

Abstract
The Bing-Neel syndrome is a rare neurological complication of Waldenström’s Macroglobulinemia which results from a direct involvement of central nervous system by malignant lymphoplasmacytic cells. The clinical suspicion of Bing-Neel syndrome may be overlooked because neurologic symptoms are heterogeneous, nonspecific and sometimes underhand. A definitive diagnosis of Bing-Neel syndrome can be confidently made using brain and spinal cord magnetic resonance imaging as well as histopathology and/or cerebrospinal fluid analysis to confirm the neoplastic infiltration of central nervous system. The detection in the cerebrospinal fluid of patients with Bing-Neel syndrome of the MYD88 (L265P) somatic mutation, which is highly recurrent in Waldenström’s Macroglobulinemia, proved useful for the diagnosis and monitoring of central nervous system involvement. Despite recommendations recently published, there is still no clear consensus on treatment of Bing-Neel syndrome, which includes systemic immunochemotherapy, intrathecal chemotherapy and brain irradiation as possible options. Ibrutinib, a Bruton kinase inhibitor approved for Waldenström’s Macroglobulinemia, has been recently added to the therapeutic armamentarium of Bing-Neel syndrome due to its ability to pass the blood-brain barrier. However, prospective clinical trials are eagerly awaited with the aim to define the optimal treatment strategy.
Here we describe four illustrative cases of Bing-Neel syndrome diagnosed and treated at our Institution and review the literature on this topic.
Keywords: Bing-Neel Syndrome, Waldenström’s Macroglobulinemia, Central Nervous System
Introduction
Waldenström’s Macroglobulinemia (WM) is a rare B-cell lymphoproliferative disorder characterized by the presence of a serum IgM paraprotein associated with bone marrow infiltration by lymphoplasmacytic lymphoma (LPL).1 Although WM is primarily localized in the bone marrow, up to 15–20% of patients has an extramedullary disease with lymphadenopathies and/or splenomegaly, while extranodal involvement is uncommon.2
The Bing-Neel syndrome (BNS) is a rare neurological complication of WM resulting from a direct infiltration of central nervous system (CNS) by lymphoplasmacytic cells that may occur at any time during the course of the disease. 3 BNS was first described in 1936 by Jens Bing and Axel Valdemar Neel who observed two women with neurological symptoms in the setting of hyperglobulinemia, in whom no evidence of myeloma was found at autopsy.4
Limited information about the incidence, clinical presentation, prognosis and treatment of BNS is currently available in the literature. Since the first description, approximately 50 patients with BNS have been reported as case reports, while in the last few years two retrospective series including 44 and 34 patients respectively have been published.5,6
During the 8th International Workshop of WM held in London in 2014 a task force on BNS was established with the aim to produce practical guidelines for the diagnosis and treatment of BNS.7
Here we present four cases of BNS diagnosed and treated at a single institution between 2012 and 2016, and review the literature on this rare complication of WM.
Description of Cases
Patient 1
This case was partially reported in a previous publication8 and here updated with a longer follow-up. A 64-year-old man was admitted to hospital for confusion, progressive cognitive decline, slurred speech, and ataxia. Cerebro-spinal fluid (CSF) analysis showed a high white blood cell (WBC) count (196/mm3), an elevated protein level (67 mg/dl) with normal glucose values (73 mg/dl). Flow cytometric analysis showed a clonal B lymphocyte population CD19+, CD20+, CD22+, SIg+, CD5+, CD23−, CD10−, FMC7+, CD79b+ representing 73% of WBC. Brain and spinal MRI showed communicating normal pressure hydrocephalus and both subtentorial and hemispheric leptomeningeal enhancement after gadolinium (Figure 1A). An IgM kappa monoclonal (M) protein was found in the serum (3 g/L). Bone marrow biopsy showed infiltration by LPL (30% of cellularity). The MYD88 (L265P) mutation was detectable by allele-specific PCR on bone marrow CD19+ mononuclear cells. Total body computed tomography (CT) scan revealed multiple bone lesions in the pelvis, and 18-Fluorodeoxyglucose Positron Emission Tomography (18-FDG-PET) showed an abnormally high uptake in the pelvis, with a maximum standardized uptake value (SUV) of 7.6. The biopsy of the largest bone lesion showed an infiltration by lymphoplasmacytic lymphoma. The final diagnosis was WM with BNS as presenting symptom. The patient was initially treated with immunochemotherapy with R-HyperC-VAD (rituximab plus high-dose methotrexate and cytarabine alternating with hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone) but treatment was withheld after the first dose of methotrexate due to acute renal failure and worsening of patient’s clinical conditions. Three weekly intrathecal administrations of methotrexate 15 mg were given. The patient was considered ineligible to further intensive chemotherapy and then switched to
Figure 1A.

FLAIR ax shows dilatation of ventricles and thickening of leptomeningeal sheaths, T1ax post gadolinium shows slightly increased interhemispheric enhancement of the meningeal sheaths (circle), T2 cor shows dilatation of ventricles, in particular of frontal and temporal horns of the laterals ventricles, rounded shaped (arrows).
Rituximab plus Bendamustine (28-day cycles with Rituximab 375 mg/m2 day 1 and Bendamustine 90 mg/m2 days 1–2) associated with intrathecal Methotrexate 15 mg on day 1 of each cycle. At the end of treatment, the patient’s clinical conditions were markedly improved. Serum paraprotein after therapy was 0.5 g/L. MRI of the brain showed the persistence of communicating hydrocephalus and almost complete disappearance of the leptomeningeal enhancement (Figure 1B). Flow cytometry on CSF was normal. Bone marrow biopsy showed the complete regression of lymphoplasmacytic infiltration, and MYD88 (L265P) mutation was undetectable on bone marrow CD19+ mononuclear cells. MRI of the pelvis showed a reduction of bone lesions, with a normal uptake at 18-FDG-PET. After 6 months, the patient underwent autologous stem cells transplantation. The patient is in complete remission, with undetectable monoclonal protein in serum and urine 21 months after transplant.
Figure 1B.

FLAIR ax confirms dilatation of ventricles, T1ax post gadolinium shows the persistence of a slight leptomeningeal enhancement (circle).
Patient 2
A 60-year-old woman was admitted to hospital because of ataxia and a distal sensitive-motor deficit to the four limbs. Electroneurography (ENG) and electromyography (EMG) showed a severe sensory-motor demyelinating polyneuropathy. Blood analyses revealed the presence of a small serum IgM kappa M protein (7.3 g/L) and presence of anti-Myelin Associated Glycoprotein (MAG) antibodies (title 1:193000). Brain and spinal MRI revealed thickening and contrast enhancement of spinal leptomeninges and roots of cauda equina, shaded enhancement of pia mater and ependyma and of bilateral internal auditory meatus (Figure 2A and 2B). The CSF analysis showed an increased WBC count (105/mmc) and a high protein level (121 mg/dl) reflecting blood-brain barrier disruption. Cytofluorimetric analysis of CSF identified the presence of monotypic CD20+ CD5+, CD23−, CD10− lymphocytes with kappa chain monoclonal restriction, representing 84% of WBC. The bone marrow biopsy showed a lymphoplasmacytic infiltration (60–70% of cellularity) consistent with an LPL. MYD88 and CXCR4 mutation status were evaluated on CD19+-selected bone marrow mononuclear cells using allele-specific PCR and Sanger sequencing respectively. The patient was found to be MYD88-mutated and CXCR4-wild type.
Figure 2A.

T1 sag shows thickening of cauda equina roots (arrow), T1 sag post gadolinium shows thickening and enhancement of spinal leptomeninges and cauda equina roots (arrow).
Figure 2B.

T1 ax after post gadolinium demonstrates bilateral internal auditory meatus enhancement (circles).
Six 28-day cycles of Rituximab (375 mg/m2 day 1) and Bendamustine (90 mg/m2 days 1–2) associated with six intrathecal injections of methotrexate (15 mg day 1) were administered. At the end of therapy, chemistry and cytofluorimetry on CSF were normal. Neurological symptoms remained stable while post-treatment MRI showed the absence of contrast enhancement in the spinal cord and cauda equina. Bone marrow biopsy was normal. These findings, taken together, were consistent with a partial response according to current guidelines.7 After three months neurological symptoms worsened. Brain and spinal MRI (Figure 2C and 2D) showed thickening of roots of cauda equina and shaded contrast enhancement of medullary cone and leptomeninges in the posterior cranial fossa. Cytofluorimetric CSF analysis detected a clonal B lymphocyte population, accounting for 44% of WBC, indicating CNS progression of the disease. Since Ibrutinib was shown to pass the blood-brain-barrier and to be active in BNS,9,10 treatment was started in March 2017.
Figure 2C.

T1 sag shows thickening of cauda equina roots (arrow); T1 sag post gadolinium shows persistent thickening and enhancement of spinal leptomeninges and roots of cauda equina (arrow).
Figure 2D.
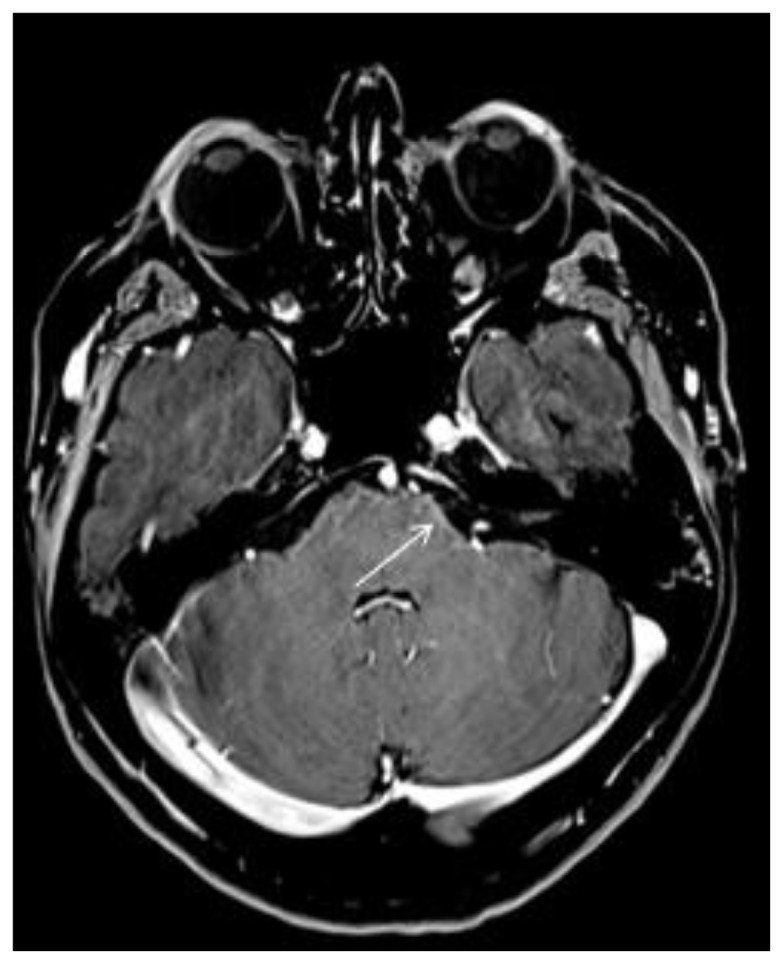
T1 ax post gadolinium shows shaded enhancement of bulbopontine leptomeninges (arrow).
Patient 3
A 68-year-old man was admitted to hospital for fatigue, weight loss, pain and motor deficit to the lower limbs. Blood cell counts were normal. Serum electrophoresis revealed the presence of an IgM kappa M protein of 17.5 g/L. Bone marrow biopsy demonstrated an LPL with a bone marrow infiltration of 60% associated with interstitial and perivascular deposits of amyloid. MYD88 L265P mutation was found by allele-specific PCR on bone marrow CD19+ mononuclear cells. Fat pad biopsy was also positive for amyloid deposits. Brain and spinal MRI detected leptomeningeal disease infiltration of the spinal cord and cauda equina (Figure 3). The CSF analysis revealed WBC count of 11/mm3 and an elevated protein level of 382 mg/dl reflecting severe disruption of the blood-brain barrier. Flow cytometric analysis of CSF showed infiltration by clonal B lymphocytes CD19+, CD20+, CD22+, CD5−, CD10−, CD23−. EMG and ENG showed demyelinating sensitive-motory polyneuropathy to upper and lower limbs. The final diagnosis was WM complicated by AL amyloidosis with initial cardiac involvement, BNS, and peripheral neuropathy.
Figure 3.

T1 sag fat and T2 sag show thickening of cauda equina roots (arrows); T1 sag and ax fat sat post gadolinium show thickening and shaded enhancement of spinal leptomeninges and roots of cauda equina (arrows).
Six cycles of Rituximab (375 mg/m2 day 1) and Bendamustine (90 mg/m2 days 1–2) with six intrathecal injections of Methotrexate (15 mg day 1) were administered. At the end of therapy, we observed a reduction >50% of M protein and bone marrow infiltration and resolution of lymphadenopathies. Spinal cord MRI showed the absence of contrast enhancement in the spinal cord and cauda equina. CSF analysis showed elevated protein level without malignant cells by flow cytometry. These findings were consistent with a complete response of BNS according to current guidelines.
Patient 4
A 38-year-old man was diagnosed with LPL associated with a serum IgG kappa M protein in 2012. At the time of diagnosis, the patient had systemic symptoms, multiple adenopathies and a bone marrow infiltration of 70%. The patient was refractory to first-line treatment with six 21-day cycles of R-CHOP and developed Rituximab intolerance after the third cycle. During salvage therapy with DHAP (Cisplatin, high dose cytarabine, and dexamethasone), the patient had a focal seizure crisis with secondary generalization. CSF analysis revealed an elevated protein level with no detectable lymphoid cells. Brain MRI showed a cortical-subcortical right temporal area with enhanced contrast, consistent with CNS parenchymal localization of lymphoma (Figure 4A), while the CT scan demonstrated progression of adenopathies. A biopsy of the brain lesion was not feasible. The patient was refractory to treatment with ICE chemotherapy (Ifosfamide, carboplatin, etoposide) and hyper-CVAD and was then treated with six 28-day cycles of Bendamustine (90 mg/m2 days 1–2) associated with six doses of intrathecal Methotrexate (day 1). At the end of treatment, the brain MRI was normal (Figure 4B), and CT scan showed regression of lymphadenopathies. Bone marrow biopsy was negative, and no M-protein was detectable in serum or urine. In conclusion, patient obtained complete remission of LPL and BNS. After 15 months the patient had an isolated CNS relapse. He was treated with high-dose cytarabine without response and then with total brain irradiation (24 Gy) which induced a clinical improvement and significant reduction of hemispheric lesions at MRI.
Figure 4A.

Proton density (PD) ax and T1-SE post gadolinium show cortical and subcortical rounded shaped lesion at the level of the right mesial temporal lobe with contrast enhancement (arrows).
Figure 4B.

Proton density (PD) ax and T1-SE post gadolinium demonstrate regression of the right temporal lesion and absence of pathological contrast enhancement. after Bendamustine treatment (arrows).
Discussion of Cases and Review of the Literature
What is the Incidence of BNS?
The exact incidence of BNS is unknown. The incidence of BNS in retrospective studies is likely to be underestimated because the awareness of this potential complication of WM has only recently increased, as witnessed by the publication of two retrospective series in the last few years.5,6 Besides the four cases reported here, two more cases had been previously diagnosed at our Institution. Therefore, 6 cases were diagnosed since 2003 (when current diagnostic criteria of WM were established) to 2016 in a series of 186 WM patients, corresponding to a prevalence of 3.2 %. Anyway, prospective observational studies are needed to address this issue.
When does BNS Occur During the Disease Course?
BNS may occur at any time during the course of the disease.3 In three of four cases here reported BNS was the first presenting symptom in patients without a previous history of WM, whereas the last patient developed CNS involvement eight months after the diagnosis of WM. In the international multicentric retrospective study conducted by Castillo, the diagnosis of BNS was concomitant with the diagnosis of WM in one-third of cases and subsequent in two thirds. In the latter scenario, the median time interval between diagnosis of WM and the diagnosis of BNS was 8.9 years.6
BNS may occur independently of a systemic progression of WM and may also present when patients are receiving WM-directed therapy, even in patients in complete remission. As CNS is a well-known “sanctuary site”, not reached by most drugs used to treat WM, the occurrence of isolated CNS progression is not unexpected.
What Are the Symptoms of BNS?
Clinical presentation of BNS is extremely heterogeneous without any specific sign or symptom: the most frequent neurological manifestations are balance disorders with ataxia (48%) or cranial nerve involvement (36%, mainly facial and oculomotor nerve). Other symptoms include headache, cognitive impairment with frontal syndrome, memory loss or dementia (27%), paresis and motor symptoms, sensory symptoms (25%) such as dysesthesia, paresthesia, psychiatric symptoms, headache (18%), cauda equina syndrome (14%), motor deficits (14%), blurred vision.5 Convulsions, hemiparesis or aphasia may occur in the tumoral form. Symptoms are gradually progressive, generally developing in weeks or months.7 Since symptoms are often nonspecific, clinical suspicion of BNS is essential. The presence of a concomitant peripheral neuropathy, as in two of the four cases here described, may be misleading and delay the diagnosis of BNS. Of note, the median time between onset of neurological symptoms and the diagnosis of BNS in the French study was 4 months (range 0–36) and more than 1 year in 20% of cases.5
When Should BNS be Suspected?
Clinical suspicion of BNS is based on the presence of neurological symptoms in patients with an already established diagnosis of WM or with an IgM monoclonal protein in the serum.
The differential diagnosis of BNS mainly includes hyperviscosity syndrome (HVS) and levels of IgM along with an evaluation of serum viscosity may be useful to distinguish HVS from BNS.11 Some neurological symptoms of BNS may mimic those of peripheral neuropathies with anti-MAG antibodies which may occur in WM and other IgM-related disorders.5 Patients with anti-MAG antibodies mostly present with a sensory ataxia and distal muscle weakness which slowly develops over the years.12 Since symptoms of BNS and those of peripheral neuropathy may be overlapping and the two conditions may coexist in the same patient, WM patients with a peripheral neuropathy should be carefully evaluated by an expert neurologist to exclude a concomitant involvement of CNS, in particular, an infiltration of cauda equina.
How many Forms of BNS Do Exist?
CNS involvement may occur in two forms: the majority of BNS patients (75% in the French study, more than 90% in the series reported by Castillo) present a diffuse form with leptomeningeal enhancement on imaging. The tumoral form is less common and is characterized by the presence of one or more parenchymal lesions, and in these cases, patients usually present with focal neurologic deficits.13 It is more challenging to diagnose because biopsy is not easily feasible in most cases. In the fourth case here reported the parenchymal lesion could not be biopsied, but the regression of CNS lesion as well as of lymphadenopathies after treatment with Bendamustine confirmed ex-post the diagnosis of BNS. In this patient, CNS involvement could be consistent with BNS resulting from a direct infiltration of lymphoplasmacytic cells, even though the serum M protein was not an IgM and therefore the diagnosis was lymphoplasmacytic lymphoma rather than WM.
Which Tests Are Necessary for the Diagnosis of BNS?
MRI of the brain and spinal cord is an essential test for the diagnosis of CNS involvement by lymphoma, and it is also recommended in case of suspected BNS due to its high sensitivity for the detection of malignant infiltration.7 In BNS, brain and spinal cord MRI is abnormal in 78% of cases generally showing enhancement and/or thickening of meningeal sheets, abnormal enhancement of cranial and spinal nerves, thickening and enhancement of cauda equina. Imaging alterations described above are supportive but not sufficient for the diagnosis of BNS,7 while the absence of MRI findings should not exclude BNS.14 However, the diagnosis of BNS in the absence of radiological abnormalities should be made with caution and only after a multidisciplinary discussion of the case.5
CSF analysis should be performed after MRI to avoid endocranial hypertension and non-specific meningeal enhancement that occurs after CSF sampling. CSF analysis may show an elevated opening pressure, elevated total protein (>100 mg/dl) reflecting the disruption of blood-brain barrier, normal or decreased glucose and increased WBC count (between 100 and 500 cells/mm3).3,7 In order to confirm the neoplastic infiltration of CSF and to exclude inflammatory or infective causes, flow-cytometric analysis of CSF is mandatory to demonstrate the presence of clonal B-cells with the same immunophenotypic features as those in bone marrow BM. Of note, while a positive test substantiates the diagnosis, negative results do not exclude BNS considering the low sensitivity of cytological testing due to the low number of neoplastic cells.3 The presence of an IgM monoclonal protein in the CSF per se does not indicate a neoplastic infiltration of CNS, because if a blood-brain-barrier disruption is present, the leakage of M-proteins from the blood into CSF may occur due to increased permeability of the barrier. Although not specific for the BNS, IgM-index calculation15 could be used to identify a proper IgM production beyond the blood-brain barrier.
Involvement of the eye is rare16,17 however it is recommended to consult a neuro-ophthalmologist in patients with visus or ocular motility impairment.
According to criteria recently proposed, a definite diagnosis of BNS requires a histological biopsy of cerebrum or meninges or the demonstration of a clonal B cell population with the same with the typical phenotype of WM in the CSF. Immunohistochemistry usually shows a malignant population expressing the same antigens of WM cells, i.e. pan-B antigens (CD19, CD20, CD79a, CD79b), in most cases also B-cell memory markers (CD27, CD52), plasma cells markers (CD138 and IgM), while CD5 and CD3 are expressed in a minority of cases.1
Differential diagnosis has to take into account primary central nervous system lymphoma (PCNSL) but also other indolent lymphomas or transformation to high-grade lymphomas which involve the CNS.7
Are Molecular Tests Essential for the Diagnosis of BNS?
Immunoglobulin gene rearrangement analysis represents an essential tool able to establish the clonal nature of the lymphoid B-cell population and the clonal relationship between CNS and BM B lymphocytes, strongly supporting the diagnosis of BNS.7 In 2012, a somatic mutation in the MYD88 gene leading to the substitution of a leucine with a proline at position 265 (MYD88 L265P) was found to be highly prevalent in WM patients.18 Poulain et al.19 recently reported for the first time the diagnostic value of MYD88 L265P mutation detection in BNS patients. They identified a MYD88 L265P mutation in the CSF and BM of all BNS cases using quantitative-PCR (q-PCR) and Sanger sequencing. Molecular testing in CNS biopsy and CSF might support the diagnosis of BNS and has recently been added to the diagnostic armamentarium. Moreover, the disappearance of MYD88 L265P mutation correlates with clinical response, suggesting a potential for monitoring response to therapy and minimal residual disease.19 However, the MYD88 L265P mutation in the CNS biopsy or CSF samples is not specific for BNS and has also been detected in one-third of patients with primary central nervous system lymphoma (PCNSL).20
What is the Prognosis of BNS?
There are no recognized prognostic factors for BNS. Simon L et al.5 in a retrospective series of 44 patients reported an overall survival rate of 71% at 5 years and 59% at 10 years after the diagnosis of BNS, while the median overall survival from the diagnosis of WM was 17.1 years.
In the series of Castillo et al.6 the estimated 3-year overall survival (OS) rate was 59%. Age >65 years, previous treatment for WM and platelet count <100 × 109/L were identified as adverse prognostic factors for survival in the univariate analysis. These findings potentially suggest that BNS occurring during the disease course may have a worse outcome compared to BNS occurring at the time of diagnosis of WM.
What Are Treatment Approaches?
Treatment approaches are not uniform, reflecting the lack of standardization for this rare entity. The choice of therapy should be based on patient condition, medical history, preference and experience of a physician.7 In the recent retrospective surveys of Simon and Castillo, the overall response rate (ORR) was 70% to first-line therapy, and no differences could be made according to treatment type. Remission has been reported either with intrathecal injection and/or systemic chemotherapies, including high-dose Methotrexate or Cytarabine which are able to penetrate the blood-brain barrier. Intrathecal treatment should be combined with systemic treatment since monotherapy with intrathecal drugs rarely induces durable responses.7
Nucleoside analogs have been demonstrated to pass the blood-brain barrier. Several previous reports suggested that fludarabine was effective in Chronic Lymphocytic Leukemia with the involvement of CNS.21,22 Vos et al. recently reported the efficacy of Fludarabine for the treatment of BNS,23 confirming its usefulness as a therapeutic option. In our experience, treatment with Rituximab-Bendamustine associated with intrathecal Methotrexate was well tolerated and effective, representing a suitable treatment option for BNS patients, especially for those who are not eligible for intensive treatment.8 Rituximab has been used in largest series mostly associated with chemotherapy; monotherapy is not advised due to its presumed low blood-brain barrier penetration. Ibrutinib, a BTK inhibitor, has recently introduced in the treatment of WM due to its efficacy in WM.2 Recent reports suggest that Ibrutinib either at the dose of 420mg or 560 mg is active and able to penetrate the blood-brain barrier24,10 and pharmacodynamic studies show CSF diffusion with a good neuromeningeal distribution.9
BNS is sensitive to radiotherapy (RT). Localized RT to affected regions (20–40 Gy) is preferred to whole brain irradiation and may be used alone or in combination with chemotherapy. Enhanced neurotoxicity has been reported mainly in the elderly,25 and cognitive impairment has been reported to occur after whole brain irradiation.26 Therefore, RT should not be considered a first-line therapy but should be reserved for patients failing other treatment options.7
Although there is no clear consensus about the role of autologous stem cell transplantation in patients with BNS, frontline intensification seems to be associated with long-term remissions.5,10 However, toxic deaths are described for autologous stem cell transplantation so that transplantation should be considered only for suitable patients.5
What is the Goal of Treatment in Patients with BNS?
Treatment should be considered in symptomatic patients with a definitive diagnosis of BNS. The goal of treatment should be to reverse clinical symptoms and increase overall survival, though a complete eradication of all malignant cells is not always possible. In fact, in some cases, the disease is still detectable on post-treatment CSF analysis, while patients become asymptomatic. Radiologic lesions may persist after successful treatment, but they do not necessarily constitute persisting disease. Therefore treatment should be guided by the clearance of patient’s symptoms.7 Neurological sequelae could determine the persistence of symptoms, due to the low regenerative ability of CNS and PNS: they must not be interpreted as treatment failure, but treatment should be continued until the best clinical result is achieved.
How Should the Neurological Response be Evaluated after Treatment?
CSF response can be monitored during and after treatment: normalization of CSF analysis indicates an adequate response. Detection of MYD88 L265P mutation using qPCR on CSF represents a promising useful molecular tool to monitor response to chemotherapy19 sequentially. Response criteria proposed in the recently published guidelines7 are reported in Table 1.
Table 1.
Response Criteria7
| Complete response (CR) |
|
| Partial response (PR) |
|
| Nonresponse (NR) |
|
| Relapse |
|
Conclusions and Open Issues
BNS is a rare and probably under-recognized complication of WM which can occur at any time during the course of the disease, even in patients who are responding to systemic therapy. BNS should be suspected early in patients with WM who develop unexplained neurological signs and symptoms. Patients with an established diagnosis of WM, manifesting any neurological symptom (including symptoms which could be consistent with peripheral neuropathy) should be promptly evaluated by a multidisciplinary team, in order to run the appropriate neurological investigations for BNS. This attitude could shorten the time for the diagnosis of BNS, potentially ameliorating outcome. MRI and CSF analysis are essential for the diagnosis. The diagnostic accuracy of BNS could be improved by the detection of MYD88 L265P mutation in CSF. However further investigations are necessary to assess the utility of this test for the diagnosis and evaluation of response. Treatment remains challenging because of lack of standardization and information about prognosis is still scanty. Therefore prospective studies are eagerly awaited with the aim of better defining treatment strategies and outcome, significantly improving our knowledge about this rare complication of WM.
Footnotes
Competing interests: The authors have declared that no competing interests exist.
References
- 1.Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom’s Macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobuliemia. Semin Oncol. 2003;30(2):110–115. doi: 10.1053/sonc.2003.50082. https://doi.org/10.1053/sonc.2003.50082. [DOI] [PubMed] [Google Scholar]
- 2.Treon SP. How I treat Waldenstrom Macroglobulinemia. Blood. 2015;126:721–732. doi: 10.1182/blood-2015-01-553974. https://doi.org/10.1182/blood-2015-01-553974. [DOI] [PubMed] [Google Scholar]
- 3.Malkani RG, Tallman M, Gottardi-Littel N, et al. Bing Neel syndrome: an illustrative case and a comprehensive review of the published literature. J Neuroncol. 2010;96(3):301–312. doi: 10.1007/s11060-009-9968-3. https://doi.org/10.1007/s11060-009-9968-3. [DOI] [PubMed] [Google Scholar]
- 4.Bing J, von Neel A. Two cases of hyperglobulinaemia with affection of the central nervous system on a toxi-infectious basis. Acta Med Scand. 1936;88:492–506. https://doi.org/10.1111/j.0954-6820.1936.tb12571.x. [Google Scholar]
- 5.Simon L, Fitsiori A, Lemal R, et al. Bing-Neel syndrome, a rare complication of Waldenström’s Macroglobulinemia: analysis of 44 cases and review of literature. A study on be-half of the French Innovative Leukemia Organization (FILO) Haematologica. 2015;100(12):1587–1594. doi: 10.3324/haematol.2015.133744. https://doi.org/10.3324/haematol.2015.133744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castillo JJ, D’Sa S, Lunn MP, et al. Central nervous system involvement by Waldenstrom macroglobulinaemia (Bing-Neel syndrome): a multi-institutional retrospective study. Br J Haematol. 2016;172:709–715. doi: 10.1111/bjh.13883. https://doi.org/10.1111/bjh.13883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Minnema MC, Kimby E, D’Sa S, et al. Guideline for the diagnosis, treatment and response criteria for Bing-Neel syndrome. Haematologica. 2017;102(1):43–51. doi: 10.3324/haematol.2016.147728. https://doi.org/10.3324/haematol.2016.147728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Varettoni M, Marchioni E, Bonfichi M, et al. Successful treatment with Rituximab and Bendamustine in a patient with newly diagnosed Waldenstrom’s Macroglobulinemia complicated by Bing-Neel syndrome. Am J Hematol. 2015;90:E152–E153. doi: 10.1002/ajh.24059. [DOI] [PubMed] [Google Scholar]
- 9.Cabannes-Hamy A, Lemal R, Goldwirt L, et al. Efficacy of Ibrutinib in the treatment of Bing-Neel syndrome. Am J Hematol. 2016;91(3):E17–19. doi: 10.1002/ajh.24279. https://doi.org/10.1002/ajh.24279. [DOI] [PubMed] [Google Scholar]
- 10.Mason C, Savona S, Rini JN, et al. Ibrutinib penetrates the blood brain barrier and shows efficacy in the therapy of Bing Neel syndrome. Br J Haematol. :2016. doi: 10.1111/bjh.14218. https://doi.org/10.1111/bjh.14218 [DOI] [PubMed] [Google Scholar]
- 11.Stone MJ, Bogen SA. Evidence-based focused review of management of hyperviscosity syndrome. Blood. 2012;119(10):2205–2208. doi: 10.1182/blood-2011-04-347690. https://doi.org/10.1182/blood-2011-04-347690. [DOI] [PubMed] [Google Scholar]
- 12.Stork AC, van der Pol WL, Franssen H, et al. Clinical phenotype of patients with neuropathy associated with monoclonal gammopathy: a comparative study and a review of the literature. J Neurol. 2014;261(7):1398–1404. doi: 10.1007/s00415-014-7354-3. https://doi.org/10.1007/s00415-014-7354-3. [DOI] [PubMed] [Google Scholar]
- 13.Grewal JS, Brar JK, Sahijdak WM, et al. Bing Neel syndrome: a case report and systematic review of clinical manifestations, diagnosis and treatment options. Clin Lymphoma Myeloma. 2009;9(6):462–466. doi: 10.3816/CLM.2009.n.091. https://doi.org/10.3816/CLM.2009.n.091. [DOI] [PubMed] [Google Scholar]
- 14.Gupta N, Gupta S, Al Ustwani O, et al. Bing-Neel syndrome in a patient with Waldenstrom’s macroglobulinemia: a challenging diagnosis in the face of normal brain imaging. CNS Neurosci Ther. 2014;20(10):945–946. doi: 10.1111/cns.12311. https://doi.org/10.1111/cns.12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zetterberg Patognomonic cerebrospinal fluid findings in Bing-Neel syndrome. J Neurooncol. 2011;104(2):615. doi: 10.1007/s11060-010-0510-4. https://doi.org/10.1007/s11060-010-0510-4. [DOI] [PubMed] [Google Scholar]
- 16.Hughes MS, Atkins EJ, Cestari DM, et al. Isolated optic nerve, chiasm, and tract involvement in Bing Neel syndrome. J Neuroophtalmol. 2014;34(4):340–345. doi: 10.1097/WNO.0000000000000138. https://doi.org/10.1097/WNO.0000000000000138. [DOI] [PubMed] [Google Scholar]
- 17.Stacy RC, Jakobiec FA, Hochberg FH, et al. Orbital involvement in Bing Neel syndrome. J Neuroophtalmol. 2010;30(3):255–259. doi: 10.1097/WNO.0b013e3181dee96c. https://doi.org/10.1097/WNO.0b013e3181dee96c. [DOI] [PubMed] [Google Scholar]
- 18.Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenstrom’s macro-globulinemia. N Engl J Med. 2012;367:826–833. doi: 10.1056/NEJMoa1200710. https://doi.org/10.1056/NEJMoa1200710. [DOI] [PubMed] [Google Scholar]
- 19.Poulain S, Boyle EM, Roumier C, et al. MYD88 L265P mutation contributes to the diagnosis of Bing Neel syndrome. Br J Haematol. 2014;167:506–513. doi: 10.1111/bjh.13078. https://doi.org/10.1111/bjh.13078. [DOI] [PubMed] [Google Scholar]
- 20.Montesinos-Rongen M, Godlewska E, Brunn A, et al. Activating L265P mutations of the MYD88 gene are common in primary central nervous system lymphoma. Acta Neuropathol. 2011 Dec;122(6):791–792. doi: 10.1007/s00401-011-0891-2. https://doi.org/10.1007/s00401-011-0891-2. [DOI] [PubMed] [Google Scholar]
- 21.Elliott MA, Letendre L, Li CY, et al. Chronic lymphocytic leukemia with symptomatic diffuse central nervous system infiltration responding to therapy with systemic fludarabine. Br J Haematol. 1999;104:689–694. doi: 10.1046/j.1365-2141.1999.01245.x. https://doi.org/10.1046/j.1365-2141.1999.01245.x. [DOI] [PubMed] [Google Scholar]
- 22.Knop S, Herrlinger U, Ernemann U, et al. Fludarabine may induce durable remission in patients with leptomeningeal involvement of chronic lymphocytic leukemia. Leuk Lymphoma. 2005;46:1593–1598. doi: 10.1080/10428190500178472. https://doi.org/10.1080/10428190500178472. [DOI] [PubMed] [Google Scholar]
- 23.Vos JM, Kersten MJ, Kraan W, et al. Effective treatment of Bing Neel syndrome with oral fludarabine: a case series of four consecutive patients. Br J Haematol. 2016;172(3):461–464. doi: 10.1111/bjh.13483. https://doi.org/10.1111/bjh.13483. [DOI] [PubMed] [Google Scholar]
- 24.Bernard S, Goldwirt L, Amorim S, et al. Activity of ibrutinib in mantle cell lymphoma patients with central nervous system relapse. Blood. 2015;126(14):1695–1698. doi: 10.1182/blood-2015-05-647834. https://doi.org/10.1182/blood-2015-05-647834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saad S, Wang TJ. Neurocognitive deficits after radiation therapy for brain malignancies. Am J Clin Oncol. 2015;38(6):634–640. doi: 10.1097/COC.0000000000000158. https://doi.org/10.1097/COC.0000000000000158. [DOI] [PubMed] [Google Scholar]
- 26.Tallet AV, Azria D, Barlesi F, et al. Neurocognitive function impairment after whole brain radiotherapy for brain metastases: actual assessment. Radiat Oncol. 2012;7:77. doi: 10.1186/1748-717X-7-77. https://doi.org/10.1186/1748-717X-7-77. [DOI] [PMC free article] [PubMed] [Google Scholar]