

Abstract
Multiple-locus variable-number of tandem-repeats analysis (MLVA) has emerged as a valuable method for subtyping bacterial pathogens and has been adopted in many countries as a critical component of their laboratory-based surveillance. Lack of harmonisation and standardisation of the method, however, has made comparison of results generated in different laboratories difficult, if not impossible, and has therefore hampered its use in international surveillance. This paper proposes an international consensus on the development, validation, nomenclature and quality control for MLVA used for molecular surveillance and outbreak detection based on a review of the current state of knowledge.
Introduction
Multiple-locus variable-number of tandem-repeats analysis (MLVA) has recently emerged as a powerful method for the subtyping of food-borne bacterial pathogens. The method is based on repetitive DNA elements organised in tandem (Figure). DNA replication errors, such as slipped-strand mispairing, generate diversity in the number of tandem repeats observed among strains of the same species [1,2]. MLVA determines the number of tandem repeats, or copy units, at multiple variable-number tandem repeat (VNTR) loci within the genome. Typically, multiplex PCR amplification of the repeat and flanking regions is followed by amplicon sizing using capillary electrophoresis. The number of repeat copy units, or allele number, at each location is calculated from the measured amplicon size. The string of alleles from multiple loci forms the MLVA profile.
Figure. Typical organisation of a variable number of tandem repeat (VNTR) locus.
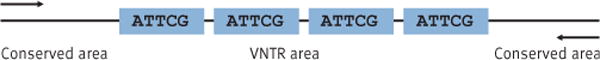
The arrows point to the annealing sites for polymerase chain reaction (PCR) primers in the conserved region flanking the repeats.
The recent development of MLVA protocols for subtyping food-borne bacterial pathogens, including Salmonella enterica serotypes Typhimurium and Enteritidis, and Shiga toxin-producing Escherichia coli (STEC) O157:H7 has facilitated the implementation and application of MLVA for the successful detection and investigation of a wide variety of food-borne disease outbreaks all over the world [3–6]. The early promise and success of MLVA triggered the independent development of multiple protocols by many different laboratories, leading to many different schemes for each organism. For example, six protocols have been described for STEC O157 [3, 7–11], six for S. Enteritidis [1, 12–16], and four for S. Typhimurium [17–20]. Differences in the choice of loci, nomenclature, amplicon sizing due to primer, platform and/or chemistry differences, and interpretation of incomplete or partial repeats have stymied and continue to stymie inter-laboratory comparisons and thus surveillance. A lack of standards for the development, validation and quality control/quality assurance of MLVA further contributes to problems in the comparison and interpretation of MLVA results.
The goal of any subtyping method is to characterise bacteria beyond the species (or subspecies) level and to group individual isolates together in a meaningful way. The ability to do this quickly and reliably is the cornerstone of laboratory-based surveillance [21]. Isolates that have indistinguishable subtypes are more likely to have originated from a common source than those with different subtypes. This concept forms the basis for applying molecular subtyping to bacterial pathogens for surveillance, outbreak detection and outbreak response.
To be suitable for laboratory-based surveillance and outbreak detection, a subtyping method should be assessed against several key performance criteria [21]: typeability, reproducibility, discriminatory power and epidemiological concordance. These criteria must be assessed using an epidemiologically relevant panel of isolates from geographically as diverse a region as where the method is to be applied. Additional criteria to assess method feasibility include speed, throughput, cost, ease of use, objectivity, versatility and portability. The importance of these criteria is further emphasised for the successful application of a subtyping method to inter-laboratory surveillance.
While no single method will have perfect performance when assessed against all criteria, MLVA performs well overall. It scores high in its performance against several key criteria including discriminatory power, robustness, portability, objectivity and throughput [21,22], but scores low in versatility, since most protocols are species or serotype specific. Comparatively, pulsed-field gel electrophoresis (PFGE), the current gold standard method for the subtyping of food-borne bacterial pathogens, scores high in discriminatory power and versatility, but medium in robustness and low in portability, objectivity and throughput [22].
The historical success of PFGE for the inter-laboratory surveillance of food- and waterborne bacterial pathogens was based on the standardisation of methodology and interpretation through an internationally coordinated approach. The future success of emerging technologies such as MLVA for inter-laboratory surveillance similarly hinges on the coordinated harmonisation of the methodology, nomenclature and interpretation.
In this paper, we describe an international consensus for the development, validation, nomenclature, and quality control for MLVA-based inter-laboratory surveillance based on a review of the current state of science. These consensus guidelines were developed following an expert consultation in Copenhagen, Denmark, in May 2011, organised by the United States (US) Centers for Disease Control and Prevention (CDC), the European Centre for Disease Prevention and Control (ECDC), the Association of Public Health Laboratories in United States, the Public Health Agency of Canada and the Statens Serum Institut, Denmark.
Method development
Selection of potential loci
The first step in the development of an MLVA method involves the selection of potential loci for inclusion in the protocol. Initial VNTR locus finding and identification is performed by querying whole genome sequences using specialised software. Some VNTR-finding software is available free of charge on the Internet, and include Tandem Repeats Finder [23] and TredD [24]. Commercial software is also available and includes GeneQuest (DnaStar Lasergene, Madison, WI, US) and CodonCode (CodonCode Corp., Dedham, MA, US). Tandem Repeats database [25] is a public repository of information on tandem repeats and also contains a variety of tools for their analysis.
There is no standardised naming of loci used in MLVA schemes. In order to create uniformity in this context, it is proposed to name the loci in relation to their positions in the prototype genome. The proposed standardised locus naming (Box 1) and its correlation with existing nomenclature for loci that overlap between most published protocols for STEC O157, and S. Typhimurium and S. Enteritidis are outlined in Tables 1–3, respectively.
Table 1.
Nomenclature for overlapping VNTR loci in published MLVA protocols for Shiga toxin-producing Escherichia coli O157:H7
| Standardised VNTR locus namea | MLVA protocol | |||||
|---|---|---|---|---|---|---|
| Noller [11] | Lindstedt [10] | Keys [9] | Cooley [3] | Kawamori [8] | Hyytiä-Trees [7] | |
| ECS271 | TR5 | Vhec3 | O157-3 | Vhec3 | VR1 | O157-3 |
| ECS1520 | TR4 | NA | O157-25 | NA | NA | O157-25 |
| ECS2862 | TR7 | NA | O157-19 | O157-19 | VR3 | O157-19 |
| ECS3490 | TR1 | Vhec4 | O157-9 | Vhec4 | VR4 | O157-9 |
| ECS3491 | TR2 | Vhec1 | O157-10 | Vhec1 | NA | NA |
| ECS5331 | TR6 | Vhec2 | O157-34 | Vhec2 | VR6 | O157-34 |
| ECS5426 | TR3 | NA | O157-17 | O157-17 | VR8 | O157-17 |
| pO15746 | NA | NA | O156-37 | O156-37 | NA | O156-37 |
| pO15754 | NA | Vhec7 | O157-36 | Vhec7 | NA | O157-36 |
MLVA: multiple-locus variable-number of tandem-repeats analysis; NA: not applicable; VNTR: variable-number tandem repeat.
Prototype genome described by Hayashi et al. [33].
Table 3.
Nomenclature for overlapping VNTR loci in published MLVA protocols for Salmonella enterica serovar Enteritidis
| Standardised VNTR locus namea | Nomenclature used in published MLVA protocol | ||||
|---|---|---|---|---|---|
| Boxrud [13] | Beranek [1] | Malorny [15], Hopkins [14] | Ross [16] | PulseNet US [12] | |
| SET533 | SE9 | NA | SENTR7 | STTR9 | PNSE9 |
| SET2073 | SE3 | NA | SE3 | N/A | PNSE3 |
| SET2504 | SE1 | ENTR13 | SENTR4 | SE1 | PNSE1 |
| SET3073 | SE5 | STTR5 | SENTR5 | STTR5 | PNSE5 |
| SET3511 | SE6 | NA | NA | STTR3 | PNSE6 |
| SET4617 | SE2 | ENTR20 | SENTR6 | SE2 | PNSE2 |
MLVA: multiple-locus variable-number of tandem-repeats analysis; NA: not applicable; US: United States; VNTR: variable-number tandem repeat.
Prototype genome described by Thompson et al. [35].
When selecting loci (Box 2), as a rule of thumb, the shorter the repeat unit, the more variation is detected in terms of copy numbers [26]. However, repeat units shorter than five bp should not be included in a subtyping system due to the limitations in sizing reproducibility in capillary electrophoresis platforms. It is critical to avoid repeat units with insertion and deletions (indels) in order to facilitate consistent sizing and allele naming using copy numbers. Low-level base variation between repeat units does not usually have a negative impact as long as the unit length is consistent. However, perfect homogeneous repeats are always better and will usually also increase polymorphism through the effect of polymerase slippage [26]. Furthermore, only loci with 100% conserved flanking sequences in the target organism should be included.
Primer design
Once loci have been identified, primers for their PCR amplification need to be designed (Box 2). There are multiple choices for primer design software, both commercial and free of charge. The shareware version of the software FastPCR [27] works well. However, more elaborate versions of commercial software, such as VisualOmp (DNA Software, Inc, Ann Arbor, MI, United States), allow for performing simulations that will check for primer interactions in multiplex reactions; such checking is not available in the free software. At the very least, primer design software should be used to verify that no secondary structures, such as hairpins or self- and cross-dimers are formed between any of the primers intended to be multiplexed in the same reaction.
When designing primers, a number of issues need to be considered. Firstly, primers should be placed as close to the VNTR array as possible since the projected fragment size should not exceed 600 bp, which is the upper limit of reproducible sizing in most capillary electrophoresis platforms. This is particularly critical for VNTR arrays with long repeat units and for arrays with shorter repeat units combined with high diversity, in which scenario dozens of repeat units may be possible. If only a few prototype genomes are available, we suggest sequencing the flanking regions of each locus in 20 strains representative of the genetic diversity of the target organism in order to ensure that the primers are placed in conserved sequence. Secondly, the intended site of the primer should be targeted so that it falls in the most accurate region of the sequence, i.e. 80–150 bp away from the sequencing primer. Thirdly, the primers for all loci should have the same annealing temperature in order to facilitate easy multiplexing of targets in the same PCR reaction. Relatively high annealing temperatures of 55 °C to 65 °C should be aimed for to enable stringent amplification conditions for specific amplification. Generally, the melting temperature for primers should be 5 °C higher than the desired annealing temperature.
Assay optimisation
Once potential loci have been selected and primers designed, it is time to optimise the assays in the laboratory setting. This process includes testing the diversity of the loci selected and optimisation of the PCR reactions. This is an iterative process that is repeated until a set of loci with appropriate diversity have been selected and PCR conditions to amplify the loci reliably have been developed. Firstly, the VNTR loci should be screened for diversity using singleplex PCR reactions against a limited panel of 10 to 20 strains that are not related to each other and have been shown to be genetically diverse using other subtyping methods. At this stage, loci showing no diversity or minimal diversity are excluded from the assay. Also loci with poor amplification, multiple amplification products or background noise should be either excluded or the primers should be re-designed at this stage.
After the initial screen, the promising VNTRs are tested against a larger panel (100–150) of isolates. This panel should contain both outbreak-related (information about patient exposures required) and epidemiologically unrelated (sporadic, i.e. different geographical locations, no temporal associations) isolates. This second screen will focus the selection process on VNTRs that generate epidemiologically relevant data. It also gives the assay developer an idea of the fragment size ranges in each locus, which is information that is needed for designing multiplex assays. Representative alleles in each locus, i.e. the smallest allele, the largest allele and at least every third in between, should be sequenced at the development phase in order to verify the copy number and to ensure that the size differences observed between different strains are due to differences in repeat unit copy numbers and not due to other genetic events.
Design of multiplex PCR reactions
Once the set of VNTR loci has passed the initial screening process, multiplex PCR reactions must be designed to enable efficient amplification of all loci in as few reactions as possible. Since the multiplex PCR reactions should be as robust as possible, no more than four or five targets should be amplified in the same reaction. Targets with overlapping fragment sizes can be differentiated using different fluorescent labels. The same label can be used multiple times in the same PCR reaction as long as there is no overlap in fragment sizes. The two main capillary electrophoresis platforms widely in use – Beckman Coulter CEQ8000/GenomeLabGeXP Genetic Analysis System (Beckman Coulter, Brea, CA, United States) and Applied Biosystems Genetic Analyzer 3130/3730/3500 (Life technologies, Carlsbad, CA, United States) – differ vastly in the fluorescent chemistries that can be used and there is no overlap in the chemistries between them. Up to four different fluorescent labels can be detected simultaneously on the Beckman Coulter platform, whereas the Applied Biosystems instruments are capable of detecting up to five different fluorescent labels from the same reaction. One of the dyes is always reserved for the DNA size standard. Since it is highly desirable that protocols could be easily converted from one platform to another by simply just re-labelling the forward primers, use of more than three fluorescent labels for targets in the same reaction is therefore not recommended.
Important parameters to consider when designing the multiplex PCR reactions are the annealing temperature, MgCl2 concentration and primer concentration. Practical tips for approaches to optimise multiplex PCR reactions can be found in the literature [28].
All targets in the multiplex reaction should be easily detectable. The desired fluorescence intensity for PCR products on the Beckman Coulter platform is 5,000– 80,000 units, on the Applied Biosystems 3130 platform 1,000–7,000 units and on the Applied Biosystems 3500 and 3730 platforms 2,000–9,000 units. Fluorescence intensity below the desirable level will result in unreliable detection of targets. Too high fluorescence intensity will cause fluorescence carry-over from one channel to another resulting in non-specific peaks that can interfere with the data analysis in downstream applications. If the same protocol is used in multiple laboratories, each laboratory typically needs to optimise the primer concentrations for their own laboratory since there are several laboratory-specific factors, such as the age of the primer stocks, the type and the calibration status of the thermocycler, which affect the amplification efficiency. Additionally, as the primer stocks age, there is a gradual drop in the fluorescence intensity, requiring further optimisation of primer concentrations over time, even within the same laboratory.
Internal validation
When a prototype of the MLVA protocol has been established, it needs to go through internal validation (Box 3). The purpose is to test the robustness and reproducibility and to establish the discriminatory power of the method when used in the laboratory (or laboratories) that developed it.
The internal validation should be comprised of two phases, which may be performed simultaneously: (i) testing of additional isolates by the protocol developers; (ii) testing of the protocol by other laboratories/ individuals within the developers’ institutions for technical performance. The number of isolates to be tested during internal validation depends on the genetic diversity of the target organism, i.e. the higher the diversity, the more isolates are needed for adequate validation. Optimally 250 to 500 isolates, in addition to those that were tested during the development phase, should be tested. If the developing laboratory does not have access to such a large culture collection, the isolates must be acquired from collaborating laboratories. Insufficiently validated protocols should not be published in the scientific literature since they almost invariably will need further optimisation by future users. By analysing a large number of isolates using the proposed protocol, the robustness of the assay can be tested, along with its ability to consistently produce profiles from all strains and generate data that are epidemiologically relevant and easy to analyse. The strains used for the validation should include well-defined sets of both outbreak-associated isolates and sporadic isolates. The outbreak-associated isolates should also include 20 to 30 isolates from the same outbreak and ideally from multiple outbreaks of different types (monoclonal vs polyclonal, short lasting vs long lasting). Multiple isolates obtained through serial passaging of the same strain may also be included to test the reproducibility of the method and in vitro stability of the loci. If desired, the sporadic isolates and one representative from each outbreak can be used to calculate the diversity index for the method [29]. If the protocol is intended for global use, geographically representative isolates around the globe should be included in the validation set. Data generated with the proposed MLVA method should be compared with the epidemiological data in order to determine concurrence. Comparisons with the gold-standard method should also be made, if a gold standard exists for the target organism. In order to determine the technical performance, the protocol should be tested using multiple different equipment brands (thermocyclers, capillary electrophoresis instruments), different lots of reagents and by multiple individuals. All null alleles (= no amplification) should be confirmed using singleplex PCR reactions in order to rule out suboptimal multiplex conditions as a cause for amplification failure.
Calibration set and allele nomenclature
Inter-laboratory comparability, as mentioned before, is of critical importance if the subtyping method is to be used for international surveillance. Determining the number of repeats using different detection platforms without sequencing all amplicons is not reliable because of use of different reagents, chemistries and detection platforms may yield slightly but sufficiently different fragment sizing results to hamper inter-laboratory comparisons [30,31]. Using different primers for amplification of the same loci will also invariably lead to lack of comparability of results generated in different laboratories. We propose to solve this problem by introducing organism-specific set of strains with well-characterised copy numbers at each locus that each laboratory implementing the method may use to calibrate the output of the protocol and detection platform they use (Boxes 4 and 5).
These strain calibration sets should be created both for existing MLVA protocols and for those developed in the future. The validation of such a calibration set for use with S. Typhimurium protocols is described in this issue of Eurosurveillance [32]. Each laboratory will use the calibration set to create a correlation table between the sequenced copy number and the observed fragment size for each allele at each locus using their preferred protocol and fragment-sizing platform. This way, the same allele type will always be assigned to the same fragment regardless of the primer sequences, reagents or capillary electrophoresis platform used to generate and size the fragment. The calibration should be repeated each time a laboratory changes any parameter in its MLVA set-up, such as using a different fluorescent dye for a primer or different type of polymer for capillary electrophoresis. The calibration set should cover representative alleles for all loci included in the new protocol, and in the case of the existing protocols, for those loci that overlap between the protocols that are already widely used. All VNTR loci should be sequenced for all isolates included in the calibration set in order to determine the actual copy number. All alleles should be included in the calibration set if the VNTR locus contains four or fewer alleles. If the VNTR locus contains five or more alleles it is proposed that at least the smallest and the largest alleles and every third allele in between should be included in the calibration set. All new alleles with unexpected fragment sizes (fragment sizes that do not fall within predicted sizes for new alleles based on the calibration set) must be sequenced, and, if needed, the calibration set should be amended.
If multiple peaks are detected in the same locus, the PCR needs to be repeated using a fresh DNA template made from a culture derived from a single colony in order to exclude the possibility of contamination, since this is the most common explanation for this phenomenon. If contamination is not the cause of the problem and the result with multiple peaks is reproducible, with the same peak always having the highest fluorescence intensity, then the allele type should be designated based on the most intense peak and the other peaks should be ignored if the locus cannot be excluded from the assay. If upon repeating the PCR the same peak does not always present with the highest fluorescence intensity, 10 colony picks should be tested from the culture. In this case, the allele type should be assigned based on the peak that has the highest fluorescence intensity in the majority of the colony picks.
External validation
When the method has passed the internal validation, it needs to be validated by the future external users. The purpose of external validation is to determine the robustness and performance of the methodology and thereby the feasibility of implementing it in multiple laboratories of end users (Box 6).
It is important that results from different laboratories in diverse geographical locations and with different skill levels are compatible and reproducible for international surveillance and outbreak detection and investigations. It is expected that different laboratories may use reagents from different suppliers. Often equipment in different laboratories is made by different manufacturers or different models from the same manufacturer are used. Although MLVA results are less prone to variability arising from subjective interpretation by trained laboratory staff, it is nevertheless important to take proficiency of data interpretation into consideration. In particular, the consistency of person-to-person interpretation of partial repeats and null alleles should be assessed, as should unpredicted results. In order to maintain consistency of results over time, quality assurance processes should also be considered after the external validation.
In selecting suitable laboratories to participate in the external validation, a survey containing questions in regard to testing capacity could be distributed to reference laboratories that have been performing PFGE or other molecular typing methods for cluster detection. Such a survey will also explore the global interest in using the method.
The aim of inter-laboratory comparison is to determine the variability of the results obtained by different laboratories using identical samples. Six to eight laboratories should be selected from different geographical locations that may have different endemic or outbreak strains with profiles determined using the gold-standard method and have the capacity to perform MLVA. These laboratories should cover the range of equipment platforms (including different manufacturers, models and analytical software) and reagents from different suppliers. It is preferable that the participating laboratories have trained microbiologists available who are knowledgeable in capillary electrophoresis for troubleshooting and interpretation of results.
The selected laboratories should initially test the calibration set of strains using the same procedures that have been internally validated to create the calibration table for standardised reporting. In addition, for comparing inter-laboratory compatibility, each laboratory needs to subtype a blinded set of at least 20 well-characterised strains supplied by the organising laboratory and covering the full spectrum of alleles at all loci, including alleles that are not present in the calibration set. The results from all the participating laboratories should be distributed and shared by the organising laboratory. The concordance is calculated for the study overall and for each individual laboratory. Discordant results must be resolved and recommendations on corrective actions to improve concordance be made. These corrective actions should be provided to future participants as part of quality assurance of the method. If the concordance was poor initially (discordant results generated for more than 5% of the isolates in more than 20% of the participating laboratories), the external validation may need to be repeated with any corrections to the protocol.
When good concordance has been achieved between the laboratories, each participant should test additional strains selected from its own culture collection that has been well characterised, ideally using the same gold-standard method, typically PFGE. These strains should be from diverse locations and epidemiological backgrounds. The number of strains will typically be between 50 and 100, depending on the diversity of the target organism. This panel should be well defined to evaluate typeability, i.e. the ability to amplify each locus, the discriminatory power and epidemiological concordance of the method [21]. It must include strains from human and non-human sources, and contain a mix of epidemiologically unrelated and related isolates. The MLVA testing should be evaluated for these criteria in comparison with the gold standard, if such a method exists.
If new alleles are encountered during the external validation, strains with these alleles should be shared with the developing laboratory for confirmation by sequencing. If necessary, the calibration set should be revised to ensure that the copy number of the new alleles can be determined reliably. The external validation laboratories should also test the strains thus added to the calibration set, to update their correlation tables.
Quality assurance
The final step before an MLVA protocol may be implemented in routine surveillance in multiple laboratories is the establishment of a quality assurance programme for future users (Box 7). Quality assurance is divided into internal and external sections.
Internal quality assurance includes the use of appropriate controls for PCR and fragment analysis, quality control of new primer lots, maintenance and calibration of instruments, such as thermocyclers and pipettors, and appropriate record keeping for monitoring reagent lots, instrument performance and run-to-run accuracy of sizing. An internal training programme should be in place as part of the human resource succession or continuity plan and for surge capacity. Newly trained personnel should be assessed for proficiency prior to assuming routine testing and then assessed annually internally. Each laboratory should also participate in external quality assurance (EQA), if available.
EQA includes initial and annual quality checks performed by a laboratory/institute that has agreed to serve as a coordinating quality assurance body for the protocol in question. When a protocol is used in an international surveillance network such as PulseNet, new participants are certified for the laboratory procedure and the correct data analysis and reporting of the results for a limited set of well-characterised strains as part of the initial quality check. Once certified, each laboratory needs to pass a proficiency test at least annually to keep their certification status [22]. Valid certification is required from each laboratory in order to be able to upload data to the PulseNet databases. In PulseNet International, the coordinating laboratory in each region is responsible for the EQA in their respective region and the US CDC performs the EQA for the coordinating laboratories. ECDC has funded an external voluntary EQA scheme for MLVA of S. Typhimurium for the public health laboratories in the European Union and European Economic Area countries. This is a new quality assessment scheme in Europe that does not provide a formal certification status but serves as ‘shelf-check’ for the participants. The first results are expected to be available in 2013.
The developing laboratory typically selects a set of strains to be used for certification and proficiency testing. The number of strains used for certification of new users and proficiency testing of current users depends on the clonality of the organism. PulseNet US’s certification sets for MLVA include eight isolates, and proficiency testing is performed by testing only a single isolate in the same test run with each laboratory’s routine isolates. The generated data are evaluated not only for correct patterns but also for the overall quality of data, e.g. non-specific peaks, primer-dimers and optimisation of PCR reactions.
Successful implementation of a new MLVA protocol may be facilitated through training of new users. This training needs to include the use of the detection platform the participants will use in their own laboratory, to make them familiar with the protocol in a setting as close as possible to the one they will use in the future.
Concluding remarks
It is our hope that the guidelines and recommendations presented here will help solve some of the problems hampering the inter-laboratory comparisons of MLVA subtyping results, provide clarification of the relationships between the multiple protocols currently available for STEC O157, S. Enteritidis and S. Typhimurium, and facilitate the development and validation of new MLVA protocols for organisms not covered by currently available protocols.
Table 2.
Nomenclature for overlapping VNTR loci in published MLVA protocols for Salmonella enterica serovar Typhimurium
| Standardised VNTR locus namea | Nomenclature used in published MLVA protocol | |||
|---|---|---|---|---|
| Lindstedt [19] | Witonski [20] | Chiou [18] | PulseNet US [17] | |
| STM2730 | STTR6 | 2730867 | ST19 | ST5 |
| STM3184 | STTR5 | 3184543 | ST25 | ST6 |
| STM3246 | STTR9 | NA | ST26 | ST7 |
| STM3629 | STTR3 | 3629542 | ST06 | ST8 |
| pSLT53 | STTR10 | NA | ST40 | STTR10 |
MLVA: multiple-locus variable-number of tandem-repeats analysis; NA: not applicable; VNTR: variable-number tandem repeat.
Prototype genome described by McClelland et al. [34].
Box 1. Standardised VNTR locus nomenclature for an MLVA protocol.
A VNTR locus is named based on its location on the chromosome on the prototype genome by the closest kilobase (kb). If located on a plasmid, the name of the plasmid is used instead of the prototype genome.
Example: the standardised name of the Salmonella enterica serovar Typhimurium VNTR locus STTR6 [18] would be STM2730, i.e. STM is the designation for the Typhimurium prototype genome LT2 and 2730 is the closest kb location for the locus STTR6 on the LT2 genome.
MLVA: multiple-locus variable-number of tandem-repeats analysis; STEC: Shiga toxin-producing Escherichia coli; VNTR: variable-number tandem repeat.
Box 2. Optimal VNTR locus and primer selection for developing an MLVA protocol.
Repeat units ≥5 base pairs
No insertions and deletions in repeat units
Perfect homogeneous repeats should be preferred
Only loci with 100% conserved flanking sequences should be used
Primers should be placed as close as possible to the VNTR unit
Primers with relatively high annealing temperatures (55 °C to 65 °C) should be used
The melting temperature should be 5 °C higher than the annealing temperature
No more than three fluorescent dyes should be used to label the primers used in the assay
MLVA: multiple-locus variable-number of tandem-repeats analysis; VNTR: variable-number tandem repeat.
Box 3. Internal validation of an MLVA prototype protocol.
Purpose: to obtain information about the robustness, reproducibility, discriminatory power and epidemiological concordance in the laboratory (or laboratories) involved in the protocol development
Comparison with gold-standard method, e.g. PFGE, if such a method is available
- Isolate selection should:
-
○include 250–500 isolates o include sporadic isolates and multiple isolates from several outbreaks, to test in vivo stability
-
○include serially passaged isolates from one strain, to test in vitro stability
-
○be representative of the intended epidemiological context, e.g. geographical region, institutions/community
-
○
MLVA: multiple-locus variable-number of tandem-repeats analysis; PFGE: pulsed-field gel electrophoresis.
Box 4. Proposed standardised allele nomenclature and reporting of allele profiles for an MLVA protocol.
Proposed standardised allele nomenclature for homogeneous VNTRs
The allele name is the actual sequenced copy number
Incomplete repeats: the copy number rounded down to the nearest complete copy number
Null alleles: the designated allele type ‘−2.0’
VNTR array missing, but the flanking region with the primer-annealing sequences present and amplifies: the designated allele type ‘0’
Proposed standardised allele nomenclature for heterogeneous VNTRs
Inclusion of loci with heterogeneous repeat units is discouraged in new protocols
Some existing protocols include heterogeneous loci, such as the locus STTR3 in the Salmonella enterica serovar Typhimurium protocol by Lindstedt et al. [19]. STTR3 consists of 27 bp and 33 bp repeat units.
- Allele type should indicate copy numbers of all different length repeat units.
-
○Example: for STTR3, the allele type 0208 corresponds to two copies of the 27 bp repeat unit and eight copies of the 33 bp repeat unit [36].
-
○
Proposed standardised reporting of allele profiles
New protocols: reported in the order the loci are located in genome. Loci located on plasmids reported last.
- Existing protocols: the currently most widely accepted reporting order for loci will be continued.
- ○ Example: the S. Typhimurium MLVA profile reported in the locus order STTR9-STTR5-STTR6-STTR10-STTR3: 3-8-13-14-0411
bp: base pair; MLVA: multiple-locus variable-number of tandem-repeats analysis; VNTR: variable-number tandem repeat.
Box 5. Calibration strain set for developing an MLVA protocol.
Purpose: a reference set of strains with diverse confirmed number of repeats at all loci to be used to create a calibration table enabling correct allele designation in the test laboratories
- Strain selection:
-
○all alleles have been confirmed by sequencing
-
○for loci with up to four alleles, all alleles must be represented
-
○for loci with five or more alleles, the smallest, the largest and at least every third allele in between must be represented
-
○
- If a new allele is identified, its copy number must be confirmed by sequencing
-
○If a strain contains a new allele outside the range of known alleles, it must be added to the calibration strain set
-
○
A new calibration table should be generated by testing the full calibration strain set when new instruments or chemistries are introduced
MLVA: multiple-locus variable-number of tandem-repeats analysis.
Box 6. External validation of an MLVA prototype protocol.
Purpose: to confirm the robustness, reproducibility, discriminatory power and epidemiological concordance, and thereby the feasibility of implementing the method in multiple laboratories representing the intended end users
- Six to eight laboratories representing the full diversity of intended end users should be selected. They should:
-
○be from different geographical locations
-
○have a full range of equipment platforms
-
○have supplies from different manufacturers
-
○
- Each laboratory should test:
-
○the calibration strain set, to create the calibration table
-
○a minimum of 20 isolates representing the full known allelic diversity at all loci. If discordant results are generated in >5% of the isolates in >20% of the participating laboratories, the protocol and of the calibration isolate set should be revisited and corrected, and the external validation repeated
-
○50–100 strains from each participating laboratory representing the local diversity of the organism
-
○
MLVA: multiple-locus variable-number of tandem-repeats analysis.
Box 7. Quality assurance and proficiency testing of an MLVA prototype protocol.
Quality assurance
Purpose: to ensure consistent high quality of the results generated
Control strains should be included for PCR and fragment analysis in each run
Multiple reference strains should be run as a quality control check when new primer lots are introduced or after any major maintenance or repair of the instrument
Records of reagent lots and accuracy of fragment sizing for control strains should be maintained for each run
An internal training programme should be in place for new personnel
Proficiency testing
If available, participation in an external quality assurance programme is mandatory
Newly trained personnel must pass an initial test for proficiency and be tested annually thereafter
Assessment of proficiency includes generation of correct allele profiles and overall quality of data, e.g. presence of non-specific peaks, primer-dimers and other PCR artifacts
MLVA: multiple-locus variable-number of tandem-repeats analysis; PCR: polymerase chain reaction.
Footnotes
Conflict of interest
None declared.
Authors’ contributions
All authors and members of the MLVA Harmonization Working Group participated in the discussions at the meeting in Copenhagen, read, commented on and approved the manuscript; Celine Nadon, Eija Trees, Lai-King Ng, Eva Møller Nielsen, Nikki Maxwell, Kristy Kubota and Peter Gerner-Smidt conceived the idea of the paper and organised the meeting in Copenhagen; Celine Nadon, Eija Trees, Lai-King Ng, Eva Møller Nielsen and Aleisha Reimer each were responsible for drafting a section of the paper; Kristy Kubota and Peter Gerner-Smidt worked the sections together into one coherent manuscript; Peter Gerner-Smidt supervised the writing process.
References
- 1.Beranek A, Mikula C, Rabold P, Arnhold D, Berghold C, Lederer I, et al. Multiple-locus variable-number tandem repeat analysis for subtyping of Salmonella enterica subsp. enterica serovar Enteritidis. Int J Med Microbiol. 2009;299(1):43–51. doi: 10.1016/j.ijmm.2008.06.002. http://dx.doi.org/10.1016/j.ijmm.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Lindstedt BA. Multiple-locus variable number tandem repeats analysis for genetic fingerprinting of pathogenic bacteria. Electrophoresis. 2005;26(13):2567–82. doi: 10.1002/elps.200500096. http://dx.doi.org/10.1002/elps.200500096. [DOI] [PubMed] [Google Scholar]
- 3.Cooley M, Carychao D, Crawford-Miksza L, Jay MT, Myers C, Rose C, et al. Incidence and tracking of Escherichia coli O157:H7 in a major produce production region in California. PloS One. 2007;2(11):e1159. doi: 10.1371/journal.pone.0001159. http://dx.doi.org/10.1371/journal.pone.0001159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heck M. Multilocus variable number of tandem repeats analysis (MLVA) - a reliable tool for rapid investigation of Salmonella Typhimurium outbreaks. Euro Surveill. 2009;14(15) pii=19177. Available from: http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19177. [PubMed] [Google Scholar]
- 5.Konno T, Yatsuyanagi J, Saito S. Application of a multilocus variable number of tandem repeats analysis to regional outbreak surveillance of Enterohemorrhagic Escherichia coli O157:H7 infections. Jpn J Infect Dis. 2011;64(1):63–5. [PubMed] [Google Scholar]
- 6.Kuhn K, Torpdahl M, Frank C, Sigsgaard K, Ethelberg S. An outbreak of Salmonella Typhimurium traced back to salami, Denmark, April to June 2010. Euro Surveill. 2011;16(19) pii=19863. Available from: http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19863. [PubMed] [Google Scholar]
- 7.Hyytiä-Trees E, Smole SC, Fields PA, Swaminathan B, Ribot E. Second generation subtyping: a proposed PulseNet protocol for multiple-locus variable-number tandem repeat analysis of Shiga toxin-producing Escherichia coli O157 (STEC O157) Foodborne Path Dis. 2006;3(1):118–31. doi: 10.1089/fpd.2006.3.118. http://dx.doi.org/10.1089/fpd.2006.3.118. [DOI] [PubMed] [Google Scholar]
- 8.Kawamori F, Hiroi M, Harada T, Ohata K, Sugiyama K, Masuda T, et al. Molecular typing of Japanese Escherichia coli O157: H7 isolates from clinical specimens by multilocus variable-number tandem repeat analysis and PFGE. J Med Microbiol. 2008;57(Pt 1):58–63. doi: 10.1099/jmm.0.47213-0. http://dx.doi.org/10.1099/jmm.0.47213-0. [DOI] [PubMed] [Google Scholar]
- 9.Keys C, Kemper S, Keim P. Highly diverse variable number tandem repeat loci in the E. coli O157:H7 and O55:H7 genomes for high-resolution molecular typing. J Appl Microbiol. 2005;98(4):928–40. doi: 10.1111/j.1365-2672.2004.02532.x. http://dx.doi.org/10.1111/j.1365-2672.2004.02532.x. [DOI] [PubMed] [Google Scholar]
- 10.Lindstedt BA, Vardund T, Kapperud G. Multiple-locus variable-number tandem-repeats analysis of Escherichia coli O157 using PCR multiplexing and multi-colored capillary electrophoresis. J Microbiol Methods. 2004;58(2):213–22. doi: 10.1016/j.mimet.2004.03.016. http://dx.doi.org/10.1016/j.mimet.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 11.Noller AC, McEllistrem MC, Stine OC, Morris JG, Jr, Boxrud DJ, Dixon B, et al. Multilocus sequence typing reveals a lack of diversity among Escherichia coli O157:H7 isolates that are distinct by pulsed-field gel electrophoresis. J Clin Microbiol. 2003;41(2):675–9. doi: 10.1128/JCM.41.2.675-679.2003. http://dx.doi.org/10.1128/JCM.41.2.675-679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.PulseNet standard operating procedure for PulseNet MLVA of Salmonella enterica serotype Enteritidis – Applied Biosystems Genetic Analyzer 3500 Platform. PulseNet USA. 2013 Available from: http://www.cdc.gov/pulsenet/PDF/se-abi-3500-508c.pdf.
- 13.Boxrud D, Pederson-Gulrud K, Wotton J, Medus C, Lyszkowicz E, Besser J, et al. Comparison of multiple-locus variable-number tandem repeat analysis, pulsed-field gel electrophoresis, and phage typing for subtype analysis of Salmonella enterica serotype Enteritidis. J Clin Microbiol. 2007;45(2):536–43. doi: 10.1128/JCM.01595-06. http://dx.doi.org/10.1128/JCM.01595-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hopkins K, Peters T, de Pinna E, Wain J. Standardisation of multilocus variable-number tandem-repeat analysis (MLVA) for subtyping of Salmonella enterica serovar Enteritidis. Euro Surveill. 2011;16(32) pii=19942. Available from: http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19942. [PubMed] [Google Scholar]
- 15.Malorny B, Junker E, Helmuth R. Multi-locus variable-number tandem repeat analysis for outbreak studies of Salmonella enterica serotype Enteritidis. BMC Microbiol. 2008;8:84. doi: 10.1186/1471-2180-8-84. http://dx.doi.org/10.1186/1471-2180-8-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ross IL, Heuzenroeder MW. A comparison of two PCR-based typing methods with pulsed-field gel electrophoresis in Salmonella enterica serovar Enteritidis. Int J Med Microbiol. 2009;299(6):410–20. doi: 10.1016/j.ijmm.2008.12.002. http://dx.doi.org/10.1016/j.ijmm.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 17.PulseNet standard operating procedure for analysis of MLVA data of Salmonella enterica serotype Typhimurium in BioNumerics – Applied BioSystems Genetic Analyzer 3130/3500 Data. PulseNet USA. 2013 Available from: http://www.cdc.gov/pulsenet/PDF/salmonella-mlva-t-abi-508c.pdf.
- 18.Chiou CS, Hung CS, Torpdahl M, Watanabe H, Tung SK, Terajima J, et al. Development and evaluation of multilocus variable number tandem repeat analysis for fine typing and phylogenetic analysis of Salmonella enterica serovar Typhimurium. Int J Food Microbiol. 2010;142(1-2):67–73. doi: 10.1016/j.ijfoodmicro.2010.06.001. http://dx.doi.org/10.1016/j.ijfoodmicro.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 19.Lindstedt BA, Vardund T, Aas L, Kapperud G. Multiple-locus variable-number tandem-repeats analysis of Salmonella enterica subsp. enterica serovar Typhimurium using PCR multiplexing and multicolor capillary electrophoresis. J Microbiol Methods. 2004;59(2):163–72. doi: 10.1016/j.mimet.2004.06.014. http://dx.doi.org/10.1016/j.mimet.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 20.Witonski D, Stefanova R, Ranganathan A, Schutze GE, Eisenach KD, Cave MD. Variable-number tandem repeats that are useful in genotyping isolates of Salmonella enterica subsp. enterica serovars Typhimurium and Newport. J Clin Microbiol. 2006;44(11):3849–54. doi: 10.1128/JCM.00469-06. http://dx.doi.org/10.1128/JCM.00469-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Belkum A, Tassios PT, Dijkshoorn L, Haeggman S, Cookson B, Fry NK, et al. Guidelines for the validation and application of typing methods for use in bacterial epidemiology. Clin Microbiol Infect. 2007;13(Suppl 3):1–46. doi: 10.1111/j.1469-0691.2007.01786.x. http://dx.doi.org/10.1111/j.1469-0691.2007.01786.x. [DOI] [PubMed] [Google Scholar]
- 22.Hyytiä-Trees E, Cooper K, Ribot E, GernerSmidt P. Recent developments and future prospects in subtyping of foodborne bacterial pathogens. Future Microbiol. 2007;2(2):175–85. doi: 10.2217/17460913.2.2.175. http://dx.doi.org/10.2217/17460913.2.2.175. [DOI] [PubMed] [Google Scholar]
- 23.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–80. doi: 10.1093/nar/27.2.573. http://dx.doi.org/10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sokol D, Benson G, Tojeira J. Tandem repeats over the edit distance. Bioinformatics. 2007;23(2):e30–5. doi: 10.1093/bioinformatics/btl309. http://dx.doi.org/10.1093/bioinformatics/btl309. [DOI] [PubMed] [Google Scholar]
- 25.Gelfand Y, Rodriguez A, Benson G. TRDB–the Tandem Repeats Database. Nucleic Acids Res. 2007;35:D80–7. doi: 10.1093/nar/gkl1013. Database issue. http://dx.doi.org/10.1093/nar/gkl1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Belkum A, Scherer S, van Alphen L, Verbrugh H. Short-sequence DNA repeats in prokaryotic genomes. Microbiol Mol Biol Rev. 1998;62(2):275–93. doi: 10.1128/mmbr.62.2.275-293.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalendar R, Lee D, Schulman AH. Java web tools for PCR, in silico PCR, and oligonucleotide assembly and analysis. Genomics. 2011;98(2):137–44. doi: 10.1016/j.ygeno.2011.04.009. http://dx.doi.org/10.1016/j.ygeno.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 28.Zangenberg G, Saiki R, Reynolds R, Multiplex PCR. Multiplex PCR: optimization guidelines. In: Innis MA, Gelfand DH, Sninsky JJ, editors. PCR applications – protocols for functional genomics. San Diego, CA: Academic Press; 1999. pp. 73–104. [Google Scholar]
- 29.Hunter PR. Reproducibility and indices of discriminatory power of microbial typing methods. J Clin Microbiol. 1990;28(9):1903–5. doi: 10.1128/jcm.28.9.1903-1905.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Valk HA, Meis JF, Bretagne S, Costa JM, Lasker BA, Balajee SA, et al. Interlaboratory reproducibility of a microsatellite-based typing assay for Aspergillus fumigatus through the use of allelic ladders: proof of concept. Clin Microbiol Infect. 2009;15(2):180–7. doi: 10.1111/j.1469-0691.2008.02656.x. http://dx.doi.org/10.1111/j.1469-0691.2008.02656.x. [DOI] [PubMed] [Google Scholar]
- 31.Hyytia-Trees E, Lafon P, Vauterin P, Ribot EM. Multilaboratory validation study of standardized multiple-locus variable-number tandem repeat analysis protocol for shiga toxin-producing Escherichia coli O157: a novel approach to normalize fragment size data between capillary electrophoresis platforms. Foodborne Pathog Dis. 2010;7(2):129–36. doi: 10.1089/fpd.2009.0371. http://dx.doi.org/10.1089/fpd.2009.0371. [DOI] [PubMed] [Google Scholar]
- 32.Larsson JT, Torpdahl M. MLVA working group, Møller Nielsen E. Proof-of-concept study for successful inter-laboratory comparison of MLVA results. Euro Surveill. 2013;18:35. doi: 10.2807/1560-7917.es2013.18.35.20566. pii=20566. Available from: http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20566. [DOI] [PubMed] [Google Scholar]
- 33.Hayashi T, Makino K, Ohnishi M, Kurokawa K, Ishii K, Yokoyama K, et al. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 2001;8(1):11–22. doi: 10.1093/dnares/8.1.11. http://dx.doi.org/10.1093/dnares/8.1.11. [DOI] [PubMed] [Google Scholar]
- 34.McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, et al. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature. 2001;413(6858):852–6. doi: 10.1038/35101614. http://dx.doi.org/10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- 35.Thomson NR, Clayton DJ, Windhorst D, Vernikos G, Davidson S, Churcher C, et al. Comparative genome analysis of Salmonella Enteritidis PT4 and Salmonella Gallinarum 287/91 provides insights into evolutionary and host adaptation pathways. Genome Res. 2008;18(10):1624–37. doi: 10.1101/gr.077404.108. http://dx.doi.org/10.1101/gr.077404.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larsson JT, Torpdahl M, Petersen RF, Sorensen G, Lindstedt BA, Nielsen EM. Development of a new nomenclature for Salmonella Typhimurium multilocus variable number of tandem repeats analysis (MLVA) Euro Surveill. 2009;14(15) pii=19174. Available from: http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19174. [PubMed] [Google Scholar]