

Abstract
The network of protein-protein interactions among the BCL-2 protein family plays a critical role in regulating cellular commitment to mitochondrial apoptosis. Anti-apoptotic BCL-2 proteins are considered promising targets for drug discovery and exciting clinical progress has stimulated intense investigations in the broader family. Here, we discuss recent developments in small molecules targeting anti-apoptotic proteins and alternative approaches to targeting BCL-2 family interactions. These studies advance our understanding of the role of BCL-2 family proteins in physiology and disease, providing unique tools for dissecting these functions. The BCL-2 family of proteins is a prime example of targeting protein-protein interactions and further chemical biology approaches will increase opportunities for novel targeted therapies in cancer, autoimmune and aging-associated diseases.
Graphical abstract
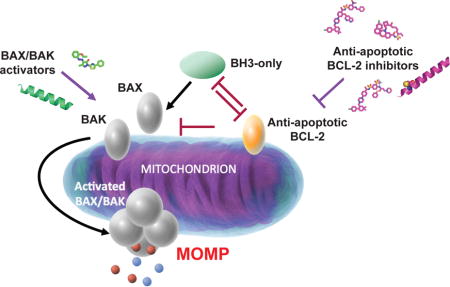
Introduction
Our understanding that regulation of cell survival and death through the mitochondrial apoptotic pathway is important in development, homeostasis, and many human diseases has led to intensive investigation of the BCL-2 family of proteins over the last two decades [1]. Protein-protein interactions of the BCL-2 family members dictate cell survival and death decisions in the mitochondrial pathway. Their deregulation can lead to imbalance of homeostasis contributing to the development of a variety of diseases [2]. Mitochondrial apoptosis is driven by the activity of the conserved BCL-2 homology domain 3 (BH3) of pro-apoptotic BCL-2 members [3,4]. Pro-apoptotic BH3-only proteins such as BIM, BID, PUMA and NOXA use their BH3 domain to inhibit anti-apoptotic BCl-2 proteins such as BCL-2, BCL-XL and MCL-1 and activate pro-apoptotic BCL-2 proteins BAX and BAK [2,5,6]. When BAX and BAK are directly activated by BH3-only proteins or released from inhibited anti-apoptotics, they use their BH3 domain to oligomerize and assemble mitochondrial pores that induce mitochondrial outer membrane permeabilization, a key event that leads to apoptosis (Figure 1) [7,8].
Figure 1. The mitochondrial pathway of apoptosis.
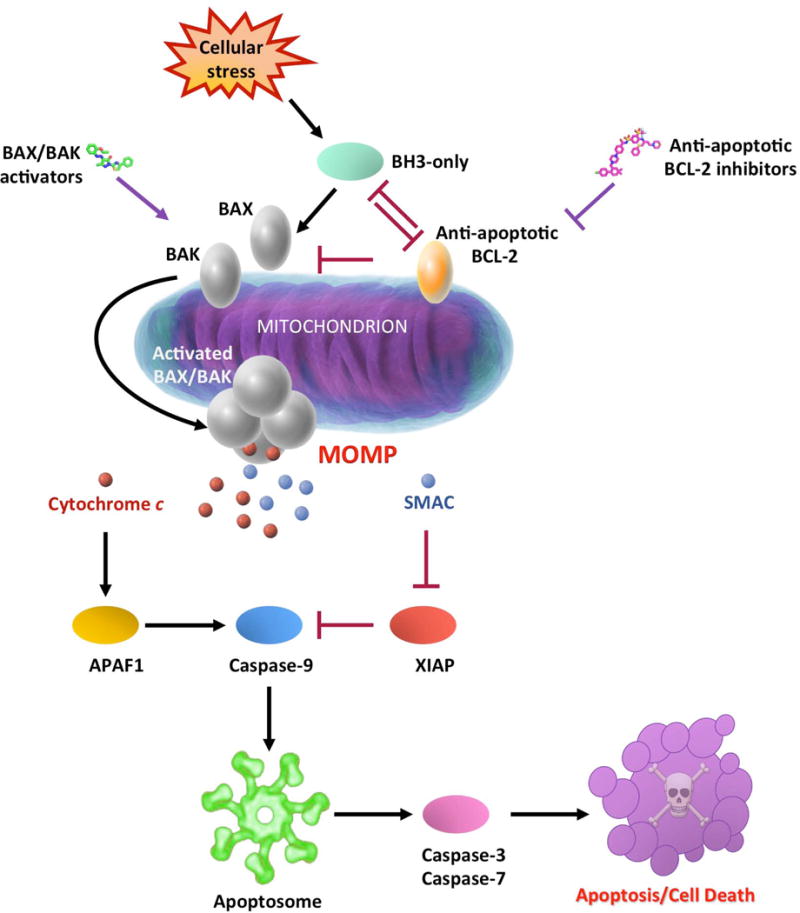
The intrinsic or mitochondrial apoptotic pathway is characterized by the mitochondrial outer membrane permeabilization (MOMP). Upon variety of stress stimuli, pro-apoptotic BH3-only proteins inhibit anti-apoptotic BCL-2 proteins and activate pro-apoptotic BAX and BAK. Activated BAX and BAK oligomerize and form pores to the mitochondrial outer membrane triggering MOMP. MOMP induces release of mitochondrial intermembrane space proteins such as cytochrome c and second mitochondria-derived activator of caspases (SMAC) into the cytosol. While SMAC boosts apoptosis by blocking caspase inhibitor X-linked inhibitor of apoptosis protein (XIAP), cytochrome c promotes apoptosis by activating the caspase cascade. Cytochrome c interacts with the apoptotic protease activating factor 1 (APAF1), leading to the activation of caspase-9 and the apoptosome assembly. Activated caspase-9 activates caspase-3 and caspase-7, leading to apoptosis. Anti-apoptotic BCL-2 inhibitors promote apoptosis by releasing sequestered BH3-only proteins and BAX and BAK from anti-apoptotic BCL-2 proteins. In contrast, BAX and BAK activators can bind directly to these pro-apoptotic proteins and activate them to promote cell death.
Anti-apoptotic BCL-2 proteins posses a hydrophobic groove, comprising conserved BCL-2 homology domain 1 (BH1) and 2 (BH2) that binds and sequesters the BH3 domains of pro-apoptotic members [9]. Structural elucidation of anti-apoptotics bound to a range of BH3 domains has led to the development of a large number of small molecules targeting the hydrophobic groove of anti-apoptotic BCL-2, BCL-XL and MCL-1 [10,11]. These small molecules, called BH3 mimetics, bind to one or more of P1-P4 sub-pockets in the BH3 groove of anti-apoptotic proteins, releasing pro-apoptotic BH3-only proteins that can activate BAX and BAK and lead to apoptosis. Here, we discuss the progress with the development of selective BH3 mimetics and the emerging approaches associated with targeting non-canonical pockets and pro-apoptotic BCL-2 proteins (Table 1). Figure 2 shows select small molecules and probes targeting the BCL-2 proteins that will be discussed in this review.
Table 1.
Characteristics of inhibitors and activators of the BCL-2 family of proteins through canonical and non-canonical interactionsa.
| Name | Target(s) | In vivo antitumor effects | Clinical Stage | References |
|---|---|---|---|---|
| AT-101 (Gossypol) | BCL-2, BCL-XL, MCL-1 | Yes | Phase IIb | 14 |
| Obatoclax | BCL-2, BCL-XL, MCL-1, BCL-W, BFL-1 | Yes | Phase IIb | 15 |
| ABT-737 | BCL-XL, BCL-2, BCL-W | Yes | 16 | |
| ABT-263 (Navitoclax) | BCL-XL, BCL-2, BCL-W | Yes | Phase IIb | 17 |
| BM-1197 | BCL-2, BCL-XL | Yes | 20 | |
| S44563 | BCL-2, BCL-XL | Yes | 21,22 | |
| BCL-32 | BCL-2, BCL-XL | Yes | 23 | |
| AZD4320 | BCL-2, BCL-XL | Yes | 24 | |
| ABT-199 (Venetoclax) | BCL-2 | Yes | FDA-Approved | 25,26 |
| S55746 (BCL201) | BCL-2 | Yes | Phase I | 27–29 |
| WEHI-539 | BCL-XL | No | 30 | |
| A-1155463 | BCL-XL | No | 31 | |
| A-1331852 | BCL-XL | Yes | 32 | |
| MCL-1-SAHBD | MCL-1 | No | 35 | |
| BimS2A | MCL-1 | No | 36 | |
| Maritoclax | MCL-1 | Yes | 37 | |
| UMI-77 | MCL-1 | Yes | 38 | |
| Compounds 4/5 | MCL-1 | No | 40 | |
| A-1210477 | MCL-1 | No | 39 | |
| S63845 | MCL-1 | Yes | 41 | |
| AMG176 | MCL-1 | Yes | Phase I | 42 |
| AZD5991 | MCL-1 | Yes | 43 | |
| BH3-SAHBA-3 | BFL-1 | No | 44 | |
| hNOXA 130G4 | BFL-1 | No | 45 | |
| BINDI | BCL-2, BCL-XL, MCL-1, BCL-W, BFL-1, BCL-B | No | 46 | |
| BDA-366 | BCL-2 | Yes | 47 | |
| NuBCP-9 | BCL-2 | Yes | 48 | |
| Compound 5 | MCL-1 | No | 49 | |
| MAIM1 | MCL-1 | No | 50 | |
| Compound 11 | MCL-1 | No | 51 | |
| BIM-SAHB | BAX | Yes | 52–54 | |
| BAM7 | BAX | No | 58 | |
| 3G11 | BAX | No | 59 | |
| BCL2-BH4 SAHB | BAX | No | 60 | |
| BID-SAHB | BAK | No | 61–62 | |
| 7D10 | BAK | No | 64 |
Canonical binders in light grey background, non-canonical/covalent binders in dark grey background, peptide/protein in italics.
completed clinical trials
Figure 2.
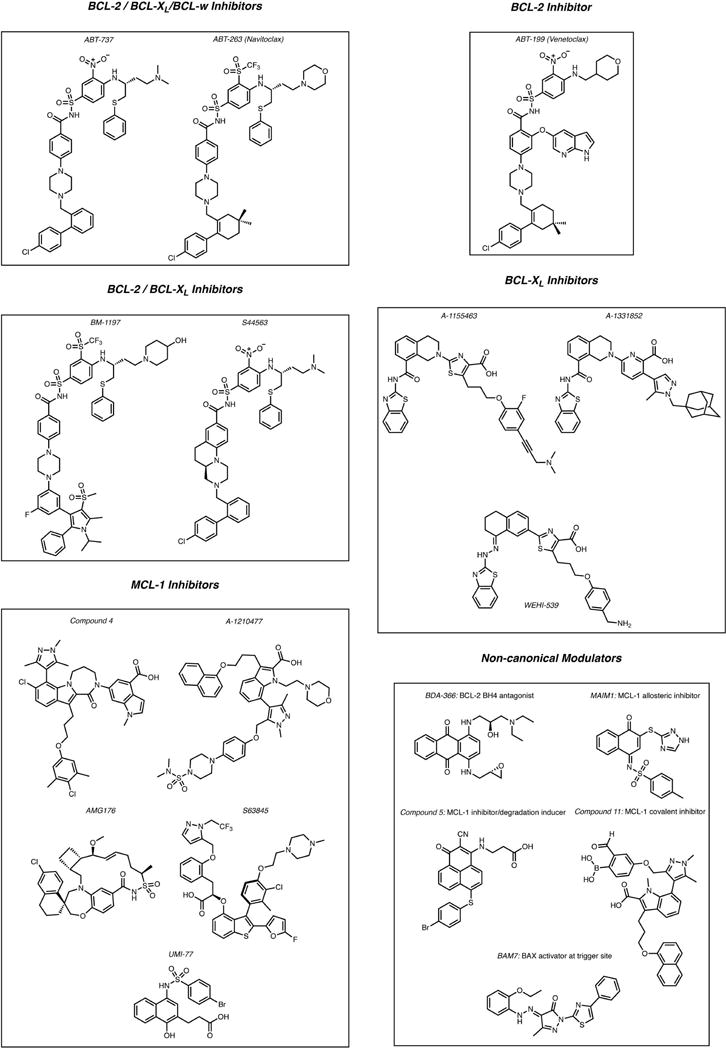
Small molecules targeting the BCL-2 family of proteins
BH3 mimetics of anti-apoptotic BCL-2 proteins
Several BH3 mimetics, either small molecules or peptidomimetics have been identified and previously reviewed [11–13]. The first generation of BH3 mimetics had limited selectivity for a specific anti-apoptotic BCL-2 protein and most of them were also found to promote cell death independently of BAX/BAK proteins. Compounds as AT-101 and Obatoclax progressed to clinical evaluation, although significant toxicities associated with off-target effects halted their development further [14,15].
The discovery of ABT-737 and its orally available analogue ABT-263 (navitoclax), demonstrated a more potent and selective profile for BAX/BAK dependent apoptosis [16,17]. ABT-737 was discovered at Abbot Laboratories using a fragment-based drug design approach by NMR and binds P1-P4 sub-pockets of BCL-XL, BCL-2 and BCL-W with subnanomolar affinity (Ki < 1nM). Navitoclax showed efficacy in clinical trials for hematological malignancies and small cell lung cancer, however, its effectiveness is limited by dose dependent BCL-XL-mediated thrombocytopenia due to platelets apoptosis [18,19].
Dual BCL-2/BCL-XL inhibitors
Based on the success of ABT-737/ABT-263, several dual BCL-2/BCL-XL inhibitors were developed using their arylsulfonamide scaffold. Wang and colleagues generated BM-1197, a dual BCL-2/BCL-XL inhibitor with subnanomolar affinity for both BCL-2/BCL-XL and >1,000-fold selectivity over MCL-1 [20]. BM-1197 exerted potent antitumor activity in small cell lung cancer cells and complete and long-term tumor regression in xenograft models after intravenous administration at daily or weekly schedule. BM-1197 caused reversible platelet reduction in mice at highly efficacious doses. Servier also generated a nanomolar affinity dual BCL-2/BCL-XL inhibitor, S44563, which exerted promising inhibitory efficacy in patient-derived uveal melanoma cell lines and xenografts and enhanced sensitivity of small cell lung cancer to radiation [21,22].
A dual inhibitor, BCL-32, with nanomolar affinity for BCL-2 (Ki = 3.3 nM) and BCL-XL (Ki = 8.5nM) was reported by AstraZeneca [23]. BCL-32 showed potent inhibitory efficacy in vivo allowing intermittent dosing schedule and full platelet recovery between doses. Another subnanomolar inhibitor of both BCL-2 and BCL-XL, AZD4320, was recently reported as a clinical candidate by AstraZeneca [24]. AZD4320 exhibited potent antitumor activity in BCL-2/BCL-XL-driven cell lines and RS4;11 ALL xenograft model. Reversible reduction of platelets counts was detected after 72 hours.
BCL-2 inhibitors
Structure-based design based on the ABT-263 co-crystal structure with BCL-2, enabled tailoring the interaction with the P4 sub-pocket of BCL-2, leading to the first potent and selective BH3 mimetic ABT-199 (Figure 3a) [25]. ABT-199 (venetoclax) has subanonomolar affinity (Ki = 0.01 nM) for BCL-2 protein inducing potent apoptosis in BCL-2-dependent patient-derived cells lines and xenografts from a variety of leukemia and lymphoma malignancies without triggering thrombocytopenia. Venetoclax became the first BH3 mimetic to be FDA-approved in April 2016, for use in patients with chronic lymphocytic leukemia (CLL) with the 17p deletion [26]. Venetoclax is currently being evaluated in multiple clinical trials as a monotherapy and combination therapy for non-Hodgkin lymphomas, acute myeloid leukemia, multiple myeloma and breast cancer.
Figure 3. Representative structures of BCL-2 family proteins in complex with modulators.
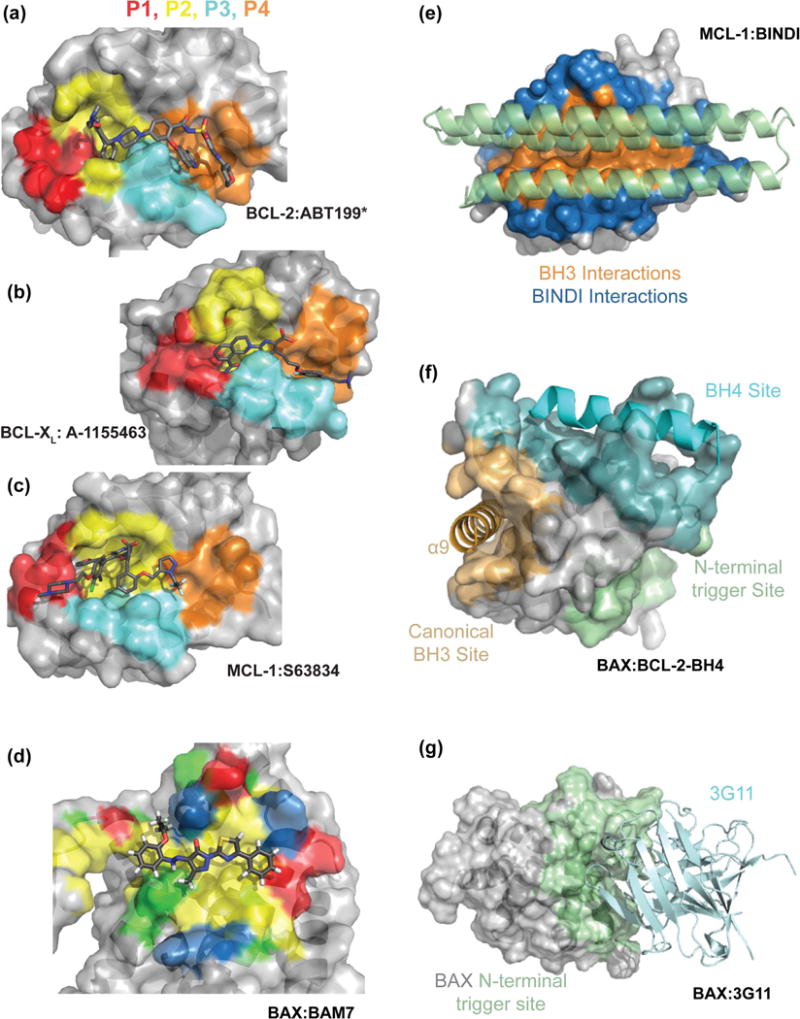
Small molecule inhibitors of anti-apoptotic members bind to the 4 sub-pockets (P1-P4) of the canonical groove, (a) BCL-2 in complex with an analogue of ABT-199* (PDB:4MAN), (b) BCL-XL in complex with A-1155463 (PDB:4QVX) and (c) MCL-1 in complex with S63845 (PDB:5LOF). (d) BAM7 binds to a hydrophobic N-terminal trigger site of BAX (Modified PDB:2K7W), surface properties illustrated as hydrophobic:yellow, hydrophilic:green, acidic:red, basic:blue. (e) BINDI helical bundles utilize both canonical groove and the surrounding surface to increase specificity and potency. MCL-1 in complex with a MCL-1 specific BINDI (PDB:5JSB). (f) A stapled peptide derived from the BH4 domain of BCL-2 binds to a surface of BAX distinct from both the canonical and trigger site of BAX [56]. (g) The antibody 3G11 binds BAX through interaction with an extended N-terminal trigger site inhibiting BAX conformational activation [55].
Following the paradigm of ABT-199 for high selectivity against one anti-apoptotic protein, monoselective BH3 mimetics for each BCL-2, BCL-XL and MCL-1 have been recently generated. Servier and Vernalis collaboration yielded a novel class of BCL-2 selective compounds based on tetrahydroisoquinoline amide substituted pyrazoles [27]. S55746 derived from this class has entered phase I trials as single agent in patients with CLL and Non-Hodgkin lymphoma and in combination with the P13Kδ inhibitor idelalisib in follicular lymphoma and mantle cell lymphoma [28,29].
BCL-XL inhibitors
Lessene et al. utilized an alpha screen competition assay to disrupt BIM BH3 interaction with BCL-XL and not MCL-1 to identify a chemical scaffold with selectivity for Bcl-XL [30]. Structure-based guided medicinal chemistry focused on specific interactions with the P2 and P4 sub-pockets led to WEHI-539, an inhibitor with subnanomolar affinity for BCL-XL (IC50 = 1.1 nM) and >400-fold selectivity against other anti-apoptotics. WEHI-539 promoted robust apoptosis induction in BCL-XL dependent small cell lung cancer cells, however, its poor physicochemical properties limits its activity in vivo.
Abbvie colleagues combined NMR fragment screening and structure-based design to optimize WEHI-539 leading to A-1155463 and A-1331852 [31,32]. A-1331852 is the most potent and selective orally available BCL-XL inhibitor (Ki = < 0.010 nM) reported with 10–50 fold improved cellular activity than A-1155463 and ABT-263 (Figure 3b). A-1331852 induced tumor regression in xenograft models of solid tumors including ovarian cancer, breast cancer and NSCLC. Moreover, A-1331852 showed improved efficacy when tested in combination with docetaxel with no apparent toxicity [31].
MCL-1 inhibitors
The structure of MCL-1 suggested that its BH3 groove is particularly shallow in P1, P2 and P4 sub-pockets compared to BCL-2 and BCL-XL [33]. Interestingly, alanine-scanning mutagenesis suggested that BH3 binding to the P4 sub-pocket is not as important as in BCl-2 and BCL-XL [34]. Earlier reports demonstrated high affinity for MCL-1 and selectivity over BCL-2 and BCL-XL with BIM BH3-based peptides, stapled peptides based on the MCL-1 BH3 domain and the natural product maritoclax [35–37].
Nikolovska-Coleska and colleagues identified UMI-77 through HTS and structure-based design, with moderate binding affinity (Ki= 490 nM) and selectivity for MCL-1 [38]. UMI-77 induces apoptosis by disrupting MCL-1/BAX and MCL-1/BAK complexes. In an in vivo pancreatic xenograft model, intravenous dosing of UMI-77 in two cycles provided evidence of tumor growth inhibition.
Abbvie and independently the Fesik lab adopted a strategy to grow P2 bound hits from fragment-based NMR screens and generate compounds based on the 2-carboxy indole core. These approaches led to larger P2-P4 binders such as A-1210477 by Abbvie [39] and compounds 4 and 5 by the Fesik lab [40], which exhibit subnanomolar affinity (Ki = 0.045 nM and Ki = 0.05 nM respectively) and selectivity >1,000-fold over BCL-2 and BCL-XL. These compounds induced single-agent mitochondrial apoptosis in MCL-1 dependent cell lines of hematologic cancers.
Servier recently reported the development of S6384 [41], with subnanomolar (Ki = 0.15 nM) binding to MCL-1 and selectivity of ~10,000 fold over BCL-2 and BCL-XL (Figure 3c). S63845 has a highly potent and broad pro-apoptotic killing activity in a variety of MCL-1-addicted hematopoietic malignancies as well as select breast and NSCLC cell lines. S63845 was highly effective in vivo in xenograft models of AML, lymphoma and multiple myeloma. Moreover, when it was combined with MEK1/2 inhibitor, trametenib, HER-2 inhibitor, lapatinib, B-RAF inhibitor, PLX-4032 and EGFR inhibitor, tarceva, it exhibited effective synergistic cytotoxicity in solid tumors [41].
AMG176 developed by Amgen is the first MCL-1 inhibitor to enter phase 1 trials for the treatment of relapsed and refractory multiple myeloma. Data disclosed recently identify AMG176 as a macrocyclic inhibitor that binds with subnanomolar affinity (Ki = 0.13 nM) around the P2 sub-pocket forming a hydrogen bond with Arg 263 of MCL-1 [42]. AMG176 induced potent apoptosis in MCL-1-addicted cell lines derived from a variety of hematologic malignancies exhibiting on-mechanism activity. AMG176 demonstrated robust efficacy in a multiple myeloma xenograft using a discontinuous dosing schedule of once or twice weekly.
AZD5991 is another rationally designed macrocyclic MCL-1 inhibitor with sub-nanomolar affinity for MCL-1 (Ki = 0.13 nM) recently disclosed by AstraZeneca [43]. AZD5991 demonstrated potent and rapid apoptosis induction in MCL-1-addicted cell lines and achieves complete tumor growth inhibition in vivo in multiple myeloma xenografts from a single intravenous dose.
BFL-1 inhibitors
Although studies have demonstrated BFL-1 upregulation as a chemoresistance factor in subsets of leukemia, lymphoma and melanoma, targeting studies have been limited. Recent efforts have been conducted towards the development of peptide-based inhibitors taking advantage of the high affinity and selective interaction of NOXA BH3 helix with BFL-1 and the reactive cysteines at each interaction partner that can form a covalent bond. Walensky and coworkers introduced to stapled peptides after NOXA and BIM BH3 domains an acrylamide moiety that reacts with BFL-1 Cys55 to generate an irreversible and highly selective BFL-1 inhibitor that enhanced apoptosis in BFL-1 driven melanoma cells [44]. Pellecchia and coworkers took a similar approach and replaced the Cys25 of NOXA BH3 with a mild Michael acceptor (Dap-2-chloroacetamide). A nanomolar (IC50 = 9 nM) BFL-1 peptide inhibitor was generated with 2–5-fold selectivity over other anti-apoptotics and cellular activity in in BFL-1 expressing melanoma and lymphoma cells [45].
Non-canonical targeting of anti-apoptotic BCL-2 proteins
Besides targeting the P1-P4 sub-pockets of the BH3 groove, additional mechanisms of BCL-2 family protein interactions have been investigated and small molecule or protein-based probes have been identified to bind non-canonical surfaces.
Berger et al. used a 3-helix bundle protein, BINDI that utilizes a BH3-like central helix with two flanking helices for additional contacts [46]. Computational mutagenesis and docking was used to develop a series of mutants capable of selectively inhibiting each of the six anti-apoptotic BCL-2 proteins. The success of this technique is attributed to the exploitation of residues outside of the canonical BH3 groove unique to each anti-apoptotic protein and formed by α3–α5 region (Figure 3e). This increase in contact surface area is translated into increased affinity and specificity. When these designed proteins were expressed in cells, they inhibited their corresponding BCL-2 protein providing unique cellular probes.
BCL-2
Using a computational docking screen to target the BH4 domain (aa 6–31) of BCL-2 protein that is essential for its anti-apoptotic function, BDA-366 was identified to bind selectively to BCL-2 and antagonize it. BDA-366 results in a BCL-2 conformational change that exposes its BH3 domain resulting in pro-apoptotic activity [47]. BDA-366 was shown to suppress lung cancer growth in lung cancer cell lines and xenografts alone and in synergy with an mTOR inhibitor. Although the mechanism for this structural transition of BCL-2 is elusive, similar conclusions were previously reported for NuBCP-9, a short Nur77-derived peptide, which bound to the loop region between the BCL-2 BH3 and BH4 domains with Kd ~ 200 nM [48]. NuBCP-9 was able to induce exposure of the BCL-2 BH3 domain, which in turn suppressed anti-apoptotic activity of BCL-XL facilitating BAX-dependent apoptosis and inhibition of tumor growth in vitro and in vivo [48].
MCL-1
Song et al. utilized NMR, limited proteolysis, and in vitro ubiquitination assays to demonstrate that subtle conformational changes in loop 2 (between α2–α3) within the BH3 binding groove of MCL-1 could influence ubiquitination and MCL-1 turnover [49]. A new MCL-1 inhibitor was identified with modest affinity, compound 5, capable of influencing the conformation of loop 2 and subsequently promoting MCL-1 degradation. In cellular assays compound 5 was able to both dissociate proteins bound to MCL-1 and lower MCL-1 levels allowing it to out perform higher affinity small molecules in the killing of MCL-1-addicted cell lines. This conformational regulation also explained the action of MCL-1 inhibitors including Maritoclax which induces the same loop 2 conformation and enhances MCL-1 turnover, while A-1210477, which fails to alter loop 2, leads to increase in MCL-1 levels in cells potentially antagonizing its efficacy [49].
Targeted covalent inhibitors combine a small molecule with specificity for a target with a covalent “warhead” which covalently binds to the target and dramatically increasing the binding energy. Lee et al. identified a small molecule to covalently bind to MCL-1 Cys286, which is both unique to MCL-1 and distant from the canonical binding groove [50]. The compound induced conformational changes and the allosteric inhibition of BH3-domain interaction with MCL-1. Another study by Akçay et al. modified a small molecule inhibitor of MCL-1 to include a strategically placed boronic acid group, which reversibly reacts with Lys234 [51]. Covalent binding to this lysine, unique to MCL-1, allowed specific increase in binding to MCL-1 over other BCL-2 family members. The resulting compound showed encouraging results in MCL-1-dependent cell lines.
Targeting of pro-apoptotic BAX and BAK
Several recent studies have significantly advanced our understanding of the different activation and regulation mechanisms of pro-apoptotic BAX and BAK. Elucidation of binding surfaces and different conformations in solution and on the membrane provides the means for selectively targeting these proteins to modulate their function.
BAX
Structural analysis of a stapled BIM BH3 helix, BIM SAHB, bound to monomeric BAX uncovered an activation site (trigger site) at the N-terminal surface of BAX [52]. This activation site regulates the trigger mechanism for conformational activation of cytosolic BAX, leading to the release of the C-terminal transmembrane α9 helix, exposure of the hydrophobic α2 helix (BH3 domain) and ultimately to mitochondrial BAX oligomer formation [53,54]. BAX without the transmembrane α9 helix additionally interacts with BID and BIM BH3 peptides at its canonical groove suggesting an additional activation mechanism for loosely attached mitochondrial BAX [55,56]. Recently, an autoinhibited dimer crystal structure of full-length BAX revealed an additional mechanism of BAX regulation that is mediated by the interaction of α9 helix from one BAX molecule with the trigger site of the second BAX molecule [57].
Gavathiotis et al. performed a computational screen and a competitive binding assay for binding small molecules to the trigger site and identified BAX activator molecule, BAM7 [58]. BAM7 directly and selectively over anti-apoptotic proteins and BAK engages the BAX trigger site promoting functional oligomerization of BAX and BAX-dependent apoptosis (Figure 3d). Thus, direct small molecule activation of BAX suggested a new paradigm for pharmacologic induction of apoptosis.
Uchime et al. identified synthetic antibodies through a phage display screen that bind with low nanomolar affinity to BAX [59]. A representative Fab 3G11 found to bind an extensive surface of BAX that includes the trigger site (helices α1/α6) and the closed α1-α2 loop (Figure 3g). 3G11 blocked BAX mitochondrial translocation and BAX-mediated cytochrome c release consistent with a mechanism of BAX conformational suppression through extensive contacts with BAX, which prevent opening of the α1-α2 loop that is essential for BAX activation [53,57].
Moreover, Barclay et al. identified a stapled peptide based on the BCL-2 BH4 domain to bind with nanomolar affinity to BAX and suppress BAX conformational activation [60]. Using NMR and paramagnetically-labeled peptides, the BCL-2 BH4 was localized on a discrete binding site formed by residues of α1, α1–α2 loop, and α2–α3 and α5–α6 hairpins on the BAX surface (Figure 3f). Residues of this binding site between the BAX trigger site and the canonical groove presumably mediate conformational BAX activation.
BAK
Structural and biochemical studies determined activating interactions of BAK by native and stapled peptides of BID BH3 through its BH3-binding groove (α3–α5) [61–63]. These studies led to the conclusion that BAK is activated through interaction with the canonical groove and opened the opportunity for pharmacological activation of BAK. However, small molecules targeting this canonical BH3-groove are still elusive. Interestingly, Kluck and colleagues identified an antibody (7D10), which bound to the α1–α2 loop of BAK, opposite to the canonical BH3-groove and promoted BAK conformational activation and oligomerization leading to MOMP [64]. The physiological relevance of this novel activation mechanism has not yet been established, although a movement of this loop on BAX to open position was determined to be essential upon BH3 trigger site activation [53]. The development of small-molecules that target the trigger site of BAX provides hope that this novel site of BAK can also be exploited pharmacologically.
Conclusions and perspectives
Significant progress has been made in targeting the BCL-2 family of proteins. The FDA-approval of ABT-199 has fueled with excitement further efforts that BH3 mimetics could have a promising future to treat cancers. The paradigm for potent and selective targeting of individual anti-apoptotic proteins to promote BAX- and BAK- dependent apoptosis has been followed with new compounds targeting both BCL-2/BCL-XL, or individual BCL-2, BCL-XL, MCL-1 and BFL-1. It remains to be seen whether these new selective BCL-2 inhibitors or their derivatives can achieve improved efficacies as monotherapies or can be combined for increased efficacy with manageable undesired effects.
Several novel approaches to target BCL-2 proteins, beyond the canonical BH3 groove binding, have been identified in recent years including allosteric or covalent small molecules, peptides and antibodies. These recent findings suggest that there are several ways to modulate the surfaces and function of these proteins. Such approaches offer an alternative path to enhanced potency and selectivity that may stimulate more efficient drug discovery. The advances in our understanding of the mechanisms of activation of BAX and BAK have already generated valuable tools and targeting strategies. The possibility of developing activators and inhibitors of apoptosis through pro-apoptotic proteins would have promising application in cancer, autoimmune and aging-associated diseases. Without doubt, this is an exciting time for the application of chemical biology and drug discovery against BCL-2 family proteins.
Highlights.
Selective BH3 mimetics for interrogating the BCL-2 protein family interactions and their development for cancer therapy
Non-canonical interactions of anti-apoptotic BCL-2 proteins offer alternative targeting from BH3 mimetics
Advances in understanding pro-apoptotic BCL-2 protein interactions leads to novel targeting opportunities
Acknowledgments
This review aims to highlight select recent studies within the allocated space and we apologize for any omissions. The authors would like to acknowledge the support from National Institute of Health (R01CA178394), the American Heart Association, the Sidney Kimmel Foundation for Cancer Research, the Gabrielle’s Angels Foundation for Cancer Research, the Alexandrine and Alexander L. Sinsheimer Foundation, the Pershing Square Sohn Cancer Research Alliance, the Melanoma Research Alliance and the Irma T. Hirschl Trust.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict Of Interest Statement
Nothing declared.
References And Recommended Reading
Papers of particular interest, published within the period of review, have been highlighted as: * of special interest ** of outstanding interest
- 1.Delbridge ARD, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016;16:99–109. doi: 10.1038/nrc.2015.17. [DOI] [PubMed] [Google Scholar]
- 2.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 3.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 4.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen H-C, Kanai M, Inoue-Yamauchi A, Tu H-C, Huang Y, Ren D, Kim H, Takeda S, Reyna DE, Chan PM, et al. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat Cell Biol. 2015;17:1270–1281. doi: 10.1038/ncb3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shamas-Din A, Brahmbhatt H, Leber B, Andrews DW. BH3-only proteins: Orchestrators of apoptosis. Biochim Biophys Acta. 2011;1813:508–520. doi: 10.1016/j.bbamcr.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 7.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luna-Vargas MPA, Chipuk JE. Physiological and pharmacological control of BAK, BAX, and beyond. Trends Cell Biol. 2016;26:906–917. doi: 10.1016/j.tcb.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, et al. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 10.Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 11.Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7:989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 12.Besbes S, Mirshahi M, Pocard M, Billard C. New dimension in therapeutic targeting of BCL-2 family proteins. Oncotarget. 2015;6:12862–12871. doi: 10.18632/oncotarget.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yap JL, Chen L, Lanning ME, Fletcher S. Expanding the Cancer Arsenal with Targeted Therapies: Disarmament of the Antiapoptotic Bcl-2 Proteins by Small Molecules. J Med Chem. 2017;60:821–838. doi: 10.1021/acs.jmedchem.5b01888. [DOI] [PubMed] [Google Scholar]
- 14.Oliver CL, Miranda MB, Shangary S, Land S, Wang S, Johnson DE. (-)-Gossypol acts directly on the mitochondria to overcome Bcl-2- and Bcl-X(L)-mediated apoptosis resistance. Mol Cancer Ther. 2005;4:23–31. [PubMed] [Google Scholar]
- 15.Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, Goulet D, Viallet J, Bélec L, Billot X, et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci U S A. 2007;104:19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16**.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. This paper describes the discovery and characterization of ABT-737. [DOI] [PubMed] [Google Scholar]
- 17.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 18.Schoenwaelder SM, Jarman KE, Gardiner EE, Hua M, Qiao J, White MJ, Josefsson EC, Alwis I, Ono A, Willcox A, et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood. 2011;118:1663–1674. doi: 10.1182/blood-2011-04-347849. [DOI] [PubMed] [Google Scholar]
- 19.Kaefer A, Yang J, Noertersheuser P, Mensing S, Humerickhouse R, Awni W, Xiong H. Mechanism-based pharmacokinetic/pharmacodynamic meta-analysis of navitoclax (ABT-263) induced thrombocytopenia. Cancer Chemother Pharmacol. 2014;74:593–602. doi: 10.1007/s00280-014-2530-9. [DOI] [PubMed] [Google Scholar]
- 20.Bai L, Chen J, McEachern D, Liu L, Zhou H, Aguilar A, Wang S. BM-1197: a novel and specific Bcl-2/Bcl-xL inhibitor inducing complete and long-lasting tumor regression in vivo. PLoS ONE. 2014;9:e99404. doi: 10.1371/journal.pone.0099404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Némati F, de Montrion C, Lang G, Kraus-Berthier L, Carita G, Sastre-Garau X, Berniard A, Vallerand D, Geneste O, de Plater L, et al. Targeting Bcl-2/Bcl-XL induces antitumor activity in uveal melanoma patient-derived xenografts. PLoS ONE. 2014;9:e80836. doi: 10.1371/journal.pone.0080836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loriot Y, Mordant P, Dugue D, Geneste O, Gombos A, Opolon P, Guegan J, Perfettini JL, Pierre A, Berthier LK, et al. Radiosensitization by a novel Bcl-2 and Bcl-XL inhibitor S44563 in small-cell lung cancer. Cell Death Dis. 2014;5:e1423. doi: 10.1038/cddis.2014.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adam A, Byth K, Secrist P, Schuller A, Reimer C, Lawson D, MacIntyre T, Powell F. A Dual Bcl-2/xL Inhibitor Induces Tumor Cell Apoptosis in a Hematopoietic Xenograft Model [Internet] Blood. 2014 [no volume] [Google Scholar]
- 24.Discovery of potent inhibitors of the anti-apoptotic Bcl-2 and Bcl-xL, resulting in the identification of a clinical candidate for the treatment of cancer (AZD4320) 250th ACS Annual Meeting. 2015 https://ep70.eventpilotadmin.com/web/page.php?page=IntHtml&project=ACS15fall&id=2265202.
- 25**.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–208. doi: 10.1038/nm.3048. This paper describes the discovery and characterization of ABT-199, the first FDA-approved BCL-2 inhibitor. [DOI] [PubMed] [Google Scholar]
- 26.Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2016;374:311–322. doi: 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Porter J, Payne A, de Candole B, Ford D, Hutchinson B, Trevitt G, Turner J, Edwards C, Watkins C, Whitcombe I, et al. Tetrahydroisoquinoline amide substituted phenyl pyrazoles as selective Bcl-2 inhibitors. Bioorg Med Chem Lett. 2009;19:230–233. doi: 10.1016/j.bmcl.2008.10.113. [DOI] [PubMed] [Google Scholar]
- 28.Dose-escalation Study of Oral Administration of S 55746 in Patients With Chronic Lymphocytic Leukaemia and B-Cell Non-Hodgkin Lymphoma - Full Text View - ClinicalTrials.gov [Internet]. [date unknown], [no volume].
- 29.Study of Safety and Efficacy of BCL201 and Idelalisib in Patients With FL and MCL - Full Text View - ClinicalTrials.gov [Internet]. [date unknown], [no volume].
- 30.Lessene G, Czabotar PE, Sleebs BE, Zobel K, Lowes KN, Adams JM, Baell JB, Colman PM, Deshayes K, Fairbrother WJ, et al. Structure-guided design of a selective BCL-X(L) inhibitor. Nat Chem Biol. 2013;9:390–397. doi: 10.1038/nchembio.1246. [DOI] [PubMed] [Google Scholar]
- 31.Tao Z-F, Hasvold L, Wang L, Wang X, Petros AM, Park CH, Boghaert ER, Catron ND, Chen J, Colman PM, et al. Discovery of a Potent and Selective BCL-XL Inhibitor with in Vivo Activity. ACS Med Chem Lett. 2014;5:1088–1093. doi: 10.1021/ml5001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32*.Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, Belmont LD, Nimmer P, Xiao Y, Ma XM, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med. 2015;7:279ra40. doi: 10.1126/scitranslmed.aaa4642. This paper describes the characterization of a BCL-XL selective inhibitor in vitro and in vivo. [DOI] [PubMed] [Google Scholar]
- 33.Lee EF, Czabotar PE, Yang H, Sleebs BE, Lessene G, Colman PM, Smith BJ, Fairlie WD. Conformational changes in Bcl-2 pro-survival proteins determine their capacity to bind ligands. J Biol Chem. 2009;284:30508–30517. doi: 10.1074/jbc.M109.040725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fire E, Gullá SV, Grant RA, Keating AE. Mcl-1-Bim complexes accommodate surprising point mutations via minor structural changes. Protein Sci. 2010;19:507–519. doi: 10.1002/pro.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stewart ML, Fire E, Keating AE, Walensky LD. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat Chem Biol. 2010;6:595–601. doi: 10.1038/nchembio.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee EF, Czabotar PE, van Delft MF, Michalak EM, Boyle MJ, Willis SN, Puthalakath H, Bouillet P, Colman PM, Huang DC, Fairlie WD. A novel bh3 ligand that selectively targets mcl-1 reveals that apoptosis can proceed without mcl-1 degradation. J Cell Biol. 2008;180:341–355. doi: 10.1083/jcb.200708096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doi K, Li R, Sung S-S, Wu H, Liu Y, Manieri W, Krishnegowda G, Awwad A, Dewey A, Liu X, et al. Discovery of marinopyrrole A (maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. J Biol Chem. 2012;287:10224–10235. doi: 10.1074/jbc.M111.334532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abulwerdi F, Liao C, Liu M, Azmi AS, Aboukameel A, Mady ASA, Gulappa T, Cierpicki T, Owens S, Zhang T, et al. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther. 2014;13:565–575. doi: 10.1158/1535-7163.MCT-12-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, Nimmer P, Jin S, Smith M, Xiao Y, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax) Cell Death Dis. 2015;6:e1590. doi: 10.1038/cddis.2014.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee T, Bian Z, Zhao B, Hogdal LJ, Sensintaffar JL, Goodwin CM, Belmar J, Shaw S, Tarr JC, Veerasamy N, et al. Discovery and biological characterization of potent myeloid cell leukemia-1 inhibitors. FEBS Lett. 2017;591:240–251. doi: 10.1002/1873-3468.12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41**.Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, Chanrion M, Kelly GL, Gong J-N, Moujalled DM, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477–482. doi: 10.1038/nature19830. This papers describes the discovery and characterization of the first MCL-1 inhibitor with potent activity in vitro and in vivo. [DOI] [PubMed] [Google Scholar]
- 42.The discovery and preclinical characterization of AMG 176: A first-in-class Mcl-1 inhibitor in clinical development for multiple myeloma. AACR Annual Meeting. 2017 http://www.abstractsonline.com/pp8/#!/4292/presentation/11027.
- 43.AZD5991: A potent and selective macrocyclic inhibitor of Mcl-1 for treatment of hematologic cancers. AACR Annual Meeting. 2017 http://www.abstractsonline.com/pp8/#!/4292/presentation/11028.
- 44.Huhn AJ, Guerra RM, Harvey EP, Bird GH, Walensky LD. Selective Covalent Targeting of Anti-Apoptotic BFL-1 by Cysteine-Reactive Stapled Peptide Inhibitors. Cell Chem Biol. 2016;23:1123–1134. doi: 10.1016/j.chembiol.2016.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barile E, Marconi GD, De SK, Baggio C, Gambini L, Salem AF, Kashyap MK, Castro JE, Kipps TJ, Pellecchia M. hBfl-1/hNOXA Interaction Studies Provide New Insights on the Role of Bfl-1 in Cancer Cell Resistance and for the Design of Novel Anticancer Agents. ACS Chem Biol. 2017;12:444–455. doi: 10.1021/acschembio.6b00962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46*.Berger S, Procko E, Margineantu D, Lee EF, Shen BW, Zelter A, Silva D-A, Chawla K, Herold MJ, Garnier J-M, et al. Computationally designed high specificity inhibitors delineate the roles of BCL2 family proteins in cancer. elife. 2016;5 doi: 10.7554/eLife.20352. This papers describes the discovery and characterization of selective three helix bundle proteins for each anti-apoptotic protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47*.Han B, Park D, Li R, Xie M, Owonikoko TK, Zhang G, Sica GL, Ding C, Zhou J, Magis AT, et al. Small-Molecule Bcl2 BH4 Antagonist for Lung Cancer Therapy. Cancer Cell. 2015;27:852–863. doi: 10.1016/j.ccell.2015.04.010. This paper describes the discovery and characterization of an allosteric BCL-2 inhibitor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kolluri SK, Zhu X, Zhou X, Lin B, Chen Y, Sun K, Tian X, Town J, Cao X, Lin F, et al. A short Nur77-derived peptide converts Bcl-2 from a protector to a killer. Cancer Cell. 2008;14:285–298. doi: 10.1016/j.ccr.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49*.Song T, Wang Z, Ji F, Feng Y, Fan Y, Chai G, Li X, Li Z, Zhang Z. Deactivation of Mcl-1 by Dual-Function Small-Molecule Inhibitors Targeting the Bcl-2 Homology 3 Domain and Facilitating Mcl-1 Ubiquitination. Angew Chem Int Ed Engl. 2016;55:14250–14256. doi: 10.1002/anie.201606543. This paper describes the discovery of dual function inhibitors of MCL-1 that bind to the BH3 groove and promote MCL-1 ubiquitination. [DOI] [PubMed] [Google Scholar]
- 50*.Lee S, Wales TE, Escudero S, Cohen DT, Luccarelli J, Gallagher CG, Cohen NA, Huhn AJ, Bird GH, Engen JR, et al. Allosteric inhibition of antiapoptotic MCL-1. Nat Struct Mol Biol. 2016;23:600–607. doi: 10.1038/nsmb.3223. This paper describes the discovery of allosteric inhibition of MCL-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akçay G, Belmonte MA, Aquila B, Chuaqui C, Hird AW, Lamb ML, Rawlins PB, Su N, Tentarelli S, Grimster NP, et al. Inhibition of Mcl-1 through covalent modification of a noncatalytic lysine side chain. Nat Chem Biol. 2016;12:931–936. doi: 10.1038/nchembio.2174. [DOI] [PubMed] [Google Scholar]
- 52**.Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu H-C, Kim H, Cheng EH-Y, Tjandra N, et al. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–1081. doi: 10.1038/nature07396. This paper describes the discovery of the BAX trigger site and BIM BH3-BAX interaction mechanism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gavathiotis E, Reyna DE, Davis ML, Bird GH, Walensky LD. BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol Cell. 2010;40:481–492. doi: 10.1016/j.molcel.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walensky LD, Gavathiotis E. BAX unleashed: the biochemical transformation of an inactive cytosolic monomer into a toxic mitochondrial pore. Trends Biochem Sci. 2011;36:642–652. doi: 10.1016/j.tibs.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55*.Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, Huang DC, et al. Bax crystal structures reveal how bh3 domains activate bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152:519–531. doi: 10.1016/j.cell.2012.12.031. This paper describes BAX crystal structures that capture conformational changes of BAX activation. [DOI] [PubMed] [Google Scholar]
- 56.Robin AY, Krishna Kumar K, Westphal D, Wardak AZ, Thompson GV, Dewson G, Colman PM, Czabotar PE. Crystal structure of bax bound to the bh3 peptide of bim identifies important contacts for interaction. Cell Death & Dis. 2015;6:e1809. doi: 10.1038/cddis.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57**.Garner TP, Reyna DE, Priyadarshi A, Chen HC, Li S, Wu Y, Ganesan YT, Malashkevich VN, Cheng EH, Gavathiotis E. An autoinhibited dimeric form of bax regulates the bax activation pathway. Mol Cell. 2016;63:485–497. doi: 10.1016/j.molcel.2016.06.010. This paper describes the crystal structure and the mechanism of the autoinhibited BAX dimer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58*.Gavathiotis E, Reyna DE, Bellairs JA, Leshchiner ES, Walensky LD. Direct and selective small-molecule activation of proapoptotic BAX. Nat Chem Biol. 2012;8:639–645. doi: 10.1038/nchembio.995. This paper describes the discovery of the first selective BAX activator molecule. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59*.Uchime O, Dai Z, Biris N, Lee D, Sidhu SS, Li S, Lai JR, Gavathiotis E. Synthetic Antibodies Inhibit Bcl-2-associated X Protein (BAX) through Blockade of the N-terminal Activation Site. J Biol Chem. 2016;291:89–102. doi: 10.1074/jbc.M115.680918. This paper describes the discovery and characterization of synthetic antibodies that bind the trigger site and inhibit soluble BAX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60*.Barclay LA, Wales TE, Garner TP, Wachter F, Lee S, Guerra RM, Stewart ML, Braun CR, Bird GH, Gavathiotis E, et al. Inhibition of Pro-apoptotic BAX by a noncanonical interaction mechanism. Mol Cell. 2015;57:873–886. doi: 10.1016/j.molcel.2015.01.014. This paper describes the discovery and characterization of BCL- BH4 peptides that inhibit BAX through a novel site. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61**.Moldoveanu T, Grace CR, Llambi F, Nourse A, Fitzgerald P, Gehring K, Kriwacki RW, Green DR. BID-induced structural changes in BAK promote apoptosis. Nat Struct Mol Biol. 2013;20:589–597. doi: 10.1038/nsmb.2563. This paper describes the structure and mechanism of the BID BH3-BAK interaction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leshchiner ES, Braun CR, Bird GH, Walensky LD. Direct activation of full-length proapoptotic BAK. Proc Natl Acad Sci U S A. 2013;110:E986–95. doi: 10.1073/pnas.1214313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brouwer JM, Westphal D, Dewson G, Robin AY, Uren RT, Bartolo R, Thompson GV, Colman PM, Kluck RM, Czabotar PE. Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol Cell. 2014;55:938–946. doi: 10.1016/j.molcel.2014.07.016. [DOI] [PubMed] [Google Scholar]
- 64*.Iyer S, Anwari K, Alsop AE, Yuen WS, Huang DCS, Carroll J, Smith NA, Smith BJ, Dewson G, Kluck RM. Identification of an activation site in Bak and mitochondrial Bax triggered by antibodies. Nat Commun. 2016;7:11734. doi: 10.1038/ncomms11734. This paper describes the characterization of antibodies that activate BAK and mitochondrial BAX. [DOI] [PMC free article] [PubMed] [Google Scholar]