

Key Points
Deletion of Arp2/3 leads to marked microthrombocytopenia due to abnormal platelet release and increased platelet clearance.
Arp2/3 is critical for platelet lamellipodia formation and spreading, but plays a minor role for platelet adhesion and hemostasis.
Abstract
Actin reorganization regulates key processes in platelet activation. Here we examined the role of the Arp2/3 complex, an essential component in actin filament branching, in platelet function. The Arpc2 gene, encoding the p34 subunit of the Arp2/3 complex, was deleted in the megakaryocyte lineage (Arpc2fl/flPF4-Cre). Deletion of the Arp2/3 complex resulted in marked microthrombocytopenia in mice, caused by premature platelet release into the bone marrow compartment and impaired platelet survival in circulation. Arpc2fl/flPF4-Cre platelets exhibited alterations in their actin cytoskeleton and their peripheral microtubule coil. Thrombocytopenia was alleviated following clodronate liposome-induced macrophage depletion in Arpc2fl/flPF4-Cre mice. Arpc2fl/flPF4-Cre platelets failed to spread and showed a mild defect in integrin activation and aggregation; however, no significant differences in hemostasis or thrombosis were observed between Arpc2fl/flPF4-Cre and control mice. Thus, Arp2/3 is critical for platelet homeostasis but plays only a minor role for vascular hemostasis.
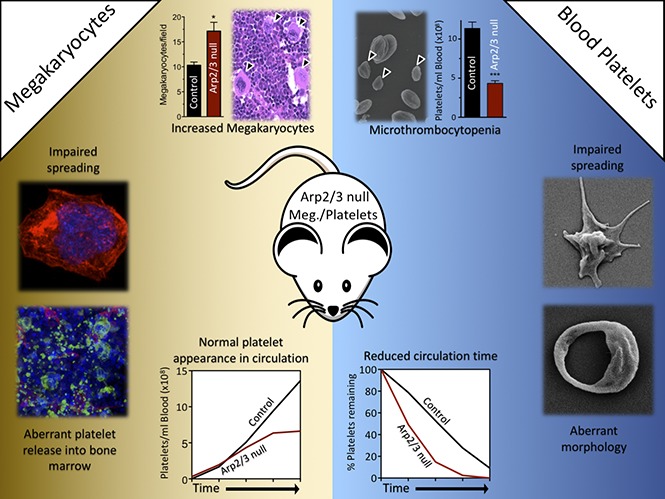
Visual Abstract
Introduction
The Arp2/3 complex is key to branched actin network formation. It consists of 7 subunits, including Arpc2, a subunit critical to proper expression of the entire complex.1 In resting cells, the Arp2/3 complex is mostly found in the cytoplasm. Upon cellular stimulation, it translocates to the periphery, where it nucleates new actin branches from existing actin filaments. The activation of Arp2/3 is dependent on nucleation promoting factors (NPFs) such as members of the WASp and WAVE/SCAR families. Consistent with its key role in actin branching, cells lacking Arp2/3 show defects in lamellipodia formation and haptotaxis.2,3
The Arp2/3 complex and NPFs of the WASp and WAVE/SCAR families are also expressed in blood platelets,4,5 anucleated cells critical for the integrity of the vascular system (hemostasis). Platelets derive from megakaryocytes, cells found in the bone marrow and the spleen. During the process of thrombopoiesis, megakaryocytes undergo rapid growth, polyploidization, and formation of the demarcation membrane system before forming proplatelet processes, which fragment into platelets.6 Cytoskeletal rearrangement is critical during megakaryocyte maturation, as evidenced by the fact that disruption of microtubules or F-actin dynamics results in profound defects in proplatelet formation.7 Once released into the bloodstream, platelets can circulate in a nonadhesive state for ∼10 and 5 days in humans and mice, respectively, unless they are consumed by the hemostatic process. Platelet homeostasis (ie, the maintenance of a defined platelet count) depends on the tight regulation of both platelet production and platelet clearance. At sites of vascular injury, platelets are activated and undergo dramatic cytoskeletal changes that are thought to be important for hemostatic plug formation.8
The role of the Arp2/3 signaling pathway in platelet homeostasis and platelet function is not well understood. Patients with mutations in WASp suffer from Wiskott-Aldrich syndrome and present with a variety of immunological disorders as well as spontaneous bleeding and bruising consequent to a marked microthrombocytopenia (∼80% of patients with platelet counts <50 × 109/L compared with healthy controls, 150-400 × 109/L)9 and a mild defect in platelet function.10 With regard to the platelet count, a milder phenotype (∼50% to 70% compared with controls) was described for mice lacking WASp.11,12 Both increased platelet clearance and aberrant platelet release into the bone marrow were shown to contribute to the thrombocytopenia observed in WASp mutant mice.12,13 Just as in humans, integrin inside-out activation and adhesion of platelets from mice lacking functional WASp are only minimally impaired.14 Hemostasis is largely intact in WASp mutant mice. Interestingly, studies in human and mouse platelets demonstrated that actin assembly and activation of the Arp2/3 complex can occur in the absence of WASp, suggesting that the Cdc42-WASp-Arp2/3 axis is not functional in these cells.15,16 The contribution of WAVE/SCAR family proteins to platelet function is also not well characterized. Genetic knockout of WAVE2, the predominant isoform in platelets and megakaryocytes, leads to early embryonic lethality in mice.17 Mice deficient in WAVE1 are viable and their platelets show a defect in spreading and integrin activation only when activated via the collagen receptor, GPVI.18 Mice deficient in Arp2/3 also die during embryonic development.19 Using blocking antibodies in platelet extracts and in permeabilized platelets, Li et al suggested that Arp2/3 is critical for actin polymerization and both filopodia and lamellipodia formation in platelets.4 The true impact of Arp2/3 on platelet homeostasis and platelet function, however, could not be established with this limited assay system.
In the present study, we generated mice lacking Arp2/3 in the megakaryocyte lineage (Arpc2fl/flPF4-Cre) to investigate its contribution to platelet homeostasis and function. Arpc2fl/flPF4-Cre mice exhibited a marked reduction in the peripheral platelet count, caused by impaired platelet release and increased platelet clearance mediated by macrophages of the reticulo-endothelial system. Surprisingly, hemostatic plug formation was intact in Arpc2fl/flPF4-Cre mice even though knockout platelets exhibited mild defects in integrin-mediated adhesion and significant defects in lamellipodia formation.
Materials and methods
Mouse strains
Mice with conditional Arpc2 alleles (Arpc2fl/fl) on a mixed genetic background20 were crossed with C57BL/6 mice expressing platelet factor 4 promoter-driven Cre-recombinase (PF4-Cre) to generate mice lacking expression of p34 in megakaryocytes and platelets (Arpc2fl/flPF4-Cre). All mouse experiments were reviewed and approved by the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill. Mice were provided with food and water ad libitum.
Platelet preparation
Blood was drawn with heparin-coated capillaries from the retro-orbital plexus into tubes containing low-molecular-weight heparin. Platelet-rich plasma was obtained by centrifugation at 100g for 5 minutes and centrifuged at 700g for 5 minutes at room temperature in the presence of 1 μg/mL prostacyclin. Pelleted platelets were resuspended in modified Tyrode’s buffer (137 mM NaCl, 0.3 mM Na2HPO4, 2 mM KCl, 12 mM NaHCO3, 5 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, and 5 mM glucose; pH 7.3) containing 1 mM CaCl2.
Platelet spreading assay
Washed platelets were adjusted to 2 × 107/mL in Tyrode’s buffer (+0.35% bovine serum albumin [BSA] +2 mM CaCl2) and seeded on human fibrinogen coated coverslips (100 μg/mL) for 10 minutes before addition of agonists. Cells were fixed with 3.7% formaldehyde after 30 minutes, permeabilized with 0.1% Triton X-100, and stained with phalloidin-594. Resting platelets were fixed in suspension and plated on poly-L-lysine–coated coverslips. Resting platelets were also stained with anti–β-tubulin (clone E7, Developmental Studies Hybridoma Bank, University of Iowa). Samples were analyzed on a Nikon Ti-U inverted microscope (Nikon Instruments Inc., Melville, NY) equipped with a Retiga EXL monochrome camera (QImaging, Surrey, Canada). Images were acquired using Slidebook 5.0 (Intelligent Imaging Innovations, Denver, CO) and a 100× oil objective. Cell surface area was quantified using Image J image analysis software.
Quantification of F-actin and G-actin
Washed platelets were resuspended at 2.5 × 107/mL in Tyrode’s buffer (0.35% BSA, 1 mM CaCl2). Platelets were activated with Par4-activating peptide (Par4p) or convulxin (Cvx) for 10 minutes before fixation in 2% formaldehyde and permeabilization in 0.5% Triton X-100. F-actin was labeled with Alexa Fluor 647-conjugated phalloidin (Thermo Fisher Scientific, Waltham, MA); G-actin was labeled with Alexa Fluor 488-conjugated deoxyribonuclease I (Thermo Fisher Scientific). Samples were analyzed on an Accuri C6 flow cytometer (BD Biosciences, Ann Arbor, MI).
Platelet depletion and recovery assay
Thrombocytopenia was induced in mice by intravenous injection of antibodies against platelet surface receptor, GPIbα (Emfret Analytics) (0.2 μg/g body weight) × (platelet count at baseline/average platelet count in control mice). Peripheral platelet counts were assessed by flow cytometry 1 hour after antibody administration and every 24 hours thereafter for 96 hours.
Platelet lifespan assays
Endogenous platelets.
Platelets were labeled in vivo by a single intravenous injection of 2.5 μg Alexa Fluor 647–conjugated anti-GPIX antibody in 100 μL phosphate-buffered saline (PBS) at t = −1 hour. Whole blood (50 μL) was drawn at t = 0 and every 24 hours thereafter, diluted, and incubated with a phycoerythrin (PE)-conjugated antibody to GPIbα for 10 minutes at room temperature. The percentage of Alexa Fluor-647–positive platelets within the PE-positive platelet population was determined. Data were normalized to t = 0 hour.
Transfused platelets.
Washed platelets were obtained from heparinized blood and labeled for 15 minutes with Alexa Fluor 647–conjugated or Alexa Fluor 488–conjugated α-GPIX antibody. Following a brief wash, labeled control and Arpc2fl/flPF4-Cre platelets were combined and 2 × 108 platelets were intravenously injected into recipient wild-type mice. Blood collection and counterstaining were done as described previously.
Phosphatidyl serine exposure assay
Washed platelets were adjusted to 3 × 108/mL in Tyrode’s buffer. Calcium stress was initiated by addition of 4 mM CaCl2. At t = 0 and every 15 minutes thereafter, a 2-μL aliquot of platelets was stained with Alexa Fluor 647–conjugated Annexin V and Alexa Fluor 488–conjugated anti-GPIX antibody and analyzed by flow cytometry.
Phagocytosis assay
Washed platelets were labeled with calcein green AM (Thermo Fisher Scientific) and incubated with cultured RAW 264.7 murine macrophage cells. After 30 minutes, the culture wells were washed with PBS and cells were harvested with a cell scraper before resuspension and analysis by flow cytometry. Increased macrophage fluorescence indicates platelet-macrophage association via direct binding or phagocytosis.
Depletion of macrophages with clodronate liposomes
Clodronate-liposomes or control PBS-liposomes (Clodronateliposomes.com, Amsterdam, Netherlands) were intravenously injected into Arpc2fl/flPF4-Cre mice (12-16 weeks) at a dose of 10 mL/kg body weight. Platelet counts were assessed every 24 hours by flow cytometry. Macrophage depletion was confirmed by detection of elevated plasma von Willebrand factor levels, as previously described.21
Quantification of platelets in bone marrow
Bone marrow was flushed from femurs into 1 mL of Dulbecco’s modified Eagle medium +10% fetal bovine serum + 2 mM EDTA and passed through a 70-μm cell strainer before centrifugation at 700g in the presence of 1 μg/mL of prostacyclin (Cayman Chemical, Ann Arbor, MI). The cell pellet was resuspended in Tyrode’s buffer (0.35% BSA, 2 mM EDTA) and adjusted to 1 × 108 cells/mL. A total of 1 × 106 cells was labeled with a PE-conjugated anti-GPIbα antibody and analyzed by flow cytometry. Data are expressed as % of total events that are GPIbα-positive and platelet sized.
Confocal immunofluorescence microscopy of bone marrow
Bone marrow was carefully flushed from femurs as a single intact tissue and immediately hybridized with Alexa Fluor 488–labeled anti-GPIX (clone Xia.B4, Emfret), Alexa Fluor 647–labeled anti-CD31 (clone MEC13.3, BioLegend), and Hoechst 33258 (Roche) for 30 minutes before washing and overnight fixation in 3.7% buffered formaldehyde. Whole bone marrow tissue was mounted and visualized on a Leica DM4000 Confocal microscope (Wetzlar, Germany).
Saphenous vein laser injury
Mice (12-14 weeks of age) were anesthetized with ketamine/xylazine (100 mg/kg and 10 mg/kg, respectively) and injected with Alexa Fluor 488–conjugated antibodies to GPIX (2.5 µg) to label circulating platelets. The saphenous vein was exposed and injured as previously described.22 Platelet accumulation at the site of laser injury was assessed by intravital microscopy using a Zeiss Examiner Z1 microscope (Zeiss, Oberkochen, Germany) equipped with a Hamamatsu Orca Flash 4.0 camera (Hamamatsu, Japan).
Study approval.
All animal experiments and protocols were reviewed, approved by, and performed in accordance with the Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
Statistics.
Statistical significance among multiple groups was assessed by 2-way analysis of variance with Bonferroni post-hoc analysis. Two-group comparisons of parametric data were compared using a 2-tailed Student t test. Statistical analyses were performed using Prism 5 software (GraphPad, La Jolla, CA). A P value of .05 or less was considered significant. All data are presented as mean ± standard error of the mean (SEM), unless otherwise indicated.
Results
Conditional deletion of Arpc2 in the megakaryocyte lineage results in microthrombocytopenia in mice
To investigate the contribution of the Arp2/3 complex to platelet and megakaryocyte function, we deleted the p34 subunit (encoded by the Arpc2 gene) of the complex in the megakaryocyte lineage (Arpc2fl/flPF4-Cre) (Figure 1A). Western blotting studies confirmed a >95% reduction in the expression of p34 in megakaryocytes and platelets, whereas the protein was still present in spleen cells (supplemental Figure 1) and liver (data not shown). Consistent with its key role in Arp2/3 complex formation and stability, platelets lacking the p34 subunit were also deficient in Arp2 and Arp3 (Figure 1B). Arpc2fl/flPF4-Cre mice showed normal viability when compared with littermate controls (not shown). As expected, megakaryocyte-specific deletion of Arpc2 did not affect peripheral blood leukocyte or red blood cell counts (supplemental Table 1). However, Arpc2fl/flPF4-Cre mice exhibited a markedly reduced peripheral platelet count (PPC; Figure 1C) and a significant reduction in platelet size (Figure 1D-E). The ratio of reticulated to nonreticulated platelets did not change (Figure 1F).
Figure 1.

Marked microthrombocytopenia in Arpc2fl/flPF4-Cre mice. (A) Schematic of gene targeting strategy. Exon 8 of the Arpc2 gene, flanked by LoxP sites, was excised by PF4 promoter-driven expression of Cre recombinase. (B) Representative immunoblot demonstrating knockdown of p34 and loss of Arp2 and Arp3. (C) Platelet count (Plts) (n = 6) and (D) size (n = 5) in peripheral blood of the indicated mice. (E) Representative scanning electron microscopy images of resting platelets. Arrows indicate abnormally small platelets in Arpc2fl/flPF4-Cre sample. (F) Percentage of reticulated platelets (n = 6-7). Blue bars, control; red bars, Arpc2fl/flPF4-Cre. Data presented as mean ± SEM, ***P < .001. ns, Not significant.
Loss of Arp2/3 affects both platelet production and survival
A reduction in the PPC can be the result of impaired platelet survival, defective platelet production, or both. We first investigated megakaryocyte function in Arpc2fl/flPF4-Cre mice. Deletion of Arpc2 in the megakaryocyte lineage led to a significant increase in megakaryocyte numbers in the spleen and the bone marrow (Figures 2A-B). Ex vivo, Arpc2fl/flPF4-Cre megakaryocytes were not significantly different from control cells with regard to the percentage of cells undergoing proplatelet formation (Figure 2C) and megakaryocyte ultrastructure (supplemental Figure 2). Interestingly, however, we observed a significant increase in CD42b-positive platelet-size particles in the bone marrow of Arpc2fl/flPF4-Cre mice (Figure 2D-E), suggestive of premature platelet release into the bone marrow compartment. As measured by their forward scatter signal, these platelets were of similar size in Arpc2fl/flPF4-Cre mice when compared with controls (data not shown). Immunofluorescence confocal imaging confirmed the marked increase of extravascular platelets in the bone marrow of Arpc2fl/flPF4-Cre mice (Figure 2F; supplemental Video 1). Given the large increase in megakaryocyte numbers, however, it seemed unlikely that the defect in proplatelet release is the only cause for the marked microthrombocytopenia observed in mice lacking the Arp2/3 complex. Consistent with this conclusion, platelet lifespan was markedly reduced in Arpc2fl/flPF4-Cre mice (24 vs 50 hours in controls; Figure 3A) and in wild-type mice transfused with Arpc2fl/flPF4-Cre platelets (Figure 3B). To address whether thrombocytopenia is predominantly the result of altered platelet production or increased platelet turnover, we compared Arpc2fl/flPF4-Cre and control mice for their ability to recover from antibody-induced severe thrombocytopenia (Figure 3C). No significant differences in platelet count recovery were observed between Arpc2fl/flPF4-Cre and control mice at 24 and 48 hours after infusion of the platelet-depleting antibody. However, although a continued increase in the PPC was observed in control mice, it plateaued in Arpc2fl/flPF4-Cre mice at t = 48 hours, a time point where the increase in platelet turnover is most prominent in mice lacking Arp2/3. The size of Arpc2fl/flPF4-Cre platelets was significantly smaller at all time points after antibody-mediated depletion (Figure 3D). Thus, these studies demonstrate that increased platelet turnover is a major contributor to the marked thrombocytopenia observed in Arpc2fl/flPF4-Cre mice.
Figure 2.

Premature platelet release into the bone marrow compartment in Arpc2fl/flPF4-Cre mice. (A) Increased megakaryocyte density in representative images of bone marrow and spleen sections stained with hematoxylin and eosin. (B) Quantification of megakaryocyte density in indicated organs (8-13 random fields analyzed per organ; n = 3). (C) Percentage of proplatelet forming bone marrow megakaryocytes observed in culture (n = 7-8). (D) Representative flow cytometry dot plots showing increased number of platelet-sized CD42b+ particles in bone marrow from Arpc2fl/flPF4-Cre mice. (E) Quantification of CD42b+ particles in bone marrow (n = 6). (F) Representative confocal fluorescence images of bone marrow from the indicated mice. Green, megakaryocytes/platelets; red, endothelial cells/vasculature; blue, nuclei; blue bars, control; red bars, Arpc2fl/flPF4-Cre. Data presented as mean ± SEM, *P < .05, ***P < .001.
Figure 3.

Arp2/3-deficient platelets are cleared from circulation at an increased rate. (A) Clearance of antibody-labeled platelets from circulation in control and Arpc2fl/flPF4-Cre mice (n = 5). (B) Clearance of antibody-labeled control and Arpc2fl/flPF4-Cre platelets cotransfused into wild-type recipient mice (n = 6). (C) Platelet count and (D) platelet size in the indicated mice at various time points following antibody-mediated depletion of circulating platelets (n = 7). Blue bars, control; red bars, Arpc2fl/flPF4-Cre. Data presented as mean ± SEM, *P < .05, **P < .01, ***P < .001.
Loss of Arp2/3 leads to macrophage-mediated clearance of platelets
We next investigated the molecular mechanisms underlying increased platelet turnover in Arpc2fl/flPF4-Cre mice. To test whether Arp2/3-deficiency leads to desialylation of platelet surface glycoproteins, we incubated washed platelets from Arpc2fl/flPF4-Cre and control mice with fluorescein isothiocyanate–labeled Ricinus communis agglutinin (RCA) I, a lectin that binds to desialylated N-acetyl glucosamine residues on membrane glycoproteins. Desialylated N-acetyl glucosamine is a known ligand for the αMβ2 integrin on the surface of macrophages and has been implicated in the enhanced clearance of chilled platelets.23,24 However, we found no evidence of increased RCA binding to platelets from Arpc2fl/flPF4-Cre mice (Figure 4A). Furthermore, no evidence for platelet-bound antibodies, and thus autoimmune thrombocytopenia, in Arpc2fl/flPF4-Cre mice could be found (Figure 4B). However, Arpc2fl/flPF4-Cre platelets were significantly more susceptible to stress-induced phosphatidyl serine exposure during storage in vitro (Figure 4C), suggesting that increased platelet clearance in Arpc2fl/flPF4-Cre mice may be mediated by macrophages of the reticulo-endothelial system (RES). Consistent with this finding, cultured RAW 264.7 murine macrophages interacted more readily with Arpc2fl/flPF4-Cre platelets in vitro (Figure 4D). To deplete splenic and liver macrophages, we intravenously injected Arpc2fl/flPF4-Cre mice with clodronate liposomes.25,26 Clodronate liposomes, but not PBS-control liposomes, led to a marked increase in circulating von Willebrand factor protein, confirming successful depletion of macrophages of the RES (data not shown). Consistent with our hypothesis, treatment with clodronate liposomes led to a significant increase in the PPC of Arpc2fl/flPF4-Cre mice (Figure 4E). When analyzed in vitro, platelets in clodronate-treated Arpc2fl/flPF4-Cre mice were comparable to those in control liposome-treated Arpc2fl/flPF4-Cre mice with regard to surface expression of important adhesion and agonist receptors (supplemental Figure 3A) and their integrin activation response to agonist stimulation (supplemental Figure 3B). Interestingly, platelets in clodronate-treated Arpc2fl/flPF4-Cre mice were significantly smaller than those in Arpc2fl/flPF4-Cre mice treated with control liposomes (Figure 4F), whereas no effect of clodronate liposomes on platelet size was observed in control mice. Furthermore, the proportion of small platelets after macrophage depletion was found to dramatically increase in clodronate treated Arpc2fl/flPF4-Cre mice compared with controls (Figure 4G). Thus, these studies provide experimental evidence that the increased platelet turnover in Arpc2fl/flPF4-Cre mice predominantly affects smaller platelets and is mediated by macrophages of the RES.
Figure 4.

Loss of Arp2/3 leads to macrophage-mediated clearance of platelets. (A) Binding of RCA-fluorescein isothiocyanate to platelets from mice treated (+neu) or not (−neu) with neuraminidase (n = 4-6). (B) Detection of platelet-associated antibodies with anti-mouse–IgG-488 (green bar, positive control). (C) Kinetics of phosphatidyl serine exposure in washed platelets incubated in the presence of 4 mM CaCl2 (n = 6). (D) Representative flow cytometry plot illustrating the interaction of RAW 264.7 macrophages and fluorescently labeled platelets. Changes in peripheral platelet count (E) and platelet size (F) in indicated mice following macrophage depletion by clodronate liposomes (lip) or treatment with PBS liposomes (n = 10). (G) Frequency analysis of circulating platelet size 72 hours after treatment with clodronate liposomes or PBS liposomes (mean distributions of 5-6 mice per group). Blue bars/lines, control; red bars/lines, Arpc2fl/flPF4-Cre. Data presented as mean ± standard deviation. *P < .05, ***P < .001. FSC-H, forward scatter height.
Deletion of Arp2/3 results in cytoskeletal changes in platelets and megakaryocytes
Arp2/3 is best known for its role in lamellipodia formation. Consistently, we observed a marked defect in platelet spreading in Arpc2fl/flPF4-Cre mice (Figure 5A-C). Compared with controls, Arpc2fl/flPF4-Cre platelets activated with Par4p, the GPVI agonist Cvx, or adenosine 5′-diphosphate, exhibited a marked defect in lamellipodia formation. A similar defect in spreading was also observed in cultured megakaryocytes isolated from Arpc2fl/flPF4-Cre mice (supplemental Figure 2B). F-actin and G-actin were quantified by flow cytometry (Figure 5D) in resting platelets and upon stimulation with Par4p and Cvx. Consistent with its role in actin nucleation, the amount of F-actin (Figure 5DI) and the ratio of F-actin to G-actin (Figure 5DIII) were significantly reduced in activated Arpc2fl/flPF4-Cre platelets, even when the reduced size of these cells was taken into account. G-actin levels where decreased only in resting Arpc2fl/flPF4-Cre platelets (Figure 5DII). Interestingly, resting Arpc2fl/flPF4-Cre platelets also exhibited marked alterations in the peripheral tubulin ring. As shown by transmission electron (Figure 5E) and immunofluorescence microscopy (Figure 5F), many Arp2/3-deficient platelets exhibited a smaller size, altered morphology, and abnormal tubulin bundles. In addition, ∼0.5% of Arpc2fl/flPF4-Cre platelets exhibited a doughnut-like morphology instead of the characteristic discoid shape (supplemental Figure 4).
Figure 5.

Altered actin dynamics in Arpc2fl/flPF4-Cre platelets. (A) Quantification of the surface area of unstimulated (resting) or stimulated platelets (500 μM Par4p, 750 ng/mL Cvx, or 10 μM adenosine 5′-diphosphate) spread on fibrinogen for 30 minutes (n = 5). (B) Representative immunofluorescent images of Cvx-stimulated platelets spread on a fibrinogen matrix and stained with phalloidin. (C) Representative scanning electron microscopy images of spread platelets on fibrinogen matrix from indicated mice, stimulated with Cvx (750 ng/mL). (D) Quantification of platelet F-actin (I), G-actin (II), and F-actin:G-actin ratio (III) at resting conditions or after stimulation with Par4p (500 μM) or Cvx (1 μg/mL) for 10 minutes before fixation (n = 5). (E) Representative transmission electron microscopy images of resting platelets from indicated mice; boxes indicate location of intracellular tubulin ring bundles. Scale bar, 200 nm. (B) Representative immunofluorescent images of tubulin rings in resting platelets from indicated mice. Blue bars, control; red bars, Arpc2fl/flPF4-Cre. Data presented as mean ± SEM. *P < .05, **P < .01, ***P < .001.
Loss of Arp2/3 has little effect on platelet activation and hemostatic plug formation
We next investigated whether loss of Arp2/3 affects platelet functions other than spreading. Flow cytometry studies using JON/A-PE, a probe that selectively binds to the activated form of αIIbβ3 integrin,27 identified only a mild defect in integrin inside-out activation in Arpc2fl/flPF4-Cre platelets (supplemental Figure 5A). Arpc2fl/flPF4-Cre platelets also showed a mild defect in α-granule secretion (supplemental Figure 5B), but not dense-granule secretion (supplemental Figure 5C). Consistent with the mild integrin activation defect, aggregation of Arpc2fl/flPF4-Cre platelets was impaired only at threshold agonist concentrations (Figure 6A). No significant differences in the expression of platelet agonist and adhesion receptors was observed, with the exception of GPIX, a consequence of its relatively high expression level and the smaller size of Arpc2fl/flPF4-Cre platelets (supplemental Figure 5D). Actin rearrangement downstream of integrin outside-in signaling also plays an important role in the process of platelet-mediated clot retraction, but the contribution of actin branching to this process is unknown. Only a minor, yet significant, delay in clot retraction was observed in Arp2/3-deficient platelets (Figure 6B; supplemental Video 2). Together, these in vitro studies suggested a minor role for Arp2/3 in platelet aggregate formation and clot stability at sites of vascular injury. To test platelet function in vivo, we subjected Arpc2fl/flPF4-Cre mice to the tail clip bleeding (Figure 6C-D) and the FeCl3-induced carotid artery thrombosis assays (Figure 6E). No significant differences in bleeding time/blood loss or time to carotid artery occlusion were observed between Arpc2fl/flPF4-Cre and control mice. We also quantified platelet adhesion during hemostatic plug formation by intravital microscopy of small injuries to the saphenous vein. No significant differences were noted for the kinetics of platelet accumulation or peak fluorescence between control and Arpc2fl/flPF4-cre mice at the site of initial injury or subsequent reinjuries (Figure 6F; supplemental Video 3).
Figure 6.

Mild platelet activation defect but intact hemostasis in Arpc2fl/flPF4-Cre mice. (A) Representative traces showing aggregation of platelet rich plasma stimulated with increasing doses of the indicated agonists. (B) Quantification of total clot area after platelet-mediated retraction (n = 3-5) and representative images of retracted clots. (C-D) Tail clip bleeding assay. Time to cessation of bleeding (C) and amount of blood loss (D) from severed tails of the indicated mice (n = 11-12). (E) Time to occlusion of carotid artery after induction of thrombosis by administration of FeCl3 solution to vessel (n = 8-9). (F) Kinetics of platelet accumulation and peak platelet density at injury site of saphenous vein after laser ablation (8 total injury sites per group; 3 mice per group). (G) Representative images of intradermal bleeding at sites of local inflammation induced by rPA reaction in platelet depleted and nondepleted Arpc2fl/flPF4-Cre mice (n = 3 mice). Blue/black bars/lines, control; red bars/lines, Arpc2fl/flPF4-Cre). Data presented as mean ± SEM. *P < .05.
Given that Arp2/3 contributes very little to integrin activation but plays a key role in platelet spreading, we wondered if Arpc2fl/flPF4-Cre mice would be more susceptible to inflammatory bleeding as seen in thrombocytopenic mice challenged by immune complex–mediated inflammation in the skin (reverse passive Arthus reaction). The work done by us and others demonstrated that, in this model, single platelets engage the extracellular matrix via immune-type receptors to prevent hemorrhage.28,29 Interestingly, compared with platelet-depleted mice, Arpc2fl/flPF4-Cre mice were protected from inflammatory bleeding (Figure 6G). These data suggest that platelet spreading does not play a significant role in the maintenance of vascular integrity at sites of mechanical injury or inflammation.
Discussion
We report here critical new data concerning the contribution of the Arp2/3 complex to peripheral platelet counts and platelet function, based on the first genetic approach to knock down Arp2/3 in megakaryocytes and platelets. The major conclusions of our studies are summarized in Figure 7: (1) Arp2/3 in platelets operates downstream of both Cdc42/WASp and Rac1/WAVE, (2) lack of the Arp2/3 complex in the megakaryocyte lineage leads to microthrombocytopenia resulting from aberrant platelet release into the bone marrow compartment and reduced platelet survival in circulation, and (3) Arp2/3 in platelets and megakaryocytes is required for actin branching and lamellipodia formation, but it seems to be dispensable for platelet adhesion and hemostatic plug formation.
Figure 7.

Schematic representation of the role of Arp2/3 in platelet function and platelet homeostasis.
From work in other cell types, Arp2/3 is known to be inactive until stimulated by NPFs, such as WASp and SCAR/WAVE, which themselves are under the control of the Rho GTPases Cdc42 and Rac1, respectively. Once activated, Arp2/3 is thought to play a critical role in actin branching and Rac1-mediated lamellipodia formation, a finding that was confirmed for platelets in studies using inhibitory antibodies.4 These latter studies also showed a defect in filopodia formation, a process thought to be controlled by Cdc42 signaling, in inhibitor-treated platelets. However, a possible role of the Cdc42-WASp-Arp2/3 axis in platelet filopodia formation is controversial because actin assembly and Arp2/3 activation can occur independent of WASp in platelets,15,16 and platelets lacking Cdc42 efficiently form filopodia.30 Our studies in Arpc2fl/flPF4-Cre mice provide compelling evidence for Arp2/3 to operate downstream of both Cdc42/WASp and Rac1/WAVE, as the phenotype of Arpc2fl/flPF4-Cre mice combines some of the phenotypes described for mice lacking WASp, WAVE1, Cdc42, or Rac1 (Figure 7). Similar to Arpc2fl/flPF4-Cre mice, both Wasp−/− and Cdc42fl/flPF4-Cre mice are characterized by marked thrombocytopenia (∼30% to 50% reduction in platelet count compared with controls), reduced platelet survival, increased numbers of megakaryocytes in bone marrow and spleen, and dysregulated platelet release into the bone marrow compartment.12,13,31 The increase in megakaryocyte numbers in the bone marrow and spleen of Arp2/3-deficient mice is likely caused by the reduced survival of platelets in circulation. As shown in mice treated with platelet-depleting antibodies or neuraminidase, megakaryocyte numbers significantly increase following increased platelet turnover and a drop in peripheral platelet count.32,33 Filopodia formation, however, is not affected in platelets lacking Cdc42, WASp, or Arp2/3.14,31 Thus, these results are consistent with (1) a critical role of the Cdc42-WASp-Arp2/3 signaling axis in platelet homeostasis and (2) previous studies that questioned the contribution of this pathway to platelet filopodia formation and actin remodeling.
We currently do not have a mechanistic explanation for why deficiency in Arp2/3 or WASp leads to smaller platelets in circulation. However, we provide here the first evidence that Arpc2fl/flPF4-Cre platelets are already smaller immediately after they are released from megakaryocytes into the circulation, whereas they are of normal size when they are released into the bone marrow compartment. One explanation for these observations may be that loss of Arp2/3 affects membrane integrity in platelets, causing premature fragmentation of these cells in the microcirculation. The existence of doughnut-shaped platelets in the blood of Arpc2fl/flPF4-Cre mice may support this conclusion. Interestingly, our studies in clodronate-treated animals demonstrate that macrophage clearance mainly affects smaller, morphologically altered platelets. These cells exhibit marked alterations in ultrastructure, including a distortion of the peripheral tubulin ring required to maintain platelets in their resting, discoid shape. No evidence for increased desialylation of platelet surface receptors or the presence of platelet-associated autoantibodies could be found. Altered tubulin structure and platelet morphology have recently been shown for mice lacking profilin-1, an actin-regulating protein that can interact with the WASp/WASp-interacting protein complex.34 Future studies will be required to elucidate how loss of Arp2/3 is linked to altered tubulin structure and if the change in tubulin observed in platelets from mice lacking Arp2/3 or profilin-1 is causative for impaired megakaryocyte development and/or increased clearance of platelets by macrophages.
Consistent with its predicted role downstream of Rac1, Arpc2fl/flPF4-Cre platelets exhibited a marked defect in lamellipodia formation. In megakaryocytes and platelets, WAVE2 is the predominant WAVE/SCAR isoform that could link Rac1 and Arp2/3.17 Unfortunately, the contribution of WAVE2 to platelet function has not been characterized, as germ line knockout mice are embryonic lethal at E = 12.5. Studies in megakaryocytes derived from Wave2−/− embryonic stem cells, however, suggest that the Rac1/WAVE2 pathway is critical for lamellipodia formation and to a lesser degree megakaryocyte development.17 Mice deficient in WAVE1, another WAVE/SCAR family member expressed in platelets, are viable. The PPC in Wave1−/− mice is normal. Knockout platelets show a defect in spreading and integrin activation only when activated via the collagen receptor, GPVI.18 Rac1fl/flPF4-Cre mice also exhibit a marked defect in platelet spreading, but no defects in platelet production and survival.31 Importantly, mice deficient in both Cdc42 and Rac1 exhibit defects in both platelet production/survival and spreading (ie, a phenotype that closely resembles that of Arpc2fl/flPF4-Cre mice). In addition to its role in spreading, Rac1 in stimulated platelets is also critical for the activation of phospholipase C and the generation of second messengers involved in integrin activation.35 It is thus not surprising that platelet activation and hemostatic plug formation are markedly impaired in Rac1fl/flPF4-Cre and Rac1/Cdc42-DKO mice. Consistent with its specific role in actin branching and platelet spreading, deletion of Arp2/3 in platelets does not cause significant defects in mice challenged in experimental models of bleeding, thrombosis, and vascular integrity at sites of inflammation. We acknowledge that the residual expression of Arp2/3 in platelets from our novel conditional knockout mice needs to be taken into account when interpreting our in vivo data. Based on western blotting studies, conditional deletion of Arpc2 in megakaryocytes leads to >95% reduction in Arp2/3 expression in circulating platelets. This marked reduction in Arp2/3 expression, however, has only a very mild effect on platelet integrin activation in vitro and hemostatic plug formation in vivo. In contrast, platelet adhesion, hemostasis, and thrombosis were significantly impaired in mice with similar reductions in CalDAG-GEFI36 or Kindlin-3,37 highly expressed proteins critical to platelet adhesion and thrombus formation. Thus, the Arp2/3 complex does not seem to be a major regulator of hemostatic plug formation at sites of vascular damage. We also did not observe bleeding at sites of inflammation in Arp2/3 mutant mice. Inflammatory bleeding in mice is observed when platelet counts drop to less than 10% of normal38 or when platelet immunoreceptor tyrosine-based activation motif signaling is impaired.28 Vascular integrity at sites of inflammation depends on the adhesion of single platelets to gaps in the endothelial lining,29 but the molecular mechanisms underlying this response are not well defined. Given this lack of mechanistic understanding and the residual Arp2/3 expression observed in platelets from Arpc2fl/flPF4-Cre mice, we cannot fully exclude the possibility that platelet Arp2/3 signaling contributes to vascular integrity in inflammation.
In summary, our studies in novel Arpc2fl/flPF4-Cre mice demonstrate that the Arp2/3 complex is important for platelet and megakaryocyte function. More specifically, we show that Arpc2fl/flPF4-Cre megakaryocytes and platelets exhibit an expected spreading defect, whereas other platelet responses critical for hemostatic plug formation were only minimally affected. Arpc2fl/flPF4-Cre platelets show marked structural changes that may contribute to their premature clearance by macrophages. The combined phenotype of microthrombocytopenia but largely intact hemostasis strongly resembles that of mice and humans with defective WASp function. Importantly, while this work was under review, the first patients with mutations in the Arpc1B subunit of the Arp2/3 complex were reported; in agreement with our murine studies, the clinical phenotype of these patients includes a marked microthrombocytopenia, impaired platelet spreading, and a largely intact hemostatic response.39
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Adarsh Rao for help quantifying megakaryocytes in hematoxylin and eosin–stained sections.
This work was supported by a Bridge Grant from the American Society of Hematology (W.B.), European Hematology Association Research Fellowship (C.C.), and the National Institutes of Health, National Heart, Lung, and Blood Institute (grants R01 HL121650 and P01 HL120846 [W.B.] and R01HL68130 [J.E.I.]), National Institute of Diabetes and Digestive and Kidney Diseases (grant K01DK111515) (K.R.M.), and National Institute of General Medical Sciences (grant R01 GM111557) (J.E.B.). K.R.M. and J.E.I. are American Society of Hematology Scholars.
Authorship
Contribution: D.S.P. and C.C. performed most of the experiments, analyzed data, and wrote the manuscript; J.E.B., J.D.R., and C.W. generated Arpc2fl/fl mice, performed western blotting studies, and helped with macrophage phagocytosis studies, platelet and megakaryocyte imaging, and the writing of the manuscript; R.P. performed hemostasis assays and analyzed data; S.P. performed platelet spreading assays; R.A.C. and A.S.W. performed proplatelet formation and analyzed data; K.O.P. assisted with experiments and was responsible for mouse colony maintenance; D.G. performed megakaryocyte function studies and imaging; R.H.L. performed and analyzed reverse passive Arthus reaction studies; B.C.C. performed and analyzed FeCl3 carotid artery thrombosis studies; K.R.M and J.E.I. performed and analyzed transmission electron microscopy experiments and helped write and edit the manuscript; and W.B. designed studies, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: J.E.I. has financial interest in and is a founder of Platelet BioGenesis, a company that aims to produce donor-independent human platelets from human-induced pluripotent stem cells at scale. J.E.I. is an inventor on this patent. The interests of J.E.I. were reviewed and are managed by the Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict-of-interest policies. The remaining authors declare no competing financial interests..
Correspondence: Wolfgang Bergmeier, University of North Carolina, 120 Mason Farm Rd, Campus Box 7260, Chapel Hill, NC 27599; e-mail: bergmeie@email.unc.edu.
References
- 1.Rotty JD, Wu C, Bear JE. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat Rev Mol Cell Biol. 2013;14(1):7-12. [DOI] [PubMed] [Google Scholar]
- 2.King SJ, Asokan SB, Haynes EM, et al. . Lamellipodia are critical for haptotactic sensing and response. J Cell Sci. 2016;129(12):2329-2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu C, Asokan SB, Berginski ME, et al. . Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell. 2012;148(5):973-987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Z, Kim ES, Bearer EL. Arp2/3 complex is required for actin polymerization during platelet shape change. Blood. 2002;99(12):4466-4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falet H, Hoffmeister KM, Neujahr R, et al. . Importance of free actin filament barbed ends for Arp2/3 complex function in platelets and fibroblasts. Proc Natl Acad Sci USA. 2002;99(26):16782-16787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Machlus KR, Italiano JE Jr. The incredible journey: from megakaryocyte development to platelet formation. J Cell Biol. 2013;201(6):785-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poulter NS, Thomas SG. Cytoskeletal regulation of platelet formation: coordination of F-actin and microtubules. Int J Biochem Cell Biol. 2015;66:69-74. [DOI] [PubMed] [Google Scholar]
- 8.Aslan JE, McCarty OJT. Rho GTPases in platelet function. J Thromb Haemost. 2013;11(1):35-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr. 1994;125(6 Pt 1):876-885. [DOI] [PubMed] [Google Scholar]
- 10.Oda A, Ochs HD. Wiskott-Aldrich syndrome protein and platelets. Immunol Rev. 2000;178(1):111-117. [DOI] [PubMed] [Google Scholar]
- 11.Snapper SB, Rosen FS, Mizoguchi E, et al. . Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity. 1998;9(1):81-91. [DOI] [PubMed] [Google Scholar]
- 12.Prislovsky A, Marathe B, Hosni A, et al. . Rapid platelet turnover in WASP(-) mice correlates with increased ex vivo phagocytosis of opsonized WASP(-) platelets. Exp Hematol. 2008;36(5):609-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sabri S, Foudi A, Boukour S, et al. . Deficiency in the Wiskott-Aldrich protein induces premature proplatelet formation and platelet production in the bone marrow compartment. Blood. 2006;108(1):134-140. [DOI] [PubMed] [Google Scholar]
- 14.Shcherbina A, Cooley J, Lutskiy MI, Benarafa C, Gilbert GE, Remold-O’Donnell E. WASP plays a novel role in regulating platelet responses dependent on alphaIIbbeta3 integrin outside-in signalling. Br J Haematol. 2010;148(3):416-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Falet H, Hoffmeister KM, Neujahr R, Hartwig JH. Normal Arp2/3 complex activation in platelets lacking WASp. Blood. 2002;100(6):2113-2122. [PubMed] [Google Scholar]
- 16.Gross BS, Wilde JI, Quek L, Chapel H, Nelson DL, Watson SP. Regulation and function of WASp in platelets by the collagen receptor, glycoprotein VI. Blood. 1999;94(12):4166-4176. [PubMed] [Google Scholar]
- 17.Eto K, Nishikii H, Ogaeri T, et al. . The WAVE2/Abi1 complex differentially regulates megakaryocyte development and spreading: implications for platelet biogenesis and spreading machinery. Blood. 2007;110(10):3637-3647. [DOI] [PubMed] [Google Scholar]
- 18.Calaminus SDJ, McCarty OJT, Auger JM, et al. . A major role for Scar/WAVE-1 downstream of GPVI in platelets. J Thromb Haemost. 2007;5(3):535-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yae K, Keng VW, Koike M, et al. . Sleeping beauty transposon-based phenotypic analysis of mice: lack of Arpc3 results in defective trophoblast outgrowth. Mol Cell Biol. 2006;26(16):6185-6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rotty JD, Wu C, Haynes EM, et al. . Profilin-1 serves as a gatekeeper for actin assembly by Arp2/3-dependent and -independent pathways. Dev Cell. 2015;32(1):54-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Romijn RA, Westein E, Bouma B, et al. . Mapping the collagen-binding site in the von Willebrand factor-A3 domain. J Biol Chem. 2003;278(17):15035-15039. [DOI] [PubMed] [Google Scholar]
- 22.Getz TM, Piatt R, Petrich BG, Monroe D, Mackman N, Bergmeier W. Novel mouse hemostasis model for real-time determination of bleeding time and hemostatic plug composition. J Thromb Haemost. 2015;13(3):417-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoffmeister KM. Glycosylation restores survival of chilled blood platelets. Science. 2003;301(5639):1531-1534. [DOI] [PubMed]
- 24.Ross GD. Regulation of the adhesion versus cytotoxic functions of the Mac-1/CR3/alphaMbeta2-integrin glycoprotein. Crit Rev Immunol. 2000;20(3):197-222. [PubMed] [Google Scholar]
- 25.Alves-Rosa F, Stanganelli C, Cabrera J, van Rooijen N, Palermo MS, Isturiz MA. Treatment with liposome-encapsulated clodronate as a new strategic approach in the management of immune thrombocytopenic purpura in a mouse model. Blood. 2000;96(8):2834-2840. [PubMed] [Google Scholar]
- 26.Casari C, Du V, Wu Y-P, et al. . Accelerated uptake of VWF/platelet complexes in macrophages contributes to VWD type 2B-associated thrombocytopenia. Blood. 2013;122(16):2893-2902. [DOI] [PubMed] [Google Scholar]
- 27.Bergmeier W, Schulte V, Brockhoff G, Bier U, Zirngibl H, Nieswandt B. Flow cytometric detection of activated mouse integrin alphaIIbbeta3 with a novel monoclonal antibody. Cytometry. 2002;48(2):80-86. [DOI] [PubMed] [Google Scholar]
- 28.Boulaftali Y, Hess PR, Getz TM, et al. . Platelet ITAM signaling is critical for vascular integrity in inflammation. J Clin Invest. 2013;123(2):908-916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gros A, Syvannarath V, Lamrani L, et al. . Single platelets seal neutrophil-induced vascular breaches via GPVI during immune-complex-mediated inflammation in mice. Blood. 2015;126(8):1017-1026. [DOI] [PubMed] [Google Scholar]
- 30.Pleines I, Eckly A, Elvers M, et al. . Multiple alterations of platelet functions dominated by increased secretion in mice lacking Cdc42 in platelets. Blood. 2010;115(16):3364-3373. [DOI] [PubMed] [Google Scholar]
- 31.Pleines I, Dütting S, Cherpokova D, et al. . Defective tubulin organization and proplatelet formation in murine megakaryocytes lacking Rac1 and Cdc42. Blood. 2013;122(18):3178-3187. [DOI] [PubMed] [Google Scholar]
- 32.Stenberg PE, Levin J, Baker G, Mok Y, Corash L. Neuraminidase-induced thrombocytopenia in mice: effects on thrombopoiesis. J Cell Physiol. 1991;147(1):7-16. [DOI] [PubMed] [Google Scholar]
- 33.Stenberg PE, Levin J, Corash L. Sustained thrombocytopenia in mice: serial studies of megakaryocytes and platelets. Exp Hematol. 1990;18(2):124-132. [PubMed] [Google Scholar]
- 34.Bender M, Stritt S, Nurden P, et al. . Megakaryocyte-specific Profilin1-deficiency alters microtubule stability and causes a Wiskott-Aldrich syndrome-like platelet defect [published correction appears in Nat Commun. 2015;6:6507]. Nat Commun. 2014;5:4746. [DOI] [PubMed] [Google Scholar]
- 35.Pleines I, Elvers M, Strehl A, et al. . Rac1 is essential for phospholipase C-gamma2 activation in platelets. Pflugers Arch. 2009;457(5):1173-1185. [DOI] [PubMed] [Google Scholar]
- 36.Piatt R, Paul DS, Lee RH, et al. . Mice expressing low levels of CalDAG-GEFI exhibit markedly impaired platelet activation with minor impact on hemostasis. Arterioscler Thromb Vasc Biol. 2016;36(9):1838-1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klapproth S, Moretti FA, Zeiler M, et al. . Minimal amounts of kindlin-3 suffice for basal platelet and leukocyte functions in mice. Blood. 2015;126(24):2592-2600. [DOI] [PubMed] [Google Scholar]
- 38.Goerge T, Ho-Tin-Noe B, Carbo C, et al. . Inflammation induces hemorrhage in thrombocytopenia. Blood. 2008;111(10):4958-4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kahr WHA, Pluthero FG, Elkadri A, et al. . Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease. Nat Commun. 2017;8:14816. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.