

Abstract
Mast syndrome (SPG21) is a childhood-onset, autosomal recessive, complicated form of hereditary spastic paraplegia (HSP) characterized by dementia, thin corpus callosum, white matter abnormalities, and cerebellar and extrapyramidal signs in addition to spastic paraparesis. A nucleotide insertion resulting in premature truncation of the SPG21 gene product maspardin underlies this disorder, likely leading to loss of protein function. In this study, we generated SPG21−/− knockout mice by homologous recombination as a possible animal model for SPG21. Though SPG21−/− mice appeared normal at birth, within several months they developed gradually progressive hind limb dysfunction. Cerebral cortical neurons cultured from SPG21−/− mice exhibited significantly more axonal branching than neurons from wild-type animals, while comprehensive neuropathological analysis of SPG21−/− mice did not reveal definitive abnormalities. Since alterations in axon branching have been seen in neurons derived from animal models of other forms of HSP as well as motor neuron diseases, this may represent a common cellular pathogenic theme.
Keywords: Maspardin, ACP33, CD4, Hereditary spastic paraplegia, Rab7
Introduction
The hereditary spastic paraplegias (HSPs) are a diverse group of neurological disorders with the cardinal feature of progressive spasticity and weakness of the lower limbs [1–7]. They are classified as “pure” if lower extremity spasticity and weakness occur in isolation or “complicated” if patients exhibit other neurological abnormalities such as cognitive impairment, distal amyotrophy, or white matter abnormalities [1]. The HSPs characteristically result from distal axonal degeneration in the long descending corticospinal tracts and ascending dorsal column fibers, which are among the longest motor and sensory axons, respectively, in the central nervous system (CNS) [5]. More recently, the identification of multiple genetic loci (SPG1-46) and 20 gene products for the HSPs has permitted a molecular genetic classification of these disorders [2–7]. Based on the proteins identified, several additional mechanisms for pathogenesis have been advanced. These include abnormal cell signaling or migration, mitochondrial abnormalities, aberrations of myelination, impaired cholesterol or neurosteroid metabolism, alterations in intracellular ER network morphology, and defects in intracellular trafficking and transport. Most known proteins appear to fall into the latter two categories—including atlastin-1 (SPG3A), spastin (SPG4), NIPA1 (SPG6), strumpellin (SPG8), KIF5A (SPG10), spastizin (SPG15), seipin (SPG17), spartin (SPG20), acidic cluster protein 33 (ACP33)/maspardin (SPG21), and receptor expression enhancing protein 1 (REEP1; SPG31) [2–10]—suggesting that defects in protein and organelle trafficking may underlie distal axonal degeneration.
Mast syndrome (SPG21; MIM 248900) is a childhood-onset, autosomal recessive, complicated HSP characterized by progressive spastic paraparesis in association with dementia, developmental defects, thin corpus callosum, white matter abnormalities, and cerebellar and extrapyramidal signs [11,12]. The early onset of SPG21 indicates that it may have a neurodevelopmental component, though it is also clearly progressive during adulthood [11,12]. All known cases of SPG21 are autosomal recessive, caused by a single nucleotide insertion (601insA) in exon 7 of the SPG21 gene (aliases MASPARDIN, ACP33, MAST, BM-019, and GLO10), resulting in a reading frame shift and subsequent premature termination (fs201–212X213) of the 308-amino acid maspardin protein [12]. Individuals heterozygous for the 601insA mutation do not exhibit any neurological symptoms. Thus, disease pathogenesis likely results from a lack of functional maspardin protein rather than a toxic gain of function, consistent with the autosomal recessive inheritance of Mast syndrome.
The ACP33/maspardin protein harbors a single known domain with limited but clear similarity to the α/β hydrolase superfamily. In maspardin, the α/β hydrolase domain comprises a characteristic nucleophile elbow and parallel β strands, and though it contains a G-X-S-X-G motif (single letter amino acid code) found in serine lipases, it lacks the full catalytic triad common to most α/β hydrolases, suggesting that it might lack enzymatic activity and instead function as a peptide-binding module mediating protein–protein interactions [12]. In fact, maspardin was originally identified as an intracellular binding protein for the cell surface glycoprotein CD4, and it has been proposed to modulate CD4 stimulatory activity [13]. Within the nucleophile elbow of the α/β hydrolase domain, Ser109 is critical for the interaction of ACP33/maspardin with CD4 [13]. However, maspardin is widely expressed in many cell types other than T cells, including cells lacking CD4 such as neurons. Furthermore, since it is broadly expressed and highly conserved phylogenetically, with orthologs present in distant species including a number of plants (Fig. 1), maspardin likely also interacts with proteins other than CD4. In fact, maspardin interacts with an aldehyde dehydrogenase super-family member, ALDH16A1, but whether this interaction relates to disease pathogenesis remains unclear [14]. Maspardin is found within both soluble and membrane fractions prepared from cells [14]. It co-localizes partially with markers for late endosomes and lysosomes [13,14] and has recently been shown to interact with the Rab7 GTPase, which is present in these organelles as well as at autophagosomes and autophagolysosomes [15].
Fig. 1.
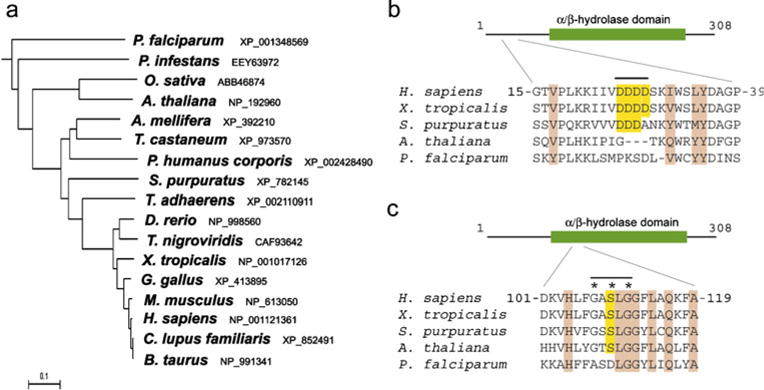
Maspardin protein phylogeny. a Phylogenetic tree. Species and GenBank protein accession numbers are shown. The tree was constructed using ClustalW (v. 1.4). Scale bar indicates number of substitutions per site. b, c Sequence alignment of maspardin-related proteins from the indicated species. The acid cluster region is highlighted in yellow (b), and the G-X-S-X-G motif at nucelophile elbow is indicated by asterisks, with the Ser residue shaded in yellow (c). Conserved residues in all species shown are shaded light brown. A schematic diagram of human maspardin is shown at the top in b and c
In this study, we show that targeted disruption of the SPG21 gene in mice using homologous recombination results in hind limb dysfunction, suggesting that these knockout mice may serve as a model to study some pathological aspects observed in Mast syndrome patients. Although morphological studies of the nervous system did not reveal definitive neuropathologic changes, primary cerebral cortical neurons cultured from SPG21−/− animals showed prominent alterations in axon branching.
Materials and methods
Generation and breeding of knockout mice
SPG21−/− mice were generated commercially (Caliper Life Sciences). For construction of the targeting vector, the mouse chromosome 9 sequence (nt # 65,660,000∼65,740,000) was retrieved from the Ensembl database (http://www.ensembl.org) and used as reference in this project. BAC clone RP24-285H11 was used for generating homologous arms and Southern probes by PCR or the RED cloning/gap-repair method. The 5′ (4.1 kb) and 3′ (5.8 kb) homologous arms were generated by RED cloning/gap repair. They were cloned into the LloxNwCD vector, with fidelity confirmed by restriction digestion and end sequencing. The final vector was obtained by standard molecular cloning methods. Aside from homologous arms, the final vector also contained a LoxP-flanked Neo expression cassette (for positive selection of embryonic stem (ES) cells) and a diphtheria toxin subunit A (DTA) expression cassette (for negative selection of ES cells). Final vector construction was confirmed by both restriction digestion and end sequencing analysis. NotI digestion was used to linearize the final vector for electroporation. 5′ and 3′ external probes were generated by PCR using proofreading TaKaRa LA Taq (Takara Bio) and tested using genomic Southern blot analysis for ES screening. They were cloned into the pCR4.0-TOPO backbone and confirmed by DNA sequencing.
For genotyping, genomic DNA was isolated from tail snips using standard procedures. Gene-specific PCR was carried out in an MJ Research PTC-200 DNA Engine Thermal Cycler (Bio-Rad) using Taq DNA polymerase (Invitrogen) and primers specific for exon 3 of SPG21 (forward: 5′-CGTGGATGACGATGACAGTA-3′), Neo cassette (forward: 5′-GCCAGCTCATTCCTCCCACTCA T-3′), and genomic reverse (5′-AAAACTAAAGG CTAGCCGGG-3′). Reactions were subjected to an initial denaturing step of 2 min at 94°C, followed by 30 cycles of 45 s at 94°C, 90 s at 60°C, and 60 s at 72°C, with a final extension of 72°C for 10 min.
Behavioral studies
For the beam-walking test, male mice of the indicated ages (2–12 months) were placed at one end of an 80-cm long, 1-cm square wooden rod elevated 30 cm above the bench with wood supports. They were allowed to walk to the goal box at the other end, with the time to traverse recorded. The number of foot slips (one or both hind limbs slipped from the beam) was recorded using a tally counter.
Histology
All animal experiments were approved by the NINDS/NIDCD Animal Care and Use Committee. Mice were perfused transcardially with 5% paraformaldehyde (PFA). Brains and spinal cords were isolated and post-fixed in 5% PFA for at least 48 h. Whole brains and large sections of spinal cord were then Golgi-stained by incubation in 3% potassium dichromate in 5% PFA for up to 1 week, followed by two washes in freshly prepared 0.75% silver nitrate and incubation in 0.75% silver nitrate for up to 1 week subsequently. Sections (50 μm thick) were then cut in distilled water on a sliding vibratome and were Nissl-stained subsequently.
Immunocytochemistry of tissue sections
Mice were transcardially perfused with 4% PFA, and skeletal muscles, spinal cords, and brains were isolated, post-fixed in 4% PFA overnight, and cryoprotected sequentially in 15% and 30% sucrose. Brains were cut coronally, and spinal cords and some muscles were cut transversely on a cryostat (14 μm thick sections); the other muscle sections (30 μm thick) were cut longitudinally. Polyclonal rabbit anti-synaptotagmin I antibodies (gift of Dr. Peter Low, Karolinska Institutet) were used to stain all section types, while polyclonal rabbit anti-GluR2/3 (3 μg/ml) (gift of Dr. Katherine Roche, NINDS, NIH), chicken anti-neurofilament-H (Clontech; 1:50,000), and rat anti-myelin basic protein (MBP) (Millipore; 1:300) were applied to brain and spinal cord sections. Additionally, polyclonal rabbit anti-neurofilament-M (Millipore; 1:300) was used to label muscle sections. After washing, each primary antibody was followed by an appropriate Alexa Fluor-conjugated secondary antibody and α-bungarotoxin (Invitrogen) where indicated for muscles. At least six sections were analyzed per animal.
For whole-mount analysis of adult hind limb muscles, mice were transcardially perfused with 4% PFA. Lateral gastrocnemius muscles were isolated, post-fixed in 4% PFA overnight, and partitioned into four longitudinal sections using a scalpel. The tissue was rinsed in phosphate-buffered saline (PBS) and incubated in 0.1 M glycine/PBS at 4°C overnight. Subsequently, tissue was permeabilized in 1% Triton X-100/PBS at room temperature for 8 h. Antibody staining was performed with anti-neurofilament-M antibody in 5% normal goat serum (NGS)/PBS at 4°C for 40 h. Samples were washed 3×20 min in PBS and incubated at 4°C overnight using Alexa Fluor-conjugated secondary antibody (1:100) and α-bungarotoxin in 5% NGS in PBS. Muscles were washed four times for 20 min in 5% NGS/PBS, rinsed in PBS, and mounted. Images were obtained with an inverted Zeiss LSM 510 META confocal microscope. Z-stack projections were made from serial scanning every 1 μm to reconstruct the neuromuscular junction (NMJ) using the tool available in the LSM software. Measurements of average cross-sectional areas and number of muscle fibers per NMJ and relative fluorescent intensities of brain and spinal cord markers were performed using ImageJ (NIH).
Neuronal culture and immunofluorescence microscopy
Primary cultures of mouse cerebral cortical neurons were prepared from P0 mice, plated at a density of ∼1.0×104/cm2 on coverslips, and maintained and immunostained as described previously [16,17]. Coverslips were mounted using Gel/Mount reagent (Biomeda). Fluorescence images were acquired using Zeiss LSM510 or Zeiss LSM710 confocal microscopes and processed with Adobe Photoshop 7.0 software.
Protein preparation, gel electrophoresis, and immunoblotting
For immunoblotting, proteins were resolved by SDS-PAGE and then electrophoretically transferred to nitrocellulose. After blocking with 5% non-fat milk/0.1% Tween 20/Tris-buffered saline (TBS, pH 7.4), antibodies (1–5 μg/ml) were added for 2.5 h at room temperature. The rabbit polyclonal anti-maspardin, anti-atlastin-1, anti-spastin, and anti-REEP1 antibodies have been described previously [10,14,16]. Rabbit polyclonal antibodies against KIF5A and KIF5B were from Abcam. Mouse polyclonal anti-ALDH16A1 antibodies (B01P) were purchased from Abnova, and goat polyclonal anti-Rab7 antibodies (C-19) were from Santa Cruz Biotechnology. Mouse monoclonal anti-β-actin antibody (clone AC-15; IgG1) was obtained from Sigma-Aldrich. Following several washes in TBS, horseradish peroxidase-conjugated secondary antibodies (1:1000; GE Healthcare Life Sciences) were added for 1.5 h in blocking buffer. Finally, after several washes with 0.1% Tween/TBS, immunoreactive proteins were revealed using Renaissance Enhanced Chemiluminescence Reagent (PerkinElmer Life Sciences). Images were acquired using the Molecular Imager ChemiDoc XRS System and Quantity One software (Bio-Rad).
BDNF treatment of neurons
Cerebral cortical neurons prepared from P0 mice as above were plated at a density of 4−5×106 per 10 cm plate and were maintained. At 7 days in vitro, 50 ng/ml BDNF in neurobasal medium was applied to neurons for 0–240 min. At the appropriate time point, cells were washed twice with PBS, then lysed with a buffer consisting of 50 mM Tris–Cl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 2% SDS, 1 mM PMSF, and complete protease inhibitor cocktail (Roche Applied Science) for 10 min on ice. Samples were subjected to SDS-PAGE and then immunoblotted using mouse monoclonal anti-TrkB (1:300; BD Biosciences) and rabbit polyclonal anti-phospho TrkA (Tyr490)/TrkB (Tyr516) (1:1,000; Cell Signaling).
Microarray analysis
Brains of SPG21−/− knockout mice and SPG21+/+ litter-mate controls (ages 6 and 15 months; n=3 for each group) were dissected. RNA was extracted, and microarray analysis using the Affymetrix GeneChip mouse genome 430 2.0 array was performed by the NIH Neuroscience Microarray Consortium (http://np2.ctrl.ucla.edu/np2/home.do) using Affymetrix GeneChip Operating Software v1.4. All RNA samples met rigorous quality control evaluations by the NIH Neuroscience Microarray Consortium that included present call rates, scaling factor, GAPDH ratio, and background analysis.
Protein content determination
Protein concentrations were determined using the Pierce BCA Protein Assay Reagent (Thermo Fisher Scientific) with bovine serum albumin as the standard.
Statistical analysis
Statistical analyses were performed using Student’s t test, assuming unequal variance, with p<0.05 considered significant.
Results
SPG21−/− mice exhibited defects in hind limb function
SPG21−/− mice were generated by homologous recombination using the targeting vector shown in Fig. 2a. Targeted disruption of exon 3 of SPG21 encoding maspardin was confirmed using PCR (Fig. 2b), and immunoblotting for ACP33/maspardin protein revealed complete absence of the protein in SPG21−/− knockout animals (Fig. 2c).
Fig. 2.
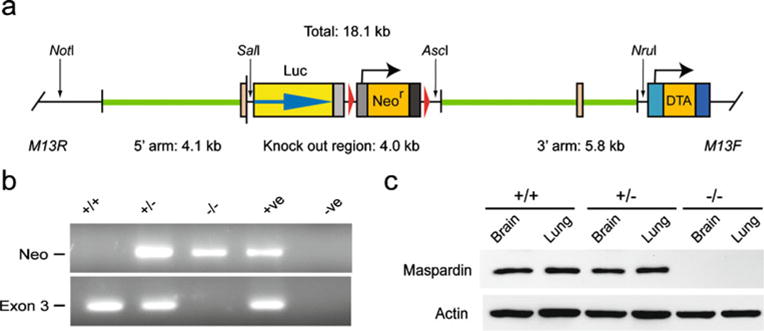
Generation of SPG21−/− knockout mice. a Schematic representation of the SPG21 gene targeting strategy. DTA, diptheria toxia A subunit. b PCR analysis of SPG21+/+, SPG21+/− and SPG21−/− mice. c Immunoblot analysis of extracts (20 μg protein/lane) from the indicated tissues isolated from mice with the indicated SPG21 alleles. Actin levels were monitored as a control for protein loading
The SPG21−/− mice bred normally, and their offspring appeared normal at birth. However, they showed slight dragging of the hind limbs at several months of age as compared with littermate controls, and this progressively worsened with increasing age. To assess formally their gait disturbance, we evaluated SPG21−/− knockout and SPG21+/+ wild-type mice at multiple ages using a narrow beam-walking test. SPG21−/− mice had a significant increase in the number of slips on the beam-walking test as early as 2–6 months of age (Fig. 3a). Later, they exhibited progressive difficulty traversing the beam, as evidenced by a prominent delay in completion of this task, particularly in animals >12 months of age (Fig. 3b). Throughout the beam-walking test, slips and dragging of the hind limbs were very prominent in the SPG21−/− mice, particularly in animals greater than 12 months of age, but not in wild-type animals (Supplementary Videos 1 and 2).
Fig. 3.
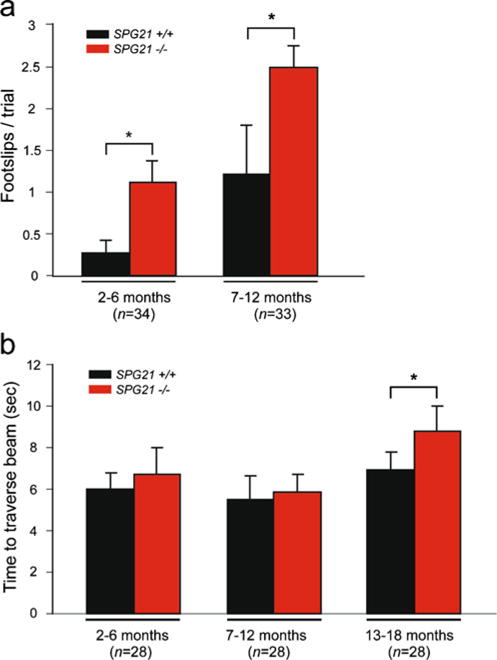
Narrow beam-walking analysis of SPG21−/− mice. a, b Mice of the indicated ages were subjected to the beam-walking test, and foot slips (a) and time to traverse (b) were recorded. Means ± S.D. are shown graphically; *p<0.05
Cultured cortical neurons from SPG21−/− mice displayed increased axonal branching
To test whether deletion of SPG21 induces morphological abnormalities in neurons, we examined cerebral cortical neurons in primary culture. Many of the studied cells displayed the morphology of pyramidal-type neurons. In a number of cell culture systems modeling the two most common HSPs, SPG3A and SPG4, abnormalities in axon elongation or branching have been observed [18–20]. We examined elongation and branching of axons from cultured cerebral cortical neurons at 3 days in vitro and noticed prominent differences. In fact, though the primary axon was the same length, there was far more branching in neurons from the SPG21−/− as compared with SPG21+/+ mice (Fig. 4a, b). The number of primary dendrites was unchanged (Fig. 4c).
Fig. 4.
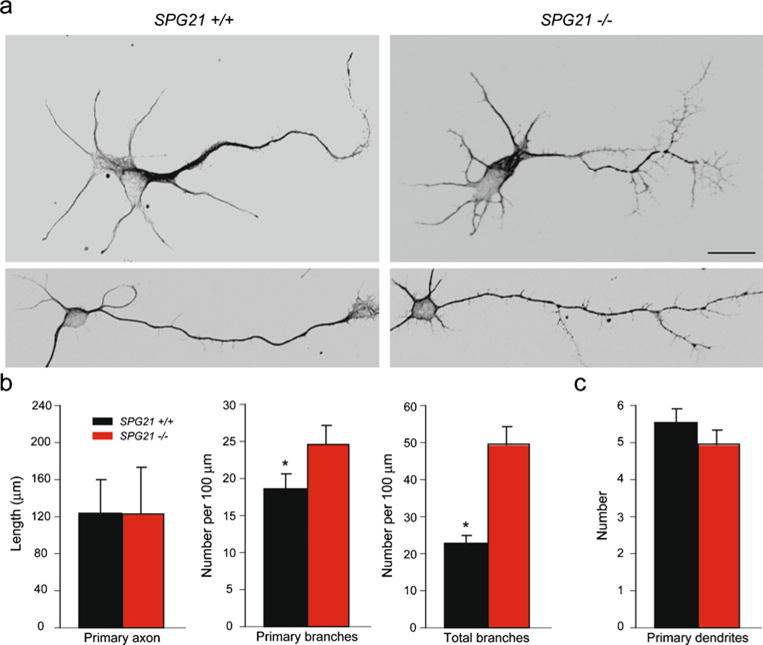
Cerebral cortical neurons from SPG21−/− mice exhibit increased axon branching. a Representative neurons at 3 days in vitro were co-stained for β-tubulin and tau to identify neuronal processes. Scale bar, 20 μm. b, c Quantifications of primary axon length and number of total axon branches (b) as well as the number of dendrites per cell (c) are shown graphically (means ± S.D.; *p<0.05)
Since differential effects on axon growth and branching have been observed in response to stimulation with different growth factors [21,22], and maspardin has been identified as a negative regulator of CD4 [13], we examined whether depletion of maspardin affected either phosphorylation of growth factor receptors in response to ligand binding or else the subsequent degradation of these receptors. We focused on BDNF, which binds the TrkB receptor, and examined both TrkB levels and TrkB phosphorylation at Tyr516. We noted no significant differences in any of these studies when studying neurons derived from SPG21−/− and SPG21+/+ mouse cortices over a period of 4 h after stimulation (data not shown).
SPG21−/− mice exhibited no gross neuropathologic abnormalities
We completed a comprehensive pathological characterization of aged SPG21−/− knockouts by investigating peripheral tissues as well as the central nervous system (ORS/VRP Mouse Phenotyping Service, National Institutes of Health, Bethesda, MD). Three SPG21−/− mice with gait disorders and three SPG21+/+ animals, including both males and females, were examined at 18–19 months of age. This examination included serum chemistries, hematology studies, organ/body weights, and comprehensive gross and histopathological analyses. Examination of the CNS and peripheral nerves did not reveal differences between the wild-type and mutant mice (n=3 of each group). Pathologic changes seen in both groups included degenerative joint disease of the knee and temporomandibular joints, mild degenerative disc disease of the spine, fibro-osseous lesions of the femur and skull, and loss of neurons from the spiral ganglia of the inner ear. These changes, however, can be observed in aged animals of any strain.
To test whether alterations in neuronal morphology observed in cultured neurons occur in adult animals with motor defects, we compared dendritic projections of selected Golgi-stained neurons in the brain and spinal cord of 12-month-old knockout and wild-type mice and found no appreciable differences in motor cortex, hippocampus, and spinal cord (Fig. 5a–c). We then verified that the total number of axons present is similar in wild-type and knockout mice (n=3 of each group). We examined the numbers and sizes of axons in ventral roots at the lumbar spinal cord level, which were not significantly changed (p>0.05, Student’s t test; n=3 of each group). The ventral roots and white matter axons in SPG21−/− and wild-type mice (n=3) did not exhibit any pathological changes suggestive of demyelination (Fig. 6), though demyelination has been observed in patients with SPG21 [12]. Quantitative immunolabeling with antibodies against myelin basic protein (MBP) in cerebral cortex and spinal cord did not reveal significant changes (Fig. 6 and Supplementary Fig. S1). No change in neurofilament-H labeling was observed, in agreement with normal morphology of the axons.
Fig. 5.
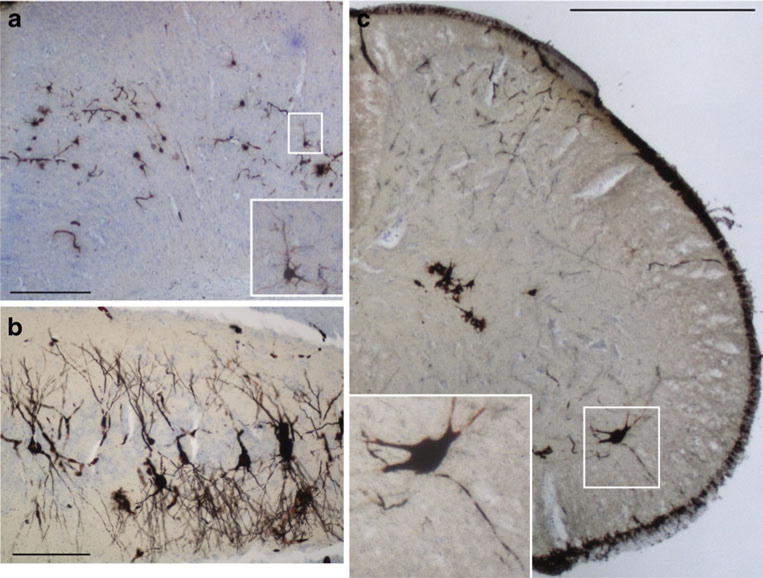
Light microscopic analysis of 40 μm thick Golgi- and Nissl-stained tissue sections from 12-month-old SPG21−/− mice. a Representative image of the cerebral motor cortex. The boxed area (enlarged in inset) shows a typical pyramidal neuron. No striking differences in neuronal morphology or layer organization were observed. Scale bar, 100 μm. b Representative image of the CA3 region of the hippocampus. Scale bar, 100 μm. c Representative image from the lumbar spinal cord. The boxed area (enlarged in inset) shows an α-motor neuron with projections. Scale bar, 500 μm
Fig. 6.
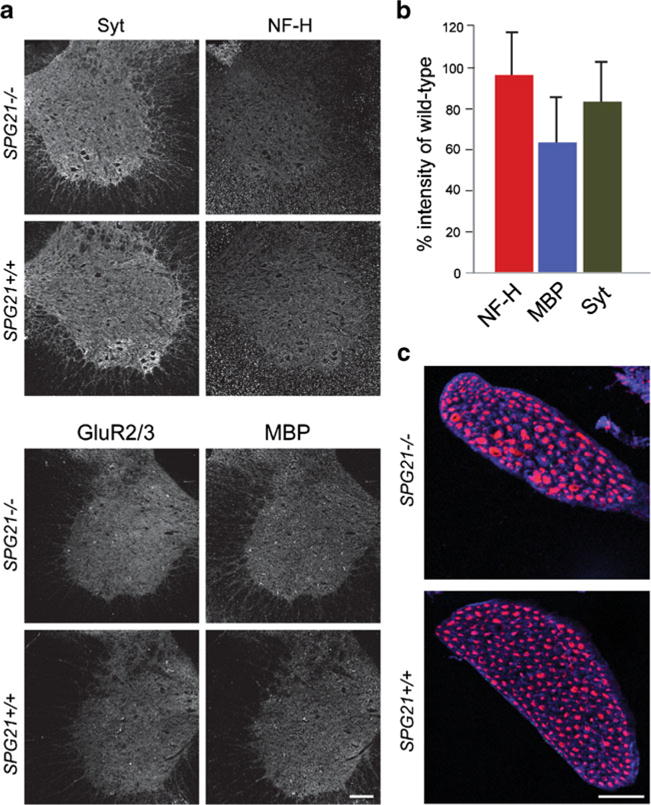
Light microscopic analysis of SPG21−/− mouse lumbar spinal cords. a Confocal images of spinal cord sections from 12-month-old mice stained for synaptotagmin I (Syt), neurofilament-H (NF-H), GluR2/3, and MBP. Scale bar, 100 μm. b Quantification of fluorescence intensity of markers for MBP, NF-H, and Syt in SPG21−/− mice expressed as a percentage of fluorescence found in tissue sections from wild-type SPG21 +/+ littermates (p>0.05, Student’s t test; n=4 for each marker). c Images of ventral roots stained for neurofilament-H (red) and MBP (blue). Scale bar, 50 μm
To test whether possible defects in axonal branching could result in formation of exuberant synaptic contacts, we stained sections from cerebral motor cortex (Supplementary Fig. S1), spinal cord (Fig. 6), and NMJs in lateral gastrocnemius muscle (Fig. 7) from 12-month-old animals with antibodies against synaptotagmin I. Brain and spinal cord sections were also stained with antibodies against the post-synaptic excitatory amino acid receptor subunits GluR2/3. In all cases, we found no significant changes in the distribution or fluorescent intensity (Fig. 6 and Supplementary Fig. S1). Lastly, axonal branching at NMJs in both SPG21−/− and wild-type SPG21+/+ animals appeared very similar, as assessed by comparing their size, number of muscle fibers innervated, and localization of presynaptic markers (Fig. 7).
Fig. 7.
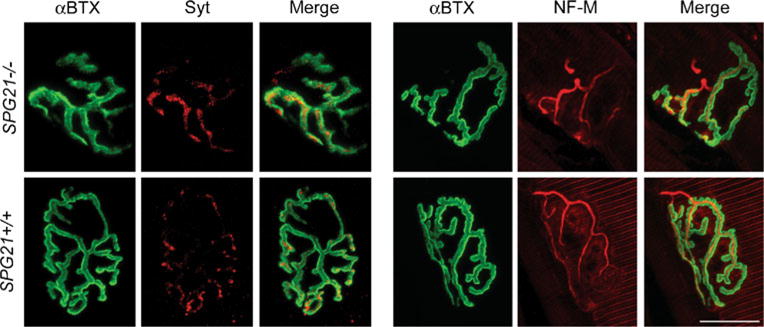
Light microscopic analysis of NMJs in 12-month-old SPG21−/− mice. Confocal immunofluorescence images of NMJs co-stained for neurofilament-M (NF-M) or synaptotagmin I (Syt; red) and α-bungarotoxin (αBTX; green). Cross-sectional areas and number of muscle fibers per NMJ were quantified in ImageJ but were not significantly different (p>0.05, Student’s t test; n=3). Two representative images are shown for each. Scale bar, 20 μm
Gene chip analysis of mRNA changes in SPG21−/− mice
To investigate whether expression of certain axonal proteins is altered, we performed a gene chip analysis of mRNA isolated from wild-type and SPG21−/− mice at 6 and 15 months of age. All mRNAs that exhibited >1.5-fold changes at each age are reported in Table 1. Interestingly, this analysis identified a prominent up-regulation in levels of KIF5B mRNA in the 15-month-old animals, prefiguring potential abnormalities in cargo trafficking. In addition, KIF5A has been shown to be mutated in another HSP, SPG10 (MIM 604187) [23]. This led us to consider whether there were any late-onset changes at the protein level for KIF5A and KIF5B. However, no significant changes were observed in SPG21−/− mice (Supplementary Fig. S2). We further probed for any changes in levels of other HSP proteins in the brain and spinal cord, including those affected in the three most common forms—spastin (SPG4), atlastin-1 (SPG3A), and REEP1 (SPG31)—as well as two proteins shown to interact with maspardin, ALDH16A1, and Rab7 [14,15]. There were no differences in levels of any of these proteins in the brain or spinal cord between wild-type and SPG21−/− animals (Supplementary Fig. S2).
Table 1.
Differential expression of mRNAs in brains from SPG21−/− compared to SPG21+/+ mice
| GenBank accession # | Symbol | Gene/protein name | Fold change | p |
|---|---|---|---|---|
| 6 months | ||||
| NM_008142 | Gnb1 | Guanine nucleotide binding protein beta 1 | −1.79 | 0.007 |
| NR_001585 | Speer7-ps1 | Spermatogenesis associated glutamate-rich protein 7, pseudogene 1 | −1.64 | 0.027 |
| NM_133347 | Dhx30 | DEAH box polypeptide 30 | −1.63 | 0.004 |
| NM_022420 | Gprc5b | G-protein coupled receptor, family C, group 5, member b | −1.62 | <0.001 |
| NM_176849 | Arglu1 | Arginine and glutamate rich 1 | −1.59 | 0.027 |
| XM_620516 | Rkhd3 | Ring finger and KH domain containing 3 | −1.54 | 0.036 |
| NM_013790 | Abcc5 | ATP-binding cassette, sub-family C (CFTR/MRP), member 5 | 1.72 | 0.045 |
| NM_019698 | Aldh18A1 | Aldehyde dehydrogenase family 18, member A1 | 1.66 | 0.024 |
| NM_010380 | H2-D1 | Histocompatibility 2, D region locus 1 | 1.55 | 0.020 |
| BC052465 | D12Ertd553e | DNA segment, ERATO Doi 553, expressed, mRNA | 1.54 | 0.032 |
| NM_011671 | Ucp2 | Uncoupling protein 2 | 1.52 | 0.042 |
| NM_030689 | Nptxr | Neuronal pentraxin receptor | 1.50 | 0.048 |
| 15 months | ||||
| NM_025287 | Spop | Speckle-type POZ protein | −1.58 | 0.049 |
| NM_021560 | Bhlhb5 | Basic helix-loop-helix family, member e22 | −1.52 | 0.031 |
| NM_011464 | Spint2 | Serine protease inhibitor, Kunitz type 2 | −1.52 | 0.003 |
| NM_008448 | Kif5b | Kinesin family member 5B | 2.70 | 0.010 |
| NM_009009 | Rad21 | RAD21 homolog (Schizosaccharomyces pombe) | 2.21 | 0.027 |
| NM_009703 | Araf | v-raf murine sarcoma 3611 viral oncogene homolog | 1.90 | 0.043 |
| NM_025827 | Lonp2 | Lon peptidase 2, peroxisomal | 1.85 | 0.024 |
| NM_026030 | Eif2s2 | Eukaryotic translation initiation factor 2, subunit 2 (beta) | 1.78 | 0.040 |
| NM_001099631 | Sh2d5 | SH2 domain containing 5 | 1.77 | 0.042 |
Discussion
A mutation in the SPG21 gene results in a shift of reading frame and premature stop codon, causing maspardin protein truncation and presumed loss of function that results in the complicated form of HSP known as Mast syndrome (SPG21). Maspardin was first identified as a CD4-binding protein and proposed as a negative regulatory factor involved in CD4-dependent T cell activation [13]. However, maspardin is a ubiquitously expressed protein, including in CD4-negative cells types, and thus likely has other functions. In this study, we generated knockout mice lacking the SPG21 gene encoding maspardin as an animal model for SPG21.
SPG21−/− animals had a clear but slowly progressive defect in motor function, with the hind limbs most affected. Some of the characteristic pathological features of human SPG21 were not clearly seen, however. In particular, we found no clear evidence of white matter disease. Even so, clear effects on axonal branching were seen when neurons were cultured from the cerebral cortex of neonatal SPG21−/− animals, with significantly more branching but no differences in primary axon length. This finding fits broadly with the proposed role of maspardin as a negative regulatory factor for CD4, since maspardin might also act as a negative regulator of other growth factor/cytokine receptors.
Since some growth factors have been described which stimulate branching of axons without increasing the primary axon length [22], as we described here, the presumptive loss of inhibition might result in positive modulation of growth factor signaling. Alternatively, based on the localization of maspardin to late endosomes and lysosomes [13,14], its co-immunoprecipitation with Rab7 [15], and the presence of a cluster of acidic amino acids at the N-terminus [13], maspardin may be involved in protein sorting in the late endosomal/lysosomal pathway. Thus, although we did not detect any changes in TrkB phosphorylation or degradation, it remains possible that ACP33/maspardin may mediate subtle changes in intracellular targeting of this or other growth factor receptors, as suggested previously for it effects on CD4 [13]. Lastly, since growth factors can regulate levels of microtubule-severing proteins in neurons and increase branching [21], this could account for the observed increase in branching. Importantly, however, we did not see an increase in protein levels of the SPG4 microtubule-severing protein spastin in the SPG21−/− mice. Another known interaction of maspardin is with the aldehyde dehydrogenase ALDH16A1 [14]. Interestingly, our comparative gene chip analysis identified a significant up-regulation in mRNA levels of another aldehyde dehydrogenase, ALDH18A1. However, very little is known about either of these two aldehyde dehydrogenase subtypes, and any pathogenic significance remains speculative.
Thus, a key factor limiting the interpretation of our data reporting an increase in axonal branching in SPG21−/− mice is that the function of maspardin itself is not well understood. Since it is a protein highly conserved throughout evolution, it likely plays a fundamental role within cells. Also, though the presumed catalytic triad of maspardin has some differences from other proteins in the esterase/lipase superfamily, it will be important to confirm that it lacks enzymatic activity. In some aspects, maspardin is reminiscent of a protein known as comparative gene identification-58 (CGI-58), which also has a short, continuous stretch of acidic amino acids at the N-terminus as well as a noncatalytic α/β-hydrolase domain [24]. CGI-58, also designated as α/β-hydrolase domain containing-5 (ABHD-5), is a lipid droplet-associated protein that activates adipose triglyceride lipase and acylates lysophosphatidic acid [24]. Since the SPG17 gene product seipin and the SPG20 gene product spartin are both associated with lipid droplets, and a number of other HSP proteins have been implicated in lipid metabolism [25–29], this pathway also could be affected. Interestingly, some animal models for amyotrophic lateral sclerosis also exhibit increased branching of axons [30], prefiguring a pathogenic link between some forms of HSP and amyotrophic lateral sclerosis. The identification of other maspardin-interacting proteins will clarify the function of maspardin and facilitate mechanistic investigations of the SPG21−/− mice.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
The authors wish to thank James Nagle and Debbie Kauffman (NINDS DNA Sequencing Facility) for DNA sequencing and Dr. Peng-Peng Zhu for generation of the phylogenetic tree. This work was supported by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke, National Institutes of Health [to C.S., J.S., H.J., C.B, and M.C.H.], the National Institute of General Medical Sciences Pharmacology Research Associate (PRAT) Program and the Swedish Research Council [grant numbers 13473, 20587 to O.S.].
Abbreviations
- CNS
Central nervous system
- DTA
Diphtheria toxin subunit A
- ES
Embryonic stem
- HSP
Hereditary spastic paraplegia
- MBP
Myelin basic protein
- NF
Neurofilament
- NGS
Normal goat serum
- NMJ
Neuromuscular junction
- PBS
Phosphate-buffered saline
- PFA
Paraformaldehyde
- TBS
Tris-buffered saline
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10048-010-0252-7) contains supplementary material, which is available to authorized users.
Contributor Information
Cynthia Soderblom, National Institutes of Health—Karolinska Institutet Graduate Partnerships Program, 171 77 Stockholm, Sweden; Department of Neuroscience, DBRM, Karolinska Institutet, 171 77 Stockholm, Sweden; Cellular Neurology Unit, Neurogenetics Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892, USA.
Julia Stadler, Cellular Neurology Unit, Neurogenetics Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892, USA.
Henri Jupille, Cellular Neurology Unit, Neurogenetics Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892, USA.
Craig Blackstone, Cellular Neurology Unit, Neurogenetics Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892, USA.
Oleg Shupliakov, Department of Neuroscience, DBRM, Karolinska Institutet, 171 77 Stockholm, Sweden.
Michael C. Hanna, Cellular Neurology Unit, Neurogenetics Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892, USA M. C. Hanna, Department of Biological and Environmental Sciences, Texas A & M University – Commerce, Commerce, TX 75428, USA.
References
- 1.Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–1155. doi: 10.1016/s0140-6736(83)92879-9. [DOI] [PubMed] [Google Scholar]
- 2.Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7:1127–1138. doi: 10.1016/S1474-4422(08)70258-8. [DOI] [PubMed] [Google Scholar]
- 3.Reid E. Science in motion: common molecular pathological themes emerge in the hereditary spastic paraplegias. J Med Genet. 2003;40:81–86. doi: 10.1136/jmg.40.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fink JK. Hereditary spastic paraplegia. Curr Neurol Neurosci Rep. 2006;6:65–76. doi: 10.1007/s11910-996-0011-1. [DOI] [PubMed] [Google Scholar]
- 5.Soderblom C, Blackstone C. Traffic accidents: molecular genetic insights into the pathogenesis of the hereditary spastic paraplegias. Pharmacol Ther. 2006;109:42–56. doi: 10.1016/j.pharmthera.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Züchner S. The genetics of hereditary spastic paraplegia and implications for drug therapy. Expert Opin Pharmacother. 2007;8:1433–1439. doi: 10.1517/14656566.8.10.1433. [DOI] [PubMed] [Google Scholar]
- 7.Stevanin G, Ruberg M, Brice A. Recent advances in the genetics of spastic paraplegias. Curr Neurol Neurosci Rep. 2008;8:198–210. doi: 10.1007/s11910-008-0032-z. [DOI] [PubMed] [Google Scholar]
- 8.Hu J, Shibata Y, Zhu P-P, Voss C, Rismanchi N, Prinz WA, Rapoport TA, Blackstone C. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell. 2009;138:549–561. doi: 10.1016/j.cell.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orso G, Pendin D, Liu S, Tosetto J, Moss TJ, Faust JE, Micaroni M, Egorova A, Martinuzzi A, McNew JA, Daga A. Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature. 2009;460:978–983. doi: 10.1038/nature08280. [DOI] [PubMed] [Google Scholar]
- 10.Park SH, Zhu P-P, Parker RL, Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J Clin Invest. 2010;120:1097–1110. doi: 10.1172/JCI40979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cross HE, McKusick VA. The Mast syndrome: a recessively inherited form of presenile dementia with motor disturbances. Arch Neurol. 1967;16:1–13. doi: 10.1001/archneur.1967.00470190005001. [DOI] [PubMed] [Google Scholar]
- 12.Simpson MA, Cross H, Proukakis C, Pryde A, Hershberger R, Chatonnet A, Patton MA, Crosby AH. Maspardin is mutated in Mast syndrome, a complicated form of hereditary spastic paraplegia associated with dementia. Am J Hum Genet. 2003;73:1147–1156. doi: 10.1086/379522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeitlmann L, Sirim P, Kremmer E, Kolanus W. Cloning of ACP33 as a novel intracellular ligand of CD4. J Biol Chem. 2001;276:9123–9132. doi: 10.1074/jbc.M009270200. [DOI] [PubMed] [Google Scholar]
- 14.Hanna M, Blackstone C. Interaction of the SPG21 protein ACP33/maspardin with the aldehyde dehydrogenase ALDH16A1. Neurogenetics. 2009;10:217–228. doi: 10.1007/s10048-009-0172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCray BA, Skordalakes E, Taylor JP. Disease mutations in Rab7 result in unregulated nucleotide exchange and inappropriate activation. Hum Mol Genet. 2010;19:1033–1047. doi: 10.1093/hmg/ddp567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu P-P, Patterson A, Lavoie B, Stadler J, Shoeb M, Patel R, Blackstone C. Cellular localization, oligomerization, and membrane association of the hereditary spastic paraplegia 3A (SPG3A) protein atlastin. J Biol Chem. 2003;278:49063–49071. doi: 10.1074/jbc.M306702200. [DOI] [PubMed] [Google Scholar]
- 17.Zheng Y-L, Li B-S, Veeranna Pant HC. Phosphorylation of the head domain of neurofilament protein (NF-M): a factor regulating topographic phosphorylation of NF-M tail domain KSP sites in neurons. J Biol Chem. 2003;278:24026–24032. doi: 10.1074/jbc.M303079200. [DOI] [PubMed] [Google Scholar]
- 18.Zhu P-P, Soderblom C, Tao-Cheng J-H, Stadler J, Blackstone C. SPG3A protein atlastin-1 is enriched in growth cones and promotes axon elongation during neuronal development. Hum Mol Genet. 2006;15:1343–1353. doi: 10.1093/hmg/ddl054. [DOI] [PubMed] [Google Scholar]
- 19.Solowska JM, Morfini G, Falnikar A, Himes BT, Brady ST, Huang D, Baas PW. Quantitative and functional analyses of spastin in the nervous system: implications for hereditary spastic paraplegia. J Neurosci. 2008;28:2147–2157. doi: 10.1523/JNEUROSCI.3159-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riano E, Martignoni M, Mancuso G, Cartelli D, Crippa F, Toldo I, Siciliano G, Di Bella D, Taroni F, Bassi MT, Cappelletti G, Ruglarli EI. Pleiotropic effects of spastin on neurite growth depending on expression levels. J Neurochem. 2009;108:1277–1288. doi: 10.1111/j.1471-4159.2009.05875.x. [DOI] [PubMed] [Google Scholar]
- 21.Qiang L, Yu W, Liu M, Solowska JM, Baas PW. Basic fibroblast growth factor elicits formation of interstitial axonal branches via enhanced severing of microtubules. Mol Biol Cell. 2010;21:334–344. doi: 10.1091/mbc.E09-09-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madduri S, Papaloïzos M, Gander B. Synergistic effect of GDNF and NGF on axonal branching and elongation in vitro. Neurosci Res. 2009;65:88–97. doi: 10.1016/j.neures.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 23.Reid E, Kloos M, Ashley-Koch A, Hughes L, Bevan S, Svenson IK, Graham FL, Gaskell PC, Dearlove A, Pericak-Vance MA, Rubinsztein DC, Marchuk DA. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10) Am J Hum Genet. 2002;71:1189–1194. doi: 10.1086/344210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schweiger M, Lass A, Zimmermann R, Eichmann TO, Zechner R. Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am J Physiol Endocrinol Metab. 2009;297:E289–E296. doi: 10.1152/ajpendo.00099.2009. [DOI] [PubMed] [Google Scholar]
- 25.Eastman SW, Yassaee M, Bieniasz PD. A role for ubiquitin ligases and spartin/SPG20 in lipid droplet turnover. J Cell Biol. 2009;184:881–894. doi: 10.1083/jcb.200808041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edwards TL, Clowes VE, Tsang HT, Connell JW, Sanderson CM, Luzio JP, Reid E. Endogenous spartin (SPG20) is recruited to endosomes and lipid droplets and interacts with the ubiquitin E3 ligases AIP4 and AIP5. Biochem J. 2009;423:31–39. doi: 10.1042/BJ20082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Windpassinger C, Auer-Grumbach M, Irobi J, Patel H, Petek E, Hörl G, Malli R, Reed JA, Dierick I, Verpoorten N, Warner TT, Proukakis C, Van den Bergh P, Verellen C, Van Maldergem L, Merlini L, De Jonghe P, Timmerman V, Crosby AH, Wagner K. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat Genet. 2004;36:271–276. doi: 10.1038/ng1313. [DOI] [PubMed] [Google Scholar]
- 28.Rainier S, Bui M, Mark E, Thomas D, Tokarz D, Ming L, Delaney C, Richardson RJ, Albers JW, Matsunami N, Stevens J, Coon H, Leppert M, Fink JK. Neuropathy target esterase gene mutations cause motor neuron disease. Am J Hum Genet. 2008;82:780–785. doi: 10.1016/j.ajhg.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsaousidou MK, Ouahchi K, Warner TT, Yang Y, Simpson MA, Laing NG, Wilkinson PA, Madrid RE, Patel H, Hentati F, Patton MA, Hentati A, Lamont PJ, Siddique T, Crosby AH. Sequence alterations within CYP7B1 implicate defective cholesterol homeostasis in motor-neuron degeneration. Am J Hum Genet. 2008;82:510–515. doi: 10.1016/j.ajhg.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kabashi E, Lin L, Tradewell ML, Dion PA, Bercier V, Bourgouin P, Rochefort D, Bel Hadj S, Durham HD, Vande Velde C, Rouleau GA, Drapeau P. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet. 2010;19:671–683. doi: 10.1093/hmg/ddp534. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.