

Abstract
Objective: To examine the potential influence of chronic inflammation on peripheral motor nerve function in vivo following spinal cord injury (SCI). Methods: This study was part of a randomized, parallel-group, controlled clinical trial. The study included 20 participants with varying levels and severities of SCI randomized (3:2) to either a treatment group, consisting of a 12-week anti-inflammatory diet program, or control group. Outcome measures were assessed at baseline, 1 month, and 3 months and consisted of measures of motor nerve conduction velocity (NCV) and amplitude as well as markers of inflammation as assessed by various pro- and anti-inflammatory cytokines. Results: Despite a significant reduction in inflammation in the treatment group, 2-way repeated measures analysis of variance (ANOVA) showed no significant Group × Time interaction for motor NCV (p = .77) or M-wave amplitude (p = .61). Further, the change in motor NCV and M-wave amplitude were not shown to be associated with the change in inflammatory mediators as assessed via a backwards elimination multiple regression analysis. Conclusion: These results suggest that at physiologically relevant concentrations, inflammatory mediators may not have a substantial influence on peripheral motor nerve conduction in vivo following SCI. Future studies may still be warranted to examine the potential for central effects.
Keywords: anti-inflammatory, chronic inflammation, motor, nerve conduction velocity, spinal cord injury
A state of chronic inflammation is commonly reported following spinal cord injury (SCI).1–5 This persistent inflammatory state, characterized by chronically elevated levels of proinflammatory cytokines, can be attributed to a number of factors that commonly arise following SCI. This population is at a heightened risk for acute secondary health complications such as pressure ulcers, urinary tract infections, and respiratory infections. In addition, the loss of motor function often leads to a more sedentary lifestyle, contributing to the risk for the development of a variety of inflammation-inducing metabolic disorders such as obesity, cardiovascular disease, and type 2 diabetes. Damage to the spinal cord may also directly result in immune and endocrine dysfunction due to the respective loss of sympathetic innervation of lymphoid organs and dysregulation of the hypothalamic-pituitary-adrenal axis.6
In addition to concerns related to immune dysregulation and the heighted risk of infection, a primary issue of SCI is the loss of varying degrees of motor and sensory function. Such losses arise from structural damage to neural tracts within the spinal cord, whereby an injury affecting all tracts would result in a motor and sensory complete injury. Previous research has demonstrated that it is possible for an injury to present clinically as motor or sensory complete despite the preservation of structurally healthy neural tracts (as shown by MRI). Such injuries are classified as discomplete. In these cases, it is possible to speculate that some form of nonstructurally based influence may be inducing deficits in these otherwise healthy tracts.7,8 As evidence exists to suggest that inflammatory mediators may possess the ability to influence ion channel kinetics and induce conduction deficits, this potential relationship may be worthy of further examination.
A study by Davies et al9 demonstrated under ex vivo conditions that highly elevated concentrations of different cytokines (eg, TNF-α) were capable of inducing conduction deficits in a reversible, dose-dependent manner. This relationship has also been demonstrated in vivo, in able-bodied humans, under conditions of acute, exercise-induced elevations of inflammatory mediators.10
Despite previous research, suggesting the potential for an inflammatory-induced channelopathy, no study has demonstrated a causal relationship in humans, in vivo, under physiologically relevant levels of chronic inflammation. If such a relationship exists, it may be possible to induce conduction improvements in motor nerves in those with SCI via the implementation of anti-inflammatory strategies. The purpose of this study was to examine the relationship between various pro- and anti-inflammatory mediators and peripheral motor nerve conduction in a population with SCI. Based on the relationship observed between acute elevations in inflammatory mediators and nerve conduction in previous studies, it was hypothesized that reducing levels of chronic inflammation would result in corresponding improvements in motor nerve conduction.
Materials and Methods
Study design and participants
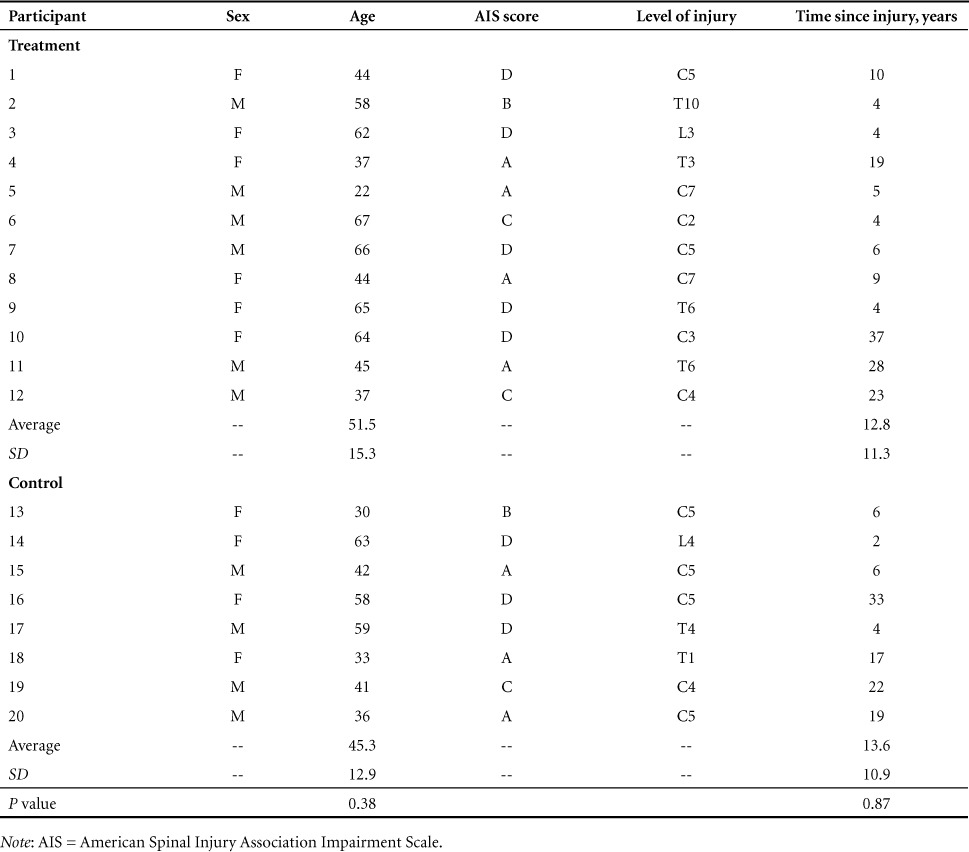
This study was performed as a component of a larger clinical trial (clinicaltrials.gov identifier: NCT02099890) that included the examination of depression,11 neuropathic pain, and cognitive impairment. Data pertaining to the change in inflammatory mediators have been previously published.11 The study was a randomized, parallel-group clinical trial. Participant recruitment occurred between September and November 2014. The study intervention was 12 weeks and included testing at baseline, 1 month, and 3 months. Participants with various levels and severities of upper motor neuron SCI, free from peripheral neuropathy, were recruited for participation in the study. Additional inclusion criteria included (1) over the age of 18 years, (2) SCI of any level or severity (American Spinal Injury Association Impairment Scale [AIS] A-D), and (3) at least 2 years post injury. Exclusion criteria included (1) any contraindications to supplements provided in the study, (2) unstable medical condition within 2 weeks prior to intervention, (3) pregnancy, and (4) breastfeeding. Participant characteristics are shown in Table 1. Twenty individuals (10 male, 10 female; age 48.7 ± 13.9 years) with chronic (4–37 years post injury) SCI (C2-L4; AIS A-D) were recruited for participation in the study. Twelve participants were randomly allocated to the treatment group and were placed on the 12-week anti-inflammatory diet intervention, while 8 were allocated to the control group and received no intervention. Informed consent was obtained from all participants. The study was registered as a clinical trial and received ethical approval from the university's research ethics board as well as the Natural Health Products Directorate of Canada.
Table 1.
Participant characteristics
Randomization
Randomization was computer generated by the primary investigator and stratified by participant gender and age using permuted blocks of 2 (male/female) and blocks of 3 (<40, 40–60, >60 years). Randomization was 3:2 to either the anti-inflammatory diet or control condition (which received no intervention).
Anti-inflammatory diet intervention
The anti-inflammatory diet intervention focused on the elimination of common food intolerances and inflammation-inducing foods, as well as the introduction of foods and supplements with established anti-inflammatory properties. Examples of foods removed from the diet included those with high glycemic indices (such as refined wheat products and refined sugars), common intolerances such as cow's milk, and foods that negatively influence cardiovascular health such as hydrogenated oils. Participants also consumed daily supplements with established anti-inflammatory benefits. Omega-3 (NOW ultra omega-3) was taken in softgel form, containing 500 mg EPA and 250 mg DHA, at a dosage of 3 per day. Chlorella (NOW chlorella) was taken in pill form, containing 1,000 mg, at a dosage of 6 per day. Antioxidants (CanPrev Antioxidant Network) were taken in pill form, containing 100 mg coenzyme Q10, 200 mg n-acetyl-cysteine, 150 mg mixed tocopherols, 100 mg DL alpha lipoic acid, 60 mg green tea extract, 5.5 mg zinc, and 100 μg selenium, at dosage of 2 per day. Curcumin (AOR InflaNOx) was taken in pill form, containing 400 mg, at a dosage of 3 per day. A vegetable-based protein powder (Progressive Vegessential) containing 27 g of protein was taken at a dosage of one scoop each morning.
The treatment and control groups were asked to complete a 7-day detailed diet record at baseline and a 3-day record at 1 month, 2 months, and 3 months during the intervention to establish baseline eating habits and assess compliance throughout the intervention. Food intake was assessed using The Food Processor software (version 10.14.2; ESHA Inc., Salem, OR). Compliance to the specific anti-inflammatory diet was also assessed by a detailed analysis of all diet records. Each food item was categorized as either a “food to consume,” a “food to avoid,” or a “neutral food” based on the parameters of the diet participants were instructed to follow. Food was categorized into servings in accordance with Canada's Food Guide. Therefore, compliance scores were based on standard servings of foods subjects were instructed to eat versus foods they were instructed to avoid. To account for differences in total energy intake, compliance scores were expressed as a ratio of the servings of foods to consume over the total servings of food (avoid + consume) multiplied by 100. The percent compliance was then generated.
Participants in the treatment group attended an information seminar that described the diet program followed by a one-on-one consultation with nutritionists during which their diet records were reviewed in detail and necessary changes were discussed. Participants received information regarding which foods to eat and avoid, a supplement intake schedule, and list of approved recipes. Participants in the treatment group received support via weekly phone calls from members of the research team and a monitored online support group whereby participants could share recipes and experiences with one another. Participants in the control group were asked to maintain their current diets throughout the duration of the study.
Measurement of serum inflammatory markers
Blood draws (20 mL) were taken from the antecubital vein of each participant at 1:00 p.m. at each of the 3 testing sessions (baseline, 1 month, and 3 months). Following extraction, the whole blood was allowed to clot for 30 minutes followed by centrifugation at 1000 × g for 15 minutes. Serum was extracted and immediately stored at −80°C until later analysis. Inflammatory mediators of interest included the pro-inflammatory cytokines IL-2, IL-1β, IL-6, TNF-α, and IFN-γ; the acute phase protein CRP; the anti-inflammatory cytokines IL-4, IL-10, and IL-1RA; and the pro-inflammatory eicosanoid PGE2. Analysis of pro- and anti-inflammatory cytokines was performed in triplicate via the Magpix Multiplex system (EMD Millipore, MA) and analyzed using Luminex software. CRP and PGE2 were analyzed in triplicate and quantified via enzyme-linked immunosorbent assay (R&D Systems, Minneapolis, MN).
Assessment of nerve conduction
Motor nerve conduction velocity (NCV) and M-wave amplitude were assessed using compound motor action potential (CMAP) recordings by stimulating the median nerve and recording the corresponding motor response of the thenar muscles. Subjects lay in a supine position with the elbow in full extension. Prior to performing the test, the electrode locations were prepared by shaving the skin, removing any dead skin cells with an abrasive gel, and disinfecting the areas with rubbing alcohol. Surface electrodes were placed in a monopolar configuration with the recording electrode directly over the motor point of the thenar muscles, the reference electrode over the tendon of the interphalangeal joint, and the ground on the palm of the hand. Stimulation of the median nerve was performed distally at the wrist between the flexor tendons of the hand and proximally on the medial side of the biceps brachii. Stimulation intensity was determined by gradually increasing the amplitude until a maximal M-wave was achieved. A stimulation amplitude of 120% of this value was then used during testing. The evoked responses were amplified using a bandwidth of 10 Hz to 1 kHz. A total of 10 trials were taken and averaged for each stimulation site and M-wave onset was used to determine latency. The amplitude of the M-wave was also assessed as an indication of the strength of the motor response. Sweep speed was 2 ms per division, sensitivity was 2 μV per division, and stimulus duration was 0.2 ms at a stimulus rate of 3 Hz. Recordings were performed on the Dantec Keypoint EMG (Dantec Medical A/S, Skovlunde, Denmark). Skin temperature was assessed with a surface probe placed over the thenar muscles to ensure consistent temperatures between testing sessions (MLT422/D skin temperature probe; AD Instruments, Colorado Springs, CO).
Statistical analysis
Two-way (Group × Time) repeated measures analysis of variance (ANOVA) were performed to investigate possible changes in scores of motor NCV and amplitude across 3 testing sessions (baseline, 1 month, 3 month). Two-way repeated measures ANOVA were also performed for the proinflammatory cytokine TNF-α and the eicosanoid PGE2. As the remaining inflammatory mediators were not normally distributed, nonparametric analyses were performed. A Friedman test of differences among repeated measures (baseline, 1 month, 3 month) for the treatment group and control group was performed. If the Friedman test resulted in a significant value, a Wilcoxon signed-rank test was then performed to provide specific information regarding which time points were significantly different from one another. Finally, a Mann-Whitney test was performed on change scores (3 month - baseline) between groups to establish whether the change experienced in inflammatory mediators significantly differed between groups. These data are expressed as means ± SD. Correlations between changes in inflammatory mediators and measures of nerve conduction were assessed by means of Pearson's r correlation. Statistical significance was set at p ≤ .05 for all tests.
Results
All participants from both the treatment and control groups completed the entire 3-month duration of the study and were included in the analysis. No adverse events were reported. The participants' overall compliance to the diet was assessed based on the average of the 3 diet records during the study (1 month, 2 months, and 3 months). One participant completed all 3 testing sessions but failed to produce the 2-month and the 3-month diet records. This participant had a dietary compliance over the first month of 92%. All other participants completed each of the required diet records and overall compliance ranged from 70% to 100%, with a mean compliance of 89%. A detailed analysis regarding specific diet adherence data will be presented elsewhere.
Change in inflammatory mediators
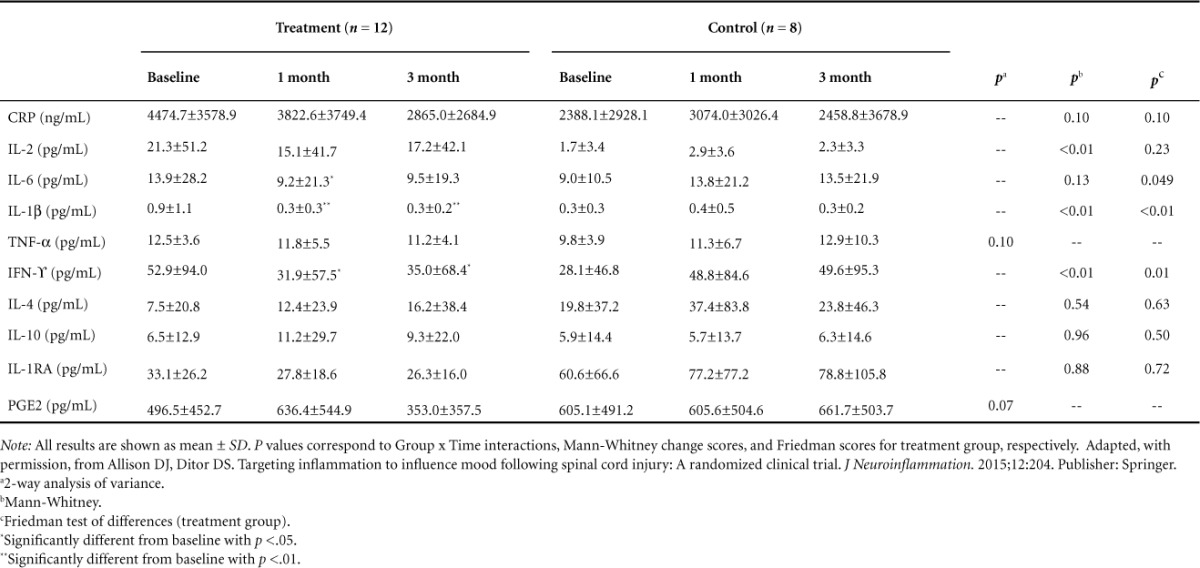
Changes in serum levels of inflammatory mediators are shown in Table 2. A Mann-Whitney test indicated that the change scores (3 month - baseline) were significantly different between the treatment group and the control group for IFN-γ (U = 13.0, p = .01), IL-1β (U = 14.0, p = .01), and IL-2 (U = 12.0, p = .01) and showed a trend for CRP (U = 27.0, p = .10). Friedman test showed that in the treatment group there was a statistically significant reduction in IFN-γ (χ2 = 8.67, p = .01), IL-1β (χ2 = 17.78, p < .01), IL-6 (χ2 = 6.17, p < .05) and a trend for CRP (χ2 = 4.5, p = .10). The Friedman test showed no statistically significant reductions for any inflammatory mediator in the control group. Post hoc analysis performed with the Wilcoxon signed-rank test showed significant reductions in the treatment group for IFN-γ from baseline to 1 month and baseline to 3 months (z = −2.275, p = .02; z = −2.510, p = .01, respectively), as well as significant reductions in the treatment group for IL-1β from baseline to 1 month and baseline to 3 months (z = −3.059, p < .01; z = −2.934, p < 0.01, respectively), a significant reduction in the treatment group for IL-6 from baseline to 1 month, and a trend from baseline to 3 months (z = −2.275, p = .02; z = −1.726, p = .08 respectively). Two-way repeated measures ANOVA were performed for the normally distributed mediator's TNF-α and PGE2 and showed trends toward Group × Time interactions (p = .10 and p = .07, respectively).
Table 2.
Changes in inflammatory mediators
Change in motor nerve conduction
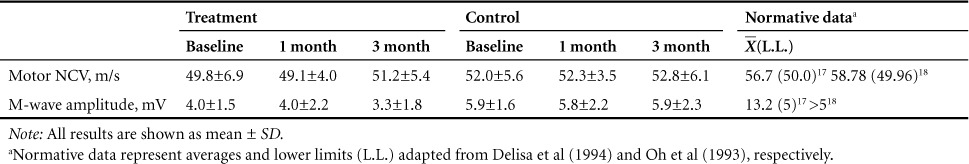
Changes in motor NCV and M-wave amplitude are shown in Table 3. No significant Group × Time interactions were observed for motor NCV (p = .77; Cohen's d = 0.26) or M-wave amplitude (p = .61; Cohen's d = 0.35).
Table 3.
Changes in motor nerve conduction velocity (NCV) and amplitudes
Relationship between inflammatory mediators and motor nerve conduction
To help elucidate a potential relationship between changes in inflammatory mediators and changes in motor NCV and M-wave amplitude, a step-wise backward elimination multiple regression analysis was performed. Due to high levels of multicollinearity, the proinflammatory cytokines IL-6 and IL-2 were excluded from the analysis. Following the removal of these analytes, all independent variables demonstrated acceptable tolerance scores (>0.20). For motor NCV, a nonsignificant regression equation was found [F(1, 16) = 3.20, p = .09)], with an R2 of .166. A nonsignificant regression equation was also found for M-wave amplitude [F(1, 16) = 3.06, p = .10], with an R2 of .161.
Discussion
The present study was the first to examine the influence of reducing chronically elevated levels of inflammation on somatic nerve conduction in vivo in humans. The intervention was successful at obtaining reductions in inflammatory mediators, yet failed to induce any changes in peripheral motor nerve conduction. As previous research has provided evidence that certain inflammatory mediators may induce channelopathy under acutely elevated concentrations,9,10 it was hypothesized that similar or even more severe effects would be observed under chronically elevated conditions. Further, if such channelopathic effects were to occur in humans, in vivo, individuals with SCI may be expected to be particularly at risk due to the high prevalence of both chronic inflammation and somatic nerve deficits.
Inflammatory mediators have been proposed to contribute to such somatic nerve deficits by interfering with neuronal membrane channels and blocking the exchange of ions across the membrane. If sodium (Na+) and potassium (K+) exchange is sufficiently blocked, normal membrane depolarization and/or repolarization may be disrupted, thereby causing a reduction in nerve excitability related to motor nerve channelopathy.9,12 Such an influence would be fitting of the dose-dependent, reversible conduction deficits observed in previous ex vivo studies9 but did not seem apparent in the current study.
Inflammatory mediators also have a well-established enhancing effect on nociceptive sensory fibers. While a potential influence on A-β fibers remains unclear, inflammatory mediators are known to sensitize fibers responsible for nociception, such as A-δ and C fibers, at various points along the nociceptive pathway. Peripherally, inflammatory mediators such as PGE2 may act on corresponding receptors on the nociceptor and induce a protein-kinase A-mediated phosphorylation of sodium channels, thereby causing peripheral sensitization.13 IL-1β and TNF-α have also been shown to increase excitability in nociceptive neurons and enhance sodium currents.14–16
Despite the significant reduction in proinflammatory mediators in the treatment group, no significant changes in motor nerve conduction were achieved; this suggests that under physiologically relevant concentrations reducing chronically elevated inflammatory mediators may not be sufficient to induce meaningful changes in motor nerve conduction. No significant Group × Time interaction was observed for motor NCV or amplitude. Further no significant correlations were observed between the changes in either index of motor conduction and the change in any inflammatory mediator. At baseline, motor NCV and amplitude values tended to be lower than what is considered normal in a healthy able-bodied population17,18; however, among all the inflammatory mediators examined, very few significant correlations were found. This may suggest that no particularly robust relationship exists between motor nerve conduction and pro- or anti-inflammatory mediators in vivo. These results are clinically relevant, as they may suggest that interventions that target inflammation may not be an effective strategy in terms of improving motor conduction in individuals with SCI.
Several potential study limitations should be noted. Although no changes in motor nerve conduction were observed in the present study, some degree of influence may still be possible under particularly severe levels of inflammation. It may also be possible that inflammatory mediators not assessed in the current study may have demonstrated a stronger relationship with measures of somatic nerve conduction. However, in terms of generalizability, our sample was representative of the SCI population in Canada regarding age, level, and severity of injury19 and the elevated levels of inflammation demonstrated were comparable to those previously reported for this population.1,3,20 In addition, the current study examined a wide array of both pro- and anti-inflammatory cytokines. As such, we feel confident that these results are representative of the average SCI population. It should also be noted that as only peripheral nerve conduction was examined, it is not possible to form any conclusions regarding potential central effects. Future research will be necessary to confirm or deny a lack of effect on central somatic nerve conduction.
Conclusion
This study demonstrated a lack of change in peripheral motor nerve conduction despite a reduction of proinflammatory mediators in a population with SCI. Such results may suggest that, when at physiologically relevant concentrations, inflammatory mediators do not have a substantial enough influence to induce meaningful alterations in peripheral motor nerve conduction in individuals with SCI. This result is of clinical significance, as it may suggest that, despite previously reported ex vivo findings, inflammatory mediators may not provide a suitable target for the enhancement of peripheral motor nerve conduction following SCI. Future, larger scale studies will, however, be required to examine the potential for central effects.
Acknowledgments
The authors declare no conflict of interest. This study was supported by the Ontario Neurotrauma Foundation. ClinicalTrials.gov ID: NCT02099890. We wish to thank Now, CanPrev, AOR, and Progressive for providing the supplements utilized in the dietary intervention.
REFERENCES
- 1. Davies AL, Hayes KC, Dekaban GA.. Clinical correlates of elevated serum concentrations of cytokines and autoantibodies in patients with spinal cord injury. Arch Phys Med Rehabil. 2007; 88 11: 1384– 1393. [DOI] [PubMed] [Google Scholar]
- 2. Hayes KC, Hull TCL, Delaney GA, . et al. Elevated serum titers of proinflammatory cytokines and CNS autoantibodies in patients with chronic spinal cord injury. J Neurotrauma. 2002; 19 6: 753– 761. [DOI] [PubMed] [Google Scholar]
- 3. Gibson AE, Buchholz AC, Martin Ginis KA.. C-Reactive protein in adults with chronic spinal cord injury: Increased chronic inflammation in tetraplegia vs paraplegia. Spinal Cord. 2008; 46 9: 616– 621. [DOI] [PubMed] [Google Scholar]
- 4. Liang H, Mojtahedi MC, Chen D, Braunschweig CL.. Elevated C-reactive protein associated with decreased high-density lipoprotein cholesterol in men with spinal cord injury. Arch Phys Med Rehabil. 2008; 89 1: 36– 41. [DOI] [PubMed] [Google Scholar]
- 5. Segal J, Gonzales E, Yousefi S, Jamshidipour L, Brunnemann S.. Circulating levels of IL-2R, ICAM-1, and IL-6 in spinal cord injuries. Arch Phys Med Rehabil. 1997; 78 1: 44– 47. [DOI] [PubMed] [Google Scholar]
- 6. Allison DJ, Ditor DS.. Immune dysfunction and chronic inflammation following spinal cord injury. Spinal Cord. 2014; 53 1: 14– 18. [DOI] [PubMed] [Google Scholar]
- 7. Sherwood AM, Dimitrijevic MR, McKay WB.. Evidence of subclinical brain influence in clinically complete spinal cord injury: Discomplete SCI. J Neurol Sci. 1992; 110 1–2: 90– 98. [DOI] [PubMed] [Google Scholar]
- 8. Yu K, Rong W, Li J, . et al. Neurophysiological evidence of spared upper motor conduction fibers in clinically complete spinal cord injury: Discomplete SCI in rats. J Neurol Sci. 2001; 189 1–2: 23– 36. [DOI] [PubMed] [Google Scholar]
- 9. Davies AL, Hayes KC, Shi R.. Recombinant human TNFα induces concentration-dependent and reversible alterations in the electrophysiological properties of axons in mammalian spinal cord. J Neurotrauma. 2006; 23 8: 1261– 1273. [DOI] [PubMed] [Google Scholar]
- 10. Allison DJ, Green LA, Gabriel DA, Roy BD, Greig Inglis J, Ditor DS.. Elevated concentrations of circulating cytokines and correlations with nerve conduction velocity in human peripheral nerves. J Neuroimmunol. 2014; 277 102: 134– 139. [DOI] [PubMed] [Google Scholar]
- 11. Allison DJ, Ditor DS.. Targeting inflammation to influence mood following spinal cord injury: A randomized clinical trial. J Neuroinflammation. 2015; 12 1: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gutmann L, Gutmann L.. Axonal channelopathies: An evolving concept in the pathogenesis of peripheral nerve disorders. Neurology. 1996; 47: 18– 21. [DOI] [PubMed] [Google Scholar]
- 13. Samad TA, Sapirstein A, Woolf CJ.. Prostanoids and pain: Unraveling mechanisms and revealing therapeutic targets. Trends Mol Med. 2002; 8 8: 390– 396. [DOI] [PubMed] [Google Scholar]
- 14. Jin X, Gereau RW.. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J Neurosci. 2006; 26 1: 246– 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Binshtok AM, Wang H, Zimmermann K, . et al. Nociceptors are interleukin-1beta sensors. J Neurosci. 2008; 28 52: 14062– 14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sommer C, Kress M.. Recent findings on how proinflammatory cytokines cause pain: Peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neurosci Lett. 2004; 361 1–3: 184– 187. [DOI] [PubMed] [Google Scholar]
- 17. DeLisa J, Lee H, Baran E, Lai K, Speilholz N.. Manual of Nerve Conduction Velocity and Clinical Neurophysiology. 3rd ed. New York: Raven Press; 1994. [Google Scholar]
- 18. Oh S. Clinical Electromyography: Nerve Conduction Studies. 2nd ed. Baltimore, MD: Williams & Wilkins; 1993. [Google Scholar]
- 19. Noonan VK, Fingas M, Farry A, . et al. Incidence and prevalence of spinal cord injury in Canada: A national perspective. Neuroepidemiology. 2012; 38 4: 219– 226. [DOI] [PubMed] [Google Scholar]
- 20. Manns PJ, McCubbin JA, Williams DP.. Fitness, inflammation, and the metabolic syndrome in men with paraplegia. Arch Phys Med Rehabil. 2005; 86 6: 1176– 1181. [DOI] [PubMed] [Google Scholar]