

 Enantioselective palladium(0)-catalyzed C–H arylations of cyclopropanes provide efficient access to dihydroquinolones, dihydroisoquinolones and the BMS-791325 indolobenzazepine core.
Enantioselective palladium(0)-catalyzed C–H arylations of cyclopropanes provide efficient access to dihydroquinolones, dihydroisoquinolones and the BMS-791325 indolobenzazepine core.
Abstract
Taddol-based phosphoramidite ligands enable enantioselective palladium(0)-catalyzed C–H arylation of cyclopropanes. The cyclized products are obtained in high yields and enantioselectivities. The reported method provides efficient access to a broad range of synthetically attractive cyclopropyl containing dihydroquinolones and dihydroisoquinolones as well as allows for an efficient enantioselective construction of the 7-membered ring of the cyclopropyl indolobenzazepine core of BMS-791325.
Introduction
Besides its occurrence in natural products,1 cyclopropane is a common structural motif in currently marketed drug or development candidates, often in conjunction with a nitrogen atom in close vicinity (Fig. 1).2 This prevalence can be attributed to its strategic use aiming to increase metabolic stability without a large increase in molecular weight or installation of fluorine atoms. In its own right, cyclopropane is an attractive scaffold providing opportunities to arrange pendant groups in a rigid and specific three-dimensional orientation in space.
Fig. 1. Marketed drugs and development candidates having an annulated cyclopropyl ring system.

A broad range of methods for the construction of cyclopropane rings have been reported and are frequently used.3 However, the direct functionalization of an existing cyclopropane group as a complementary strategy remains underdeveloped. These comprise mainly stoichiometric metalations with Grignard reagent or organo-lithium bases.4 Sparteine could be used as chiral modifier for enantioselective deprotonations.5 Some transition-metal catalyzed cyclopropane C–H functionalizations have been reported.6,7 However, related enantioselective reactions are very scarce.8,9 Yu reported directed palladium(ii)-catalyzed processes for enantioselective C–H arylations of cyclopropyl carboxamides8b and cyclopropylmethylamines.8c We have reported enantioselective C–H functionalization of N-cyclopropylmethyl trifluoromethanesulfonamides to access tetrahydroquinolines.8a This initial proof of concept prompted us to exploit this reaction principle further with the aim to develop a rapid access to synthetically versatile chiral building blocks (Scheme 1). We have focused on implementing amides as connectors,7e,g,10 which would not only significantly simplify the access to the required substrates 1 and 3, but also largely enhance the utility of the arising products. Moreover, different ring sizes and positions of the nitrogen heteroatom were envisioned. Herein we report an enantioselective Pd0-catalyzed C–H arylation strategy enabling the access to chiral cyclopropylquinolones 2, cyclopropylisoquinolones 4 and also cyclopropylindolobenzazepines.
Scheme 1. Rapid enantioselective assembly of dihydroquinolones and dihydroisoquinolones by Pd0-catalyzed C–H functionalization.

Results and discussion
Enantioselective synthesis of cyclopropane containing dihydroquinolones
Dihydroquinolones are common structures among natural products and bioactive compounds.11 Cyclopropyl substituted congeners are not oxidizable to the corresponding aromatic quinolones. A rapid C–H functionalization access to this compound class would represent a significant advantage compared to the N-triflyl tetrahydroquinoline product range.8a Although the N-triflyl group was removable under strongly reducing conditions, it represented a limiting factor. Moreover, the envisioned substrates would be readily accessible via an amide bond forming reaction from simple 2-bromoanilines and a cyclopropyl carboxylic acid building block. In addition, the scope would be significantly broadened by having the additional flexibility of a second substituent R2 on the amide nitrogen atom. In this case, the interference of R2 with the regio- and enantioselectivity of the C–H activation is an unknown variable. Especially the potential of arene containing groups competing with the cyclopropane C–H bonds for the activation has to be considered. Therefore, we aimed also to explore these selectivities and derive some guidelines. To our delight, model substrate 1a could be selectively activated and cyclized to dihydroquinolone 2a in 95% yield with a 98 : 2 er employing L2 and a similar set of conditions as previously developed for the N-triflyl tetrahydroquinolines (Scheme 2). This finding underscores the robustness and reliability of the Taddol phosphoramidites for Pd0-catalyzed asymmetric activations.12
Scheme 2. Enantioselective dihydroquinolone synthesis.

Concerning the scope of the reaction, we found that the size of the alkyl chain of the amide substituent R2 did not influence the reaction outcome and dihydroquinolinones 2d–2g were all obtained in high yields and selectivities (Scheme 3). However, a secondary amide substrate (1h) did not cyclize and was almost fully recovered after the reaction. This suggests that the free N–H locks the molecule in an unfavorable conformation for the C–H bond activation. A range of different substituents R1 are tolerated on the cyclopropane ring. For example, methyl, benzyl and phenyl groups have negligible influence on the reaction outcome and provide dihydroquinolones 2b–2d decorated with different quaternary stereocenters. Trimethylsilyl substituted cyclopropane 1o afforded dihydroquinolinone 2o in low yield and moderate selectivity. Spirooxindole 5 was formed as the major product presumably by an intramolecular Hiyama–Denmark coupling. Substrates 1i–1m having R3 substituents on the aniline moiety gave the corresponding dihydroquinolinones with the same efficiency. However, a pyridyl substrate (1n) with the bromide group in the ortho-position reacted very sluggishly. Of note, an aromatic chloride substituent (1m) was tolerated by the employed palladium catalyst allowing further orthogonal functionalizations of 2m by cross-coupling chemistry.
Scheme 3. Scope of the enantioselective cyclopropanecarboxamide cyclization. Reaction conditions: 1 (0.10 mmol), Cs2CO3 (0.15 mmol), Pd(dba)2 (2.00 μmol), L2 (4.00 μmol), PivOH (30 μmol), 0.30 M in mesitylene at 130 °C for 12 h. Yields of isolated products 2; er's determined by HPLC with a chiral stationary phase. a [(η3-cinnamyl)Pd(Cp)] instead of Pd(dba)2. b With Cs2CO3 (0.20 mmol), Pd(dba)2 (10.0 μmol), L2 (20.0 μmol), PivOH (50 μmol).

Substrate 1f having a p-methoxybenzyl (PMB) protecting group as the R1 substituent on the nitrogen atom gave exclusively the cyclopropane activation product 2f (Scheme 4). This is very remarkable as activation of the C(sp2)–H group of the PMB group would also proceed by the same sized 7-membered palladacycle (6 vs. 7). Generally the activation of arene C–H groups is believed to proceed more easily, making the result unexpected. One might explain this observation by a conformational bias of the amide tether favoring cyclopropane C(sp3)–H activation for this substrate class.
Scheme 4. Cyclopropane C(sp3)–H activation over arene C(sp2)–H activation in 1f. Conditions: 2.0 mol% [(η3-cinnamyl)Pd(Cp)], 4.0 mol% L2, 0.3 equiv. PivOH (30 μmol), 1.5 equiv. Cs2CO3, mesitylene, 130 °C, 12 h.

When substrate 1p possessing two aryl bromide groups was subjected to the reaction conditions, we obtained selectively pentacyclic dihydroquinolinone 2p in excellent yield and enantioselectivity (Scheme 5). To the best of our knowledge, this represents the only example of a tandem C–H arylation process involving an enantioselective step.
Scheme 5. Enantioselective double C–H arylation process.

As secondary amides fail to undergo the enantioselective cyclization, we aimed to access this product type by deprotection of 2f (Scheme 6). The PMB group of 2f could be efficiently removed with TFA using anisole as scavenger, giving amide 2h in 90% yield without ring-opened byproducts of the donor–acceptor cyclopropane moiety. Finally, the absolute configuration of dihydroquinolone 2c was established by the optical rotation of its corresponding amine 9. 9 was independently prepared from 10, whose absolute configuration had been previously established by X-ray crystallographic analysis.8a Both have a comparable optical rotation (+307 prepared from 10 and +303 prepared from 2f), thus the same absolute configuration was attributed to all lactams 2.
Scheme 6. PMB group cleavage and determination of the absolute configuration.

Enantioselective C–H functionalization of aminocyclopropanes
The dihydroquinolones 2 possess an electron-rich aromatic ring. Consequently, such compounds are vulnerable towards metabolic oxidation at the para-position with respect to the nitrogen atom. Structurally related dihydroisoquinolones 4 having an inverted amide structure (Scheme 7) are valuable building blocks13 and possess an electron-poorer and more stable arene core. Moreover, the required aminocyclopropane precursors for the substrate synthesis are conveniently accessible by Kulinkovich–Szymoniak reactions.14 Therefore, a similar enantioselective C–H functionalization access to the cyclopropane bearing dihydroisoquinolones 4 would be of high value. However, such aminocyclopropanes behave very differently in Pd(0)-catalyzed C–H bond functionalization reactions, leading to a destruction of the valuable cyclopropane moiety by C–C bond cleavage (Scheme 7). For instance, Fagnou reported the conversion of aminocyclopropane 11 into dihydroquinoline 13.7i Along the same lines, Charette found that 3a is cyclized to a mixture of dihydrobenzapepinones 14.7g These reports point towards the strong influence of the amino group on the ring opening. Interestingly, Charette observed some amounts of the intact cyclopropane product under different conditions. Futhermore, Fagnou reported that an independently synthesized cyclopropane product proved to be stable under the reaction conditions. These two findings made us confident that a carefully chosen ligand on the palladium might prevent the ring-opening pathway and would instead provide the desirable cyclization product with an intact cyclopropane moiety.
Scheme 7. Reports on cyclopropane ring-opening under Pd0/PdII catalysis.

To explore the influence of different phosphines ligands and reaction conditions on the product distribution, we chose unsubstituted aminocyclopropane 3a as a model substrate (Table 1). The bulky and electron-rich tBu3P leads almost exclusively to ring-opened products 14a and 14b which is in full agreement with the findings of Charette (entry 1). Tricyclohexylphosphine and triphenylphosphine mostly suppressed the ring-opening pathway, leading to a mixture of the desired dihydroisoquinolone 4a and spiro-cyclic product 15 arising from a methine C–H activation7e,f (entries 2 and 3). No ring-opened product was observed with xylyl ligand L2 providing 4a in 63% yield (entry 4). tButyl ligand L3 proved to be very unselective, giving a mixture of all four products (entry 5). Overall, there is a trend that less bulky ligands (PCy3 vs. tBu3P and L2 vs. L3) favor conservation of the cyclopropyl unit.
Table 1. Influence of the ligand on the product distribution a .
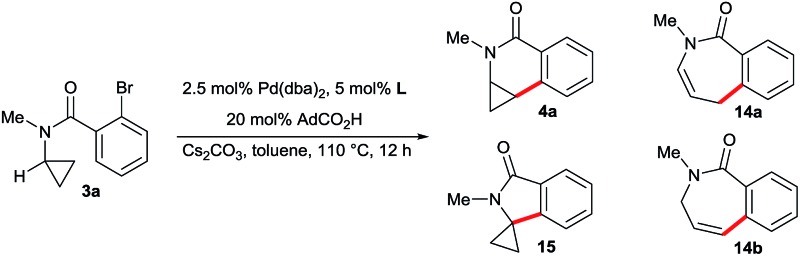
| |||||
| Entry | L | 4a b (%) | 15 b (%) | 14a b (%) | 14b b (%) |
| 1 | tBu3P | 4 | <1 | 59 | 21 |
| 2 | PCy3 | 58 | 30 | 8 | — |
| 3 | PPh3 | 40 | 40 | 11 | — |
| 4 | L2 | 63 | 33 | <1 | — |
| 5 | L3 | 12 | 40 | 28 | 8 |
aReaction conditions: 0.05 mmol 3a, 2.5 mol% Pd(dba)2, 5.0 mol% L, 20 mol% AdCO2H, 1.5 equiv. Cs2CO3 0.25 M in toluene, 110 °C, 12 h.
bDetermined by 1H-NMR with an internal standard.
With the above proven suitability of the Taddol phosphoramidites as ligands for the preservation of the cyclopropane moiety during the C–H functionalization process, we fine-tuned the catalyst composition for optimal enantioselectivity (Table 2). The parent ligand L1 is not competent, giving 4b only in trace amounts (entry 1). The already (for the above-described functionalization of amides 2) successful xylyl version L2 gave dihydroisoquinolone 4b in almost quantitative yield and very good enantioselectivity of 93.5 : 6.5 (entry 2). Ligands having larger aryl or amino groups are inferior (entries 3–5). As expected from the CMD mechanism,15 the carboxylic acid has a critical impact on the reaction outcome. In its absence, only traces of product are formed (entry 6). Acetic or xanthene carboxylic acid are detrimental for the enantioselectivity (entries 7 and 8) and pivalic acid gives a marginally reduced er (entry 9). Notably, the reaction works equally well using K2CO3 instead of Cs2CO3 (entry 10). The catalyst loading was reduced to 2.5 mol% Pd and 4b could be isolated in 93% yield with identical enantioselectivity (entry 11).
Table 2. Optimization of the enantioselective aminocyclopropane activation a .
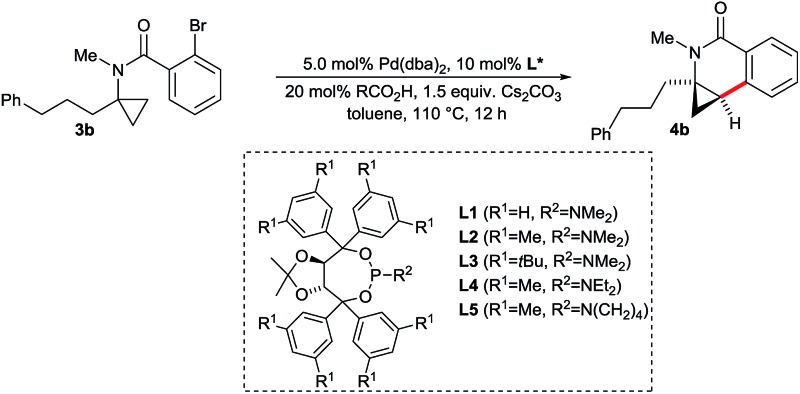
| ||||
| Entry | L* | RCO2H | % yield of 4b b | er c |
| 1 | L1 | AdCO2H | 2 | n.d. |
| 2 | L2 | AdCO2H | 99 | 93.5 : 6.5 |
| 3 | L3 | AdCO2H | 55 | 85.5 : 14.5 |
| 4 | L4 | AdCO2H | 94 | 90 : 10 |
| 5 | L5 | AdCO2H | 99 | 93 : 7 |
| 6 | L2 | — | 3 | n.d. |
| 7 | L2 | AcOH | 86 | 60 : 40 |
| 8 | L2 | XanthCO2H | 25 | 77 : 23 |
| 9 | L2 | PivOH | 92 | 92.5 : 7.5 |
| 10 d | L2 | AdCO2H | 99 | 93.5 : 6.5 |
| 11 e | L2 | AdCO 2 H | 96 (93) | 93.5 : 6.5 |
aReaction conditions: 0.05 mmol 3b, 5.0 mol% Pd(dba)2, 10 mol% L*, 20 mol% RCO2H, 1.5 equiv. Cs2CO3 0.1 M in toluene, 110 °C, 12 h.
bDetermined by 1H-NMR with an internal standard (isolated yield in parentheses).
cer values were determined by HPLC with a chiral stationary phase.
dWith K2CO3 instead of Cs2CO3.
eWith 2.5 mol% Pd(dba)2, 5.0 mol% L2, 0.25 M in toluene.
Next, we evaluated the scope for the aminocyclopropane activation and cyclization to dihydroisoquinolones with L2 under the optimized conditions (Scheme 8). First, a variety of amide substituents R2 were checked. C–H groups of methyl, alkyl and benzyl groups are neither activated nor do significantly interfere with the efficiency of the cyclopropane activation. For substrates having no additional substituent R1 on the cyclopropane ring, larger groups R2 reduce the amount of the spirocyclic by-product (4a, 4c and 4d). A secondary amide (3e) is not a competent substrate and the starting material was recovered. Presumably, the unfavourable conformation of a secondary amide16 precludes the activation of the cyclopropane. The reaction was then evaluated for tolerance of the cyclopropane substituent R1. A broad variety of groups with potentially activatable C–H bonds such as linear alkyl (4b), branched alkyl (4g), aryl (4h, 4i) and benzyl (4j) selectively react at the desired cyclopropane unit, maintaining excellent yields and high enantioselectivities. Moreover, aminocyclopropanes bearing common versatile functional groups such as ester (4k) or nitrile (4l) are compatible with the process. The substitution pattern R3 of the aryl portion can be varied as well, as exemplified by p-Cl (4m) and m-MeO (4n) groups. Heteroaryl bromides were also evaluated. In this respect, pyridyl containing substrate 3o performs very well. When running the reaction at 10-fold increased scale, the product 4o was isolated in 94% yield and 95.5 : 4.5 er. In contrast, thiophene substrate 3p proved to be more reluctant and required a higher reaction temperature resulting in a somewhat eroded yield and enantioselectivity of 4p.
Scheme 8. Scope of the enantioselective aminocyclopropane cyclization. Reaction conditions: 3 (0.10 mmol), Cs2CO3 (0.15 mmol), Pd(dba)2 (2.50 μmol), L2 (5.00 μmol), AdCO2H (20.0 μmol), 0.25 M in toluene at 110 °C for 12 h. Yields of isolated products 4; er's determined by HPLC with a chiral stationary phase. a With Pd(dba)2 (5.00 μmol) and L2 (10.0 μmol) for 12 h. b 24 h reaction. c With Pd(dba)2 (5.00 μmol) and L2 (10.0 μmol) in mesitylene at 130 °C for 12 h. d 1.0 mmol scale.

To provide cyclized products of secondary amides such as 4e which are not directly accessible, cleavage of the PMB group was targeted. It could be efficiently removed by heating the lactams in TFA with anisole as carbenium ion scavenger (Scheme 9). The secondary amides 4e and 4q were obtained in virtually quantitative yield. The p-chloro phenyl substituted lactam 4q gave single crystals suitable for X-ray crystallographic analysis,† unambiguously providing its absolute configuration. The same preference as previously observed for the dihydroquinolones 2 was found.
Scheme 9. Removal of the PMB group and X-ray crystal structure of product 4q.

An enantioselective C–H functionalization route for the BMS-791325 ring system
The attractive properties of the cyclopropane moiety as rigid17 and metabolically more stable surrogates of olefins18 made them sought after design elements in pharmaceuticals.2 In this respect, Bristol Myers Squibb's indolobenzazepine BMS-791325, a hepatitis C virus NS5B replicase inhibitor,2f is a prime example (Scheme 10). The replacement of the double bond by a cyclopropane removed a potentially reactive Michael acceptor. Moreover it introduced a conformational constraint across the indolobenzazepine ring system leading to productive interactions between the cyclopropyl group and the NS5B protein.19 Owing to its favorable pharmacological properties, BMS-791325 is undergoing clinical evaluation in conjunction with the NS5A inhibitor daclatasvir and the NS3/4A protease inhibitor asunaprevir. From the chemical point of view, the structure of BMS-791325 has interesting and noteworthy features. The highest concentration of molecular complexity resides in its chiral trisubstituted cyclopropane ring comprising also a quaternary stereogenic center. In addition, it is part of the saturated and largest ring system of the molecule. We hypothesized that the pentacyclic core of 15 might be accessed by an enantioselective intramolecular cyclopropane arylation of substrate 16 (Scheme 10). This would allow the use of an achiral indole substrate. In turn, the required heavily substituted indole 16 could be assembled by a Rh(iii)-catalyzed C–H functionalization from simple acetanilide 17 and alkyne 18. Such a consecutive C–H functionalization strategy represents a streamlined synthesis of 15 and allows also for the rapid synthesis of analogs.
Scheme 10. A consecutive C–H activation strategy for BMS-791325.

The synthesis started with a rhodium(iii)-catalyzed indole synthesis20 linking acetanilide 17 and alkyne 18 (Scheme 11). Both represent a highly challenging substrate combination. In consequence, the hindered nature of the alkyne in addition to regioselectivity issues (H vs. H′ activation and regioselectivity of the alkyne incorporation) required a modification of the original Fagnou conditions.20b A viable solution was found using the 1,3-di-tert-butyl cyclopentadienyl (Cpt)21 instead of Cp* in combination with catalytic amounts of copper(ii) acetate and oxygen as terminal oxidant. This system provided indole 19 in 68% isolated yield without any H′ activation and an alkyne regioselectivity of 15 : 1 in favor of 19. Subsequently, the N-acetyl group of the indole was cleaved under acidic conditions. The installation of the required hindered cyclopropylcarboxylate moiety was efficiently achieved with chloro-substituted acrylate 20.22 In the presence of cesium carbonate in DMF, a smooth conjugate addition/intramolecular electrophilic trapping provides the desired C–H activation substrate 16 in excellent yield at ambient temperature. The hypothesized enantioselective cyclopropane activation was subsequently tested with our above described catalyst system. Pleasingly, the cyclization efficiently took place, providing ent-15 in 73% yield and an enantiomeric ratio of 92.5 : 7.5 when L2 was used. We found that the selectivity could be further enhanced using Taddol phosphonite L6, giving ent-15 in 80% yield and 94.5 : 5.5 er.‡ To the best of our knowledge, this transformation represents the first example of enantioselective Pd(0)-catalyzed C(sp3)–H arylation to form a seven-membered ring. Subsequently, we could show that the ethyl esters can be differentiated. The ester adjacent to the cyclopropane moiety was selectively saponified employing Bu4POH.
Scheme 11. Enantioselective synthesis of the BMS-791325 ring system.

Conclusions
In summary, we have reported an enantioselective palladium(0)-catalyzed C–H arylative cyclization strategy of cyclopropanes. The method enables rapid access to a broad range of versatile dihydroquinolone and dihydroisoquinolone building blocks in high yields and enantioselectivities using simple Taddol-based phosphoramidite ligands. Notably, in contrast to previous work, the cyclopropane group remains intact during the functionalization of aminocyclopropane substrates. Moreover, the method is not limited to the construction of 6-membered ring systems and was successfully applied for the efficient enantioselective construction of the 7-membered ring of the cyclopropyl indolobenzazepine core of BMS-791325.
Acknowledgments
This work is supported by the European Research Council under the European Community's Seventh Framework Program (FP7 2007–2013)/ERC Grant agreement no 257891 and the Swiss National Science Foundation (no. 155967). We thank Dr R. Scopelliti for X-ray crystallographic analysis of compound 4q.
Footnotes
‡The absolute configuration was determined based on the optical rotation of a reported derivative, see ESI† for details.
References
- (a) de Meijere A., Kozhushkov S. I., Schill H. Chem. Rev. 2006;106:4926. doi: 10.1021/cr0505369. [DOI] [PubMed] [Google Scholar]; (b) Gnad F., Reiser O. Chem. Rev. 2003;103:1603. doi: 10.1021/cr010015v. [DOI] [PubMed] [Google Scholar]
- (a) Gootz T. D., Zaniewski R., Haskell S., Schmieder B., Tankovic J., Girard D., Courvalin P., Polzer R. J. Antimicrob. Agents Chemother. 2006;40:2691. doi: 10.1128/aac.40.12.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Augeri D. J., Robl J. A., Betebenner D. A., Magnin D. R., Khanna A., Robertson J. G., Wang A., Simpkins L. M., Taunk P., Huang Q., Han S.-P., Abboa-Offei B., Cap M., Xin L., Tao L., Tozzo E., Welzel G. E., Egan D. M., Marcinkeviciene J., Chang S. Y., Biller S. A., Kirby M. S., Parker R. A., Hamann L. G. J. Med. Chem. 2005;48:5025. doi: 10.1021/jm050261p. [DOI] [PubMed] [Google Scholar]; (c) Micheli F., Cavanni P., Andreotti D., Arban R., Benedetti R., Bertani B., Bettati M., Bettelini L., Bonanomi G., Braggio S., Carletti R., Checchia A., Corsi M., Fazzolari E., Fontana S., Marchioro C., Merlo-Pich E., Negri M., Oliosi B., Ratti E., Read K. D., Roscic M., Sartori I., Spada S., Tedesco G., Tarsi L., Terreni S., Visentini F., Zocchi A., Zonzini L. J. Med. Chem. 2000;53:4989. doi: 10.1021/jm100481d. [DOI] [PubMed] [Google Scholar]; (d) Krattenmacher R. Contraception. 2000;62:29. doi: 10.1016/s0010-7824(00)00133-5. [DOI] [PubMed] [Google Scholar]; (e) Lin T. I., Lenz O., Fanning G., Verbinnen T., Delouvroy F., Scholliers A., Vermeiren K., Rosenquist A., Edlund M., Samuelsson B., Vrang L., De Kock H., Wigerinck P., Raboisson P., Simmen K. Antimicrob. Agents Chemother. 2009;53:1377. doi: 10.1128/AAC.01058-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Gentles R. G., Ding M., Bender J. A., Bergstrom C. P., Grant-Young K., Hewawasam P., Hudyma T., Martin S., Nickel A., Regueiro-Ren A., Tu Y., Yang Z., Yeung K.-S., Zheng X., Chao S., Sun J.-H., Beno B. R., Camac D. M., Chang C.-H., Gao M., Morin P. E., Sheriff S., Tredup J., Wan J., Witmer M. R., Xie D., Hanumegowda U., Knipe J., Mosure K., Santone K. S., Parker D. D., Zhuo X., Lemm J., Liu M., Pelosi L., Rigat K., Voss S., Wang Y., Wang Y.-K., Colonno R. J., Gao M., Roberts S. B., Gao Q., Ng A., Meanwell N. A., Kadow J. F. J. Med. Chem. 2014;57:1855. doi: 10.1021/jm4016894. [DOI] [PubMed] [Google Scholar]
- (a) Lebel H., Marcoux J.-F., Molinaro C., Charette A. B. Chem. Rev. 2003;103:977. doi: 10.1021/cr010007e. [DOI] [PubMed] [Google Scholar]; (b) Pellissier H. Tetrahedron. 2008;64:7041. [Google Scholar]
- (a) Eaton P. E., Lee C.-H., Xiong Y. J. Am. Chem. Soc. 1989;111:8016. [Google Scholar]; (b) Zhang M.-X., Eaton P. E. Angew. Chem., Int. Ed. 2002;41:2169. [PubMed] [Google Scholar]
- Lauru S., Simpkins N. S., Gethin D., Wilson C. Chem. Commun. 2008:5390. doi: 10.1039/b810441g. [DOI] [PubMed] [Google Scholar]
- For selected recent reviews on C–H functionalization: ; (a) Chen X., Engle K. M., Wang D.-H., Yu J.-Q. Angew. Chem., Int. Ed. 2009;48:5094. doi: 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Colby D. A., Bergman R. G., Ellman J. A. Chem. Rev. 2010;110:624. doi: 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lyons T. W., Sanford M. S. Chem. Rev. 2010;110:1147. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Le Bras J., Muzart J. Chem. Rev. 2011;111:1170. doi: 10.1021/cr100209d. [DOI] [PubMed] [Google Scholar]; (e) McMurray L., O'Hara F., Gaunt M. J. Chem. Soc. Rev. 2011;40:1885. doi: 10.1039/c1cs15013h. [DOI] [PubMed] [Google Scholar]; (f) Gutekunst W. R., Baran P. S. Chem. Soc. Rev. 2011;40:1976. doi: 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]; (g) Wencel-Delord J., Droge T., Liu F., Glorius F. Chem. Soc. Rev. 2011;40:4740. doi: 10.1039/c1cs15083a. [DOI] [PubMed] [Google Scholar]; (h) Cho S. H., Kim J. Y., Kwak J., Chang S. Chem. Soc. Rev. 2011;40:5068. doi: 10.1039/c1cs15082k. [DOI] [PubMed] [Google Scholar]; (i) Liu C., Zhang H., Shi W., Lei A. Chem. Rev. 2011;111:1780. doi: 10.1021/cr100379j. [DOI] [PubMed] [Google Scholar]; (j) Yamaguchi J., Yamaguchi A. D., Itami K. Angew. Chem., Int. Ed. 2012;51:8960. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]; (k) Wencel-Delord J., Glorius F. Nat. Chem. 2013;5:369. doi: 10.1038/nchem.1607. [DOI] [PubMed] [Google Scholar]; (l) Rouquet G., Chatani N. Angew. Chem., Int. Ed. 2013;52:11726. doi: 10.1002/anie.201301451. [DOI] [PubMed] [Google Scholar]; (m) Kuhl N., Schröder N., Glorius F. Adv. Synth. Catal. 2014;356:1443. [Google Scholar]; (n) De Sarkar S., Liu W., Kozhushkov S. I., Ackermann L. Adv. Synth. Catal. 2014;356:1461. [Google Scholar]; (o) Segawa Y., Maekawa T., Itami K. Angew. Chem., Int. Ed. 2015;54:66. doi: 10.1002/anie.201403729. [DOI] [PubMed] [Google Scholar]; (p) Daugulis O., Roane J., Tran L. D. Acc. Chem. Res. 2015;48:1053. doi: 10.1021/ar5004626. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Yang L., Huang H. Chem. Rev. 2015;115:3468. doi: 10.1021/cr500610p. [DOI] [PubMed] [Google Scholar]; (r) Cheng C., Hartwig J. F. Chem. Rev. 2015 doi: 10.1021/cr5006414. [DOI] [PubMed] [Google Scholar]
- (a) Kim J., Sim M., Kim N., Hong S. Chem. Sci. 2015;6(6):3611. doi: 10.1039/c5sc01137j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liskey C. W., Hartwig J. F. J. Am. Chem. Soc. 2013;135:3375. doi: 10.1021/ja400103p. [DOI] [PubMed] [Google Scholar]; (c) Parella R., Gopalakrishnan B., Babu S. A. Org. Lett. 2013;15:3238. doi: 10.1021/ol4012212. [DOI] [PubMed] [Google Scholar]; (d) Roman D. S., Charette A. B. Org. Lett. 2013;15:4394. doi: 10.1021/ol401931s. [DOI] [PubMed] [Google Scholar]; (e) Ladd C. L., Roman D. S., Charette A. B. Org. Lett. 2013;15:1350. doi: 10.1021/ol4003338. [DOI] [PubMed] [Google Scholar]; (f) Saget T., Perez D., Cramer N. Org. Lett. 2013;15:1354. doi: 10.1021/ol400380y. [DOI] [PubMed] [Google Scholar]; (g) Ladd C. L., Roman D. S., Charette A. B. Tetrahedron. 2013;69:4479. [Google Scholar]; (h) Wasa M., Chan K. S. L., Zhang X.-G., He J., Miura M., Yu J.-Q. J. Am. Chem. Soc. 2012;134:18570. doi: 10.1021/ja309325e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Rousseaux S., Liegault B., Fagnou K. Chem. Sci. 2012;3:244. [Google Scholar]; (j) Yoo E. J., Wasa M., Yu J.-Q. J. Am. Chem. Soc. 2010;132:17378. doi: 10.1021/ja108754f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Kubota A., Sanford M. S. Synthesis. 2011;22:2579. doi: 10.1055/s-0030-1260087. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Wasa M., Engle K. M., Yu J.-Q. J. Am. Chem. Soc. 2010;132:3680. doi: 10.1021/ja1010866. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Wang D.-H., Wasa M., Giri R., Yu J.-Q. J. Am. Chem. Soc. 2008;130:7190. doi: 10.1021/ja801355s. [DOI] [PubMed] [Google Scholar]
- (a) Saget T., Cramer N. Angew. Chem., Int. Ed. 2012;51:12842. doi: 10.1002/anie.201207959. [DOI] [PubMed] [Google Scholar]; (b) Wasa M., Engle K. M., Lin D. W., Yoo E. J., Yu J.-Q. J. Am. Chem. Soc. 2011;133:19598. doi: 10.1021/ja207607s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chan K. S. L., Fu H.-Y., Yu J.-Q. J. Am. Chem. Soc. 2015;137:2042. doi: 10.1021/ja512529e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reviews on asymmetric C–H functionalizations: ; (a) Giri R., Shi B.-F., Engle K. M., Maugel N., Yu J.-Q. Chem. Soc. Rev. 2009;38:3242. doi: 10.1039/b816707a. [DOI] [PubMed] [Google Scholar]; (b) Wencel-Delord J., Colobert F. Chem. - Eur. J. 2013;19:14010. doi: 10.1002/chem.201302576. [DOI] [PubMed] [Google Scholar]; (c) Saget T., Cramer N. Pure Appl. Chem. 2014;86:265. [Google Scholar]; (d) Zheng C., You S.-L. RSC Adv. 2014;4:6173. [Google Scholar]; (e) Ye B., Cramer N. Acc. Chem. Res. 2015; 48:1308. doi: 10.1021/acs.accounts.5b00092. [DOI] [PubMed] [Google Scholar]
- (a) Rousseaux S., Gorelsky S. I., Chung B. K. W., Fagnou K. J. Am. Chem. Soc. 2010;132:10692. doi: 10.1021/ja103081n. [DOI] [PubMed] [Google Scholar]; (b) Yan J.-X., Li H., Liu X.-W., Shi J.-L., Wang X., Shi Z.-J. Angew. Chem., Int. Ed. 2014;53:4945. doi: 10.1002/anie.201402562. [DOI] [PubMed] [Google Scholar]
- (a) Harmata M., Hong X. Org. Lett. 2007;9:2701. doi: 10.1021/ol0710358. [DOI] [PubMed] [Google Scholar]; (b) Uchida R., Imasato R., Shiomi K., Tomoda H., Ōmura S. Org. Lett. 2005;7:5701. doi: 10.1021/ol052458h. [DOI] [PubMed] [Google Scholar]; (c) Turner K. L., Baker T. M., Islam S., Procter D. J., Stefaniak M. Org. Lett. 2005;8:329. doi: 10.1021/ol052730n. [DOI] [PubMed] [Google Scholar]; (d) Carling R. W., Leeson P. D., Moore K. W., Smith J. D., Moyes C. R., Mawer I. M., Thomas S., Chan T., Baker R. J. Med. Chem. 1993;36:3397. doi: 10.1021/jm00074a021. [DOI] [PubMed] [Google Scholar]; (e) Hayashi H., Miwa Y., Miki I., Ichikawa S., Yoda N., Ishii A., Kono M., Suzuki F. J. Med. Chem. 1992;35:4893. doi: 10.1021/jm00104a016. [DOI] [PubMed] [Google Scholar]
- (a) Albicker M. R., Cramer N. Angew. Chem., Int. Ed. 2009;48:9139. doi: 10.1002/anie.200905060. [DOI] [PubMed] [Google Scholar]; (b) Saget T., Cramer N. Angew. Chem., Int. Ed. 2013;52:7865. doi: 10.1002/anie.201303816. [DOI] [PubMed] [Google Scholar]; (c) Pedroni J., Boghi M., Saget T., Cramer N. Angew. Chem., Int. Ed. 2014;53:9064. doi: 10.1002/anie.201405508. [DOI] [PubMed] [Google Scholar]; (d) Liu L., Zhang A.-A., Zhao R.-J., Li F., Meng T.-J., Ishida N., Murakami M., Zhao W.-X. Org. Lett. 2014;16:5336. doi: 10.1021/ol502520b. [DOI] [PubMed] [Google Scholar]; (e) Liu L., Zhang A.-A., Wang Y., Zhang F., Zuo Z., Zhao W.-X., Feng C.-L., Ma W. Org. Lett. 2015;17:2046. doi: 10.1021/acs.orglett.5b00122. [DOI] [PubMed] [Google Scholar]; (f) Lin Z.-Q., Wang W.-Z., Yan S.-B., Duan W.-L. Angew. Chem., Int. Ed. 2015;54:6265. doi: 10.1002/anie.201500201. [DOI] [PubMed] [Google Scholar]
- (a) Fisher M. J., Gunn B. P., Harms C. S., Kline A. D., Mullaney J. T., Scarborough R. M., Skelton M. A., Um S. L., Utterback B. G., Jakubowski J. A. Bioorg. Med. Chem. Lett. 1997;7:2537. doi: 10.1021/jm9701076. [DOI] [PubMed] [Google Scholar]; (b) Byler K. G., Brito-Arias M., Marquez-Navarro A., Nogueda-Torres B., Torres-Bustillos L. G., Martínez-Mayorga K. Bioorg. Med. Chem. 2012;20:2587. doi: 10.1016/j.bmc.2012.02.046. [DOI] [PubMed] [Google Scholar]; (c) Johnson M. C., Hu Q., Lingardo L., Ferre R. A., Greasley S., Yan J., Kath J., Chen P., Ermolieff J., Alton G. J. Comput.-Aided Mol. Des. 2011;25:689. doi: 10.1007/s10822-011-9456-7. [DOI] [PubMed] [Google Scholar]
- Bertus P., Szymoniak J. Synlett. 2007:1346. [Google Scholar]
- (a) Ackermann L. Chem. Rev. 2011;111:1315. doi: 10.1021/cr100412j. [DOI] [PubMed] [Google Scholar]; (b) Lafrance M., Fagnou K. J. Am. Chem. Soc. 2006;128:16496. doi: 10.1021/ja067144j. [DOI] [PubMed] [Google Scholar]; (c) Saget T., Lémouzy S., Cramer N. Angew. Chem., Int. Ed. 2012;51:2238. doi: 10.1002/anie.201108511. [DOI] [PubMed] [Google Scholar]
- Gonzalez-de-Castro A., Broughton H., Martinez-Perez J. A., Espinosa J. F. J. Org. Chem. 2015;80:3914. doi: 10.1021/acs.joc.5b00236. [DOI] [PubMed] [Google Scholar]
- (a) Armstrong P. D., Cannon G. J., Long J. P. Nature. 1968;220:65. doi: 10.1038/220065a0. [DOI] [PubMed] [Google Scholar]; (b) Stammer S. H. Tetrahedron. 1990;46:2231. [Google Scholar]; (c) Shimamoto K., Ofune Y. J. Med. Chem. 1996;39:407. doi: 10.1021/jm9502908. [DOI] [PubMed] [Google Scholar]; (d) Sekiyama T., Hatsuya S., Tanaka Y., Uchiyama M., Ono N., Iwayama S., Oikawa M., Suzuki K., Okunishi M., Tsuji T. J. Med. Chem. 1998;41:1284. doi: 10.1021/jm9705869. [DOI] [PubMed] [Google Scholar]; (e) Martin S. F., Dwyer M. P., Hartmann B., Knight K. S. J. Org. Chem. 2000;65:1305. doi: 10.1021/jo991288h. [DOI] [PubMed] [Google Scholar]
- Bender D. M., Peterson J. A., McCarthy J. R., Gunaydin H., Takano Y., Houk K. N. Org. Lett. 2008;10:509. doi: 10.1021/ol702892e. [DOI] [PubMed] [Google Scholar]
- Zheng X., Hudyma T. W., Martin S. W., Bergstrom C., Ding M., He F., Romine J., Poss M. A., Kadow J. F., Chang C.-H., Wan J., Witmer M. R., Morin P., Camac D. M., Sheriff S., Beno B. R., Rigat K. L., Wang Y.-K., Fridell R., Lemm J., Qiu D., Liu M., Voss S., Pelosi L., Roberts S. B., Gao M., Knipe J., Gentles R. G. Bioorg. Med. Chem. Lett. 2011;21:2925. doi: 10.1016/j.bmcl.2011.03.067. [DOI] [PubMed] [Google Scholar]
- (a) Huestis M. P., Chan L., Stuart D. R., Fagnou K. Angew. Chem., Int. Ed. 2011;50:1338. doi: 10.1002/anie.201006381. [DOI] [PubMed] [Google Scholar]; (b) Stuart D. R., Alsabeh P., Kuhn M., Fagnou K. J. Am. Chem. Soc. 2010;132:18326. doi: 10.1021/ja1082624. [DOI] [PubMed] [Google Scholar]
- (a) Hyster T. K., Dalton D. M., Rovis T. Chem. Sci. 2015;6:254. doi: 10.1039/c4sc02590c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hyster T. K., Rovis T. Chem. Commun. 2011;47:11846. doi: 10.1039/c1cc15248c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hyster T. K., Rovis T. Chem. Sci. 2011;2:1606. doi: 10.1039/C1SC00235J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachia M., Iriart S., Baalouch M., De Mesmaeker A., Beaudegnies R. Tetrahedron Lett. 2011;52:3219. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.