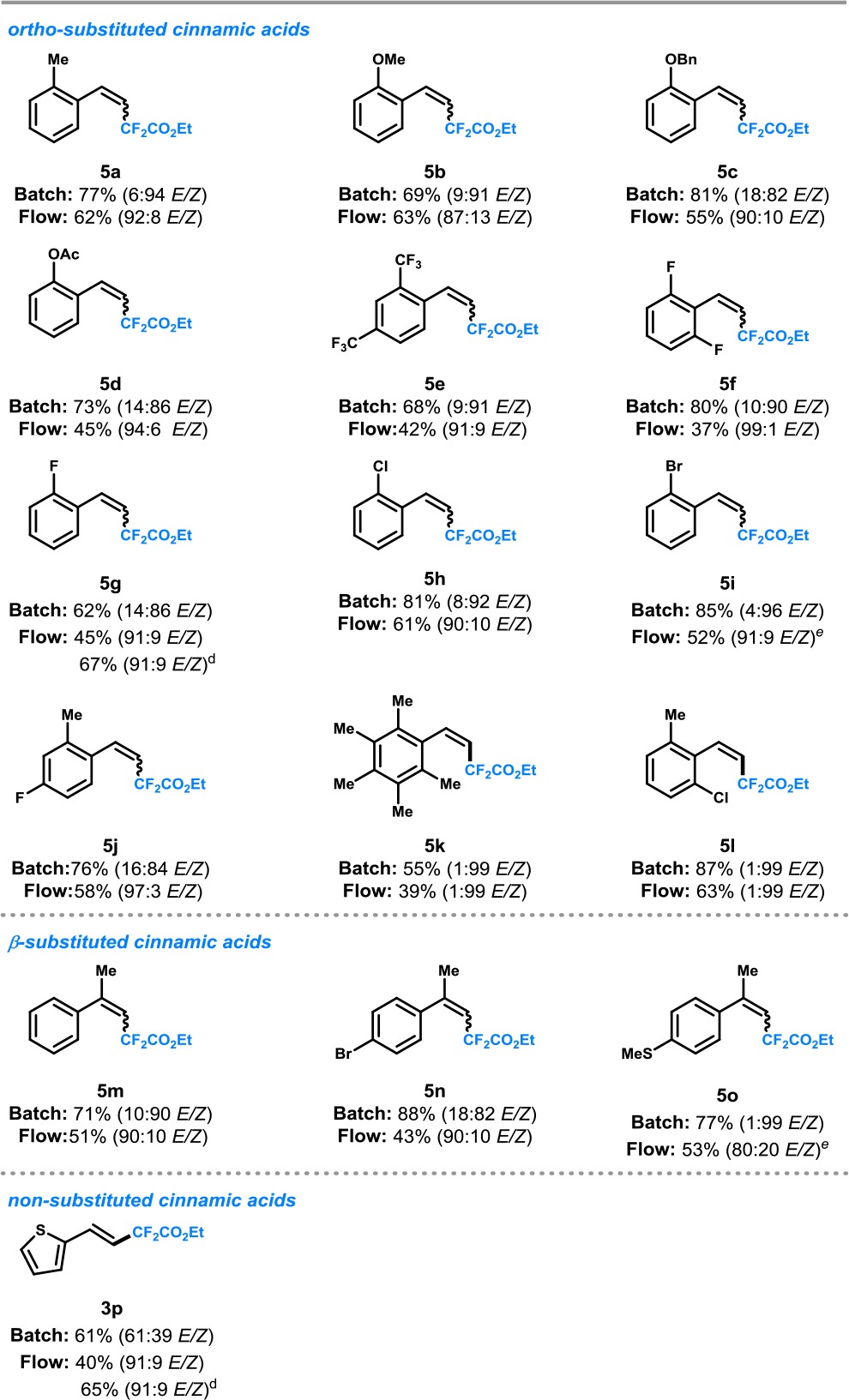
Reaction conditions in batch: fac-Ir(ppy)3 (3 mol %), o-cinnamic acid 4 (0.2 mmol), ethyl bromodifluoroacetate 2 (0.6 mmol), NaHCO3 (0.4 mmol), H2O(3.0 mmol), 1,4-dioxane (0.4 mL, 0.5 M), blue LEDs (3.12 W), room temperature, argon atmosphere, stirred for 24 h.
Reaction conditions in continuous flow: fac-Ir(ppy)3 (1 mol %), o-cinnamic acid 4 (1.0 mmol), ethyl bromodifluoroacetate 2 (3.0 mmol), 2,6-lutidine (2.0 mmol), 1,4-dioxane/EtOH (v/v 5:1, 6.7 mL, 0.15 M), blue LEDs (3.12 W), room temperature, argon atmosphere, residence time: 15 min.
Reported yields are those of isolated compounds; E/Z values are determined by 1H NMR of isolated products.
Yield based on one time starting material recycle.
10 min residence time.
Next, a diverse set of ortho-substituted cinnamic acids were examined in both batch and flow (Table 4). Cinnamic acids bearing electron-neutral (5a), electron-donating (5b–d) and electron-withdrawing substituents (5e) could be difluoromethylated in high Z-selectivity in batch, while the corresponding E-isomer could be readily accessed via continuous-flow processing. Also, o-halogenated cinnamic acids were competent substrates (5f–i). Enhanced selectivity for the Z-isomer was observed with increasing steric bulk (F < Cl < Br). Interestingly, when both ortho positions were occupied with bulky groups (e.g., 5k,l), a high Z-selectivity was observed which could not be revoked via continuous-flow processing. This observation highlights the need for sterical bulk in the ortho-position to access the Z-stereoisomer. Furthermore, in the case of β-methyl-substituted cinnamic acids, a similar trend in the selectivity was observed due to the steric effect of the β-substituent (5m–o). Finally, substrates with a low E-selectivity in batch (e.g., substrate 3p) could be obtained in flow with an improved stereoselectivity.
To further demonstrate the utility of our protocol, we sought to demonstrate its potential for the decarboxylative difluoromethylation of aryl propiolic acids. A small tweak of the reaction conditions (i.e., CsOAc as a base) resulted in the formation of the desired compounds in modest but synthetically useful yields (Table 5). Our protocol was successfully applied to ortho-, meta-, and para-substituted aryl propiolic acids (7a–l). These findings are noteworthy because, to the best of our knowledge, this is the first time that propiolic acids are used as substrates for photocatalytic decarboxylation chemistry.6a,6b
Table 5. Decarboxylative Difluoromethylation of Aryl Propiolic Acidsa,b.
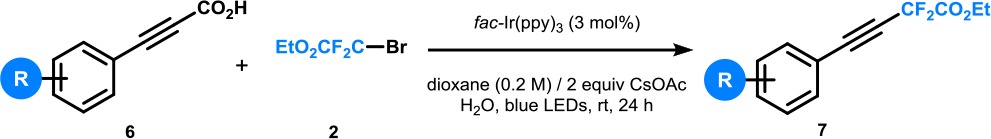
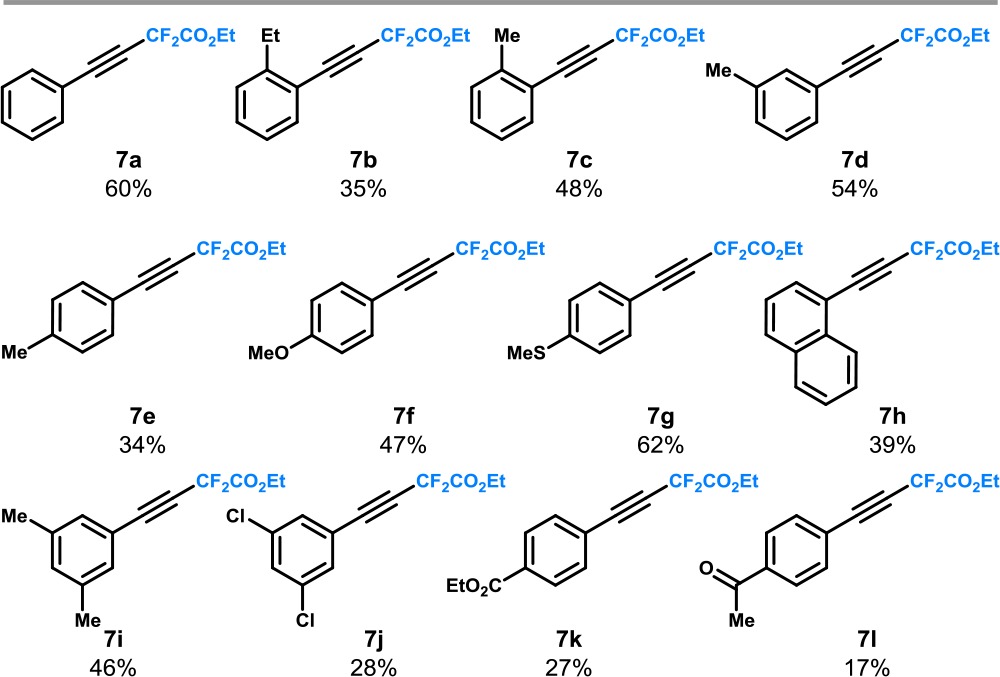
Reaction conditions: aryl propiolic acid 6 (0.2 mmol, 1.0 equiv), ethyl bromodifluoroacetate 2 (0.6 mmol, 3.0 equiv), fac-Ir(ppy)3 (3 mol %), CsOAc (0.4 mmol, 2.0 equiv), H2O(2 mmol, 10 equiv), 1,4-dioxane (1.0 mL), argon, blue LEDs (3.12 W), 24 h.
Reported yields are those of isolated compounds.
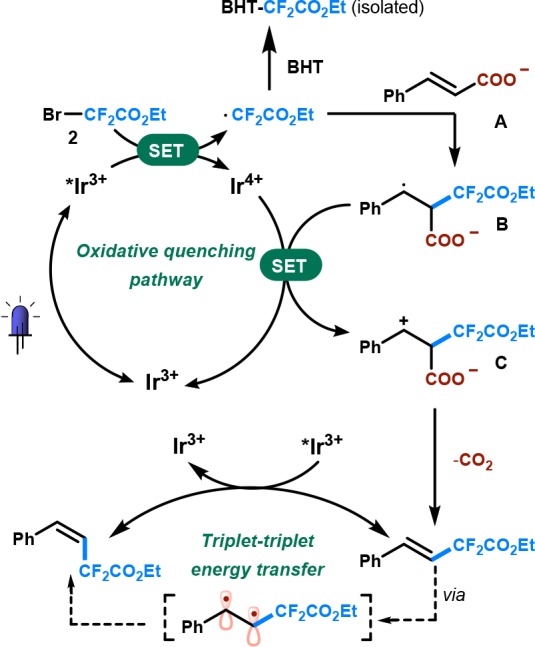
Based on the above results, we suggest a plausible mechanism for the developed transformation as outlined in Scheme 2. Photoexcitation of fac-Ir(ppy)3 upon blue irradiation results in a metal-to-ligand charge transfer (MLCT) excited state. Photoluminescence quenching experiments showed that the photoexcited fac-[Ir3+(ppy)3]* (E1/2red [*Ir3+/Ir4+] = −1.72 V vs SCE) can be quenched by 2 (Ered = −0.57 V vs SCE)12c at a rate constant of 1.84 × 108 M–1 s–1. Our investigations also showed that the excited photocatalyst can be quenched by cinnamate A though at a lower rate constant of 1.24 × 108 M–1 s–1. However, radical trapping experiments showed that only the key intermediate •CF2CO2Et could be captured by BHT (2,6-di-tert-butyl-4-methylphenol) suggesting the feasibility of this oxidative quenching pathway. Next, intermolecular π-addition of the radical to A produces the benzylic radical B. Single-electron transfer from radical B to fac-[Ir(ppy)3]+ affords carbocation C and facilitates subsequent CO2 extrusion to yield the E stereoisomer. The formation of E isomer as both the thermodynamically and kinetically preferred isomer is supported by kinetic experiments (see the Supporting Information). The Z isomer is subsequently formed due to a triplet–triplet energy transfer process with fac-[Ir3+(ppy)3]* (τ0 = 1.9 μs5a and ET = 2.41 eV12a,12b).
Scheme 2. Proposed Catalytic Cycle for the Decarboxylative Difluoromethylation.
In this work, we have introduced a simple yet effective photocatalytic decarboxylative protocol to prepare difluoromethylated styrenes and phenylacetylenes. In contrast to previously described methods, this procedure does not require additional metal catalysts or hypervalent iodine reagents to facilitate CO2 extrusion. The generality of our protocol is demonstrated by the broad substrate scope (difluoromethylated styrenes, 31 E-selective examples and 15 Z-selective; difluoromethylated phenylacetylenes, 12 examples). Ortho-substituted cinnamic acids give the less stable Z-selective products. The thermodynamically favored E-stereoisomer could be readily obtained in continuous-flow through accurate control of the reaction time. Having access to both stereoisomers simply by changing the reactor is a unique and powerful approach and provides opportunities for other photocatalytic transformations.
Acknowledgments
X.J.W. and T.N. acknowledge the European Union for a Marie Curie ITN Grant (Photo4Future, Grant No. 641861). We also acknowledge the Dutch Science Foundation (NWO) for a VIDI grant for T.N. (SensPhotoFlow, No. 14150). We thank Ir. Koen P. L. Kuijpers and Cecilia Bottecchia for their help with the mechanistic part of this manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.7b03019.
Experimental procedures, mechanistic studies, and NMR spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Zhou Y.; Wang J.; Gu Z.; Wang S.; Zhu W.; Acena J. L.; Soloshonok V. A.; Izawa K.; Liu H. Chem. Rev. 2016, 116, 422–518. 10.1021/acs.chemrev.5b00392. [DOI] [PubMed] [Google Scholar]; b Yerien D. E.; Bonesi S.; Postigo A. Org. Biomol. Chem. 2016, 14, 8398–8427. 10.1039/C6OB00764C. [DOI] [PubMed] [Google Scholar]; c Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. J. Med. Chem. 2015, 58, 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]; d Purser S.; Moore P. R.; Swallow S.; Gouverneur V. Chem. Soc. Rev. 2008, 37, 320–330. 10.1039/B610213C. [DOI] [PubMed] [Google Scholar]; e Mueller K.; Faeh C.; Diederich F. Science 2007, 317, 1881–1886. 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- a Alonso C.; Martínez de Marigorta E.; Rubiales G.; Palacios F. Chem. Rev. 2015, 115, 1847–1935. 10.1021/cr500368h. [DOI] [PubMed] [Google Scholar]; b Lu Y.; Liu C.; Chen Q.-Y. Curr. Org. Chem. 2015, 19, 1638–1650. 10.2174/1385272819666150615235605. [DOI] [Google Scholar]; c Merino E.; Nevado C. Chem. Soc. Rev. 2014, 43, 6598–6608. 10.1039/C4CS00025K. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Furuya T.; Kamlet A. S.; Ritter T. Nature 2011, 473, 470–477. 10.1038/nature10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Meanwell N. A. J. Med. Chem. 2011, 54, 2529–2591. 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]; b Erickson J. A.; McLoughlin J. I. J. Org. Chem. 1995, 60, 1626–1631. 10.1021/jo00111a021. [DOI] [Google Scholar]
- Chatterjee T.; Iqbal N.; You Y.; Cho E. J. Acc. Chem. Res. 2016, 49, 2284–2294. 10.1021/acs.accounts.6b00248. [DOI] [PubMed] [Google Scholar]
- a Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lu Z.; Yoon T. P. Angew. Chem., Int. Ed. 2012, 51, 10329–10332. 10.1002/anie.201204835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wei Y.; Hu P.; Zhang M.; Su W. Chem. Rev. 2017, 117, 8864–8907. 10.1021/acs.chemrev.6b00516. [DOI] [PubMed] [Google Scholar]; b Jin Y.; Fu H. Asian J. Org. Chem. 2017, 6, 368–385. 10.1002/ajoc.201600513. [DOI] [Google Scholar]; c Xuan J.; Zhang Z.-G.; Xiao W.-J. Angew. Chem., Int. Ed. 2015, 54, 15632–15641. 10.1002/anie.201505731. [DOI] [PubMed] [Google Scholar]; d Rodriguez N.; Goossen L. J. Chem. Soc. Rev. 2011, 40, 5030–5048. 10.1039/c1cs15093f. [DOI] [PubMed] [Google Scholar]
- For selected papers on metal-catalyzed and -mediated decarboxylative synthesis of difluoromethylated olefins, see:; a Li G.; Wang T.; Fei F.; Su Y.-M.; Li Y.; Lan Q.; Wang X.-S. Angew. Chem., Int. Ed. 2016, 55, 3491–3495. 10.1002/anie.201511321. [DOI] [PubMed] [Google Scholar]; b Chen Q.; Wang C.; Zhou J.; Wang Y.; Xu Z.; Wang R. J. Org. Chem. 2016, 81, 2639–2645. 10.1021/acs.joc.6b00031. [DOI] [PubMed] [Google Scholar]; c Li G.; Cao Y.-X.; Luo C.-G.; Su Y.-M.; Li Y.; Lan Q.; Wang X.-S. Org. Lett. 2016, 18, 4806–4809. 10.1021/acs.orglett.6b02216. [DOI] [PubMed] [Google Scholar]; d Pannecoucke X.; Poisson T. Synlett 2016, 27, 2314–2326. 10.1055/s-0035-1562784. [DOI] [Google Scholar]; e Li Z.; Cui Z.; Liu Z.-Q. Org. Lett. 2013, 15, 406–409. 10.1021/ol3034059. [DOI] [PubMed] [Google Scholar]; f He Z.; Luo T.; Hu M.; Cao Y.; Hu J. Angew. Chem., Int. Ed. 2012, 51, 3944–3947. 10.1002/anie.201200140. [DOI] [PubMed] [Google Scholar]
- For selected papers on photocatalytic decarboxylative perfluoroalkylation of olefins, see:; a Zhang H.-R.; Chen D.-Q.; Han Y.-P.; Qiu Y.-F.; Jin D.-P.; Liu X.-Y. Chem. Commun. 2016, 52, 11827–11830. 10.1039/C6CC06284A. [DOI] [PubMed] [Google Scholar]; b Xu P.; Abdukader A.; Hu K.; Cheng Y.; Zhu C. Chem. Commun. 2014, 50, 2308–2310. 10.1039/C3CC48598F. [DOI] [PubMed] [Google Scholar]
- For hypervalent iodine-assisted decarboxylation, see:; Huang H.; Jia K.; Chen Y. ACS Catal. 2016, 6, 4983–4988. 10.1021/acscatal.6b01379. [DOI] [Google Scholar]; b Huang H.; Jia K.; Chen Y. Angew. Chem., Int. Ed. 2015, 54, 1881–1884. 10.1002/anie.201410176. [DOI] [PubMed] [Google Scholar]; c Huang H.; Zhang G.; Chen Y. Angew. Chem., Int. Ed. 2015, 54, 7872–7876. 10.1002/anie.201502369. [DOI] [PubMed] [Google Scholar]
- While the Z isomer is thermodynamically favored, high Z selectivities remain challenging via photoredox catalysis; see:; a Straathof N. J. W.; Cramer S. E.; Hessel V.; Noel T. Angew. Chem., Int. Ed. 2016, 55, 15549–15553. 10.1002/anie.201608297. [DOI] [PubMed] [Google Scholar]; b Honeker R.; Garza-Sanchez R. A.; Hopkinson M. N.; Glorius F. Chem. - Eur. J. 2016, 22, 4395–4399. 10.1002/chem.201600190. [DOI] [PubMed] [Google Scholar]; c Roh G.-b.; Iqbal N.; Cho E. J. Chin. J. Chem. 2016, 34, 459–464. 10.1002/cjoc.201500919. [DOI] [Google Scholar]; d Yu C.; Iqbal N.; Park S.; Cho E. J. Chem. Commun. 2014, 50, 12884–12887. 10.1039/C4CC05467A. [DOI] [PubMed] [Google Scholar]; e Iqbal N.; Jung J.; Park S.; Cho E. J. Angew. Chem., Int. Ed. 2014, 53, 539–542. 10.1002/anie.201308735. [DOI] [PubMed] [Google Scholar]; f Wei X.-J.; Yang D.-T.; Wang L.; Song T.; Wu L.-Z.; Liu Q. Org. Lett. 2013, 15, 6054–6057. 10.1021/ol402954t. [DOI] [PubMed] [Google Scholar]; g Nguyen J. D.; D’Amato E. M.; Narayanam J. M. R.; Stephenson C. R. J. Nat. Chem. 2012, 4, 854–859. 10.1038/nchem.1452. [DOI] [PubMed] [Google Scholar]
- For photocatalytic Z-selective alkene synthesis, see:; a Singh A.; Fennell C. J.; Weaver J. D. Chem. Sci. 2016, 7, 6796–6802. 10.1039/C6SC02422J. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Metternich J. B.; Gilmour R. J. Am. Chem. Soc. 2016, 138, 1040–1045. 10.1021/jacs.5b12081. [DOI] [PubMed] [Google Scholar]; c Fabry D. C.; Ronge M. A.; Rueping M. Chem. - Eur. J. 2015, 21, 5350–5354. 10.1002/chem.201406653. [DOI] [PubMed] [Google Scholar]; d Metternich J. B.; Gilmour R. J. Am. Chem. Soc. 2015, 137, 11254–11257. 10.1021/jacs.5b07136. [DOI] [PubMed] [Google Scholar]; e Singh K.; Staig S. J.; Weaver J. D. J. Am. Chem. Soc. 2014, 136, 5275–5278. 10.1021/ja5019749. [DOI] [PubMed] [Google Scholar]; f Lin Q.-Y.; Xu X.-H.; Qing F.-L. J. Org. Chem. 2014, 79, 10434–10446. 10.1021/jo502040t. [DOI] [PubMed] [Google Scholar]; For a recent review on this topic, see:; Metternich J. B.; Gilmour R. Synlett 2016, 27, 2541–2552. 10.1055/s-0036-1588621. [DOI] [Google Scholar]
- a Flamigni L.; Barbieri A.; Sabatini C.; Ventura B.; Barigelletti F. Top. Curr. Chem. 2007, 281, 143–203. 10.1007/128_2007_131. [DOI] [Google Scholar]; b Chen F.-C.; He G.; Yang Y. Appl. Phys. Lett. 2003, 82, 1006–1008. 10.1063/1.1544658. [DOI] [Google Scholar]; c Jung J.; Kim E.; You Y.; Cho E. J. Adv. Synth. Catal. 2014, 356, 2741–2748. 10.1002/adsc.201400542. [DOI] [Google Scholar]
- For selected reviews describing the benefits of flow on photochemistry, see:; a Photochemical processes in continuous-flow reactors; Noel T., Ed.; World Scientific Publishing: London, 2017; 271 pp. [Google Scholar]; b Cambié D.; Bottecchia C.; Straathof N. J. W.; Hessel V.; Noel T. Chem. Rev. 2016, 116, 10276–10341. 10.1021/acs.chemrev.5b00707. [DOI] [PubMed] [Google Scholar]; c Plutschack M. B.; Correia C. A.; Seeberger P. H.; Gilmore K. Top. Organomet. Chem. 2015, 57, 43–76. 10.1007/3418_2015_155. [DOI] [Google Scholar]
- Su Y.; Kuijpers K.; Koenig N.; Shang M.; Hessel V.; Noel T. Chem. - Eur. J. 2016, 22, 12295–12300. 10.1002/chem.201602596. [DOI] [PubMed] [Google Scholar]
- For enhanced selectivity in continuous-flow microreactors and the rationale behind it, see:; a Noel T.; Su Y.; Hessel V. Top. Organomet. Chem. 2015, 57, 1–42. 10.1007/3418_2015_152. [DOI] [Google Scholar]; b Hartman R. L.; McMullen J. P.; Jensen K. F. Angew. Chem., Int. Ed. 2011, 50, 7502–7519. 10.1002/anie.201004637. [DOI] [PubMed] [Google Scholar]; c Yoshida J.-i.; Nagaki A.; Iwasaki T.; Suga S. Chem. Eng. Technol. 2005, 28, 259–266. 10.1002/ceat.200407127. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.