

Summary
We describe a mechanism by which the anti-apoptotic B-cell lymphoma 2 (Bcl-2) protein is down-regulated to induce apoptosis. ARTS (Sept4_i2) is a tumor suppressor protein that promotes cell death through specifically antagonizing XIAP (X linked Inhibitor of Apoptosis). ARTS and Bcl-2 reside at the outer mitochondrial membrane in living cells. Upon apoptotic induction, ARTS brings XIAP and Bcl-2 into a ternary complex allowing XIAP to promote ubiquitylation and degradation of Bcl-2. ARTS binding to Bcl-2 involves the BH3 domain of Bcl-2. Lysine 17 in Bcl-2 serves as the main acceptor for ubiquitylation and a Bcl-2 K17A mutant has increased stability and is more potent in protection against apoptosis. Bcl-2 ubiquitylation is reduced in both XIAP and Sept4/ARTS deficient MEFs, demonstrating that XIAP serves as an E3-ligase for Bcl-2 and ARTS is essential for this process. Collectively, these results suggest a distinct model for the regulation of Bcl-2 by ARTS-mediated degradation.
Keywords: Apoptosis, Mitochondria, Bcl-2, Caspase, XIAP, Ubiquitin, Protein degradation, E3-ligase
eTOC Blurb
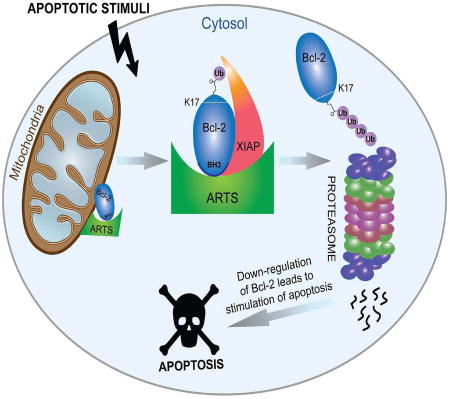
Many cancers avoid cell death (apoptosis) by expressing high levels of apoptosis inhibitors, such as Bcl-2. Thus, Bcl-2 is a major target for cancer therapy. Edison et al. describe a mechanism by which ARTS protein promotes proteasome-mediated degradation of Bcl-2 and thereby stimulates cell death.
Introduction
Apoptosis plays a major role in development, tissue homeostasis, and as a defense mechanism against unwanted and potentially dangerous cells (Fuchs and Steller, 2015; Malin and Shaham, 2015). When deregulated, apoptosis can result in various pathologies, including cancer (Fuchs and Steller, 2011). Caspases, a family of cysteine proteases, are the central executioners of apoptosis (Boyce et al., 2004). In living cells, caspases are inhibited by the Inhibitor of Apoptosis (IAP) proteins (Gyrd-Hansen and Meier, 2010). To stimulate apoptosis the function of IAPs needs to be overcome. This is achieved by IAP-antagonists such as Smac/Diablo (Du et al., 2000; Verhagen et al., 2000) and ARTS (Gottfried et al., 2004b). ARTS is localized at the mitochondrial outer membrane (MOM) (Edison et al., 2012b). Upon induction of apoptosis, ARTS accumulates in the cytosol, directly binds and antagonizes XIAP, causing activation of caspases and cell death (Bornstein et al., 2011; Edison et al., 2012b; Gottfried et al., 2004b).
XIAP is the only known direct inhibitor of proteolytic caspase activity (Bergmann et al., 2003; Eckelman et al., 2006; Vasudevan and Ryoo, 2015). XIAP can directly bind and inhibit caspases 3, 7 and 9 via its three Baculoviral IAP Repeats (BIR) domains (Bergmann et al., 2003; Shi, 2002). In addition, it contains an Ubiquitin-associated (UBA) domain, which enables the binding of polyubiquitin conjugates via lysine 63 (Gyrd-Hansen et al., 2008; Rajalingam and Dikic, 2009) and a RING domain that bestows E3-ligase activity (Schile et al., 2008). XIAP functions as an E3-ligase for several pro-apoptotic proteins such as caspases, SMAC, AIF and ARTS (Bornstein et al., 2012; Galban and Duckett, 2010; Schile et al., 2008).
The mitochondrial pathway of apoptosis is regulated by Bcl-2 family members (Cory et al., 2003; Gross et al., 1999a). This family is composed of pro- and anti-apoptotic proteins that share up to four conserved Bcl-2 homology (BH) domains, and form complexes by binding to their common BH3 domain (Adams and Cory, 1998; Youle and Strasser, 2008). Importantly, many cancers are characterized by high levels of Bcl-2 (Adams and Cory, 1998; Castle et al., 1993; Krajewska et al., 1996; Robertson et al., 1996; Youle and Strasser, 2008). Therefore, Bcl-2 antagonists are promising anti-cancer drugs and Venclexta was recently approved for the treatment of chronic lymphocytic leukemia.
Degradation of intracellular proteins in eukaryotic cells occurs via the Ubiquitin Proteasome System (UPS). This involves the ordered action of a ubiquitin-activating enzyme (E1), ubiquitin conjugating enzymes (E2) and E3 ubiquitin ligases, which recognize and transfer ubiquitin to the target proteins (Ciechanover et al., 1980; Glickman and Ciechanover, 2002). It was previously reported that Bcl-2 can be degraded by the UPS, but the underlying molecular mechanism has remained obscure (Dimmeler et al., 1999; Kassi et al., 2009; Wang et al., 2008). In this study, we describe a mechanism by which cells are sensitized towards apoptosis through UPS-mediated degradation of Bcl-2. We show that the pro-apoptotic XIAP-antagonist ARTS is required for down-regulation of Bcl-2 levels upon induction of apoptosis. ARTS directly binds to the BH3 domain of Bcl-2 and enables the formation of a XIAP-ARTS-Bcl-2 complex. XIAP functions as an E3-ligase for Bcl-2 which binds and ubiquitylates Lysine 17 in Bcl-2. Collectively, our data indicate that ARTS acts as a distinct Bcl-2 antagonist that brings Bcl-2 into a ternary complex with XIAP and thereby stimulates UPS-mediated degradation of Bcl-2 to promote apoptosis.
Results
Bcl-2 protein levels are down-regulated by the UPS during apoptosis
Levels of Bcl-2 decrease during apoptosis, but the underlying mechanism remains to be elucidated. (Dimmeler et al., 1999; Kassi et al., 2009; Wang et al., 2008). We confirmed that Bcl-2 levels are decreased upon induction of apoptosis as early as 1–2 hours following staurosporine (STS) and etoposide treatment (Figures 1A, 1B, 2B). To examine whether the observed reduction in Bcl-2 is due to degradation via the UPS, HeLa cells were pre-treated with MG132, a potent proteasome inhibitor, and with STS to induce apoptosis. Accumulation of Bcl-2 was observed upon treatment with MG132, suggesting that induction of apoptosis results in UPS-mediated degradation of Bcl-2 (Figure 1C). Next, we investigated whether in vivo ubiquitylation of Bcl-2 occurs upon induction of apoptosis. Both MEFs and HeLa cells pretreated with MG132 showed accumulation of poly-ubiquitylated Bcl-2 upon induction of apoptosis (Figure 1D). The appearance of poly-ubiquitylated Bcl-2 was correlated with decreased Bcl-2 levels in apoptotic cells (Figure 1D). This suggests that Bcl-2 is down-regulated through UPS-mediated degradation during apoptosis.
Figure 1. Bcl-2 protein levels are down-regulated by the ubiquitin-proteasome system during apoptosis.

A. Apoptosis was induced in HeLa and immortalized MEFs using STS for the indicated times, and endogenous Bcl-2 was detected by Western blot analysis. B. Apoptosis was induced in HeLa, BT-549 and COS-7 cells with etoposide. NT (No Treatment). In primary MEFs apoptosis was induced with 100 µM etoposide for 5 h. A decrease in endogenous Bcl-2 levels was seen upon treatment with both STS and etoposide. C. Apoptosis was induced in HeLa cells using STS in the presence or absence of 20 µM of MG132. Western and densitometry analyses revealed decreased levels of Bcl-2 with STS treatment, and stabilization of Bcl-2 upon MG132 treatment. This suggests that Bcl-2 levels are down-regulated via the UPS. D. WT MEFs and HeLa cells were transiently transfected with Bcl-2, XIAP and ubiquitin and treated with 20 µM MG132 for 6 h and with 1.75 µM STS. IP with anti-Bcl-2 was followed by Western Blotting with anti-ubiquitin antibodies. *Represents the IG heavy chain. Poly-ubiquitylated forms of Bcl-2 appeared in apoptotic cells and correlated with decreased Bcl-2 levels.
Figure 2. ARTS is required for down-regulation of Bcl-2 levels in the cytosol.

AI. HeLa ARTS knockdown (ARTS KD) cells and Sept4/ARTS KO MEFs show significantly higher levels of steady-state Bcl-2 protein when compared with WT cells. This suggests that ARTS plays an important role in regulating Bcl-2 levels. B. WT and ARTS KD HeLa cells were treated with 1.75 µM STS. Western Blot analyses demonstrate that while decreased Bcl-2 levels were seen in apoptotic WT HeLa cells, Bcl-2 levels in ARTS KD HeLa cells remained unchanged. C. Western Blot analyses of cytosolic fractions of BT-549, HeLa WT and HeLa ARTS KD cells reveal that endogenous Bcl-2 is found in the cytosol of WT STS-treated cells. In contrast, a strong inhibition in translocation of Bcl-2 to the cytosol was seen in ARTS KD HeLa cells. This suggests that ARTS is required for the proper translocation of Bcl-2 from mitochondria to the cytosol upon apoptotic induction. D. Immuno-fluorescence (IF) was performed on HeLa and a stable Bcl-2 knockdown (Bcl-2 KD) cells. The fraction of cells with cytosolic staining of ARTS is represented in the bar chart. While only a small portion of WT untreated (NT) HeLa cells show the presence of ARTS in the cytosol, a significant increase in cells containing cytosolic ARTS was seen following STS treatment. In contrast, the majority of HeLa Bcl-2 KD NT cells exhibit cytosolic ARTS (four fold higher than WT HeLa cells), and, only a slight increase in cells with cytosolic ARTS is seen after STS treatment. (* * p-value ≤ 0.01). See also supplemental Figure S1. E. Cytosolic and mitochondrial fractions of WT MEFs and Bcl-2 KO MEFs were analyzed by WB analysis with COX IV as a mitochondrial and GAPDH as a cytosolic marker. In Bcl-2 KO MEFs, the majority of ARTS was in the cytosol. This suggests that Bcl-2 is involved in localizing ARTS to mitochondria. F. IF of WT MEFs and Sept4/ARTS KO MEFs transiently transfected with GFP-Bcl-2. Cellular localization of Bcl-2 was quantified and the fraction of cells with cytosolic Bcl-2 is shown in the bar charts. While a significant increase in cytosolic Bcl-2 was seen in apoptotic WT MEFs, the levels of cytosolic Bcl-2 in Sept4/ARTS KO MEFs remained unchanged. See also supplemental Figure S1. G. Subcellular fractionation of HeLa cells was followed by in vivo ubiquitylation of each fraction. IgG represents the control cells incubated with non-specific IgG. Poly-ubiquitylated forms of Bcl-2 were seen only in the cytosolic fraction. * Represents the IG heavy chain. This indicates that ubiquitylation of Bcl-2 occurs in the cytosol and that ARTS is required for translocation of Bcl-2 to the cytosol during apoptosis.
ARTS is required for down-regulation of Bcl-2 levels in the cytosol
High levels of ARTS are sufficient to promote apoptosis in a variety of cell lines, and inactivation of ARTS protects against apoptosis (Edison et al., 2012b; Garcia-Fernandez et al., 2010; Lotan et al., 2005). To determine whether ARTS is required for reducing Bcl-2 protein levels we used HeLa cells in which ARTS expression was knocked-down with shRNA (“ARTS KD HeLa cells”, (Edison et al., 2012b) and MEFs from Sept4/ARTS KO mice (Garcia-Fernandez et al., 2010; Kissel et al., 2005). All these cells exhibited a significant increase in the steady state levels of endogenous Bcl-2 (Figure 2A). This indicates that ARTS restricts Bcl-2 levels in vivo and may therefore function as a Bcl-2 antagonist. Similarly, a decrease in Bcl-2 was seen in HeLa cells upon treatment with STS, while the levels of Bcl-2 in ARTS KD HeLa cells remained unchanged (Figure 2B). Thus, ARTS is required for down-regulation of Bcl-2 upon induction of apoptosis.
Both ARTS and Bcl-2 are localized at the MOM (Edison et al., 2012b; Volkmann et al., 2014). Shortly following induction of apoptosis, ARTS translocates to the cytosol where it binds XIAP and initiates apoptosis (Edison et al., 2012b). This translocation occurs prior to mitochondrial outer membrane permeabilization (MOMP), an event that allows the release of other pro-apoptotic factors such as Smac/Diablo and cytochrome c from the inner membrane space of the mitochondria (Edison et al., 2012b). Cellular fractionation assays showed a concomitant appearance of Bcl-2 and ARTS in the cytosol of apoptotic cells (Figure 2C). This translocation of both Bcl-2 and ARTS to the cytosol occurred very rapidly after 15 min of addition of STS in HeLa cells (Figure 2C). ARTS is localized at the MOM, yet it does not contain a trans-membrane domain (Edison et al., 2012b). We investigated whether Bcl-2 plays a role for the localization of ARTS to the MOM. Immunofluorescence (IF) staining showed that while ARTS was seen in the cytosol in 18% of WT HeLa cells, ARTS was cytosolic in the majority of Bcl-2 KD cells (78%, Figure 2D, supplemental Figure S1A). Similarly, the vast majority of ARTS in Bcl-2 KO MEFs was found in the cytosol with no detectable levels in mitochondria (Figure 2E). This suggests that the mitochondrial localization of ARTS depends, at least in part, on Bcl-2. Furthermore, ARTS seems to be important for the translocation of Bcl-2 to the cytosol during apoptosis. While a significant increase in cytosolic Bcl-2 is found in STS-treated WT MEFs, the levels of cytosolic Bcl-2 in apoptotic Sept4/ARTS KO MEFs remain unchanged (Figure 2F, supplemental Figure S1B). Finally, we observed accumulation of poly-ubiquitylated Bcl-2 in the cytosol with a concomitant decrease of Bcl-2 protein in the mitochondrial fraction (Figure 2G). Taken together, these results indicate that ARTS is required for the ubiquitylation, translocation and degradation of Bcl-2.
ARTS is required for the formation of a ternary complex with Bcl-2 and XIAP
Since ARTS binds directly to XIAP we examined the possibility that ARTS can bind to both XIAP and Bcl-2 and form a ternary complex. Immunoprecipitation (IP) experiments with an anti-ARTS monoclonal antibody suggest that ARTS can indeed form a complex with both XIAP and Bcl-2 (Figures 3AI, II). Furthermore, while strong binding of ARTS to Bcl-2 was seen both in untreated and apoptotic cells, an increase in binding to XIAP was observed upon induction of apoptosis (Figure 3AII). This implies that while ARTS and Bcl-2 bind to each other under normal conditions at the MOM, upon induction of apoptosis they can form a ternary complex with XIAP that leads to ubiquitylation and degradation of Bcl-2. Moreover, using an in vitro binding assay with recombinant proteins we observed a significant and strong formation of a ternary complex only when recombinant ARTS was added (Figure 3B).
Figure 3. ARTS is required for formation of a ternary complex with Bcl-2 and XIAP.

AI. IP of ARTS in COS-7 cells transiently transfected with 6-Myc-ARTS using a monoclonal anti-ARTS antibody (Sigma-Aldrich). Endogenous ARTS was immunoprecipitated from HeLa and BT-549 cells. Western blot analyses show that in COS-7, HeLa and BT-549 cells, Bcl-2 and XIAP co- precipitate with ARTS. AII. HeLa cells were treated with 1.75 µM STS and IP of ARTS was performed as described in AI. While binding of ARTS to Bcl-2 was seen in non-treated cells (NT), binding of ARTS to XIAP and Bcl-2 is increased in STS treated cells. B. Recombinant His-ARTS, Bcl-2 and GST-XIAP were incubated overnight at 4 °C. Pulldown of GST-XIAP shows that binding of Bcl-2 to XIAP depends on ARTS. C. To assess proximity of proteins, consistent with complex formation, we used BiFC. HeLa cells were transiently transfected with ARTS, Bcl-2 and XIAP fused to parts of YFP YFP-Venus. Jun and bFos, known to form heterodimers, served as a positive control (p.c.). Jun and bFosdelZIP lacking the carboxyl-terminal half of the bFos were used as a negative control (n.c.). The fluorescent signal indicating the proximity of each pair of proteins was measured by flow cytometry. Mean fluorescence intensity (MFI). FACS results were normalized to the readings of transfection efficiency reporter (pdsRED). The values represent mean values ± SE of three independent experiments (*, p ≤ 0.05; **, p ≤ 0.01). The Y-axis represents the ratio between YFP fluorescence (reflecting binding of a pair of proteins) and the red fluorescence (marking the transfected cells). FACS analyses reveal that ARTS can bind to both Bcl-2 and to XIAP. Yet, only background levels of fluorescence were seen with XIAP and Bcl2, suggesting that these two proteins do not bind each other. These results indicate that ARTS, XIAP and Bcl-2 form a ternary complex, and that ARTS is required for the formation of this complex. See also supplemental Figure S2.
Next, we used the BiFC, split-venus assay (Li et al., 1998). The proximity between each pair of proteins was measured by flow cytometry. These results confirm our in vitro binding data (Figure 3B) showing that while ARTS can bind directly to both XIAP and Bcl-2, the latter two proteins did not bind each other directly (Figure 3C, Supplemental figure S2). Together, these results show that ARTS binds directly to XIAP and Bcl-2 and enables the formation of a ternary complex in which ARTS serves as an adaptor to bring XIAP and Bcl-2 into close proximity to allow degradation of Bcl-2 by XIAP (see graphical abstract).
ARTS interacts with the BH3 domain of Bcl-2
To more precisely define the interaction of ARTS and Bcl-2 we used peptide array binding assays and structure-function analysis with deletion mutants of Bcl-2. First, to identify the ARTS-binding motif in Bcl-2 we used the peptide array method. The array was screened for binding with recombinant (His)6-Lipo-TEV (HLT)-tagged ARTS protein. Bcl-2 peptides that showed binding to HLT-ARTS were concentrated around the BH3 binding cleft, the same binding region which binds to the BH3 domain of other Bcl-2 family members (Figure 4A, Supplemental tables S1, S2). Next, IP experiments were performed with Bcl-2 WT and four constructs with deletions of different BH domains (BH-1,2,3,4). IP assays revealed a significant decrease in binding of ARTS to the Bcl-2 mutant lacking the BH3 domain (Bcl-2delBH3) (Figure 4B). Moreover, BiFC assays also showed strongly reduced interaction of Bcl-2delBH3 with ARTS compared to WT Bcl-2 (Figure 4C, Supplemental Figure S3). Additionally, Bcl-2delBH3 showed a major reduction in its ability to undergo ubiquitylation compared to WT Bcl-2 upon induction of apoptosis (Figure 4D, supplemental Figure S4). Furthermore, the BH3-mimetic ABT-199 can attenuate the interaction of Bcl-2 and ARTS, indicating that the BH3-binding motif to which ABT-199 binds in Bcl-2 is important for the interaction with ARTS (Supplemental Figure S6). Taken together, these results indicate that the BH3 domain of Bcl-2 is important for the interaction with ARTS, and that this interaction is required to promote ubiquitylation and degradation of Bcl-2 during apoptosis.
Figure 4. ARTS binds to the BH3 domain of Bcl-2.

A. A custom designed Bcl-2 derived peptide array (CelluSpot™) was incubated with recombinant (His)6-Lipo-TEV (HLT-) tagged ARTS. The peptide array was also incubated with recombinant HLT-tag alone which served as a negative control. The arrays were probed with the indicated antibodies. Lower panel depicts the Bcl-2 peptides which bind to ARTS. These peptides are schematically represented across the Bcl-2 domains; the black lines indicate strong binding and the grey lines a weak to moderate binding. B. Bcl-2 expression vectors; full length and four constructs deleting each BH domain in Bcl-2 are illustrated at the top (FLD- Flexible Loop Domain, TM-Transmembrane domain). These vectors were co-transfected with 6myc-ARTS into Bcl-2 KO MEFs. IP of ARTS was performed in MEFs transfected with Bcl-2-BH deletions, WT Bcl-2 (positive control) and empty vector (negative control). A significant reduction in binding of ARTS to Bcl-2delBH3 was seen. See also supplemental Tables S1 and S2. C. BIFC assays were performed as described in Figure 3C. HeLa cells were co-transfected with vectors expressing ARTS, Bcl-2 and Bcl-2delBH3 fused to either VN or VC parts of the YFP Venus fragment (ARTS-VN, Bcl-2-VC. Bcl-2delBH3 –VC). A 47% decrease in the proximity between ARTS and Bcl-2delBH3 was seen compared to WT Bcl-2. Jun/bFos served as positive control (p.c.), and Jun/bFosdel ZIP as negative control (n.c.). Results are shown as mean + S.E. of three independent experiments. (*, p ≤ 0.05; **, p ≤ 0.01). See also supplemental Figure S3. D. In vivo ubiquitylation of Bcl-2 WT and Bcl-2delBH3. MEFs Bcl-2 KO cells were transfected with Flag-Bcl-2WT or Flag-Bcl-2delBH3. Cells were treated with MG132 and STS for one hour or left untreated. Proteins were immunoprecipitated with anti-ubiquitin. Poly-ubiquitylated forms of Bcl-2 were detected using an anti-Bcl-2 antibody. While WT Bcl-2 undergoes ubiquitylation following treatment with STS, no ubiquitylation of Bcl-2delBH3 was seen. See also supplemental Figure S4.
XIAP serves as an E3-ligase for Bcl-2
Since ARTS, XIAP and Bcl-2 can form a complex, we investigated whether the decrease in Bcl-2 levels upon induction of apoptosis depends on the catalytic activity of XIAP. For this purpose, we over-expressed either XIAP or a mutant plasmid lacking its RING domain (XIAPdelRING) (Schile et al., 2008), which abolishes its E3-ligase function, together with Bcl-2 in HeLa cells. While exogenous XIAP reduced the levels of Bcl-2, Bcl-2 levels remained high in cells overexpressing XIAPdelRING (Figure 5AI). Furthermore, increased endogenous Bcl-2 levels were seen in primary XIAPdelRING MEFs compared to WT MEFs (Figure 5AII). This suggests that the E3-ligase catalytic activity of XIAP is required for regulating the levels of Bcl-2, and that XIAP functions as the physiological E3-ligase for Bcl-2. We also used a mutant version of ARTS deleted at its unique C-terminal part (Cdel-ARTS) that cannot bind to XIAP (Gottfried et al., 2004a). Sept4/ARTS KO MEFs expressing WT ARTS exhibited decreased levels of Bcl-2 upon apoptotic induction, whereas cells transfected with the Cdel-ARTS had increased levels of Bcl-2 (Figure 5B). Next, in vitro ubiquitylation assays were performed with recombinant Bcl-2 and XIAP together with E1, E2-UbcH5b and ubiquitin. These experiments show that ubiquitylation of Bcl-2 occurs only when XIAP was present (Figure 5C). Furthermore, in vitro ubiquitylation assays with Bcl-2 and three other E3-ligases (cIAP1, Parkin, or Siah2) revealed that only XIAP was capable to ubiquitylate Bcl-2 (Figure 5D, Supplemental Figure S5). Finally, we compared in vivo ubiquitylation of Bcl-2 in WT and XIAPdelRING MEFs (Figure 5E) and in WT and XIAP knock-out (KO) MEFs (Figure 5F). A strong reduction in accumulation of poly-ubiquitylated forms of Bcl-2 was seen in XIAP XIAPdelRING and XIAP KO MEFs (Figure 5E, F respectively). These results indicate that XIAP ubiquitylates Bcl-2 in a direct and specific manner. Furthermore, the catalytic activity of XIAP is necessary for UPS-mediated degradation of Bcl-2 upon induction of apoptosis. In these in vitro assays, the presence of recombinant XIAP and Bcl-2 is sufficient to ubiquitylate Bcl-2, presumably due to the high concentration of these proteins. In contrast, in living cells ARTS is required for bringing XIAP and Bcl-2 into sufficiently close proximity to allow for XIAP-mediated ubiquitylation of Bcl-2 (see below). Collectively, these data show that XIAP serves as a physiological E3-ligase for Bcl-2.
Figure 5. XIAP serves as the E3 ligase of Bcl-2.

AI. HeLa cells were transiently transfected with Bcl-2 or co-transfected with Bcl-2 and XIAP or with Bcl-2 and XIAPdelRing. AII. MEFs were produced from WT mice and from XIAPdelRing 14-day old mouse embryos. WB and densitometry analyses reveal a significant accumulation of Bcl-2 in HeLa cells co-transfected with XIAPdelRing and in XIAPdelRing MEFs. B. Sept4/ARTS KO MEFs were transfected with WT ARTS and a mutant version of ARTS lacking its unique C-terminus (Cdel-ARTS) which cannot bind to XIAP. Sept4/ARTS KO MEFs transfected with WTARTS exhibit decreased levels of Bcl-2 following apoptotic induction (UV), while MEFs transfected with the Cdel-ARTS have increased levels of Bcl-2. C. In vitro ubiquitylation assays were performed by incubating recombinant Bcl-2 with recombinant XIAP, E1, E2-UbcH5b and ubiquitin. In the control reactions the indicated components were excluded. Ubiquitylation of Bcl-2 was seen only upon addition of XIAP. D. In vitro ubiquitylation assays with purified Bcl-2 and recombinant XIAP, cIAP1, Parkin, or Siah2. Only addition of XIAP resulted in the ubiquitylation of Bcl-2. See also Supplemental Figure S5 E. In vivo ubiquitylation in WT and XIAPdelRing MEFs. MEFs were co-transfected with ARTS, Bcl-2 and HA-ubiquitin. Cells were treated with 20 µM MG-132 for 6h and concomitantly treated with 1.75 µM STS and IP was performed with an anti-Bcl-2 antibody. *Represents the heavy chain of the antibody. While significant ubiquitylation of Bcl-2 occurred after treatment with STS in WT MEFs, no ubiquitylation of Bcl-2 was seen in XIAPdelRing MEFs. F. In vivo ubiquitylation assays using WT and XIAP KO MEFs. No ubiquitylation of Bcl-2 was seen in XIAP KO MEFs. Collectively these results show that XIAP serves as the specific E3-ligase for Bcl-2 and is required for its degradation.
ARTS is required for UPS-mediated degradation of Bcl-2 by XIAP
In order to investigate the requirement of ARTS for XIAP-mediated ubiquitylation of Bcl-2, we performed several in vitro and in vivo ubiquitylation assays. We observed a significant increase in the appearance of ubiquitylated forms of Bcl-2 upon addition of recombinant ARTS (Figure 6A). To examine whether ARTS is required for the early, pre-MOMP ubiquitylation and degradation of Bcl-2, we examined in vivo ubiquitylation of Bcl-2 as early as 30 minutes following STS treatment, which is several hours prior to MOMP (Adrain et al., 2001; Edison et al., 2012b; Gao et al., 2001). Significant accumulation of poly-ubiquitylated forms of Bcl-2 was seen 30 minutes after induction of apoptosis (Figure 6B). This indicates that ubiquitylation of Bcl-2 starts prior to MOMP and release of cytochrome c and Smac/Diablo.
Figure 6. ARTS is required for ubiquitylation of Bcl-2 by XIAP.

A. In vitro ubiquitylation assays were performed by incubating recombinant Bcl-2 with XIAP and ARTS, E1, E2-UbcH5b and ubiquitin in the presence of ATP at 37 °C for 1 hr. A significant increase in the appearance of ubiquitylated forms of Bcl-2 was seen in the presence of ARTS. B. HeLa WT or HeLa ARTS KD cells were transiently transfected with Bcl-2, XIAP and ubiquitin and treated with 20 µM MG132 for 6 h and STS for 0.5 h. Bcl-2-ubiquitin conjugates were seen in the apoptotic WT HeLa cells, but not in ARTS KD HeLa cells. C. In vivo ubiquitylation assays of Sept4/ARTS KO MEFs transfected with either empty vector or ARTS expression vector, followed by treatment with MG132 and STS for 3 h. *Represents the IgG heavy chain. While no ubiquitylation of Bcl-2 was observed in the Sept4/ARTS KO MEFs, transfection of ARTS restored their ability to generate poly-ubiquitylated forms of Bcl-2 upon induction of apoptosis.
To further establish that Bcl-2 ubiquitylation and degradation is dependent on the presence of ARTS, in vivo ubiquitylation assays were performed using Sept4/ARTS KO MEFs. While no ubiquitylation of Bcl-2 was observed in the Sept4/ARTS KO MEFs, transfections of ARTS into these MEFs restored their ability to ubiquitylate and degrade Bcl-2 upon induction of apoptosis (Figure 6C). These results demonstrate that ARTS is required for the ubiquitylation of Bcl-2.
Lysine 17 (K17) is the main ubiquitin acceptor in Bcl-2
Bcl-2 contains four lysine residues (Figure 7A). To specifically identify the lysine in Bcl-2 which is ubiquitylated by XIAP we performed mass spectrometry analysis on lysates of HeLa cells transfected with a GFP-Bcl-2 construct. IP experiments with an anti-GFP antibody showed specific ubiquitylation of Lysine 17 in Bcl-2 (Figure 7B). We conclude that K17 of Bcl-2 is an acceptor for XIAP-mediated ubiquitylation. To examine the role of K17 we mutated it to Alanine (K17A). In addition, we also generated a Bcl-2 mutant in which all lysines were mutated (no K). Protein levels of both mutant forms of Bcl-2 were increased as compared to WT under apoptotic conditions (Figure 7C). Furthermore, three different markers of apoptosis (cleaved PARP, cleaved caspase-9, and cleaved caspase-3) showed a consistent decrease in cells expressing both Bcl-2 mutant forms as compared to WT Bcl-2 (Figure 7C). Finally, expression of either Bcl-2 K17A or Bcl-2 No K was more effective in preventing caspase-3 cleavage than WT Bcl-2 upon induction of apoptosis (Figure 7D). Since mutation of either K17 or all lysines showed similar results, we conclude that Lysine 17 is the main acceptor for ubiquitylation of Bcl-2. Collectively, these results suggest a distinct model for the regulation of Bcl-2 by ARTS-mediated degradation (see graphical abstract). According to this model, ARTS brings XIAP and Bcl-2 into a ternary complex that stimulates ubiquitylation and degradation of Bcl-2, thereby lowering the threshold of cells to undergo apoptosis.
Figure 7. Lysine 17 (K17) is the main ubiquitin acceptor in Bcl-2.

A. Schematic representation of Bcl-2 highlighting its four lysine residues. B. MS/MS spectrum spanning the ubiquitylation site in Bcl-2. The trypsin cleaved peptides were enriched for Gly-Gly peptides using Pilot Ubiquitin Remnant Motif (K-ε-GG) kit and subjected to liquid chromatography-mass spectrometry separation. Precursor ion (brown) represents the uncleaved GFP-Bcl-2. Lines represent the relative abundance of detected peptides. The lines in black represent peptides that are not cleaved products of the precursor ion (y1–y6) and precursor ion of the un-fragmented peptide (brown). Lines in green (y1–y8) represent the cleaved GFP-Bcl-2 peptides and their corresponding amino acid sequence. KGG (red) represents an ubiquitylated lysine. This analysis identified Lysine 17 in Bcl-2 as the acceptor for XIAP-mediated ubiquitylation. C. MEFs Bcl-2 KO and HeLa cells were transfected with expression vectors for WT Bcl-2 (WT), Bcl-2 containing a substitution mutation of Lysine 17 into Alanine (K17A), and Bcl-2 in which all Lysines were changed to Alanine (No K). Increased levels of mutant Bcl-2 (K17A, No K) were seen upon apoptotic induction. This was accompanied by a decrease in apoptosis, as shown with three different apoptotic markers. Densitometry analyses are shown in the lower panel. D. Bcl-2 KO MEFs and HeLa cells were transfected with Bcl-2 as in (C) together with a GFP-cleaved caspase-3 reporter. Bcl-2 KO MEFs and HeLa cells expressing lysine mutants had significantly less cleaved caspase 3-positive cells compared to WT Bcl-2. These results suggest that Lysine 17 is the main acceptor for ubiquitylation of Bcl-2.
Discussion
Bcl-2 is a key cell death regulator (Vaux et al., 1988). Most efforts to understand the regulation of this protein have focused on interactions with other Bcl-2 family members (Adams and Cory, 2007; Chipuk et al., 2010; Youle and Strasser, 2008). Here, we investigated the mechanism by which Bcl-2 protein levels are regulated at the onset of apoptosis. We show that XIAP serves as an E3-ligase for Bcl-2 to promote its degradation, and that ARTS brings XIAP into a ternary complex with Bcl-2 (graphical abstract). This study focuses specifically on the mechanism by which ARTS and XIAP mediate proteasomal degradation of Bcl-2. It is possible that other Bcl-2 family members are regulated in a similar fashion, but this point remains to be investigated. This work reveals that ARTS functions as a distinct Bcl-2-antagonist which directly binds to the BH3 cleft of Bcl-2. By reducing Bcl-2 levels ARTS acts upstream of MOMP to promote initiation of caspase activation and cell death.
Mitochondrial regulation of apoptosis
Mitochondrial apoptosis involves MOMP and the release of cytochrome c (Adrain et al., 2001; Edison et al., 2012b; Gao et al., 2001; Rehm et al., 2006; Strasser et al., 2011; Youle and Strasser, 2008). Bcl-2 family members can control apoptosis through their pore forming abilities which enable MOMP. During MOMP, mitochondrial pro-apoptotic proteins, including Smac/Diablo and cytochrome c, are released into the cytosol and stimulate caspase activation (Edison et al., 2012b; Strasser et al., 2011; Youle and Strasser, 2008).
We previously reported that ARTS is important for the initiation of apoptosis upstream of MOMP, and that ARTS is required for the proper release of cytochrome c and Smac/Diablo (Edison et al., 2012b). The current study extends these results and further suggests a mechanism by which ARTS can promote MOMP by stimulating UPS-mediated degradation of Bcl-2. Additionally, this study also explains several other previous observations. First, ARTS resides in the MOM in living cells, but it does not contain any known transmembrane domain. Since Bcl-2 is inserted into the MOM and ARTS can bind directly to Bcl-2, this association offers a possible explanation for the localization of ARTS to the MOM. Consistent with this idea, ARTS is mostly in the cytosol in Bcl-2 KD cells. Upon induction of apoptosis, both ARTS and Bcl-2 translocate to the cytosol (Figure 2C, 2F) and are subsequently degraded. Importantly, we show that XIAP acts as an E3-ligase for Bcl-2 to promote its degradation. On the surface, these results are unexpected since they suggest a pro-apoptotic activity of XIAP. The general accepted view is that XIAP has anti-apoptotic function. This is based on the ability of XIAP to inhibit caspases in vitro, because over-expression of XIAP can inhibit cell death, and because genetic inactivation of XIAP in mice facilitates apoptosis in at least some paradigms (Garcia-Fernandez et al., 2010; Jost et al., 2009; Schile et al., 2008). However, none of these observations are inconsistent with the proposed pro-apoptotic function of XIAP via targeting Bcl-2. First, the available genetic evidence reveals only the net effect of XIAP-overexpression/inactivation, and any pro-apoptotic roles of this protein may be masked by a more prominent anti-apoptotic function of XIAP. Second, dual functions have been reported for other proteins regulating cell death, including Bak, Bax and Bcl-2 (Darding and Meier, 2012; Kirsch et al., 1999; Lewis et al., 1999). We reconcile these diverse findings by proposing that XIAP has two opposing functions for the regulation of apoptosis. First, in its “classic” function XIAP behaves as an anti-apoptotic protein by binding to and inhibiting caspases. However, upon induction of apoptosis when ARTS promotes the formation of a ternary complex with Bcl-2, the function of XIAP is “switched” to become pro-apoptotic since it promotes degradation of Bcl-2. According to this model, ARTS acts as a switch to change the activity of XIAP from anti-apoptotic to pro-apoptotic (graphical abstract). Since complete loss of XIAP function sensitizes at least certain cells towards apoptosis, we conclude that in the paradigms studies so far the anti-apoptotic function of XIAP is more dominant than its pro-apoptotic function (Edison et al., 2012b; Fuchs et al., 2013; Garcia-Fernandez et al., 2010). However, our work raises the intriguing possibility that there may be conditions where the net-effect of eliminating XIAP may produce the opposite outcome.
Several key observations support this model. First, down-regulation of Bcl-2 occurs already 30–60 minutes after induction of apoptosis by STS, whereas the release of cytochrome c and Smac/Diablo occurred only after 3 hours (Figure 1D and 2B). Furthermore, translocation of Bcl-2 to the cytosol and ubiquitylation of Bcl-2 were observed as early as 30 minutes after STS induction (Figures 2CII, 2F). Moreover, ARTS KD HeLa cells exhibit a significant inhibition in MOMP and the release of cytochrome c and Smac/Diablo (Edison et al., 2012b). Finally, inactivation of ARTS by either KD in HeLa cells or deletion of the Sept4/ARTS in mice can inhibit apoptosis (Edison et al., 2012b; Fuchs et al., 2013; Garcia-Fernandez et al., 2010).
It appears that ARTS can promote MOMP through more than one mechanism. First, ARTS stimulates cleavage of Bid, which is known to promote MOMP (Edison et al., 2012b; Gross et al., 1999b; Li et al., 1998; Lovell et al., 2008). Second, reduced levels of Bcl-2 protein are expected to facilitate MOMP through de-repression of Bax and Bak. Furthermore, upon apoptotic induction, components of the fission machinery co-localize with Bax, suggesting that MOMP is linked to mitochondrial fragmentation (Delivani et al., 2006; Martinou and Youle, 2011). We have shown that the BH3 domain of Bcl-2 is important for binding to ARTS (Figure 4 A, B, C, D and Supplemental Figure S4). This is surprising as the BH3 domain in Bcl-2 is thought to mainly interact with BH3 domains of other Bcl-2 members. Using a peptide array method, we found that ARTS binds to the same Bcl-2-BH3 binding pocket as the pro-apoptotic Bax protein (Figure 4A). These results are further supported by our finding that the BH3-mimetic ABT-199 can attenuate the interaction of Bcl-2 and ARTS (Supplemental Figure S6).
Intriguingly, it was previously suggested that “in addition to the post-mitochondrial apoptosome pathway, there are initiator caspases perhaps controlled fairly directly by Bcl-2, which act upstream of organelle damage to activate Bax and Bak, perhaps via cleavage of a Bid-like protein that interacts with them” (Cory et al., 2003). Our results support this postulate and provide evidence that ARTS acts upstream of MOMP by releasing pre-apoptosome active caspases through de-repression. This non-lethal amount of active caspase 9 can cleave Bid and promote MOMP (Edison et al., 2012b). In addition, ARTS also directly antagonizes Bcl-2 by promoting its degradation. Support for the importance of controlling Bcl-2 stability in the regulation of apoptosis comes from the identification of Lysine 17 as the main ubiquitin acceptor in Bcl-2. Because mutating either K17 or all lysines gave virtually identical results (Figure 7C, D), we conclude that Lysine 17 is the main acceptor for ubiquitylation of Bcl-2.
Implications for cancer
Bcl-2 is over-expressed in different hematological malignancies and solid tumors (Kelly and Strasser, 2011; Youle and Strasser, 2008) Given the oncogenic potential of Bcl-2, its antagonists are expected to act as tumor suppressors (Cory and Adams, 2002). Here we show that ARTS can antagonize Bcl-2 by promoting the degradation of this protein. Significantly, ARTS functions as a tumor suppressor protein as ARTS expression is frequently lost in acute lymphoblastic leukemia patients, lymphoma and hepatocellular carcinoma, and deletion of Sept4/ARTS in mice accelerates tumorigenesis (Elhasid et al., 2004; Garcia-Fernandez et al., 2010). Our study points to a distinct connection between ARTS and Bcl-2 in malignancies as silencing of ARTS may contribute to increased levels of Bcl-2.
Anticancer agents such as ABT-737, ABT-263 and ABT-199 that directly target Bcl-2-like pro-survival proteins by mimicking the BH3 domain have been developed (Baell and Huang, 2002; Fesik, 2005; Oltersdorf et al., 2005; Rutledge et al., 2002) Interestingly, we found that the BH3-mimetic ABT-199 can disrupt the interaction of Bcl-2 and ARTS indicating that the BH3-binding motif to which ABT-199 binds in Bcl-2 is important for the interaction with ARTS (Supplemental Figure S6). This activity of ABT-199 is expected to reduce degradation of Bcl-2, which has the potential to cause increased resistance towards apoptosis. However, because ABT-199 causes cell killing it is clear that this compound potently inactivates the anti-apoptotic activity of Bcl-2, even if present at somewhat elevated levels. Since ARTS antagonizes both XIAP and Bcl-2, these features may be useful for developing compounds that target both Bcl-2 and XIAP for cancer therapy.
Experimental Procedures
Antibodies
In all our assays we used the monoclonal anti-ARTS antibody (A4471, Sigma Aldrich) which is the only currently commercially available antibody directed against the unique C-terminus of ARTS binding specifically to ARTS (Sept4_i2) and not to the other Septin 4 protein product- Sept4_i1 (H5, PNUTL). The entire antibody list is detailed in the supplemental experimental procedures.
Mammalian cell cultures and treatments
COS-7 (monkey fibroblast-like kidney cells) and HeLa (human cervical carcinoma) cells were grown in Dulbecco’s modified Eagle medium (DMEM) with 4.5 g/l D-glucose. Media were supplemented with 10% heat inactivated fetal calf serum (FCS), 100 U/ml penicillin, 100 µg/ml streptomycin, 1 mM sodium pyruvate and 2 mM glutamine (Biological Industries). Knocked-down (KD) ARTS HeLa stable cell line and Bcl-2-HeLa KD lines were established by using short hairpin RNAs (shRNAs) as described in (Edison et al., 2012a). Cells were grown in the presence of 0.5 mg/ml G418 (Sigma). The WT and XIAPdelRing deficient MEFs were prepared from 14-day old WT and XIAPdelRing deficient mouse embryos as described in (Schile et al., 2008). To induce apoptosis COS-7, HeLa, MEFs, BT-549 (human breast epithelial carcinoma) cells were incubated with staurosporine (STS) (Sigma-Aldrich) (1.75 µM for HeLa, 0.6 µM for BT-549, 1.75 µM for MEFs and 1.25µM for COS-7) for different time periods or with Etoposide (Sigma Aldrich) (200µM for HeLa, BT-549, COS-7, and immortalized MEFs and 100µM for primary MEFs) for different time periods. Proteasome inhibition: the cells were incubated with 20µM MG132 for 6 h.
Constructs & transient transfection of cells
The entire constructs list is detailed in the supplemental experimental procedures. For transient transfections the following reagents were used according to the manufacturer’s instructions: jetPEI™ (Polyplus), Transfectol (GeneChoice) Turbofect™ (Thermo), PolyJet (SignaGen).
Western blot analysis
Western blot analysis was performed as described in (Lotan et al., 2005). Visualization was performed using LAS4000 Luminescent Image Analyzer (Fujifilm) and densitometry analysis was performed by TotalLab TL100 graphic software.
Cell fractionation assay
Subcellular fractionation was done using a digitonin-based method as described in (Adrain et al., 2001). Briefly, cells were harvested and centrifuged at 300g for 10min, washed in TBS 2.5mM pH 7.5, and repelleted. Cells were permeabilized for 5min on ice with cytosolic extraction buffer (CEB) (250 mM sucrose, 70 mM KCl, 137 mM NaCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4, pH 7.2, 1× complete protease inhibitor cocktail, Roche) containing freshly prepared digitonin (200 µg/ml, D-5628, Sigma-Aldrich). Cytosolic fraction was isolated by collecting the supernatant after centrifugation at 1000 g for 5 min at 4 °C. The mitochondrial fraction was washed in CEB buffer with digitonin, CEB buffer and twice with PBS and resuspended in whole cell extraction buffer (WCE) (25mM Hepes pH 7.7, 0.3M NaCl, 1.5mM Mg Cl22, 0.2mM EDTA, 0.1% Triton X-100, 100µg/ml PMSF, 1× complete protease inhibitor cocktail, Roche).
Immuno-Fluorescence assay (IF)
Immuno-Fluorescence assays were performed as detailed in the supplemental experimental procedures. Briefly, cells were seeded in 24-wells. Following apoptotic induction (1.75 µM STS), cells were fixed with 4% paraformaldehyde. Nuclei were stained with DAPI (157574, MP Biomedicals), mitochondria were visualized by MitoTracker™ Red CMXRos (M-7512, ThermoFischer Scientific), IF staining’s were performed with anti-ARTS or anti Bcl-2 antibodies followed by FITC conjugated fluorescent secondary antibody. Image analysis was carried out using fluorescent microscope (Nikon 50i). 300 cells from each sample were analyzed for cellular localization of ARTS or Bcl-2. Diffused staining of the protein, not co-localized with MitoTracker was determined as cytosolic. Dotted staining of the protein, co-localized to the MitoTracker staining was determined as localized to the mitochondria.
GFP-Caspase-3 Cleavage reporter assay
MEFs Bcl2 KO and HeLa cells were seeded on 13 mm round slides previously coated with fibronectin (5 mg/ml, Biological Industries). Cells were co-transfected with 0.25 µg DNA of GC3AI plasmid and 0.25 µg DNA of Bcl-2 WT, Bcl-2 K17A, Bcl-2 No K for 40 hours. GC3AI is a switch-on GFP-based reporter for caspase-3 cleavage (a kind gift from Prof. Binghui Li). Apoptosis was induced with 0.9 µM STS for 2 h. Cells were then fixed with 4% paraformaldehyde in PBS for 20 min at room temperature, washed with PBS and stained with DAPI (#157574, MP Biomedicals). 300 cells were counted in each slide; the percent of positive GFP cells exhibiting cleaved caspase-3 was calculated out of Bcl-2 transfected cells.
Binding assays
In vitro pull-down binding assays
Bacterially expressed His-ARTS, Bcl-2 and GST-XIAP (5 µg each) were co-incubated at 4 °C overnight in 60 µl binding buffer (20 mM Tris, pH7.6, 1 mM DTT). GST-XIAP was pulled down after incubation (at 4 °C for 2 h) with 20 µl Glutathion-Sepharose 4B in the same buffer. After removal of unbound proteins, the resin was washed three times with the binding buffer. XIAP and associated proteins were electrophoresed and detected by western blotting with anti-ARTS (A4471, Sigma-Aldrich), anti-Bcl-2 (sc-492, N-19, Santa Cruz Biotechnology) and anti-XIAP (610763, BD TransductionLaboratories).
Co-immunoprecipitation
Co-immunoprecipitation was performed as previously described (Gottfried et al., 2004b). Briefly, lysates were prepared in WCE. 10 µg of anti-ARTS antibody or 2 µg of anti-Bcl-2 or anti-XIAP antibody was added to the protein lysate and the samples were left rotating over night at 4 °C. Next day, agarose beads conjugated to protein A/G (Santa Cruz Biotechnology) were added for 4 h. Samples were centrifuged at 4000 rpm at 4 °C for 5 min and washed five times with WCE Proteins were eluted from beads following 5 min of boiling in sample buffer and separated on 12.5% SDS–PAGE gel, followed by western blot analysis.
BiFC assay
The split-Venus, BiFC system (Li et al., 1998) was used to evaluate close proximity indicating possible direct binding between pairs of proteins. The assay was performed as detailed in the supplemental experimental procedures. Briefly, the proteins were fused either to the N-terminal part of the Venus-YFP (VN) or the C-terminal part (VC). All Venus fragments were fused to the C-terminal sequences of these proteins. Jun and bFos pair was used as positive control (p.c.), Jun and bFosdeltaZIP pair was used as negative control (n.c.). A vector encoding dsRed was used as a transfection efficiency marker. Venus and dsRed fluorescence were analyzed by flow cytometry as detailed in the supplemental experimental procedures.
Protein expression and purification
Protein expression procedures for both peptide array and in vitro Ubiquitylation assays are detailed in the supplemental experimental procedures.
Peptide array screening
Peptide arrays (CelluSpots™) were custom made and purchased from INTAVIS Bioanalytical Instruments AG (Köln, Germany) based on the sequence of Human Bcl-2 and ARTS. The designed arrays included 33 Bcl-2 peptides and 41 ARTS peptides (Supplementary table 1) that are 9–21 residues long, partially overlapping and acetylated at their N-termini and attached to a cellulose membrane via their C-termini through an amide bond. The array was screened for binding following incubation with the recombinant HLT-ARTS protein and Bcl-2 protein minus C- terminus (827-BC-050, R&D systems). First, the peptide array was washed with Hepes Buffer (HB) (25 mM Hepes pH 7.4, 150 mM NaCl) and HB supplemented with 0.05% Tween 20 (HB-T). The array membrane was incubated for 4 h in blocking solution (BS) containing 2.5% BSA (Sigma-Aldrich) in HB-T. The blocking was followed by two washing steps in HB-T and two washing steps with HB. Purified HLT-tagged ARTS protein and His-tagged Bcl-2 protein were diluted in BS to final concentrations of ~ 2 µM and incubated with the array overnight at 4 °C, in the presence of 2 mM DTT. The binding was detected by incubation with either anti-ARTS or anti-Bcl-2 and anti-His antibodies in BS. Binding of antibodies to the peptide array were visualized as described in the western blotting section.
In vivo ubiquitylation assay
Cells were transiently transfected with different constructs as indicated in text and figure legends and treated with 20 µM MG132 (Sigma-Aldrich) for 6 hours. The medium was aspirated and the cells were washed with 1xPBS at room temperature. The cells were scraped in 100 µl of lysis buffer (1% SDS, 1 mM EDTA in PBS) containing protease inhibitor cocktail (Complete, Roche), 5 mM N-ethylmaleimide (NEM) and 5 mM iodoacetamide to preserve ubiquitin chains. The cells were homogenized by passing through a 25-gauge needle (20 times), and boiled for 10 min after vigorous vortexing. 400 µl of renaturation buffer (2% TritonX100, 0.5% Na Deoxycholate, 1% BSA,1mM EDTA in PBS) containing protease inhibitor cocktail (Complete, Roche), 5 mM NEM and 5 mM iodoacetamide was added to the lysate. Following 15 min of centrifugation (10,000 g, 4 °C), the supernatant was transferred into a clean Eppendorf tube. TUBE immunoprecipitation was performed as described above with 2 µg anti-Ubiquitin antibody (sc-8017, P4D1, Santa Cruz Biotechnology). Poly-ubiquitylated forms of Bcl-2 were detected using anti-Bcl2 antibody (ab32124, Abcam).
In vitro ubiquitylation assay
In vitro ubiquitylation was assayed as previously described (Kim et al., 2007). Briefly, purified Bcl-2 was incubated with E1, UbcH5b, Ub and appropriate E3 in the conjugation buffer (20 mM Tris-Cl, pH7.6, 100 mM KCl, 5 mM MgCl2 and 1 mM DTT) containing 2 mM ATP at 37 °C for 1 h. In a control reaction, a component indicated was excluded. Changes of the molecular weight of Bcl-2 by ubiquitylation were detected by western blotting using anti-Bcl2 antibody (sc-492 N-19, Santa Cruz Biotechnology).
Mass-spectrometry analysis of ubiquitylated Bcl-2
Ubiquitylated Bcl-2 isolation
Hela cells were transfected with 3 µg of Bcl-2-GFP together with 3 µg of HA-Ubiquitin. 24 hours post transfection the cells were treated with 20µM MG132 for six hours. Apoptosis was induced using 1.75 µM STS for 30 min. The cells were counted and then lysed with CHAPS-buffer (10 mM Hepes pH 7.5, 1% CHAPS, 150 mM NaCl,) buffer containing protease inhibitor cocktail (Roche). The lysates were precipitated with α-GFP magnetic beads (MBL International) for 18 hours. For eight million cells 40 µl antibody-beads slurry was used. Beads were washed three times with CHAPS buffer. Detection of ubiquitylated Bcl-2 lysines is detailed in the supplemental experimental procedures. Briefly, the anti-GFP beads–GFP-Bcl2 conjugates were digested with trypsin (Promega Corporation). Digested peptides were enriched for Glycine-Glycine peptides with the PTMScan® Pilot Ubiquitin Remnant Motif (K-ε-GG) Kit (Cell Signaling Technology) according to the manufacturer’s instructions. Peptide sequences were identified by liquid chromatography followed by mass spectrometry. The analysis of the mass spectrometry data detected the K-GG containing peptides.
Statistical analysis
Densitometry analyses of Western blot results were performed using TotalLab TL100 graphic software. Image Analysis was performed using the Imaris Image Analysis Software. At least 300 cells were counted for each IF sample. Standard errors were obtained from three to four biologically independent experiments. Indicated p-values were calculated using Student's t-test (* = p ≤ 0.05, **= p ≤ 0.001).
Supplementary Material
Highlights.
ARTS binds directly to both XIAP and Bcl-2 bringing them into a ternary complex.
ARTS bridges between XIAP and Bcl-2 allowing XIAP to serve as an E3-ligase for Bcl-2.
ARTS interacts with the BH3 domain of Bcl-2.
ARTS functions as a distinct Bcl-2 antagonist by binding and leading to its degradation.
Acknowledgments
We thank Hermann Steller, Suzanne Cory, David Huang, Binghui Li and Reuven Stein for generously providing us with constructs and cell lines used in this manuscript. We thank Hermann Steller for thoughtful discussion on the manuscript and Gal Sevi Karniel for excellent graphical work. This work was supported by funds the NIH-NIGMS grant R01GM051923 (to H.T.K) and by the BSF (US Israel Binational Science Foundation) grant #2003085 (to S.L), ISF (Israel Science Foundation), Grant #1264/06 and 822/12 (to S.L) and INCPM-ISF grant #2376/15 (to S.L), the Charles Wolfson Charitable Trust, England, and by a generous award from the Hymen Milgrom Trust (to S.L).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, N.E and S.L.; Methodology, H.T.K., and S.L.; Investigation, N.E., Y.C., N.P., D.M, N.K., N.C., D.M., M.K., T.H.R., J.K., and H.T.K.; Writing – Original Draft, S.L.; Writing-Review & Editing, H.T.K. and S.L.; Funding Acquisition, S.L.; Resources, H.T.K., S.L; Supervision, S.L.
References
- Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- Adams JM, Cory S. Bcl-2-regulated apoptosis: mechanism and therapeutic potential. Curr Opin Immunol. 2007;19:488–496. doi: 10.1016/j.coi.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrain C, Creagh EM, Martin SJ. Apoptosis-associated release of Smac/DIABLO from mitochondria requires active caspases and is blocked by Bcl-2. EMBO J. 2001;20:6627–6636. doi: 10.1093/emboj/20.23.6627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell JB, Huang DC. Prospects for targeting the Bcl-2 family of proteins to develop novel cytotoxic drugs. Biochemical pharmacology. 2002;64:851–863. doi: 10.1016/s0006-2952(02)01148-6. [DOI] [PubMed] [Google Scholar]
- Bergmann A, Yang AY, Srivastava M. Regulators of IAP function: coming to grips with the grim reaper. Curr Opin Cell Biol. 2003;15:717–724. doi: 10.1016/j.ceb.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Bornstein B, Edison N, Gottfried Y, Lev T, Shekhtman A, Gonen H, Rajalingam K, Larisch S. X-linked Inhibitor of Apoptosis Protein promotes the degradation of its antagonist, the pro-apoptotic ARTS protein. Int J Biochem Cell Biol. 2012;44:489–495. doi: 10.1016/j.biocel.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Bornstein B, Gottfried Y, Edison N, Shekhtman A, Lev T, Glaser F, Larisch S. ARTS binds to a distinct domain in XIAP-BIR3 and promotes apoptosis by a mechanism that is different from other IAP-antagonists. Apoptosis. 2011 doi: 10.1007/s10495-011-0622-0. [DOI] [PubMed] [Google Scholar]
- Boyce M, Degterev A, Yuan J. Caspases: an ancient cellular sword of Damocles. Cell Death Differ. 2004;11:29–37. doi: 10.1038/sj.cdd.4401339. [DOI] [PubMed] [Google Scholar]
- Castle VP, Heidelberger KP, Bromberg J, Ou X, Dole M, Nunez G. Expression of the apoptosis-suppressing protein bcl-2, in neuroblastoma is associated with unfavorable histology and N-myc amplification. Am J Pathol. 1993;143:1543–1550. [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Heller H, Elias S, Haas AL, Hershko A. ATP-dependent conjugation of reticulocyte proteins with the polypeptide required for protein degradation. Proc Natl Acad Sci U S A. 1980;77:1365–1368. doi: 10.1073/pnas.77.3.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22:8590–8607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- Darding M, Meier P. IAPs: guardians of RIPK1. Cell Death Differ. 2012;19:58–66. doi: 10.1038/cdd.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delivani P, Adrain C, Taylor RC, Duriez PJ, Martin SJ. Role for CED-9 and Egl-1 as regulators of mitochondrial fission and fusion dynamics. Mol Cell. 2006;21:761–773. doi: 10.1016/j.molcel.2006.01.034. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Breitschopf K, Haendeler J, Zeiher AM. Dephosphorylation targets Bcl-2 for ubiquitin-dependent degradation: a link between the apoptosome and the proteasome pathway. J Exp Med. 1999;189:1815–1822. doi: 10.1084/jem.189.11.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Eckelman BP, Salvesen GS, Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 2006;7:988–994. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edison N, Reingewertz TH, Gottfried Y, Lev T, Zuri D, Maniv I, Carp MJ, Shalev G, Friedler A, Larisch S. Peptides Mimicking the Unique ARTS-XIAP Binding Site Promote Apoptotic Cell Death in Cultured Cancer Cells. Clin Cancer Res. 2012a;18:2569–2578. doi: 10.1158/1078-0432.CCR-11-1430. [DOI] [PubMed] [Google Scholar]
- Edison N, Zuri D, Maniv I, Bornstein B, Lev T, Gottfried Y, Kemeny S, Garcia-Fernandez M, Kagan J, Larisch S. The IAP-antagonist ARTS initiates caspase activation upstream of cytochrome C and SMAC/Diablo. Cell Death Differ. 2012b;19:356–368. doi: 10.1038/cdd.2011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhasid R, Sahar D, Merling A, Zivony Y, Rotem A, Ben-Arush M, Izraeli S, Bercovich D, Larisch S. Mitochondrial pro-apoptotic ARTS protein is lost in the majority of acute lymphoblastic leukemia patients. Oncogene. 2004;23:5468–5475. doi: 10.1038/sj.onc.1207725. [DOI] [PubMed] [Google Scholar]
- Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer. 2005;5:876–885. doi: 10.1038/nrc1736. [DOI] [PubMed] [Google Scholar]
- Fuchs Y, Brown S, Gorenc T, Rodriguez J, Fuchs E, Steller H. Sept4/ARTS regulates stem cell apoptosis and skin regeneration. Science. 2013;341:286–289. doi: 10.1126/science.1233029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs Y, Steller H. Programmed Cell Death in Animal Development and Disease. Cell. 2011;147:1–17. doi: 10.1016/j.cell.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs Y, Steller H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol. 2015;16:329–344. doi: 10.1038/nrm3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galban S, Duckett CS. XIAP as a ubiquitin ligase in cellular signaling. Cell Death Differ. 2010;17:54–60. doi: 10.1038/cdd.2009.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao CF, Ren S, Zhang L, Nakajima T, Ichinose S, Hara T, Koike K, Tsuchida N. Caspase-dependent cytosolic release of cytochrome c and membrane translocation of Bax in p53-induced apoptosis. Exp Cell Res. 2001;265:145–151. doi: 10.1006/excr.2001.5171. [DOI] [PubMed] [Google Scholar]
- Garcia-Fernandez M, Kissel H, Brown S, Gorenc T, Schile AJ, Rafii S, Larisch S, Steller H. Sept4/ARTS is required for stem cell apoptosis and tumor suppression. Genes & development. 2010;24:2282–2293. doi: 10.1101/gad.1970110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- Gottfried Y, Rotem A, Lotan R, Steller H, Larisch S. The mitochondrial ARTS protein promotes apoptosis through targeting XIAP. The EMBO Journal. 2004a;23:1627–1635. doi: 10.1038/sj.emboj.7600155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottfried Y, Rotem A, Lotan R, Steller H, Larisch S. The mitochondrial ARTS protein promotes apoptosis through targeting XIAP. EMBO J. 2004b;23:1627–1635. doi: 10.1038/sj.emboj.7600155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999a;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem. 1999b;274:1156–1163. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- Gyrd-Hansen M, Darding M, Miasari M, Santoro MM, Zender L, Xue W, Tenev T, da Fonseca PC, Zvelebil M, Bujnicki JM, et al. IAPs contain an evolutionarily conserved ubiquitin-binding domain that regulates NF-kappaB as well as cell survival and oncogenesis. Nat Cell Biol. 2008;10:1309–1317. doi: 10.1038/ncb1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyrd-Hansen M, Meier P. IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer. 2010;10:561–574. doi: 10.1038/nrc2889. [DOI] [PubMed] [Google Scholar]
- Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC, Bouillet P, Thomas HE, Borner C, Silke J, et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature. 2009;460:1035–1039. doi: 10.1038/nature08229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassi E, Sourlingas TG, Spiliotaki M, Papoutsi Z, Pratsinis H, Aligiannis N, Moutsatsou P. Ursolic acid triggers apoptosis and Bcl-2 downregulation in MCF-7 breast cancer cells. Cancer Invest. 2009;27:723–733. doi: 10.1080/07357900802672712. [DOI] [PubMed] [Google Scholar]
- Kelly PN, Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 2011;18:1414–1424. doi: 10.1038/cdd.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HT, Kim KP, Lledias F, Kisselev AF, Scaglione KM, Skowyra D, Gygi SP, Goldberg AL. Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J Biol Chem. 2007;282:17375–17386. doi: 10.1074/jbc.M609659200. [DOI] [PubMed] [Google Scholar]
- Kirsch DG, Doseff A, Chau BN, Lim DS, de Souza-Pinto NC, Hansford R, Kastan MB, Lazebnik YA, Hardwick JM. Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. J Biol Chem. 1999;274:21155–21161. doi: 10.1074/jbc.274.30.21155. [DOI] [PubMed] [Google Scholar]
- Kissel H, Georgescu MM, Larisch S, Manova K, Hunnicutt GR, Steller H. The Sept4 septin locus is required for sperm terminal differentiation in mice. Dev Cell. 2005;8:353–364. doi: 10.1016/j.devcel.2005.01.021. [DOI] [PubMed] [Google Scholar]
- Krajewska M, Krajewski S, Epstein JI, Shabaik A, Sauvageot J, Song K, Kitada S, Reed JC. Immunohistochemical analysis of bcl-2, bax, bcl-X, and mcl-1 expression in prostate cancers. Am J Pathol. 1996;148:1567–1576. [PMC free article] [PubMed] [Google Scholar]
- Lewis J, Oyler GA, Ueno K, Fannjiang YR, Chau BN, Vornov J, Korsmeyer SJ, Zou S, Hardwick JM. Inhibition of virus-induced neuronal apoptosis by Bax. Nat Med. 1999;5:832–835. doi: 10.1038/10556. [DOI] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Lotan R, Rotem A, Gonen H, Finberg JP, Kemeny S, Steller H, Ciechanover A, Larisch S. Regulation of the proapoptotic ARTS protein by ubiquitin-mediated degradation. J Biol Chem. 2005;280:25802–25810. doi: 10.1074/jbc.M501955200. [DOI] [PubMed] [Google Scholar]
- Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, Andrews DW. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell. 2008;135:1074–1084. doi: 10.1016/j.cell.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Malin JZ, Shaham S. Cell Death in C. elegans Development. Current topics in developmental biology. 2015;114:1–42. doi: 10.1016/bs.ctdb.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21:92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Rajalingam K, Dikic I. Inhibitors of apoptosis catch ubiquitin. Biochem J. 2009;417:e1–3. doi: 10.1042/BJ20082215. [DOI] [PubMed] [Google Scholar]
- Rehm M, Huber HJ, Dussmann H, Prehn JH. Systems analysis of effector caspase activation and its control by X-linked inhibitor of apoptosis protein. EMBO J. 2006;25:4338–4349. doi: 10.1038/sj.emboj.7601295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson LE, Plunkett W, McConnell K, Keating MJ, McDonnell TJ. Bcl-2 expression in chronic lymphocytic leukemia and its correlation with the induction of apoptosis and clinical outcome. Leukemia. 1996;10:456–459. [PubMed] [Google Scholar]
- Rutledge SE, Chin JW, Schepartz A. A view to a kill: ligands for Bcl-2 family proteins. Current opinion in chemical biology. 2002;6:479–485. doi: 10.1016/s1367-5931(02)00352-6. [DOI] [PubMed] [Google Scholar]
- Schile AJ, Garcia-Fernandez M, Steller H. Regulation of apoptosis by XIAP ubiquitin-ligase activity. Genes Dev. 2008;22:2256–2266. doi: 10.1101/gad.1663108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell. 2002;9:459–470. doi: 10.1016/s1097-2765(02)00482-3. [DOI] [PubMed] [Google Scholar]
- Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30:3667–3683. doi: 10.1038/emboj.2011.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan D, Ryoo HD. Regulation of Cell Death by IAPs and Their Antagonists. Current topics in developmental biology. 2015;114:185–208. doi: 10.1016/bs.ctdb.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Volkmann N, Marassi FM, Newmeyer DD, Hanein D. The rheostat in the membrane: BCL-2 family proteins and apoptosis. Cell Death Differ. 2014;21:206–215. doi: 10.1038/cdd.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Chanvorachote P, Toledo D, Stehlik C, Mercer RR, Castranova V, Rojanasakul Y. Peroxide is a key mediator of Bcl-2 down-regulation and apoptosis induction by cisplatin in human lung cancer cells. Mol Pharmacol. 2008;73:119–127. doi: 10.1124/mol.107.040873. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.