

Abstract
We examined the role of the mechanistic target of rapamycin (mTOR) pathway in delayed diazoxide (DZ)-induced preconditioning of cultured rat primary cortical neurons. Neurons were treated for three days with 500 μM DZ or feeding medium and then exposed to three hours of continuous normoxia in DMEM with glucose or with three hours of oxygen-glucose deprivation (OGD) followed by normoxia and feeding medium. The OGD decreased viability by 50 percent, depolarized mitochondria and reduced mitochondrial respiration, whereas DZ treatment improved viability and mitochondrial respiration, and suppressed reactive oxygen species production, but did not restore mitochondrial membrane potential after OGD. Neuroprotection by DZ was associated with increased phosphorylation of protein kinase B (Akt), mTOR, and the major mTOR downstream substrate, S6 Kinase (S6K). The mTOR inhibitors rapamycin and Torin-1, as well as S6K-targeted siRNA abolished the protective effects of DZ. The effects of DZ on mitochondrial membrane potential and reactive oxygen species production were not affected by rapamycin. Preconditioning with DZ also changed mitochondrial and non-mitochondrial oxygen consumption rates. We conclude that in addition to reducing ROS production and mitochondrial membrane depolarization, DZ protects against OGD by activation of the Akt-mTOR-S6K pathway and by changes in mitochondrial respiration.
Keywords: Diazoxide preconditioning, mTOR pathway, rapamycin, Torin-1, oxygen glucose deprivation, S6 Kinase, ischemic stroke
Introduction
Ischemic stroke is a leading cause of morbidity and mortality. Currently, the only Food and Drug Administration-approved treatment for ischemic stroke is the use of tissue plasminogen activator (tPA) (Hacke et al. 2008). However, tPA therapy is beneficial only when given within a three-hour window after the onset of a stroke and is associated with risk of hemorrhagic stroke when administered to older patients (Floris et al. 2014). Recent studies have indicated that tPA administration given beyond the traditional three-hour window still has beneficial effects (Ahmed et al. 2010; Elijovich and Chong 2010; Hacke et al. 2004; Wahlgren et al. 2008), but the delay induced by missing the three-hour window increases the risk of neuronal injury. The time limitations for initiation of tPA, the increased chance of neuronal damage with delayed treatment, and the risk of administration to an older patient population illustrate the urgent need for new therapies.
Elucidating protective neural mechanisms may offer insights for developing novel treatments, used singly or in conjunction with tPA that may be effective for high-risk stroke patients. Preconditioning protects cells, and can be induced by the transient exposure of cells to a sub-lethal stimulus or pharmacological agents before a subsequent, potentially lethal stimulus (Busija et al. 2008). There are many preconditioning approaches. One proven approach is the opening of mitochondrial ATP-sensitive potassium channels (mitoKATP) with selective pharmacological agents (Busija and Katakam 2014).
Our laboratory has shown that opening mitoKATP channels pharmacologically with drugs such as diazoxide (DZ) (Domoki et al. 2005; Kis et al. 2003b; Kis et al. 2004; Lenzser et al. 2005; Mayanagi et al. 2007a; Nagy et al. 2004; Rajapakse et al. 2003; Shimizu et al. 2002) and BMS-191095 (Busija et al. 2005; Gaspar et al. 2008b; Kis et al. 2004; Mayanagi et al. 2007b) can protect the brain and cerebral vasculature against ischemic damage (Domoki et al. 1999; Kis et al. 2003b). However, mechanisms of protection are not completely understood and additional information is necessary to exploit this potential therapy.
The mTOR pathway has recently emerged as a master regulator of cell metabolism, autophagy, and mitochondrial metabolism (Laplante and Sabatini 2012). Most investigators have shown that mTOR activation is important for cell survival during ischemia (Chen et al. 2012; Mao et al. 2013; Wang et al. 2012; Xie et al. 2013), but the involvement of the mTOR pathway in neuronal preconditioning by DZ is unclear. Activation of S6K, a downstream target of mTOR, is known to increase cell survival by upregulating Blastomyces adhesin 1, mouse double minute 2 homolog, protein kinase C, and phosphorylates translational factors such as eukaryotic translation initiation factor 4B, eukaryotic elongation factor 2-kinase, and ribosomal protein S6 which cause de novo protein synthesis (Fenton and Gout 2011).
We hypothesized that DZ-induced preconditioning results from activation of the mTOR pathway. We exposed primary rat neuronal cell cultures to an in vitro model of stroke: transient oxygen-glucose deprivation (OGD). Pharmacological inhibitors of mTOR as well as siRNA-targeting S6K were used to determine the mechanisms of DZ-induced preconditioning. We also addressed the effects of DZ-induced preconditioning on mitochondrial membrane potential, reactive oxygen species (ROS) production, and mitochondrial oxygen consumption rate (OCR).
Methods
Animals
Thirty, pregnant, E-18, Sprague Dawley rats and their embryos (average of 10 per dam) were obtained from Harlan (Indianapolis, IN). Their use for these studies was approved by the Tulane University School of Medicine Animal Care and Ethics Committee. Animals were given access to food and water ad libitum.
Rat Primary Cortical Neuronal Cell Culture
Rats were deeply anesthetized with isoflurane and decapitated. Primary rat cortical neurons were isolated from E-18 rat fetuses as described previously (Deadwyler et al. 1993; Kis et al. 2003b). After digestion and trituration, isolated cells were plated onto poly-D-lysine coated plates or dishes in a plating medium consisting of 60% DMEM, 20% F-12 HAM, 20% horse serum, and L-glutamine (0.5 mM). Cultures were maintained in a humidified 5% CO2 incubator. After cell attachment, the plating medium was replaced with Neurobasal medium supplemented with B27 (2%), L-glutamine (0.5 mM), 2-mercaptoethanol (55 μM), and KCl (25 mM). Positive immunostaining for microtubule-associated protein-2 and negative immunostaining for glial fibrillary acidic protein verified that the cultures consisted of more than 99% neurons.
Materials
Diazoxide (500 μM, Sigma Aldrich, St Louis, MO, USA) was used to induce preconditioning, as described previously (Kis et al. 2003b). Rapamycin (R, 400 nM, Sigma Aldrich) and Torin-1 (2 nM, Tocris Biosciences, Bristol, United Kingdom) were used to inhibit the mTOR pathway. Diazoxide was dissolved in 0.1M NaOH and the other drugs were dissolved in dimethyl sulfoxide (DMSO). Vehicle controls were performed for each type of experiment. S6K targeted siRNA was purchased from Invitrogen (Grand Island, NY, USA) and dissolved in RNAse free water. For mitochondrial respiration measurements, 2 μM oligomycin (ATP synthase inhibitor, Seahorse Biosciences, Billerica, MA, USA), 3.5 μM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, an ionophore, Seahorse Biosciences), 1.5 μM antimycin (complex III inhibitor, Seahorse Biosciences), and 1.5 μM rotenone (a complex I inhibitor, Seahorse Biosciences) were all dissolved in DMSO.
Experimental Design
Protocols are presented in Figure 1A. Primary cortical neurons were cultured for 7 days in vitro (DIV). On the 7th day, cells were treated with (1) DZ, (2) R, (3) DZ+R, (4) DZ+Torin-1, or (5) DZ+S6K-targeted siRNA (DZ+S6K siRNA) for 3 consecutive days. The S6K-targeted siRNA was added for 4 h before treating with DZ. On DIV 10, neurons were either subjected to OGD or maintained at normoxia, as described below. Mitochondrial membrane potential and ROS measurements were conducted immediately after OGD. Lysates were collected after 2 h of reoxygenation for Western blotting and viability assays and mitochondrial respiration assays were performed 24 h after reoxygenation.
Figure 1.

(Panel A) Schematic illustration of experimental design for experiments conducted in cultured rat neurons. To validate acute effects of diazoxide (DZ), mitochondrial membrane potential (Panel B) and ROS production (Panel C) were measured in normoxic neurons after 15 min DZ application. As expected, DZ depolarized mitochondria and increased ROS production. n = 6 repetitions of both experiments from 3 independent cultures. 288 wells per treatment for mitochondrial membrane potential measurement and 6 dishes for each group measuring ROS. * = p < 0.05 vs. untreated neurons. Data are expressed as mean ± SEM.
To document the effectiveness of DZ, we demonstrated that acute application of DZ to cultured neurons depolarized mitochondria (Fig. 1B) and increased ROS production (Fig. 1C), as previously described (Busija et al. 2004; Kis et al. 2003b; Lenzser et al. 2005; Mayanagi et al. 2007a; Rajapakse et al. 2002).
Oxygen Glucose Deprivation
Briefly, 96-well cell culture plates and dishes were rinsed with PBS and the culture medium was replaced with glucose-free DMEM (Wappler et al. 2013). Cultured neurons were placed in a Shel Lab Bactron Anaerobic Chamber (Sheldon Manufacturing, Cornelius, OR, USA) containing anaerobic mixed gas (AMG; 5% CO2 - 5% H2 - 90% N2) at 37°C for 3 h. The 5% H2 in the AMG removed remaining traces of oxygen-forming water on a platinum catalyst. The oxygen level was continuously monitored with an infrared gas analyzer (Illinois Instruments, Ingleside, IL, USA) and maintained at <0.1% during the experiments. Control cell cultures were treated identically and incubated in glucose-containing DMEM (4.5 mg/ml) in a 5% CO2 cell culture incubator. OGD was terminated by removing the cell culture plates and dishes from the anoxic chamber and replacing the glucose-free DMEM with regular culture medium containing glucose. The cells were then maintained in a 5% CO2 incubator until determination of cell viability.
Quantification of cellular viability assay
Twenty-four hours after OGD, cell viability was assessed using the tetrazolium-based CellTiter 96 AQueous One Solution Assay (Promega, Madison, WI, USA). Twenty microliters of solution were added to the culture wells. Cultures were incubated for 1 h at 37 °C followed by measurement of absorbance at λabs = 492 nm with a FLUOstar OPTIMA microplate reader (BMG Labtech GmbH, Offenburg, Germany). Results were compared with paired cultures exposed to the same neurotoxic stimulus on the same day and cell viability was expressed as the percentage of the corresponding control culture (untreated and not exposed to the lethal insult) using the following formula:
Mitochondrial respiration of neurons
Oxygen consumption rate was measured using a Seahorse XFe 24 analyzer (Fig. 2A), as described previously (Yao et al. 2011a; Yao et al. 2011b; Yao et al. 2013). Specially designed, poly-D-lysine coated 24-well plates were used to measure mitochondrial respiration in the cultured neurons. Fluorescent sensors measured the oxygen and proton fluxes present in the microchamber above the cells, which allowed calculation of the OCR. Neurons were washed twice with Assay Medium (Seahorse Biosciences) supplemented with 5mM glucose (Sigma Aldrich) and 2mM pyruvate (Sigma Aldrich). Mitochondrial electron transport chain function was altered using serial injections of the drugs oligomycin, FCCP, and antimycin plus rotenone (Fig. 2A). Each assay was normalized to and expressed as a percentage of the baseline value.
Figure 2.

(Panel A) Schematic illustration showing protocol for measuring mitochondrial respiration in the form of OCR via serial injections of oligomycin, FCCP, and antimycin/rotenone in a Seahorse XFe 24 analyzer (adapted from http://www.seahorsebio.com). (Panel B): Representative tracings showing OCR in untreated (dark bars) and DZ-treated (open bars) neurons 24 h after OGD. DZ preconditioning significantly increased OCR when normalized for baseline in response to oligomycin (Panel C), FCCP (Panel D), and antimycin/rotenone (Panel E) injections 24 h after return to normoxia after OGD. The “control” designation refers to normoxic neurons not previously exposed to OGD. n = 4 repetitions of the experiment from 4 independent cultures, 80 wells per treatment. # = p < 0.05 vs. untreated OGD neurons. Data are expressed as mean ± SEM.
Western blots
Proteins were harvested by scraping neurons from the dishes in ice-cold NP40 lysis buffer (Invitrogen, Grand Island, NY, USA) supplemented with proteinase and phosphatase inhibitors and then homogenized and stored for protein estimation (Sigma Aldrich) (each 5 μl/mL). Cell lysates were resolved using standard denaturing conditions: cells were homogenized and equal amounts of protein (30 μg) from the whole cell lysates were incubated with sodium dodecyl sulfate (SDS)/β-mercaptoethanol sample buffer at 100°C for 5 min. Protein samples were separated by electrophoresis on a 4–20% SDS-PAGE gradient gel and proteins were transferred onto a nitrocellulose membrane and incubated in a Tris- buffered saline containing 0.1% Tween 20 with 5% Bovine Serum Albumin blocking solution for 1 h at room temperature followed by incubation with primary antibodies overnight at 4°C in the blocking solution. The membranes were washed three times in Tween 20 and incubated for 1 h in the blocking buffer with goat anti-rabbit IgG (1:5000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) or goat anti-mouse IgG (1:5000, Santa Cruz) conjugated to horseradish peroxidase.
Protein levels were determined using the following primary antibodies: phosphorylated Akt Ser473 (p-AktSer473, 60 KDa, Cell Signaling, Danvers, MA), total Akt (60 KDa, Cell Signaling), phosphorylated mTOR Ser2448 (p-mTOR Ser2448, 298 KDa, Cell Signaling), total mTOR (289 KDa, Cell Signaling), phosphorylated S6K Thr389 (p-S6K Thr389, 70 and 85 KDa, Cell Signaling), and total S6K (70 and 85 KDa, Cell Signaling). Chemiluminescence and autoradiography were used to visualize the final reaction of the primary antibodies and their respective horseradish peroxidase conjugated secondary antibody. β-actin was used as a house-keeping protein to ensure equal loading. Western blots for Akt and mTOR showed a single band at the appropriate molecular weight whereas two bands at appropriate molecular weights were present for S6K. We included both bands for S6K in our analyses.
For quantitative analysis, the immunobands were scanned and band intensities were quantified using Image J 1.3.1 software. Band intensities were normalized to β-actin (1:5000, Sigma Aldrich) and the control group’s normalized protein levels were considered as 100%.
siRNA transfection
Lipofectamine RNAiMax was used with silencer select siRNA targeting S6K (cat #4390771, ID: 29105) to transfect neurons on DIV 7. Cells were washed twice with Opti-MEM media. Lipofectamine RNAiMax and siRNA were dissolved in Opti-MEM. Recommended dilutions of the siRNA (25 – 50 pmol) and lipofectamine mixtures were incubated at room temperature for 5 min and then added to cultured neurons for 4 h before resuming pharmacological treatment with DZ. Lipofectamine RNAiMax with Opti-MEM was used as a vehicle control. The negative control (cat #4390843) was non-targeting siRNA with comparable chemical modifications as the S6K targeted siRNA. To confirm knock downs, we performed Western blots after 72 h of transfection on total Akt, mTOR, and S6K and conducted viability assays on normoxic neurons and 24 h after OG.
Mitochondrial membrane potential (ΔΨm) (Rhodamine Assay)
The ΔΨm was analyzed using the Rhodamine 123, as previously described (Carvalho et al. 2014). Neuronal cultures in 96-well plates were loaded in the dark with Rhodamine (0.025 nM) in a 5% CO2 incubator (for cells maintained at normoxia) or in the OGD chamber (for cells undergoing OGD). After 45 min loading, the existing DMEM was replaced with phenol-free DMEM with glucose (normoxic control) or without glucose (OGD). Rhodamine fluorescence was measured with a FLUOstar OPTIMA microplate reader (λex = 505 nm). Data were expressed as a percentage of the intensity of the untreated control culture as follows:
Electron paramagnetic resonance (EPR)
For these experiments, neurons were cultured in 30 mm dishes and pre-treated with DZ, DZ+R, or regular medium. For measurement of ROS, we used the spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH, Noxygen Science Transfer & Diagnostics, Elzach, Germany), as described previously (Katakam et al. 2014; Mrakic-Sposta et al. 2012). A 10 mM CMH solution was prepared in Krebs-Hepes buffer (KHB) containing 25 μM deferoxamine methane-sulfonate salt (DF) chelating agent and 5 μM sodium diethyldithio-carbamate trihydrate (DETC) at pH 7.4. Dishes were washed with KHB containing DF and DETC and allowed to stabilize in the temperature and gas controller Bio III unit for 20 min and then CMH was added (200 μmol/L). After 15 min, 20 μL of the supernatant was transferred into the EPR capillary tube (Noxygen Science Transfer & Diagnostics), and placed in an E-scan spectrometer (Bruker, Billerica, MA) for data acquisition. The reaction of the probe with the neuronal ROS generated a spectrum which was sequentially recorded for 5 min to calculate the ROS production rate. The EPR signal was proportional to the unpaired electron numbers and transformed into micromoles per min (μmol · min−1). Neuronal lysates were collected from each dish for protein analysis and normalization as described in the “Western blots” section.
Statistical Analysis
All data are expressed as mean ± standard error of the mean (mean ± SEM) and analyzed using ANOVA (for experiments using more than 2 groups) and the Tukey post hoc test or a t-test (for experiments using 2 groups). A p < 0.05 was considered statistically significant. Information concerning sample sizes is presented in the figure legends.
Results
Viability of neurons
There were no differences in OCR between DZ treated and control neurons under normoxic conditions without OGD. Oxygen consumption rate was consistently higher in DZ compared with untreated neurons 24 h after OGD (Fig. 2 B, C, D, E). After normalization of values for OCR to baseline values for DZ treated and control neurons, DZ-treated neurons subjected to OGD showed significantly elevated OCR 24 h post-OGD following oligomycin (Fig. 2C), FCCP (Fig. 2D), and antimycin/rotenone (Fig. 2E) when compared to untreated neurons 24 h post-OGD.
Neurons showed significantly reduced viability 24 h after exposure to 3 h OGD when compared with untreated, normoxic cells (from 102.6±1.9% to 50.2 ± 2.2%, Fig. 3A).
Figure 3.
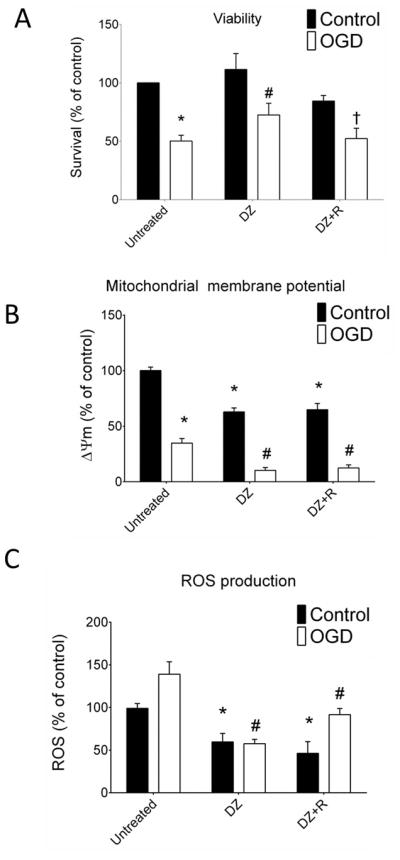
(Panel A) DZ treatment led to increased neuronal viability 24 h after 3 h OGD compared with untreated neurons. The “control” designation refers to normoxic neurons not previously exposed to OGD. Co-application of R with DZ blocked the protective effects exerted by DZ. n = 6 independent cultures, 96 wells per treatment. (Panel B): DZ reduced mitochondrial membrane potential during normoxia and after OGD. DZ+R also chronically depolarized the mitochondria similar to DZ alone. R in the absence of DZ failed to depolarize the mitochondria during normoxia. Exposure to OGD resulted in mitochondrial depolarization in all groups and DZ treatment with or without R enhanced the depolarization. The profile of mitochondrial membrane potential in the neurons treated with R was similar to that of untreated neurons. n = 6 independent cultures, 96 wells per treatment. (Panel C): DZ treatment alone or co-application with R reduced ROS production during normoxia and this effect was maintained after OGD. Thus, DZ+R treated neurons behaved similarly to DZ treated neurons and showed reduced ROS production. n = 6 repetitions of the experiment from 6 independent cultures, 96 wells per treatment. * = p < 0.05 vs. untreated normoxic neurons, # = p < 0.05 vs. untreated OGD neurons. Data are expressed as mean ± SEM.
Diazoxide treatment caused a significant increase in the viability of neurons 24 h after OGD (72.5% ± 4.1, p < 0.05, Fig. 3A). Mitochondrial membrane potential was significantly reduced by OGD (34.8% ± 4.2, p<0.05 Fig. 3B) when compared with normoxia. Diazoxide treatment reduced membrane potential significantly during normoxia (63.4% ± 3.9, p<0.05) and after OGD (10.3% ± 2.6, p < 0.05, Fig. 3B) and also reduced ROS production during normoxia (59.7% ± 9.9, p<0.05) and following OGD significantly (57.6% ± 4.9, p < 0.05, Fig. 3C).
Mechanisms of protection by DZ
Neither DZ nor DZ+R produced significant effects on baseline viability. Inhibition of the mTOR pathway with co-application of R abolished the protective effect of DZ on neuronal viability after OGD (Fig. 3A). However, co-application of R alone did not prevent mitochondrial depolarization or suppression of ROS production by DZ after OGD (Fig. 3B–C). Application of Torin-1 alone in untreated, control neurons decreased viability to 75.3 ± 1.8% compared with neurons not receiving Torin-1 (data not shown). Following OGD, viability in DZ+Torin-1 treated neurons was reduced (40.7 ± 2.7%). Despite the fall in baseline viability with DZ + Torin-1, the percentage difference in viability following OGD (48%) was similar to that observed in untreated neurons (50%), and less than in neurons treated with DZ alone.
Oxygen-glucose deprivation decreased phosphorylation/total protein levels of Akt, mTOR, and S6K significantly in untreated neurons (Fig. 4A–C). Treatment of the neurons with DZ did not change phosphorylation of these proteins under control conditions, but increased phosphorylated/total levels of Akt (73.4% ± 7.5, p < 0.05), mTOR (171.9% ± 33.9, p < 0.05), and S6K (290.5% ± 72.4, p < 0.05) after OGD significantly when compared with untreated neurons after OGD (Fig. 4A–C). Co-application of R with DZ blocked the increase in phosphorylation of Akt, mTOR, and S6K seen with DZ alone after OGD (Fig. 4A–C).
Figure 4.

DZ administration increased Akt, mTOR and S6K phosphorylation in cultured neurons. (Panel A): Akt phosphorylation decreased during OGD and this effect was attenuated by DZ administration. The “control” designation refers to normoxic neurons not previously exposed to OGD. Co-administration of R with DZ decreased Akt phosphorylation in control cells and prevented the preservation of Akt phosphorylation after OGD. (Panel B): Phosphorylation of mTOR decreased following OGD in untreated neurons but was increased by DZ treatment after OGD. In contrast, co-application of R with DZ decreased mTOR phosphorylation under control conditions and did not increase mTOR phosphorylation following OGD. (Panel C): Phosphorylation of S6K fell during OGD in untreated neurons but increased in the DZ treated neurons. Rapamycin administration blocked increased S6K phosphorylation in DZ treated neurons. n = 4 repetitions of the experiments from 4 independent cultures, 8 dishes per group. * = p < 0.05 vs. untreated normoxic neurons, # = p < 0.05 vs. untreated OGD neurons. Data are expressed as mean ± SEM.
The validity of the S6K targeted siRNA was measured using western blots 72 h after treatment. The S6K siRNA significantly reduced total levels of S6K but not the total levels of mTOR or Akt (Fig. 5 A). Additionally, the S6K-targeted siRNA reduced neuronal viability significantly at normoxia (Fig 5 B). S6K knock-down alone reduced neuronal viability at baseline (74.1% ± 4.2). Co-application of DZ with S6K siRNA showed a similar reduction at baseline (78.8% ± 7.3). After OGD, neurons with S6K knock down alone as well as with DZ+S6K siRNA treatment showed reduced viability (41.6 ± 4.1% and 38.5 ± 5.7%, respectively) (Fig. 6 A). When corrected for baseline effects, the difference in viability in DZ+S6K siRNA treated neurons was approximately 49% which was similar to that observed in untreated neurons exposed to OGD as well as DZ+R neurons exposed to OGD (Fig. 3A). The siRNA transfection reduced phosphorylated/total S6K protein level to 84.4 ± 2.5% of baseline during normoxia and co-treatment with DZ+S6K siRNA had a similar effect in reducing phosphorylated S6K (87.3 ± 13.9%) (data not shown). After OGD, phosphorylation of S6K in the knock down alone group was reduced to 49.5 ± 3.2% and was reduced to 52.8± 15.4% in the DZ+S6K siRNA group (Fig. 6 B).
Figure 5.

Confirmation of S6K knockdown using S6K-targeted siRNA. (Panel A): Knockdown of S6K using siRNA decreased total S6K level significantly (* = p < 0.05), but not total mTOR and Akt levels. n = 3 repetitions of the experiment from 3 independent cultures, 6 dishes per group. * = p < 0.05 vs. untreated normoxic neurons. (Panel B): S6K siRNA decreased viability at normoxia in neurons significantly in untreated and DZ-treated neurons. n = 3 repetitions of the experiment from 3 independent cultures, 144 wells per treatment. * = p < 0.05 vs. DZ - treated normoxic neurons. Data are expressed as mean ± SEM.
Figure 6.

S6K-targeted siRNA blocked DZ-induced preconditioning in neurons. (Panel A): Knock down of S6K blocked survival of neurons after OGD. n = 3 repetitions of the experiment from 3 independent cultures, 144 wells per treatment. (Panel B): Administration of S6K siRNA blocked increased phosphorylation of S6K normally seen with DZ administration after OGD in neurons. Both bands for phosphorylated S6K were included in the determinations of phosphorylated/total S6K values. Upper panel shows original Western blots and lower panel represents summarized data. The DZ viability and S6K phosphorylation data after OGD have been shown previously in Figures 3A and 4C. These data are presented as grey bars to indicate prior use. n = 3 repetitions of the experiment from 3 independent cultures, 6 dishes per group. # = p < 0.05 vs. DZ - treated OGD neurons. Data are expressed as mean ± SEM.
Discussion
There are two significant findings from our study. First, we found that increased cell survival after OGD is associated with altered mitochondrial and non-mitochondrial oxygen consumption in DZ-preconditioned neurons. Second, we found that the mTOR pathway, culminating with S6K phosphorylation, plays an important role in mediating DZ-induced preconditioning. Although previous studies have suggested that mitochondrial depolarization and decreased ROS production are important mediators of DZ-induced preconditioning during OGD, our study indicates a more significant involvement of the Akt-mTOR-S6K pathway.
Although it is well-established that DZ protects mitochondrial integrity and calcium handling after anoxia (Busija and Katakam 2014), this is the first report showing elevated OCR in response to DZ treatment 24 h after an anoxic event, indicating a more robust electron transport chain function in preconditioned neurons and/or increased non-mitochondrial oxygen consumption. During normoxia, vehicle and DZ treated neurons behaved similarly and showed the normal profile observed in most cell types after administration of oligomycin, FCCP, and antimycin/rotenone. Thus, in both groups during normoxia, oligomycin reduced OCR by more than 60%, FCCP increased OCR by more than 60%, and antimycin/rotenone decreased OCR by 80%. Since OCR values were almost identical in the vehicle and DZ treated neurons during normoxia, despite a dramatically reduced mitochondrial membrane potential and ROS production in the DZ group, we concluded that membrane potential and ROS availability have minimal effects on OCR in healthy neurons. However, OGD changed the OCR profiles for both treatment groups. First, vehicle-treated neurons showed a 50% reduction in OCR following oligomycin whereas DZ-treated neurons showed a smaller drop in OCR. Therefore, at baseline, ATP synthesis may be driven by oxidative phosphorylation, but after an ischemic insult there is an uncoupling effect which is more pronounced in the DZ preconditioned neurons. A similar uncoupling effect was reported previously by mitoKATP activation (Costa et al. 2006; Pamenter et al. 2008). Second, adding FCCP, which allowed us to determine the maximal respiratory capacity, showed reductions in OCR for vehicle and DZ-treated neurons after OGD compared with normoxia. However, the reduction in OCR was less in the DZ-treated neurons, thereby indicating that OGD exhausts the maximal capacity of neurons and DZ treatment allows for a partial preservation of the maximal capacity. Third, antimycin/rotenone administration reduced OCR less after OGD than in normoxic neurons, indicating increased involvement of non-mitochondrial mechanisms. Therefore, after OGD, neurons appear to consume more oxygen via non-mitochondrial enzymes, especially in DZ treated neurons.
We considered the involvement of oxygen consuming reactions such as those involving nitric oxide synthase (NOS), cyclooxygenase (COX), and heme-oxygenase (HO). Specifically, our lab has shown that the depolarization of mitoKATP channels using DZ leads to activation of the NOS pathway in cultured endothelial cells and cerebral blood vessels (Katakam et al. 2013; Rutkai et al. 2014). Additionally, Alcindor et al (2004) concluded that COX-2 mediates the protection imparted by DZ in pharmacological preconditioning in a canine model of myocardial infarction. Similarly, Zeng and colleagues (2012) found that DZ pre-treatment enhanced HO-1 expression which led to increased resistance to liver ischemia-reperfusion injury in rodents. There is some literature suggesting cross-talk between mitochondria and NADPH oxidases (Bedard and Krause 2007; Dikalov 2011; Nazarewicz et al. 2013) and between vasorelaxant effects of DZ and cytochrome P450 (Oyekan et al. 1994). Santos and colleagues (2002) showed that in the ischemic heart, DZ pre-treatment allowed more efficient energy transfer between mitochondrial and non-mitochondrial enzymes upon reperfusion. A reduction in oxygen consumption has also been shown in Alzheimer’s disease (Yao et al. 2011a), ischemia-reperfusion injury (Porter et al. 2014), and hypoxic insult of cardiomyocytes (Neary et al. 2014). Finally, Kelly et al. (2014) reported that the inhibition of the PI3K-mTOR pathway decreases OCR in several cancer cell lines, supporting our premise that this axis is important for DZ-induced preconditioning. Further studies are needed to confirm that mitochondrial respiration is enhanced and the mTOR pathway is activated in vivo by pre- and post-conditioning with DZ.
Increased viability of neurons by DZ treatment after OGD was associated with increased phosphorylation of Akt, mTOR, and S6K, which are essential components of the cellular protection pathway. Thus, DZ treatment resulted in increased levels of phosphorylation compared with vehicle treated neurons of Akt, mTOR, and S6K after OGD. Furthermore, by using two structurally and mechanistically distinct inhibitors of mTOR, R and Torin-1, which counteracted the effects of DZ treatment, we have provided pharmacological evidence that the mTOR-S6K signaling pathway is essential for DZ to exert its protective effects against the anoxic insult of neuronal cells. To supplement and verify our findings with pharmacological inhibitors, we used a siRNA approach designed to impair S6K. Similar to our pharmacological approaches, we found that siRNA against S6K prevented DZ from increasing neuronal viability as well as reduced S6K phosphorylation after OGD. Thus, our three lines of evidence using viability assays, protein expression, and siRNA knockdowns provide strong support for a critical role of the Akt-mTOR-S6K pathway in protecting neurons against anoxic stress in DZ treated neurons.
Activation of the mTOR pathway has been implicated in several disorders of the nervous system. Li and colleagues (Li et al. 2010b) showed that mTOR activation is involved in the beneficial effects of ketamine in the acute treatment of depression. In addition, Kumar et al. (2013) reported that mTOR activation promoted remyelination of nerves in a multiple sclerosis mouse model. Moreover, the mTOR complex and its main downstream kinase, S6K, are thought to be key players in the outcome of an ischemic insult to neurons and astrocytes as shown in both in vitro and in vivo models (Chen et al. 2012; Mao et al. 2013; Pastor et al. 2009; Shi et al. 2011; Wang et al. 2012; Xie et al. 2013). Our results are in agreement with several other studies which have demonstrated that neuroprotective mechanisms such as ischemic preconditioning (Kis et al. 2003a), and agents such as phosphatase and tensin homolog deleted on chromosome 10 (PTEN) inhibitor BPV, melatonin, dehydroepiandrosterone, and atorvastatin can activate the mTOR pathway (Jin et al. 2012; Koh 2008; Li et al. 2010a; Mao et al. 2013). Significantly, previous studies in our laboratory showed that BMS-191095 (Gaspar et al. 2008b) and NS1619 (Gaspar et al. 2008a), two distinct preconditioning agents, also activate the Akt pathway.
Acute treatment with DZ depolarized mitochondria and increased ROS production whereas chronic treatment with DZ maintained the depolarized state of mitochondria but reduced ROS production during normoxia and OGD conditions: results that concur with our previous findings. A new finding is that co-application of R with DZ in the preconditioning paradigm did not prevent mitochondrial depolarization or reduced ROS production while eliminating neuronal protection. Thus, inhibiting ROS production or maintaining depolarized membrane potentials at the onset of OGD probably are not sufficient alone to ensure neuronal survival. Activation of pro-survival signaling pathways, such as those involving Akt-mTOR-S6K, also appear to be required. Additionally, the fact that DZ-preconditioned neurons showed increased OCR but unchanged ROS production indicates more energy production vs. free-radical generation.
In conclusion, our findings show that the change in mitochondrial respiration induced by DZ-induced preconditioning is an important component of neuronal survival after OGD and support the hypothesis that the mTOR pathway is activated after DZ-induced preconditioning (Fig. 7). Associated with changes in membrane potential and ROS production, our data suggest changes in electron transport chain activity and the Akt-mTOR-S6K signaling cascade contributes to the increased survival of neurons with DZ preconditioning. The current therapeutic option, tPA, has limited use (Kirkman et al. 2014). Our studies in neurons using a DZ-preconditioning model against OGD suggest that the mTOR pathway and mitochondria are potential therapeutic targets for high-risk patients.
Figure 7.

Schematic illustration showing the main signaling events occurring during DZ preconditioning in neurons. DZ administration initiates preconditioning by depolarizing mitochondria and increasing ROS production. Protection against OGD develops following the activation of the Akt-mTOR-S6K signaling cascade resulting in increased viability of neurons and preservation of OCR following OGD.
Acknowledgments
The authors thank Nancy Busija, M.A. for editing the manuscript. We also thank Dana Liu and Korey Walter for technical assistance.
Funding: This work was supported by National Institutes of Health grants (D.W.B: HL-077731and HL093554), Louisiana Board of Regents Support Fund-Research Competitiveness Subprogram (PVK: LEQSF(2014-17)-RD-A-11), American Heart Association National Center NRCP Scientist Development Grant (PVK: 14SDG20490359), and American Heart Association Post-Doctoral Fellowship Grant (IR: 15POST23040005).
List of abbreviations used in text
- mTOR
mammalian/mechanistic target of rapamycin
- p-mTOR
phosphorylated mTOR
- t-mTOR
total mTOR
- DZ
diazoxide
- R
rapamycin
- T
Torin-1
- OGD
oxygen glucose deprivation
- PI3K
PI3 Kinase
- Akt
Protein Kinase B
- p-Akt
phosphorylated Akt
- t-Akt
total Akt
- S6K
S6 Kinase
- p-S6K
phosphorylated S6K
- t-S6K
total S6K
- siRNA
silencing RNA
- DMEM
Dulbecco’s Modified Eagle Medium
- PBS
phosphate buffered saline
- tPA
tissue plasminogen activator
- OCR
oxygen consumption rate
- mitoKATP
mitochondrial ATP sensitive potassium channel
- ΔΨm
mitochondrial membrane potential
- ROS
reactive oxygen species
- DMSO
dimethyl sulfoxide
- FCCP
carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone
- A/R
antimycin and rotenone
- CMH
1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine
- KHB
Kreb’s Hepes Buffer
- DF
defferoxamine
- DETC
diethyldithio-carbamate trihydrate
- NOS
nitric oxide synthase
- COX
cyclooxygenase
- HO
heme-oxygenase
- NaOH
sodium hydroxide
Footnotes
The authors have no conflicts of interest to declare.
References
- Ahmed N, Wahlgren N, Grond M, Hennerici M, Lees KR, Mikulik R, Parsons M, Roine RO, Toni D, Ringleb P SITS investigators. Implementation and outcome of thrombolysis with alteplase 3–4.5 h after an acute stroke: an updated analysis from SITS-ISTR. Lancet Neurol. 2010;9:866–874. doi: 10.1016/S1474-4422(10)70165-4. [DOI] [PubMed] [Google Scholar]
- Alcindor D, Krolikowski JG, Pagel PS, Warltier DC, Kersten JR. Cyclooxygenase-2 mediates ischemic, anesthetic, and pharmacologic preconditioning in vivo. Anesthesiology. 2004;100:547–554. doi: 10.1097/00000542-200403000-00013. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Busija DW, Gaspar T, Domoki F, Katakam PV, Bari F. Mitochondrial-mediated suppression of ROS production upon exposure of neurons to lethal stress: mitochondrial targeted preconditioning. Adv Drug Deliv Rev. 2008;60:1471–1477. doi: 10.1016/j.addr.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busija DW, Katakam P, Rajapakse NC, Kis B, Grover G, Domoki F, Bari F. Effects of ATP-sensitive potassium channel activators diazoxide and BMS-191095 on membrane potential and reactive oxygen species production in isolated piglet mitochondria. Brain Res Bull. 2005;66:85–90. doi: 10.1016/j.brainresbull.2005.03.022. [DOI] [PubMed] [Google Scholar]
- Busija DW, Katakam PV. Mitochondrial Mechanisms in Cerebral Vascular Control: Shared Signaling Pathways with Preconditioning. J Vasc Res. 2014;51:175–189. doi: 10.1159/000360765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busija DW, Lacza Z, Rajapakse N, Shimizu K, Kis B, Bari F, Domoki F, Horiguchi T. Targeting mitochondrial ATP-sensitive potassium channels--a novel approach to neuroprotection. Brain Res Brain Res Rev. 2004;46:282–294. doi: 10.1016/j.brainresrev.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Carvalho C, Katz PS, Dutta S, Katakam PV, Moreira PI, Busija DW. Increased Susceptibility to Amyloid-beta Toxicity in Rat Brain Microvascular Endothelial Cells under Hyperglycemic Conditions. J Alzheimers Dis. 2014;38:75–83. doi: 10.3233/JAD-130464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Qu Y, Tang B, Xiong T, Mu D. Role of mammalian target of rapamycin in hypoxic or ischemic brain injury: potential neuroprotection and limitations. Rev Neurosci. 2012;23:279–287. doi: 10.1515/revneuro-2012-0001. [DOI] [PubMed] [Google Scholar]
- Costa AD, Quinlan CL, Andrukhiv A, West IC, Jaburek M, Garlid KD. The direct physiological effects of mitoK(ATP) opening on heart mitochondria. Am J Physiol Heart Circ Physiol. 2006;290:H406–15. doi: 10.1152/ajpheart.00794.2005. [DOI] [PubMed] [Google Scholar]
- Deadwyler SA, Hampson RE, Bennett BA, Edwards TA, Mu J, Pacheco MA, Ward SJ, Childers SR. Cannabinoids modulate potassium current in cultured hippocampal neurons. Receptors Channels. 1993;1:121–134. [PubMed] [Google Scholar]
- Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med. 2011;51:1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domoki F, Kis B, Nagy K, Farkas E, Busija DW, Bari F. Diazoxide preserves hypercapnia-induced arteriolar vasodilation after global cerebral ischemia in piglets. Am J Physiol Heart Circ Physiol. 2005;289:H368–73. doi: 10.1152/ajpheart.00887.2004. [DOI] [PubMed] [Google Scholar]
- Domoki F, Perciaccante JV, Veltkamp R, Bari F, Busija DW. Mitochondrial potassium channel opener diazoxide preserves neuronal-vascular function after cerebral ischemia in newborn pigs. Stroke. 1999;30:2713–8. doi: 10.1161/01.str.30.12.2713. discussion 2718–9. [DOI] [PubMed] [Google Scholar]
- Dos Santos P, Kowaltowski AJ, Laclau MN, Seetharaman S, Paucek P, Boudina S, Thambo JB, Tariosse L, Garlid KD. Mechanisms by which opening the mitochondrial ATP- sensitive K(+) channel protects the ischemic heart. Am J Physiol Heart Circ Physiol. 2002;283:H284–95. doi: 10.1152/ajpheart.00034.2002. [DOI] [PubMed] [Google Scholar]
- Elijovich L, Chong JY. Current and future use of intravenous thrombolysis for acute ischemic stroke. Curr Atheroscler Rep. 2010;12:316–321. doi: 10.1007/s11883-010-0121-8. [DOI] [PubMed] [Google Scholar]
- Fenton TR, Gout IT. Functions and regulation of the 70kDa ribosomal S6 kinases. Int J Biochem Cell Biol. 2011;43:47–59. doi: 10.1016/j.biocel.2010.09.018. [DOI] [PubMed] [Google Scholar]
- Floris R, Cozzolino V, Meschini A, Garaci F, Konda D, Marziali S, Sallustio F, Di Legge S, Claroni G, Fanucci E, Simonetti G, Stanzione P. Efficacy of systemic thrombolysis within 4.5 h from stroke symptom onset: a single-centre clinical and diffusion-perfusion 3T MRI study. Radiol Med. 2014;10:767–774. doi: 10.1007/s11547-014-0394-z. [DOI] [PubMed] [Google Scholar]
- Gaspar T, Katakam P, Snipes JA, Kis B, Domoki F, Bari F, Busija DW. Delayed neuronal preconditioning by NS1619 is independent of calcium activated potassium channels. J Neurochem. 2008a;105:1115–1128. doi: 10.1111/j.1471-4159.2007.05210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar T, Snipes JA, Busija AR, Kis B, Domoki F, Bari F, Busija DW. ROS-independent preconditioning in neurons via activation of mitoK(ATP) channels by BMS-191095. J Cereb Blood Flow Metab. 2008b;28:1090–1103. doi: 10.1038/sj.jcbfm.9600611. [DOI] [PubMed] [Google Scholar]
- Hacke W, Donnan G, Fieschi C, Kaste M, von Kummer R, Broderick JP, Brott T, Frankel M, Grotta JC, Haley EC, Jr, Kwiatkowski T, Levine SR, Lewandowski C, Lu M, Lyden P, Marler JR, Patel S, Tilley BC, Albers G, Bluhmki E, Wilhelm M, Hamilton S ATLANTIS Trials Investigators ECASS Trials Investigators, NINDS rt-PA Study Group Investigators. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet. 2004;363:768–774. doi: 10.1016/S0140-6736(04)15692-4. [DOI] [PubMed] [Google Scholar]
- Hacke W, Kaste M, Bluhmki E, Brozman M, Davalos A, Guidetti D, Larrue V, Lees KR, Medeghri Z, Machnig T, Schneider D, von Kummer R, Wahlgren N, Toni D ECASS Investigators. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359:1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- Jin Y, Sui HJ, Dong Y, Ding Q, Qu WH, Yu SX, Jin YX. Atorvastatin enhances neurite outgrowth in cortical neurons in vitro via up-regulating the Akt/mTOR and Akt/GSK-3beta signaling pathways. Acta Pharmacol Sin. 2012;33:861–872. doi: 10.1038/aps.2012.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katakam PV, Gordon AO, Sure VN, Rutkai I, Busija DW. Diversity of Mitochondria-Dependent Dilator Mechanisms in Vascular Smooth Muscle of Cerebral Arteries from Normal and Insulin Resistant rats. Am J Physiol Heart Circ Physiol. 2014;4:H493–503. doi: 10.1152/ajpheart.00091.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katakam PV, Wappler EA, Katz PS, Rutkai I, Institoris A, Domoki F, Gaspar T, Grovenburg SM, Snipes JA, Busija DW. Depolarization of mitochondria in endothelial cells promotes cerebral artery vasodilation by activation of nitric oxide synthase. Arterioscler Thromb Vasc Biol. 2013;33:752–759. doi: 10.1161/ATVBAHA.112.300560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly CJ, Hussien K, Fokas E, Kannan P, Shipley RJ, Ashton TM, Stratford M, Pearson N, Muschel RJ. Regulation of O2 consumption by the PI3K and mTOR pathways contributes to tumor hypoxia. Radiother Oncol. 2014;111:72–80. doi: 10.1016/j.radonc.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkman MA, Citerio G, Smith M. The intensive care management of acute ischemic stroke: an overview. Intensive Care Med. 2014;5:640–53. doi: 10.1007/s00134-014-3266-z. [DOI] [PubMed] [Google Scholar]
- Kis A, Yellon DM, Baxter GF. Second window of protection following myocardial preconditioning: an essential role for PI3 kinase and p70S6 kinase. J Mol Cell Cardiol. 2003a;35:1063–1071. doi: 10.1016/s0022-2828(03)00208-6. [DOI] [PubMed] [Google Scholar]
- Kis B, Nagy K, Snipes JA, Rajapakse NC, Horiguchi T, Grover GJ, Busija DW. The mitochondrial K(ATP) channel opener BMS-191095 induces neuronal preconditioning. Neuroreport. 2004;15:345–349. doi: 10.1097/00001756-200402090-00027. [DOI] [PubMed] [Google Scholar]
- Kis B, Rajapakse NC, Snipes JA, Nagy K, Horiguchi T, Busija DW. Diazoxide induces delayed pre-conditioning in cultured rat cortical neurons. J Neurochem. 2003b;87:969–980. doi: 10.1046/j.1471-4159.2003.02072.x. [DOI] [PubMed] [Google Scholar]
- Koh PO. Melatonin prevents ischemic brain injury through activation of the mTOR/p70S6 kinase signaling pathway. Neurosci Lett. 2008;444:74–78. doi: 10.1016/j.neulet.2008.08.024. [DOI] [PubMed] [Google Scholar]
- Kumar S, Patel R, Moore S, Crawford DK, Suwanna N, Mangiardi M, Tiwari-Woodruff SK. Estrogen receptor beta ligand therapy activates PI3K/Akt/mTOR signaling in oligodendrocytes and promotes remyelination in a mouse model of multiple sclerosis. Neurobiol Dis. 2013;56:131–144. doi: 10.1016/j.nbd.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzser G, Kis B, Bari F, Busija DW. Diazoxide preconditioning attenuates global cerebral ischemia-induced blood-brain barrier permeability. Brain Res. 2005;1051:72–80. doi: 10.1016/j.brainres.2005.05.064. [DOI] [PubMed] [Google Scholar]
- Li L, Xu B, Zhu Y, Chen L, Sokabe M, Chen L. DHEA prevents Abeta25–35-impaired survival of newborn neurons in the dentate gyrus through a modulation of PI3K-Akt-mTOR signaling. Neuropharmacology. 2010a;59:323–333. doi: 10.1016/j.neuropharm.2010.02.009. [DOI] [PubMed] [Google Scholar]
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010b;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L, Jia J, Zhou X, Xiao Y, Wang Y, Mao X, Zhen X, Guan Y, Alkayed NJ, Cheng J. Delayed administration of a PTEN inhibitor BPV improves functional recovery after experimental stroke. Neuroscience. 2013;231:272–281. doi: 10.1016/j.neuroscience.2012.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayanagi K, Gaspar T, Katakam PV, Busija DW. Systemic administration of diazoxide induces delayed preconditioning against transient focal cerebral ischemia in rats. Brain Res. 2007a;1168:106–111. doi: 10.1016/j.brainres.2007.06.071. [DOI] [PubMed] [Google Scholar]
- Mayanagi K, Gaspar T, Katakam PV, Kis B, Busija DW. The mitochondrial K(ATP) channel opener BMS-191095 reduces neuronal damage after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2007b;27:348–355. doi: 10.1038/sj.jcbfm.9600345. [DOI] [PubMed] [Google Scholar]
- Mrakic-Sposta S, Gussoni M, Montorsi M, Porcelli S, Vezzoli A. Assessment of a standardized ROS production profile in humans by electron paramagnetic resonance. Oxid Med Cell Longev. 2012;2012:973927. doi: 10.1155/2012/973927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy K, Kis B, Rajapakse NC, Bari F, Busija DW. Diazoxide preconditioning protects against neuronal cell death by attenuation of oxidative stress upon glutamate stimulation. J Neurosci Res. 2004;76:697–704. doi: 10.1002/jnr.20120. [DOI] [PubMed] [Google Scholar]
- Nazarewicz RR, Dikalova AE, Bikineyeva A, Dikalov SI. Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. Am J Physiol Heart Circ Physiol. 2013;305:H1131–40. doi: 10.1152/ajpheart.00063.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary MT, Ng KE, Ludtmann MH, Hall AR, Piotrowska I, Ong SB, Hausenloy DJ, Mohun TJ, Abramov AY, Breckenridge RA. Hypoxia signaling controls postnatal changes in cardiac mitochondrial morphology and function. J Mol Cell Cardiol. 2014;74:340–352. doi: 10.1016/j.yjmcc.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyekan AO, McGiff JC, Rosencrantz-Weiss P, Quilley J. Relaxant responses of rabbit aorta: influence of cytochrome P450 inhibitors. J Pharmacol Exp Ther. 1994;268:262–269. [PubMed] [Google Scholar]
- Pamenter ME, Shin DS, Cooray M, Buck LT. Mitochondrial ATP-sensitive K+ channels regulate NMDAR activity in the cortex of the anoxic western painted turtle. J Physiol. 2008;586:1043–1058. doi: 10.1113/jphysiol.2007.142380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor MD, Garcia-Yebenes I, Fradejas N, Perez-Ortiz JM, Mora-Lee S, Tranque P, Moro MA, Pende M, Calvo S. mTOR/S6 kinase pathway contributes to astrocyte survival during ischemia. J Biol Chem. 2009;284:22067–22078. doi: 10.1074/jbc.M109.033100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter GA, Urciuoli WR, Brookes PS, Nadtochiy SM. SIRT3 deficiency exacerbates ischemia-reperfusion injury: implication for aged hearts. Am J Physiol Heart Circ Physiol. 2014;306:H1602–9. doi: 10.1152/ajpheart.00027.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapakse N, Kis B, Horiguchi T, Snipes J, Busija D. Diazoxide pretreatment induces delayed preconditioning in astrocytes against oxygen glucose deprivation and hydrogen peroxide-induced toxicity. J Neurosci Res. 2003;73:206–214. doi: 10.1002/jnr.10657. [DOI] [PubMed] [Google Scholar]
- Rajapakse N, Shimizu K, Kis B, Snipes J, Lacza Z, Busija D. Activation of mitochondrial ATP-sensitive potassium channels prevents neuronal cell death after ischemia in neonatal rats. Neurosci Lett. 2002;327:208–212. doi: 10.1016/s0304-3940(02)00413-5. [DOI] [PubMed] [Google Scholar]
- Rutkai I, Katakam PV, Dutta S, Busija DW. Sustained Mitochondrial Functioning in Cerebral Arteries after Transient Ischemic Stress in the Rat: A potential target for therapies. Am J Physiol Heart Circ Physiol. 2014;7:H958–66. doi: 10.1152/ajpheart.00405.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi GD, OuYang YP, Shi JG, Liu Y, Yuan W, Jia LS. PTEN deletion prevents ischemic brain injury by activating the mTOR signaling pathway. Biochem Biophys Res Commun. 2011;404:941–945. doi: 10.1016/j.bbrc.2010.12.085. [DOI] [PubMed] [Google Scholar]
- Shimizu K, Lacza Z, Rajapakse N, Horiguchi T, Snipes J, Busija DW. MitoK(ATP) opener, diazoxide, reduces neuronal damage after middle cerebral artery occlusion in the rat. Am J Physiol Heart Circ Physiol. 2002;283:H1005–11. doi: 10.1152/ajpheart.00054.2002. [DOI] [PubMed] [Google Scholar]
- Wahlgren N, Ahmed N, Davalos A, Hacke W, Millan M, Muir K, Roine RO, Toni D, Lees KR SITS investigators. Thrombolysis with alteplase 3–4.5 h after acute ischaemic stroke (SITS-ISTR): an observational study. Lancet. 2008;372:1303–1309. doi: 10.1016/S0140-6736(08)61339-2. [DOI] [PubMed] [Google Scholar]
- Wang C, Wang Z, Zhang X, Zhang X, Dong L, Xing Y, Li Y, Liu Z, Chen L, Qiao H, Wang L, Zhu C. Protection by silibinin against experimental ischemic stroke: up-regulated pAkt, pmTOR, HIF-1alpha and Bcl-2, down-regulated Bax, NF-kappaB expression. Neurosci Lett. 2012;529:45–50. doi: 10.1016/j.neulet.2012.08.078. [DOI] [PubMed] [Google Scholar]
- Wappler EA, Institoris A, Dutta S, Katakam PV, Busija DW. Mitochondrial dynamics associated with oxygen-glucose deprivation in rat primary neuronal cultures. PLoS One. 2013;8:e63206. doi: 10.1371/journal.pone.0063206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie R, Wang P, Ji X, Zhao H. Ischemic postconditioning facilitates brain recovery after stroke by promoting Akt/mTOR activity in nude rats. J Neurochem. 2013;5:723–32. doi: 10.1111/jnc.12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Chen S, Cadenas E, Brinton RD. Estrogen protection against mitochondrial toxin-induced cell death in hippocampal neurons: antagonism by progesterone. Brain Res. 2011a;1379:2–10. doi: 10.1016/j.brainres.2010.11.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Chen S, Mao Z, Cadenas E, Brinton RD. 2-Deoxy-D-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer’s disease. PLoS One. 2011b;6:e21788. doi: 10.1371/journal.pone.0021788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Zhao L, Mao Z, Chen S, Wong KC, To J, Brinton RD. Potentiation of brain mitochondrial function by S-equol and R/S-equol estrogen receptor beta-selective phytoSERM treatments. Brain Res. 2013;1514:128–141. doi: 10.1016/j.brainres.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Z, Huang HF, He F, Wu LX, Lin J, Chen MQ. Diazoxide attenuates ischemia/reperfusion injury via upregulation of heme oxygenase-1 after liver transplantation in rats. World J Gastroenterol. 2012;18:1765–1772. doi: 10.3748/wjg.v18.i15.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]