

Abstract
Case
A 19‐year‐old man had nausea, diarrhea, and general malaise the day before requesting emergency transport to his former primary physician. The patient became restless and had tonic seizures after admission. The patient was transferred to our hospital as there had been no improvement in his level of consciousness. On arrival, cranial computed tomography showed generalized swelling, and blood ammonia concentration was ≥500 μg/dL. After admission, seizures recurred continually despite careful monitoring and increased doses of sedatives and antiseizure drugs. Dilated pupils and a flat electroencephalogram were evident on day 4 of admission, and the patient died on day 11. Ornithine transcarbamylase deficiency was diagnosed at postmortem based on fractionation of blood and urine amino acids and orotic acid.
Outcome
Urea cycle disorders need to be treated urgently to prevent irreversible neurological damage when accompanied by hyperammonemia.
Conclusion
In cases of hyperammonemia, early dialysis should be considered to reduce the ammonia level.
Keywords: Endocrinology, metabolism
Introduction
It is rare to encounter patients with urea cycle disorder in the emergency room or intensive care unit. According to the 2010 report issued by the Japan Intractable Diseases Information Center, the prevalence of urea cycle disorders is 1 per 8,000 individuals, among which ornithine transcarbamylase (OTC) deficiency is the most common and has a prevalence of 1 per 14,000. The disorder normally presents in neonates or young children, but approximately 20% of all OTC deficiency presents after the age of 6 years.1 Although extremely rare, if OTC deficiency presents after the age of 18 years accompanied by disturbance of consciousness due to hyperammonemia, it should be treated urgently and appropriately to prevent irreversible neurological damage.
Here, we report a case of acute cerebral edema due to hyperammonemia and convulsive status epilepticus in a patient with OTC deficiency.
Case
Patient: 19‐year‐old man
In early November 2013, the man visited a nearby clinic because of his poor physical condition with symptoms of nausea and dizziness. Gastroenteritis was suspected. His condition improved in approximately 1 week, but 3 weeks later, the day before admission to our hospital, the man developed progressively worsening nausea and diarrhea and became unresponsive by the late afternoon. He was transported by an emergency vehicle to his former primary care physician and was admitted for general malaise but cranial computed tomography (CT) and hematological testing showed no abnormalities. After admission, he began to exhibit restless behavior, such as shouting loudly and slamming his body against the wall, as well as tonic seizures at approximately 2.00 am the next morning. Despite the resolution of seizures after i.v. administration of 5 mg diazepam and no signs of abnormalities on cranial CT, magnetic resonance imaging, or encephalography, the patient remained in a coma with a Japan Coma Scale score of 200. He was transferred to our hospital, which is a tertiary emergency medical institution, for a complete clinical examination.
Medical history
The previously healthy patient had been taking 1 mg/day oral betamethasone for leukoplakia that had developed 2 months earlier.
On arrival at our institution, the patient had a Japan Coma Scale score of 200, Glasgow Coma Scale score of 6 (E1V1M4), body temperature of 38.6°C, blood pressure of 155/82 mmHg, heart rate of 132 b.p.m., respiratory rate of 38 breaths/min, and tetanic contractions of arms and legs indicating convulsive status epilepticus. No signs of neck stiffness or Babinski reflex on either side were noted.
Findings of investigations
Arterial blood gas findings (fraction of inspired oxygen = 0.40) were pH 7.465, PaCO2 of 20.4 mmHg, PaO2 of 144 mmHg, bicarbonate 14.2 mmol/L, lactate 9.5 mmol/L, and a base excess of −0.6 mmol/L.
Hematological findings were: white blood cell count of 19,200/μL, red blood cell count of 580 × 10−4/μL, hemoglobin 17.2 g/dL, hematocrit 50.0%, and platelet count of 22.0 × 10−4/μL.
Biochemical findings were: total bilirubin 1.2 mg/L, aspartate aminotransferase 57 IU/L, alanine aminotransferase12 IU/L, lactate dehydrogenase 305 IU/L, creatine kinase 497 IU/L, blood urea nitrogen 8.5 mg/dL, creatinine (Cr) 0.93 mg/dL, sodium 150 mEq/L, potassium 4.4 mEq/L, chloride 112 mEq/L, glucose 176 mg/dL, and ammonia >500 μg/dL (>294 μmol/L).
Cranial CT on admission revealed narrowing of cerebral sulcus and swelling/edema‐like changes in the brain parenchyma (Fig. 1A).
Figure 1.
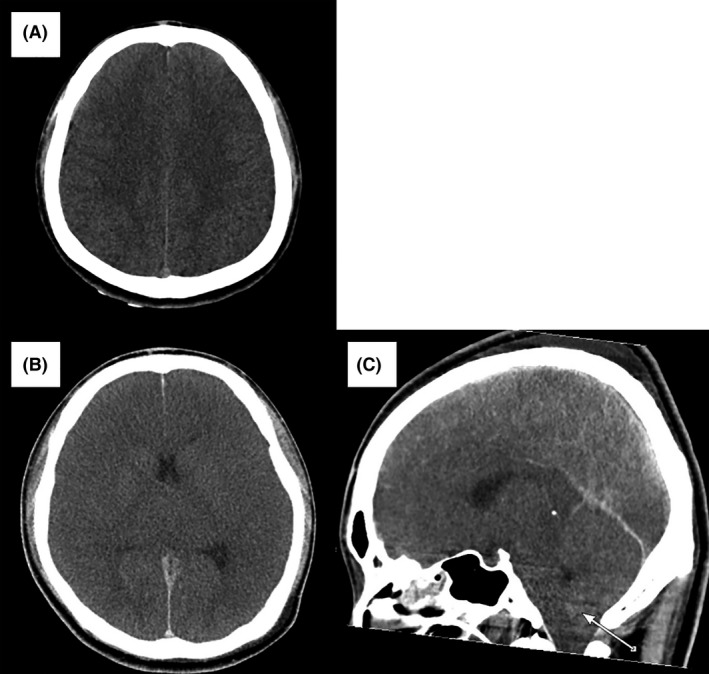
Cranial computed tomography findings in a 19‐year‐old man with hyperammonemia due to ornithine transcarbamylase deficiency. A, On day 1 of hospitalization, the corticomedullary junction was clear, but the brain was slightly edematous. B, C, On day 3, cranial computed tomography findings were remarkable with unclear corticomedullary junction, generalized brain edema, and cerebral herniation (arrow).
Cerebrospinal fluid had an initial pressure of 28 cm H2O, a clear appearance, <5/μL cells, 22 mg/dL protein, and 103 mg/dL glucose.
Treatment course
Because of convulsive status epilepticus, 5 mg diazepam was given i.v. to successfully treat the rigid arms and legs seen on arrival. However, due to prolonged disturbance of consciousness and clinical finding of the tongue falling back in the throat, we carried out tracheal intubation and admitted the patient to the intensive care unit. Figure 2 shows the treatment course during the first 30 h of hospital admission. Seizures recurred despite i.v. treatment with phenobarbital and phenytoin as well as concurrent and continuous i.v. midazolam, propofol, and thiopental. Seizures resolved completely after 12 h of treatment with the maximal doses of these drugs. Even though no increase in cell number was observed in the cerebrospinal fluid, acyclovir was given for possible herpes viral encephalitis, vitamin B1 and B6 for potential vitamin B1 and B6 deficiency, and fructose‐added glycerol for the prevention of cerebral edema. Furthermore, the replacement of extracellular fluid and the intestinal infusion of lactulose were carried out to treat hyperammonemia. The patient's blood ammonia level reduced to 258 μg/dL (151 μmol/L) 24 h after admission, but he developed mydriasis shortly thereafter. A gradual reduction in sedatives did not trigger seizures but termination of spontaneous respiration occurred. As shown in Figure 1, generalized cerebral edema and cerebral herniation were observed on cranial CT on day 3 of hospitalization, followed by a near flat electroencephalogram and loss of brain stem reflexes on day 4. Despite our effort to sustain life through the administration of vasopressors, the patient died on day 11.
Figure 2.
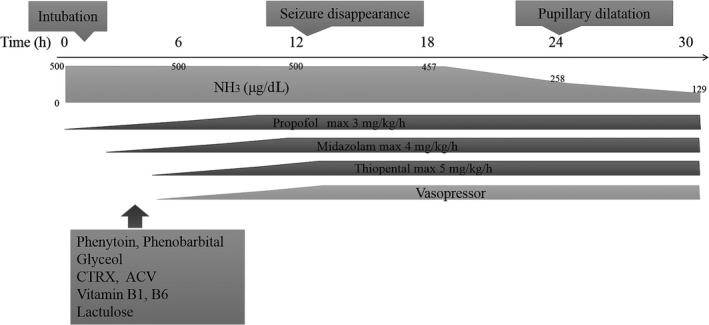
Treatment course of a 19‐year‐old man with hyperammonemia due to ornithine transcarbamylase deficiency, showing the initial 30 h of hospital admission. Seizures completely disappeared 12 h after starting treatment after increased medication doses. Thereafter, blood ammonia concentration gradually decreased. ACV, aciclovir; CTRX, ceftriaxone.
Blood and urine amino acids and orotic acid analyses were carried out using samples collected on day 10, and results revealed elevated levels of urinary orotic acid (28.0 mmol/mol Cr [normal range, 0.4–1.2 mmol/mol Cr]) and blood glutamine (12,850 nmol/mL [418.0–739.8 nmol/mL]) but no significant increase in other proteins in the urea cycle: citrulline (91.7 nmol/mL [17.9–48.0 nmol/mL]), arginine (242.4 nmol/mL [31.8–149.5 nmol/mL]), or argininosuccinic acid (0.89 μmol/L [<1.5 μmol/L]). Based on these findings, OTC deficiency was diagnosed at postmortem. Furthermore, no fatty liver or cirrhosis was observed in the postmortem autopsy, and gene analysis of the liver tissue collected and frozen during examination revealed a single base substitution (c.1033T>C; p.Tyr345His) in exon 10 of the OTC gene, which has been shown to cause late‐onset OTC deficiency.2
Discussion
A urea cycle disorder is a collective term for disorders caused by a defect in the urea cycle, which is a metabolic pathway responsible for the excretion of ammonia produced from nitrogen that is generated during the degradation of amino acids. Ornithine transcarbamylase is an enzyme involved in the synthesis of citrulline from ornithine and carbamoyl phosphate that are produced from ammonia in liver mitochondria. Although OTC deficiency is an X‐linked recessive disorder, 10% of female carriers are symptomatic.3
Clinically, OTC deficiency manifests as gastrointestinal symptoms such as nausea, anorexia, and diarrhea, and neurological symptoms such as loss or alteration of consciousness, restlessness, and seizure, all of which are caused by hyperammonemia. Some patients with OTC deficiency have a history of chronic nausea, sleep disturbance, developmental delay, or mental disorder. Disease onset characterized by hyperammonemia results from hypercatabolism due to infection, starvation, surgery, injury, and steroid administration.
When patients present with typical symptoms of the disorder, elevated blood ammonia concentration, normal anion gap, normal blood glucose level, but no hepatic lesion, a urea cycle disorder is suspected and diagnosed by plasma and urine amino acid analysis, urine organic (orotic) acid analysis, and gene analysis4 (Fig. 3). Excess ammonia in the blood enters the brain through the blood–brain barrier and is converted into glutamine. Because excess glutamine causes osmotic stress and swelling in the brain, leading to neurological damage, the primary treatment of OTC deficiency5 is to reduce blood ammonia levels by limiting the intake of protein and by treatment with 250 mg/kg sodium benzoate, 300 mg/kg sodium phenylbutyrate, and 400 mg/kg arginine as a loading dose as well as daily dose. Although no clear criteria have been established for starting dialysis in patients with OTC deficiency, physicians should use disturbance of consciousness due to hyperammonemia or ammonia levels elevated above 360 μmol/L as indices to start dialysis to prevent neurological damage.1 It has also been suggested that if ammonia concentration stays high for ≥8 h, dialysis should be considered because prolonged exposure to ammonia results in poor neurological outcome,6 suggesting that rapid and appropriate treatment is required. It is particularly important for patients with adult‐onset OTC deficiency to work with professional medical teams that include pediatricians specialized in urea cycle disorders.7
Figure 3.
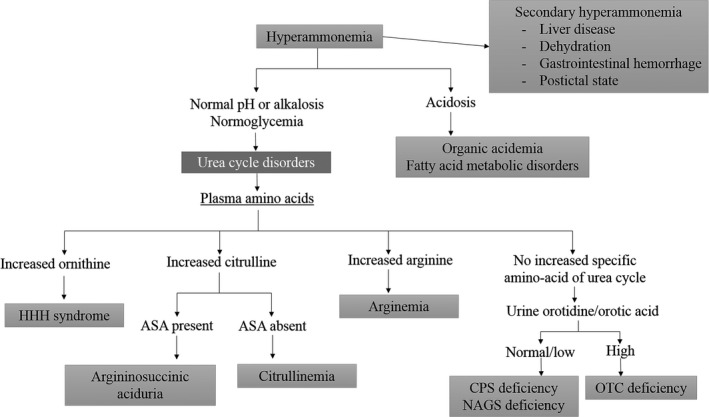
Differential diagnosis of hyperammonemia4. ASA, argininosuccinic aciduria; CPS, carbamoyl phosphate synthetase; HHH, hyperornithinemia–hyperammonemia–homocitrullinuria; NAGS, N‐acetyl glutamate synthetase; OTC, ornithine transcarbamylase.
This study offers three key points. First, this is a case of late‐onset OTC deficiency that presented at the age of 19 years, which is extremely rare when taking into consideration the statistics on urea cycle disorders mentioned above. The treatment of leukoplakia with steroids started 2 months prior to the hospital visit likely contributed to the onset of the disease. Furthermore, the high hematocrit and hypernatremia on arrival indicate dehydration from gastrointestinal symptoms, which might have exacerbated hyperammonemia, triggering convulsive status epilepticus and acute cerebral edema.
Second, it was difficult to differentiate hyperammonemia. The OTC deficiency was diagnosed at postmortem using a sample collected on day 10 of hospitalization. In patients with OTC deficiency, the urinary concentration of orotic acid is elevated because excess carbamoyl phosphate is converted to orotic acid through an alternate metabolic pathway. Similarly, in the present patient, urinary orotic acid concentration was high with no significant increase in the concentration of citrulline, arginine, or argininosuccinic acid, leading to the diagnosis of OTC deficiency (Fig. 3).
The final key point is that we failed to initiate dialysis for rapid and unexplained hyperammonemia. The first rule about secondary hyperammonemia is to treat the cause. Removal of ammonia by dialysis is an effective way to treat primary hyperammonemia. However, in reality, it is difficult to decide whether dialysis is indicated without a past history of the disease. It is particularly difficult to determine whether convulsive status epilepticus is the cause of hyperammonemia or primary hyperammonemia is the cause of convulsive status epilepticus. In the study by Liu et al., ammonia levels returned to original levels 3 h after seizure.8 However, if seizures recur continually, it is difficult to determine the indication for dialysis. Although blood ammonia levels were measured every 6 h (Fig. 1), dialysis was not carried out in the present patient because our priority was to treat seizures. Seizures disappeared 1 h after hospital admission owing to increased doses of antiseizure drugs, and the blood concentration of ammonia tended to decrease, which led us to think that the seizures contributed to hyperammonemia. However, the pupils subsequently dilated and the patient suffered irreversible neurological damage. When patients have unexplained hyperammonemia, physicians should consider the possibility of primary hyperammonemia and continuously monitor blood ammonia levels. According to previous studies, the rapid initiation of dialysis improved the survival rate of patients with hyperammonemia (≥360 μmol/L) lasting over 8 h.1, 4
Depending on the length of exposure, neuroimaging findings of patients with hyperammonemia show cerebral edema in CT and hypoxic ischemic encephalopathy in magnetic resonance imaging.9 However, such findings may be transient in some cases. In this study, CT scan on arrival revealed cerebral edema, which was thought to be reversible. On day 3 of hospitalization, when mydriasis occurred, cranial CT revealed exacerbated brain swelling and cerebral herniation. In the end, we failed to save the patient's life.
Conclusion
We recently experienced a case of late‐onset OTC deficiency that presented at the age of 19 years, in which elevated blood ammonia levels caused convulsive status epilepticus and acute cerebral edema. When patients present with unexplained hyperammonemia, physicians should monitor ammonia levels continuously and urgently consider dialysis.
Conflict of Interest
None.
Acknowledgments
We thank Dr. Tomiko Kuhara of the Japan Clinical Metabolomics Institute and Dr. Makoto Yoshino of the Cognitive and Molecular Research Institute of Brain Diseases, Kurume University, for their cooperation in the diagnosis of this patient. This article is based on a study first reported in Nihon KyuKyu Igakukai Zasshi 2014; 25: 833–8.
[Correction added on 29 June 2016, after first online publication: The corresponding author's email address has been updated.]
References
- 1. Kido J, Nakamura K, Mitsubuchi H et al Long‐term outcome and intervention of urea cycle disorders in Japan. J. Inherit. Metab. Dis. 2012; 35: 777–85. [DOI] [PubMed] [Google Scholar]
- 2. Yamaguchi S, Brailey LL, Morizono H et al Mutations and polymorphisms in the human ornithine transcarbamylase (OTC) gene. Hum. Mutat. 2006; 27: 626–32. [DOI] [PubMed] [Google Scholar]
- 3. Maestri NE, Brusilow SW, Clissold DB et al Long‐term treatment of girls with ornithine transcarbamylase deficiency. N. Engl. J. Med. 1996; 335: 855. [DOI] [PubMed] [Google Scholar]
- 4. Summar M. Current strategies for the management of neonatal urea cycle disorders. J. Pediatr. 2001; 138(1 Suppl): S30. [DOI] [PubMed] [Google Scholar]
- 5. Hoshino K, Nishisako R, Tateishi K et al A case of effective high dialysate flow rate continuous hemodialysis for hyperammonemia due to ornithine transcarbamylase deficiency. Journal of the Japanese Society of Intensive Care Medicine 2013; 20: 25–8. [Google Scholar]
- 6. Enns GM, Berry SA, Berry GT et al Survival after treatment with phenylacetate and benxoate for urea‐cycle disorders. N. Engl. J. Med. 2007; 356: 2282–92. [DOI] [PubMed] [Google Scholar]
- 7. Iyer H, Sen M, Prasad C et al Coma, hyperammonemia, metabolic acidosis, and mutation: lessons learned in the acute management of late onset urea cycle disorders. Hemodial. Int. 2012; 16: 95–100. [DOI] [PubMed] [Google Scholar]
- 8. Liu KT, Su CS. Postictal transient hyperammonemia. Am. J. Emerg. Med. 2008;26:388e1–2. [DOI] [PubMed] [Google Scholar]
- 9. Gropman A. Brain imaging in urea cycle disorders. Mol. Genet. Metab. 2010; 100(Suppl 1): S20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]