

Abstract
Aging-related diseases show a marked sex bias. For example, women live longer than men yet have more Alzheimer’s disease and osteoporosis, whereas men have more cancer and Parkinson’s disease. Understanding the role of sex will be important in designing interventions and in understanding basic aging mechanisms. Aging also shows sex differences in model organisms. Dietary restriction (DR), reduced insulin/IGF1-like signaling (IIS) and reduced TOR signaling each increase life span preferentially in females, in both flies and mice. Maternal transmission of mitochondria to offspring may lead to greater control over mitochondrial functions in females, including greater life span and a larger response to diet. Consistent with this idea, males show greater loss of mitochondrial gene expression with age.
Keywords: sex, aging, X chromosome, dosage compensation, mitochondria, Strehler-Mildvan
Sex is a critical factor in regulation of gene expression and life span across species
Women consistently exhibit greater longevity than men, making sex (see Glossary)[1] one of the strongest genetic predictors of human life span. Aging-related diseases also show a marked sex bias, for example, men have greater cardiovascular disease, cancer, Parkinson’s disease, and stroke, while women have greater Alzheimer’s disease, autoimmune disease, and osteoporosis [2]. Here the mechanisms for sex differences in gene expression are explored for mammals and model organisms, with the goal of relating sex differences in gene expression to sex differences in life span and aging phenotypes. Understanding mechanisms for sex-specific gene expression will be important in designing effective aging interventions, and may be important for understanding the basic mechanisms of aging.
Sexual differentiation, including dosage compensation, shows partial conservation of mechanisms across species (Text Box 1), including Drosophila (Text Box 2), C. elegans (Text Box 3) and mammals (Text Box 4) (Figure 1)[3, 4]. In each species, sexual differentiation and dosage compensation result in cell-autonomous chromatin structures and gene expression patterns that require active maintenance in the adult. In addition, sex-specific hormones, including steroid-related hormones, regulate developmental transitions, further sexual differentiation, reproduction, and life span.
Box 1. Common features of sexual differentiation in Drosophila, C. elegans and mammals.
Sexual differentiation, including dosage compensation, can be divided into several common steps: (1) a chromosomal counting mechanism involving sequence elements that mark the X chromosomes and autosomes, (2) activation of a binary switch gene in females and not in males, (3) targeting of transcriptional regulatory factors to the X chromosomes through mechanisms involving X-linked sequence elements, (4) up-regulation of X chromosome gene expression in males, or in both males and females, depending on the species [5], (5) down-regulation of X chromosome gene expression specifically in females. The sometimes opposing modes for regulation of X chromosome gene expression in females are thought to yield a net dosage compensation relative to males. At the same time, certain genes can escape dosage compensation mechanisms in the female to varying degrees, leading to greater expression of such X-linked genes in females relative to males. (6) Promotion of male sexual differentiation by Y-linked gene(s) for species where Y is present, (7) regulation of transcriptional and signaling cascades to yield cell-autonomous sexual differentiation, including a conserved doublesex-like gene important for specification of male cell fate in each species, and (8) production of sex-specific, steroid-related hormones that further regulate sexual differentiation (non-cell-autonomous mechanisms).
Box 2. Drosophila sexual differentiation and dosage compensation.
In Drosophila, the male is X/Y and the female is X/X. Early in embryogenesis the X/A ratio is counted by a mechanism involving multiple X chromosome-specific sequence elements. The X/A ratio regulates the on/off state of a binary switch gene Sxl, so that Sxl is on in females and off in males (Figure 1). In females, Sxl prevents the formation of the male-specific-lethal (MSL) transcriptional regulatory complex by repressing expression of the critical MSL subunit MSL-2; this mechanism thereby reduces X chromosome gene expression in females. In males, msl-2 is expressed, and the resultant MSL complex is targeted to the X chromosome via unique X-linked sequence elements. The MSL mediates dosage compensation in males through mechanisms that are not yet entirely clear, but that may include up-regulation of X chromosome gene expression as well as down-regulation of autosomal gene expression by titration of transcription factors away from the autosomes [4, 5]. Sxl also regulates most of the rest of sexual differentiation. In females, Sxl regulates the alternative splicing of the transformer (tra) gene, so that tra is on and is expressed in females and is off in males. Tra in turn regulates alternative splicing of transcription factor genes fruitless (fru) and doublesex (dsx), such that a female-specific isoform of each is expressed in females. In males, fru and dsx undergo a default splicing pattern to produce male-specific isoforms. The sex-specific Fru and Dsx transcription factors then regulate a large part of sex-specific structural, functional, and behavioral differentiation. Tra also regulates female-specific gene expression and differentiation through mechanisms that do not involve regulation of alternative splicing. For example, Tra-ON regulates the expression of a female-specific protein isoform of the mitochondrial protease Lon [6]. Finally, Sxl regulates sex-specific expression of additional targets including Notch. The resultant sex-specific autonomous cell differentiation then produces sex-specific hormones, including the steroid ecdysone, which further regulates sexual differentiation, reproduction, metabolism, and life span. Tra continues to be required to maintain sexual identify of cells in the adult, as over-expression or knockdown of Tra expression in the adult is sufficient to transform cells of the gut, fat-body, and other tissues to the opposite sex [6, 7]. In Drosophila, Y chromosome linked genes are required for normal spermatogenesis in the male.
Box 3. Mammalian sexual differentiation and dosage compensation.
In mammals, the male is X/Y and the female is X/X. During embryogenesis, a chromosomal counting mechanism regulates the on/off state of the X-linked binary switch gene Xist, such that Xist is in the on state and is expressed from only one of the two X chromosomes in females, and is off in males (Figure 1). Xist encodes a lncRNA that recruits multiple transcriptional regulatory factors into complexes that coat the X chromosome, resulting in cis-inactivation of the expression of most (but not all) genes on that X chromosome to regulate dosage compensation. Mammals also show evidence of up-regulation of X chromosome gene expression to regulate dosage compensation, with relatively greater X gene expression observed in brain tissue [5]. Also during embryogenesis the key sexual differentiation genes Dmrt1 (doublesex and mab-3 related transcription factor 1) and Foxl2 (forkhead box L2) function in an antagonistic manner to produce a bi-potential gonad [8]. In the absence of further regulatory input in females this will result in differentiation of a female gonad. In males, the Y-linked gene Sry activates the sexual differentiation gene Sox-9 which tilts the balance towards male development by repressing the ovary-promoting genes including Foxl2, and activating testis-promoting genes including Dmrt1. The cell autonomous differentiation of the male gonad cells then produces sex-specific hormones FGF9, PDG2, PDGF and DHH which promote further proliferation and differentiation of male germ cells and steroidogenic cells in the gonad. Finally, the male gonad produces sex-specific steroid hormones including testosterone and the female gonad produces sex-specific steroid hormones including estrogen that further regulate sexual differentiation and reproduction. Studies in mice reveal that the balance between Dmrt1 and Foxl2 activities continues to be required in the adult to maintain sexual identity of gonadal cells, as inhibition of either gene in the adult can result in trans-differentiation to the other sex [8]. In mammals Y-linked genes in addition to Sry are required for normal spermatogenesis.
Box 4. C. elegans sexual differentiation and dosage compensation.
In C. elegans the male is X/0 (one copy of the X chromosome) and the hermaphrodite is X/X (two copies of the X chromosome). The X/A ratio is counted by a mechanism involving multiple X chromosome-specific and autosome-specific sequence elements [9]. The X/A ratio regulates the on/off state of a binary switch gene Xol-1, so that Xol-1 is off in females and on in males (Figure 1). Xol-1 in turn regulates the on/off state of a second binary switch gene called Sdc-2, such that Sdc-2 is on in females and off in males. SDC-2 acts with zinc-finger transcription factors SDC-1 and SDC-3 to inhibit expression of sexual differentiation gene her-1. HER-1 otherwise promotes male cell differentiation by regulating a transcriptional regulatory cascade including the conserved doublesex-like gene Mab-3. Dosage compensation in C. elegans is proposed to proceed through up-regulation of X chromosome gene expression relative to the autosomes in both males and hermaphrodites, combined with down-regulation of X chromosome gene expression specifically in the hermaphrodite. The possible mechanisms for X chromosome up-regulation in both males and hermaphrodites are not well understood. In contrast, the mechanisms for X chromosome down-regulation in hermaphrodites is well studied, and is mediated by the targeting of the Dosage Compensation Complex (DCC) to the X chromosomes to mediate chromosome compaction and transcriptional down-regulation. The on/off state of Sdc-2 controls the activity of the DCC by encoding a key subunit. The DCC is composed of a chromosome condensin complex, as well DPY-30 which is also a component of the MLL/COMPASS histone methyltransferase complex, SDC-2 and SDC-3 which function in targeting the complex to the X chromosome, and DPY-21 which regulates growth and metabolism downstream of TOR signaling. The steroid-related hormone dafachronic acid and its receptor DAF-12 regulate developmental transitions and adult life span [10].
Figure 1. Outline of sex determination and dosage compensation pathways.

The sex determination and dosage compensation pathways are outlined for Drosophila, human and C. elegans, with an emphasis on aspects that are similar between the species. Large ovals indicate X chromosomes, small ovals indicate Y chromosomes. Arrows indicate activation, T-bars indicate repression. Dashed arrows and T-bars indicate activation or repression of X chromosome gene expression by the respective dosage compensation machineries. The conserved doublesex-related gene is indicated in red font, and is involved in specification of male cell differentiation in each species. Please note that “Tra” in Drosophila is not homologous to “Tra” in C. elegans. Please see text for further details.
Sex gene dosage and aging
In addition to being the determining factor for sexual identity of the animal, the sex chromosome composition has many potential consequences for gene expression during aging and the regulation of life span. For example, the presence of a single X chromosome in males of Drosophila, C. elegans and mammals means that any X-linked recessive mutant phenotype will be expressed in the male (the “unprotected X”), whereas in females the second X chromosome may contribute a wild-type copy of the gene, and therefore the recessive phenotype will not be expressed. This may be one contributing factor for the greater life span of females relative to males observed for certain species and genotypes [11]. Consistent with this idea, bird species utilize the ZW sex determination system (males ZZ and females ZW), and males tend to live longer than females. In contrast, in C. elegans, males are generally reported to be longer-lived than the hermaphrodite [10]; this fact does not rule out an important role for the unprotected X, but does indicate that it is not the main determinant for sex differences in life span in C. elegans.
Another implication of sex chromosome dosage for gene expression and aging involves incomplete dosage compensation. In both mouse and human a significant number of X-linked genes “escape” from X chromosome inactivation, thereby increasing the expression level for these genes in females relative to males. The degree of escape varies depending on the tissue, developmental stage and age [12]. Escape from X inactivation can have both beneficial and deleterious consequences. For example, escape from X inactivation of two critical immune regulatory genes, CD40LG and OGT, is implicated in mediating the observed immune advantage of women relative to men, but may also play a role in the greater susceptibility of women to autoimmune disease [13]. Escape from X inactivation of genes involved in stress responses, such as G6PD and XIAP, has been implicated in mediating the generally greater stress resistance of female cells relative to male cells [2], and greater X-linked gene expression may be one mechanism for greater female control over mitochondrial functions (discussed further below). However, escape from X inactivation is also implicated in diseases, including mental disorders and sex-biased cancers. Recently the inactive X chromosome in human females has been shown to be subject to hyper-mutation in cancers, apparently due to replication stress imposed by its unique heterochromatic structure and late S-phase replication timing in aberrantly proliferating cells [14]; escape from X inactivation of these mutated X-linked genes may contribute to the progression of female-biased cancers.
Finally, mammalian X chromosome inactivation is essentially random with regard to inactivation of the maternal versus paternal X chromosome in the developing female somatic tissues. This leads to a genetic mosaic where X linked mutations are expressed in some cells/tissues and not others.
Uni-parental mitochondrial inheritance
In the majority of eukaryotic species, the mitochondria (and their endogenous mitochondrial genomes) are transmitted to offspring preferentially from one parent. In heterogametic species (with egg and sperm) the mitochondria are transmitted preferentially through the egg. This situation has several implications for sex-specific gene expression and life span regulation. The ubiquity of this phenomenon across species indicates that it is a result of strong selective pressure, and several non-exclusive hypotheses have been put forward to explain why uniparental mitochondrial transmission is selectively advantageous [2]. One possibility is that this limits the horizontal spread of deleterious mitochondrial genes and cytoplasmic parasites in the population, because such elements can only be transmitted vertically from one parent to offspring. A second possibility is that this helps avoid genetic conflicts between different mitochondrial alleles in the zygote. A third possibility is that this avoids damage to the mitochondrial genome that might be greater in the more metabolically active sperm. Finally, it is possible that uniparental mitochondrial transmission may help drive evolution, including evolution of the sexes [15]. In addition to these generally beneficial effects, uniparental mitochondrial transmission may also have negative effects that contribute to, or create, the aging phenotype. Because the mitochondrial genes are only being transmitted from the mother, natural selection can only act to optimize mitochondrial gene alleles and nuclear-mitochondrial genetic interactions for function in females. This is expected to lead to enhanced control over mitochondrial functions in females relative to males, and may be one reason for the greater stress resistance and life span often observed for females. However, these female-optimized mitochondrial alleles may function non-optimally in males, thereby contributing to disease in males (the Frank and Hurst hypothesis), a situation sometimes referred to as “mother’s curse”. Consistent with these ideas, mitochondrial genome mutations often cause greater disease in men than in women, and recent studies in Drosophila report mitochondrial genome alleles that contribute to decreased life span preferentially in males [16].
Because natural selection cannot act to optimize mitochondrial gene function in males, the expectation is that natural selection will act on nuclear gene alleles to select for alleles that can function in males to compensate for the less-than-optimal mitochondrial gene alleles. These male-optimized nuclear gene alleles will then be inherited in the next generation by females where they may function non-optimally in the female genetic background, thereby promoting further female-specific selection on both nuclear and mitochondrial alleles. These conflicting selective pressures in male and female may therefore maintain gene alleles in the population that are relatively beneficial to one sex and relatively deleterious to the other sex, or even deleterious in different ways to both sexes, i.e., gene alleles exhibiting sexual antagonistic pleiotropy (SAP). These alleles are expected to have deleterious effects centered around mitochondrial functions, and thus may contribute to the mitochondrial maintenance failure that is observed in aging across species [2, 17]. The potential benefit of this ongoing selection for genetic diversity is that it may promote the evolution of the sexes and promote evolution in general [15].
In summary, the genetic forces resulting from uni-parental mitochondrial transmission are expected to maintain deleterious nuclear gene alleles in the population that contribute to mitochondrial maintenance failure and aging in both males and females. At the same time, these forces are expected to lead to relatively greater control over mitochondrial maintenance and function in females relative to males. Recent observations are consistent with the idea of greater female control over mitochondrial functions. Stress adaptation is the ability of a mild stress challenge to make cells or animals more resistant to subsequent, more toxic levels of that stress [18]. It has recently been demonstrated that female Drosophila, but not male Drosophila, are able to adapt to hydrogen peroxide stress [6], and this ability correlated with female-specific expression of protein isoforms of the conserved mitochondrial Lon protease [6]. It is possible that this observation may extend to mammalian cells, as adaptation has been documented for female cells, but results for male cells remain unclear [18]. In mammals, female cells are more resistant to oxidative stress than are male cells, consistent with the greater resistance of women to ischemic heart disease and ischemic stroke relative to men. In the future, it will be important to conduct side-by-side comparisons of male and female mammalian cells and tissues for ability to adapt to oxidative stress, including the possible role of Lon.
Sex-specific selective pressures
In addition to the sex-specific selective pressures resulting from uniparental mitochondrial transmission discussed above, there are additional sex-specific selective pressures that can lead to SAP. For example, different microbial flora composition or sex-specific selective pressures such as childbirth. In addition, the sexes will exert different selective pressures on each other, through sexual selection, for many traits including behaviors and morphology. Opposing sex-specific selective pressures acting on the same gene are sometimes referred to as “intra-locus sexual conflict” or “sexual antagonism” [11]. These sex-specific selective pressures are also expected to maintain alleles in the population that function optimally in one sex and suboptimally in the other sex, or that function sub-optimally in both sexes, i.e., alleles exhibiting SAP, and these alleles are expected to contribute to aging and life span phenotypes [2, 11, 15].
Common themes for gene expression changes during aging
Genome-wide analyses indicate that the patterns of gene expression change during aging include many features that are specific to the species and/or the specific tissue [19]. However, four themes have emerged that are common to several species. These four themes are consistent with a failure in mitochondrial maintenance such as might result from the genetic forces discussed above [2, 20]. The first theme is a down-regulation of genes encoding the structural and functional components of the mitochondria. The down-regulation of mitochondrial genes is consistent with decreased mitochondrial turnover, which in turn is expected to result in longer-lived and more damage-prone mitochondria. The second theme is an up-regulation of the innate immune response (inflammation), including antimicrobial peptide genes and additional targets of the conserved NF-κB signaling pathway [21]. The mitochondria may favor this response by releasing pro-inflammatory molecules including mitochondrial DNA fragments, formyl-peptides, and increased ROS. The third theme is an oxidative stress response, often including GST and cytochrome p450 genes, consistent with increased mitochondrial production of ROS [20, 21]. Finally, the fourth theme is a proteotoxicity response, characterized by up-regulation of basal heat shock protein gene expression, including HSPs targeted to the cytoplasm and mitochondria [22, 23]. The decreased production of ATP by abnormal mitochondria is expected to lead to decreased protein turnover and therefore longer-lived and more damage-prone proteins, and this damage will be exacerbated by the increased production of ROS.
Sex-specific changes in gene expression during aging
Several tissues have been examined for changes in gene expression during aging with a comparison between the sexes. The mammalian liver is a highly sex-specific tissue and one of the first for which gene expression changes during aging were characterized. Genome-wide analysis of gene expression in rat liver across adult aging time points revealed up-regulation of inflammatory markers including acute phase response, complement system, immune response, xenobiotic metabolism, oxidative stress response and cell death signaling. Dynamic sex-specific changes were observed for genes of Phase I xenobiotic metabolism, circadian rhythm regulation and energy metabolism [24]. Male patterns diverged from female during adulthood followed by convergence at late ages, consistent with an observed “feminization” of male rat and mouse liver gene expression patterns at late age, that may correlate with age-related reductions in steroid metabolism [25]. Interestingly, the sex differences in expression of liver steroid hormone metabolism genes and xenobiotic detoxification genes is reduced or eliminated in several long-lived mouse models, including the Ames dwarf mouse and growth hormone receptor mutant mice, consistent with a role for liver steroid hormone metabolism in limiting normal life span [2, 26].
Analysis of gene expression in mouse brain confirms down-regulation of mitochondrial metabolism genes and upregulation of immune response genes as a common feature of late aging across brain regions [27]. Comparison of transcriptomes of human male and female brain aging revealed relatively greater down-regulation of mitochondrial genes in the male, and relatively greater up-regulation of immune response genes in the female [28]. Proteomic analysis of aging in monkey heart tissue revealed greater decrease in mitochondrial metabolism pathways in males relative to females [29]. Similarly, transcriptome analysis of aging rat heart showed greater decrease in mitochondrial genes in males relative to females [30]. Transcriptome analysis of human and mouse heart revealed greater expression of X-linked genes in females relative to males [31].
Taken together these data are generally consistent with the four themes in aging gene expression patterns discussed above. In addition, the observed preferential down-regulation of mitochondrial genes in males relative to females with age supports the idea of better female control over mitochondrial function and maintenance.
Sex-specific life span interventions
Several types of interventions have been reported to increase life span across multiple species. These interventions include reduced Insulin/IGF-like signaling (IIS), dietary restriction (DR), reduction of TOR pathway signaling, and reduction of mitochondrial ETC gene expression [32–36]. Studies in C. elegans suggest that most if not all of these life span extending interventions may require function of the autophagy pathway [37, 38]. Because the autophagy pathway degrades mitochondria (mitophagy) these observations are consistent with the hypothesis that reduced mitochondrial turnover is a common mechanism of aging [2, 39]. Consistent with this conclusion, both reduced IIS and reduced TOR signaling have been shown to favor mitochondrial turnover in C. elegans [35].
A female bias is often reported for the life span increase caused by reduced IIS, DR and reduced TOR pathway signaling, consistent with sex differences in the underlying mechanisms that limit life span. DR preferentially increases median life span in females in both Drosophila and mouse [40, 41]. Inhibition of IIS also preferentially increases female median life span in Drosophila and mouse. For example, in Drosophila, mutation of IIS component Chico preferentially increased median life span in females [42]. In mouse, inhibition of IIS by heterozygous mutation of IGF-1, or mutation of IRS1 was reported to increase median life span preferentially in females [36]. Inhibition of TOR pathway signaling with the drug rapamycin increases life span preferentially in females in both Drosophila and mouse [43, 44]. Similarly, in mice, mutation of ribosomal protein S6 kinase 1 (S6K1), a TOR pathway target, or over-expression of TOR inhibitor TSC1[45] increased median life span in females but not in males.
Male biased life span interventions are also observed. In Drosophila, increased life span caused by heat stress hormesis is generally greater in males than in females [46]. Interestingly, most reports of increased life span upon over-expression of heat shock protein genes in Drosophila are for males, with few if any results reported for females [22]. Also in Drosophila, mutation or chemical inhibition of the anti-apoptotic mitochondrial protein dTSPO increased life span in males but not females [47]. In mice, over-expression of the Sirt6 gene, which is implicated in DNA repair pathways, increases median life span in males but not females [48]. Also in mice, null mutation of the PKA regulatory subunit gene RIIβincreased median and maximum life span in males but not females [49]. Feeding mice the anti-inflammatory drugs aspirin or NDGA, or feeding mice the drugs acarbose or 17-α-estradiol each increased life span preferentially in males [50]. Eight different NSAIDs were analyzed for effects in Drosophila, and of these 6 showed a preferential life span increase in males, whereas two, including aspirin, had greater benefit in females [51]. Whereas no strong consensus emerges for male-biased effects across species, it is possible there is a trend towards greater efficacy of stress response pathway and anti-inflammatory interventions.
Taken together the data support the idea that life span in females is more limited by diet and nutrient-responsive pathways that modulate mitochondrial turnover. This would be consistent with the greater female control over mitochondrial functions and maintenance predicted by the genetic and selective forces discussed above, as well as the generally greater investment of resources into offspring made by females (Figure 2). In contrast, male life span may reflect a more linear trajectory of mitochondrial maintenance failure, and thereby be more limited by chronic stress, including the chronic oxidative, inflammatory and proteotoxic stress associated with mitochondrial maintenance failure (Figure 2).
Figure 2. Model for sex differences in life span regulation.
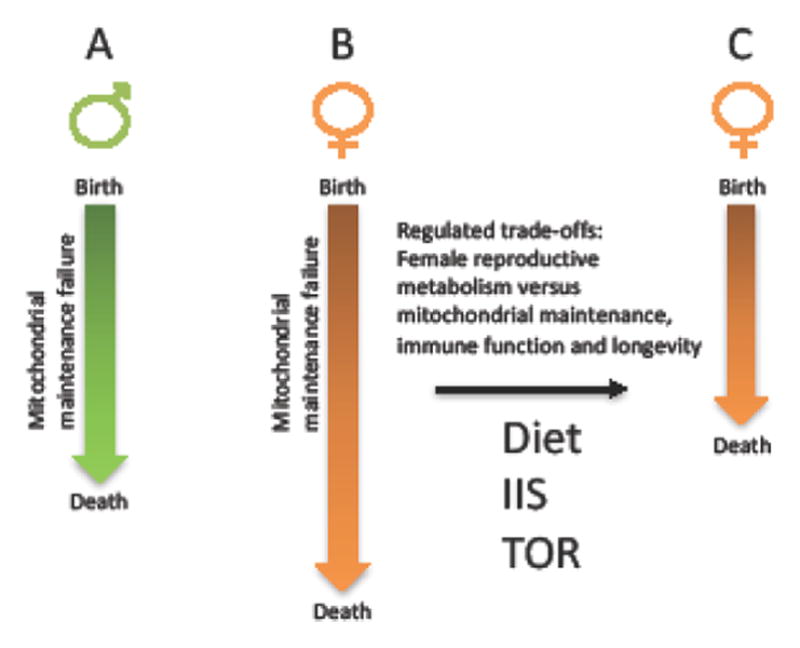
Block arrows indicate life span. A. In males, mitochondrial maintenance failure follows a relatively linear trajectory. B. In females, the “protected X” and the optimized control over mitochondrial function and maintenance allows for a more gradual mitochondrial maintenance failure, and a longer potential life span. C. In females, the greater degree of control over mitochondrial function and turnover allows for more dynamic responses to the environment, including diet and reproductive status. This enables greater magnitude trade-offs between reproductive metabolism versus mitochondrial maintenance and immune function in females relative to males, sometimes resulting in shorter life span in females relative to males. Depending upon where the female physiology falls in the continuum between B and C, the same dietary or genetic intervention might have the same effect in males and females, a female-specific effect, or even an opposite effect in males and females.
Hormone signaling between the sexes can reduce life span
Recently several large-magnitude alterations in median life span have been reported for C. elegans and Drosophila, where hormones produced in one sex act in the other sex to reduce life span. In C. elegans, hormones produced by males have been shown to greatly reduce hermaphrodite life span, through mechanisms that involve activation of IIS pathways in the hermaphrodite [52, 53]. In Drosophila, males produce a seminal peptide hormone called Sex Peptide that is introduced into females upon mating. Recent studies show that Sex Peptide can reduce female life span by over 50% [54]. Strikingly, feeding the steroid hormone antagonist mifepristone to the mated females could block this effect, leading to increases in female life span of over 100%, and thereby implicating steroid hormone signaling in female life span regulation by Sex Peptide. Conversely, in Drosophila a female pheromone is reported to decrease male life span through mechanisms that involve activation of neuropeptide signaling in the male [55]. Because the genes that encode these hormones (and their biosynthetic pathways) act in one sex to reduce life span in the other sex, they are examples of genes exhibiting SAP. Overall these results are consistent with the conclusion that hormonal signaling in general, and steroid hormone signaling in particular, are conserved mechanisms that limit life span across species, most likely through regulation of trade-offs between reproductive metabolism versus mitochondrial and somatic maintenance [2, 56].
Sexual dimorphism in gut function
Gut dysfunction is observed in aging Drosophila, including increased microbial load and increased and aberrant stem cell proliferation [57]. These changes are greater in females relative to males, correlating with the greater food intake and greater contribution of macromolecules to gametes in females. These changes are stimulated by mating in females through a mechanism regulated by male Sex Peptide and the hormone JH [58]. DR delayed gut deterioration in females, and increased life span to a greater extent in females relative to males [7]. Strikingly, transformation of male gut cells to female by forced expression of Tra conferred greater age-related gut deterioration to the male, and increased response to DR and rapamycin. These observations point to sex differences in gut physiology and stem cell proliferation as one determinant of sex differences in life span in Drosophila. Consistent with this idea, antibiotics are reported to increase life span in female Drosophila [54, 57], but not in males [59]. Some studies report a correlation between Drosophila female mortality and a loss of intestinal barrier integrity, as indicated by leakage of dietary dye out of the gut (the “SMURF” assay) [7, 57], however this is not always observed, for example this was not observed with the increased female mortality caused by mating [54]. Gut is also a critical tissue for DR and genetic life span interventions in C. elegans hermaphrodites [60, 61], and increased microbial load with age has been reported [62]; however male/female comparisons are limited. The gut microbiome is increasingly implicated in modulation of human disease, including aging-related diseases. The gut microbiome differs between men and women [63], and women have more functional gastrointestinal disorders than do men [64]. Notably, female mammals exhibit expansion and remodeling of the gut in response to the increased nutritional demands of lactation, similar to the expansion and remodeling of the gut observed in female Drosophila in response to mating, male Sex Peptide, and the nutritional demands of increased egg production. In the future, it will be of interest to determine if sex differences in gut function and microbiome might contribute to sex differences in human life span.
Increased median life span is not always indicative of slower aging
Aging in biological systems is more correctly called “senescence”, however the term “aging” is often used for convenience, as is done here. There is currently no universally accepted definition of aging (senescence), however life span is most often used as the measure of aging. Increased median life span is often interpreted as an indication of a successful intervention in aging. However, traditionally, aging is defined by the exponential increase in mortality rate with age (Gompertz parameter b). Because median life span can be increased by a decrease in the initial mortality rate (Gompertz parameter a) or a decrease in Gompertz parameter b, it is possible for an intervention to increase median life span without decreasing “aging”. DR in Drosophila was found to increase median life span by decreasing Gompertz parameter a [41]. Gompertz parameter b was proportionally increased by the DR intervention, such that maximum life span was not increased [65]. The inverse relationship between Gompertz parameters a and b is called the Strehler-Mildvan relationship, according to its first report [66]. It has been suggested that the Strehler-Mildvan relationship can sometimes result from an artifact of the model-fitting process, when the Gompertz equation is used instead of the Gompertz-Makeham equation [67], and when the magnitude of life span change is small [68]. However, recent reports demonstrate that in Drosophila, numerous interventions that cause large-magnitude life span changes exhibit a robust Strehler-Mildvan relationship when modelled using the Gompertz-Makeham equation, including DR, mating, mifepristone feeding, antibiotic treatment and the effect of male Sex Peptide [54, 65]. The inverse relationship between Gompertz parameters a and b observed using Gompertz-Makeham modelling has also been called the compensation law of mortality [67]. Whereas the biological interpretation of the Strehler-Mildvan relationship remains controversial, one possibility is that it indicates the preferential survival of the most-frail individuals in the cohort in response to the intervention [65]; because these frail individuals are more susceptible to aging, increasing their survival by lowering the initial mortality rate then results in an increased rate of aging for the cohort. These data indicate that DR in Drosophila does not decrease aging rate. Meta-analyses of rodent (mouse and rat, sexes combined) DR interventions have suggested that both parameters a and b are reduced [69, 70], or that parameter b is reduced independent of alterations in parameter a [71]. A meta-analysis of life-span extending mutations (excluding DR interventions) in mice and C. elegans suggested that increased life span in mutant mice was usually associated with decreased parameter a, whereas increased life span in C. elegans was usually associated with decreased parameter b [72].
Notably, genetic and dietary interventions in aging can sometimes have opposite effects on mortality rates and life span in males versus females. In Drosophila, because nutrient optima for median life span differed between males and females, the same alteration in diet had opposite effects on Gompertz mortality rate parameters in males versus females across a range of nutrient concentrations [65]. Similarly, a hypomorphic mTOR mutation in mouse was found to increase female median life span by decreasing parameter a, whereas parameter b tended to be increased, indicating that mTOR mutation does not decrease aging in females [65]. Notably, both p53 mutation in Drosophila and hypomorphic mTOR mutation in mouse tended to have opposite effects on mortality rate parameters in males versus females [65]. In a panel of recombinant inbred mouse strains, the effect of DR was found to increase median life span in some genotypes but decrease median life span in other genotypes, and for certain genotypes the change in life span was in opposite direction in males versus females [73]. Also in mice, a heterozygous mutation of the IGF-1 receptor increased median life span in females, and tended to decrease maximum life span in males [74]. In contrast, a heterozygous mutation of the mouse ETC protein gene Risp decreased median life span in males but tended to increase maximum life span in females [75]. Finally, in C. elegans a high-glucose diet was reported to decrease hermaphrodite life span but slightly increase male life span [76]; and similarly, mutation of the Daf-12 steroid-like hormone receptor was reported to decrease hermaphrodite life span but slightly increase male life span [10]. Taken together these data demonstrate that males and females can sometimes respond in opposite directions to the same dietary or genetic intervention, consistent with sex-specific and sometimes sexually antagonistic regulation of life span. The results underscore the importance of analyzing males and females separately, as well as the utility of Gompertz-Makeham equation modeling to discern effects on health (parameter a) versus aging (parameter b). One possible explanation for these results is that opposite effects in male and female are observed depending upon the signaling state of the female, and the degree to which trade-offs between reproductive metabolism versus mitochondrial maintenance and longevity are already activated in the female by the dietary environment and genetic background (Figure 2).
A unifying model for sex differences in aging involving mitochondrial maintenance failure
Down-regulation of mitochondrial turnover during aging is consistent with a phased increase in mitochondrial abundance and ATP production, as well as increased ROS and accumulation of damaged and abnormal mitochondria [2, 77]. Because ROS (like IIS) promotes cell differentiation [78, 79], including sexual differentiation, one hypothesis is that decreased mitochondrial turnover and increased ROS may be selected for in part due to favorable effects on sexual differentiation and reproduction [2]. For example, when receptor tyrosine kinases involved in cell differentiation are activated by ligand, this activates membrane-bound NADPH oxidase, which in turn results in a localized burst of hydrogen peroxide (Figure 3) [80]. The hydrogen peroxide inactivates protein phosphatases, which would otherwise de-phosphorylate the receptor and downstream signaling factors. In this way hydrogen peroxide normally stimulates growth factor signaling. Because IIS signaling can reduce mitochondrial turnover [81], potentially these (sexual) differentiation pathways will be stimulated by increased mitochondrial hydrogen peroxide production, resulting in positive feedback (Figure 3). Similarly, in Drosophila and C. elegans, increased abundance of denatured proteins, such as would result from mitochondrial maintenance failure, has recently been shown to titrate proteolytic pathway factors resulting in decreased degradation of the insulin-like receptor and increased IIS [82], again consistent with positive feedback effects on IIS.
Figure 3. Model for positive feedback regulation of growth factor signaling, sexual differentiation and aging.
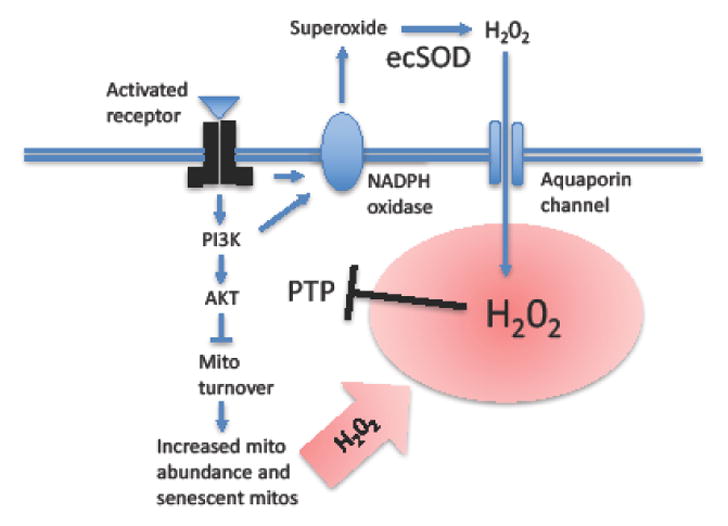
Activation of growth factor receptor by ligand (for example, IIS) activates a signaling cascade involving tyrosine phosphorylation of the receptor and downstream signaling factors. The protein tyrosine phosphatase (PTP) normally limits signaling by removing the phosphate residues from the receptor and downstream signaling pathway factors including AKT. The activated receptor and/or activated PI3K activate nearby membrane-associated NADPH oxidase enzyme. The NADPH oxidase catalyzes the production of extracellular superoxide. Extracellular superoxide dismutase enzyme (ecSOD) then converts the superoxide into hydrogen peroxide (H2O2). The H2O2 enters the cell through the aquaporin water channel, thereby creating a localized burst of hydrogen peroxide near the receptor and downstream signaling factors (indicated with red-color background). The H2O2 reversibly inactivates the PTP, thereby amplifying the magnitude of signaling through the pathway. The signaling pathway down-regulates mitochondrial turnover, thereby creating increased mitochondrial abundance, ATP production and ROS production, including H2O2 (indicated in red) that support growth and sexual differentiation. The mitochondrial H2O2 further contributes to the inactivation of PTP, potentially creating a positive feedback loop. The ultimate consequences of reduced mitochondrial turnover are longer-lived and more damage-prone mitochondria, mitochondrial maintenance failure and aging. Mito = mitochondria.
A potential unifying model for sex differences in life span regulation is that in males mitochondrial maintenance failure follows a more linear trajectory leading to mortality (Figure 2A). In contrast, in females, the “protected X” and the optimized mitochondrial function allows for a more gradual mitochondrial maintenance failure, leading to longer potential life spans in females (Figure 2B). At the same time, the greater control of females over mitochondrial functions and turnover allows for greater response to diet and environment, including mating effects, such that females have the potential for greater trade-offs between reproductive metabolism and life span than do males. Therefore, the greater degree of mitochondrial regulation in the female may sometimes result in female life span that is shorter than that observed for the male, depending on environment and signaling conditions (Figure 2C).
Conclusions and future directions
In the future, it will be important to further investigate the mechanisms for sex-dimorphism in aging and life span regulation, to facilitate possible future sex-specific aging and health interventions. This includes the potential role of dynamic regulation of dosage compensation and X-linked gene expression in females in enabling enhanced control over stress responses and mitochondrial function (see Outstanding Questions).
Outstanding Questions box.
Will the female-specific ability to adapt to hydrogen peroxide stress recently observed in Drosophila be found to extend to mammalian cells? The results could be relevant to understanding the greater resistance of women to ischemic heart disease and ischemic stroke.
What is the biological mechanism for the “Strehler-Mildvan” relationship, where an intervention increases median life span but not maximum life span? Does this indicate the rescue of “frail” or “low vitality” individuals that are also faster-aging?
Do females (two X chromosomes) make use of the dosage compensation machinery to regulate X chromosome gene expression across a greater dynamic range than is possible in males (one X chromosome)? Does this contribute to the greater stress resistance and life span of females?
DR, experimental down-regulation of IIS and experimental down-regulation of TOR each increase life span to a greater extent in females than in males, in both fly and mouse. Is this because females make greater trade-offs between reproductive metabolism and longevity than do males, and these pathways promote those trade-offs?
Do male-biased drug interventions indicate an underlying sex difference in activity of aging regulatory pathway(s)? To what extent might this result from different/less efficient handling of dietary drugs in males leading to greater bioavailability?
Do sex differences in the gut microbiome contribute to sex differences in life span and aging-related disease?
It is worth noting that laboratory studies of longevity do not generally account for (sex specific) selective pressures that may exist in the wild, such as microbiome diversity, competition for mates, fluctuating environments, etc., and thus may be incomplete in some ways. In mouse, the sex chromosome content and presence/absence of male-determining Sry gene have been elegantly manipulated in gonad-intact and gonad-ablated animals. These experiments have revealed effects of gonadal hormones versus other sex chromosome effects, for phenotypes including growth, metabolism, and stroke sensitivity with age [1, 83]. In the future, it will be of interest to use these powerful mouse models to further investigate sex-specific regulation of aging and life span.
Gompertz-Makham modeling of mortality rates provides a useful tool to help distinguish interventions that affect health of the animal versus interventions that alter aging rate. The likely role of sex-specific hormones in regulating trade-offs that promote aging (and reduced health) in both males and females provides promising targets for future sex-specific interventions.
Trends Box.
Sexual differentiation pathways include cell-autonomous mechanisms that continue to be required in the adult to maintain cellular sexual identity.
Maternal-only transmission of mitochondrial genes and additional genetic conflicts between male and female may keep deleterious gene alleles in the population that create the aging phenotype.
Gene expression changes during aging are consistent with mitochondrial maintenance failure: decreased mitochondrial gene expression, inflammation, oxidative stress response and proteotoxicity response.
Maternal-only transmission of mitochondrial genes may result in relatively greater female control over mitochondria, leading to longer life span, greater stress resistance and greater response to diet in females.
Sex-dimorphic hormones, including steroid hormones, may promote aging by regulating tradeoffs between reproductive metabolism versus mitochondrial maintenance.
Acknowledgments
This work was supported by grants from the Department of Health and Human Services to JT (AG011833 and R56AG049629).
Glossary
- Sex
is defined by the sex chromosome complement of the animal. For example, XX female and XY male in Drosophila and humans, XX hermaphrodite and X0 male in C. elegans, and ZZ male and ZW female in birds. In certain species sex is also regulated by environmental factors such as temperature.
- Sexual differentiation
refers to the development and maintenance of all differences between male and female, including gene expression, cellular differentiation, physiology, anatomical structures, behaviors, and life span.
- Sex differences/sex-specific differences
refers to all the differences between male and female resulting from sexual differentiation. Sex differences include endpoints that are unique to the sexes, such as sperm in males and oocytes in females; also called “Type I” sex differences. Sex differences also include endpoints that exist on a continuum where the average is different between male and female, such as greater basal metabolic rate in men compared to women; also referred to as sex-bias or “Type II” sex differences.
- Sexual dimorphism
traditionally refers to morphological differences between males and females. More recently the term has been used to refer to all differences between males and females (sex differences).
- Sex convergence/divergence
refers to a phenotype that is the same in male and female, however the molecular/physiological underpinnings are different in male and female (sex convergence). Alternatively, a sex difference might be expressed only in response to a stress (sex divergence).
- Dosage compensation
is a component of sexual differentiation, and refers to the mechanisms for equalizing gene expression levels between the sexes.
- Sexual antagonistic pleiotropy (SAP)
refers to genes that respond to sex-specific selective pressures (also called intra-locus sexual conflict), resulting in gene alleles that are beneficial to one sex and detrimental to the other sex, or detrimental (in different ways) to both sexes. For example, selection for greater activity of a gene in one sex and selection for reduced activity of that gene in the other sex might lead to maintenance of an allele of intermediate activity that is non-optimal for either sex.
- Gompertz equation parameters a and b
The Gompertz equation describes the survival of a cohort of animals in terms of the initial mortality rate, which is indicative of animal health, including vulnerability to environmental challenges such as pathogens (Gompertz parameter a), and the exponential acceleration in mortality rate with age (senescence) (Gompertz parameter b). The related Gompertz-Makeham equation includes an additional parameter (parameter c) describing age-independent mortality.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McCarthy MM, et al. Sex differences in the brain: the not so inconvenient truth. J Neurosci. 2012;32:2241–2247. doi: 10.1523/JNEUROSCI.5372-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tower J. Mitochondrial maintenance failure in aging and role of sexual dimorphism. Archives of biochemistry and biophysics. 2015;576:17–31. doi: 10.1016/j.abb.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ercan S. Mechanisms of x chromosome dosage compensation. Journal of genomics. 2015;3:1–19. doi: 10.7150/jgen.10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birchler JA. Parallel Universes for Models of X Chromosome Dosage Compensation in Drosophila: A Review. Cytogenet Genome Res. 2016;148:52–67. doi: 10.1159/000445924. [DOI] [PubMed] [Google Scholar]
- 5.Deng X, et al. Evidence for compensatory upregulation of expressed X-linked genes in mammals, Caenorhabditis elegans and Drosophila melanogaster. Nat Genet. 2011;43:1179–1185. doi: 10.1038/ng.948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pomatto LC, et al. The Mitochondrial Lon Protease Is Required for Age-Specific and Sex-Specific Adaptation to Oxidative Stress. Curr Biol. 2017;27:1–15. doi: 10.1016/j.cub.2016.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Regan JC, et al. Sex difference in pathology of the ageing gut mediates the greater response of female lifespan to dietary restriction. eLife. 2016;5:e10956. doi: 10.7554/eLife.10956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang S, et al. Sex determination and maintenance: the role of DMRT1 and FOXL2. Asian journal of andrology. 2017 doi: 10.4103/1008-682X.194420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farboud B, et al. Molecular antagonism between X-chromosome and autosome signals determines nematode sex. Genes Dev. 2013;27:1159–1178. doi: 10.1101/gad.217026.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCulloch D, Gems D. Sex-specific effects of the DAF-12 steroid receptor on aging in Caenorhabditis elegans. Ann N Y Acad Sci. 2007;1119:253–259. doi: 10.1196/annals.1404.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maklakov AA, Lummaa V. Evolution of sex differences in lifespan and aging: causes and constraints. Bioessays. 2013;35:717–724. doi: 10.1002/bies.201300021. [DOI] [PubMed] [Google Scholar]
- 12.Balaton BP, Brown CJ. Escape Artists of the X Chromosome. Trends Genet. 2016;32:348–359. doi: 10.1016/j.tig.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 13.Wang J, et al. Unusual maintenance of X chromosome inactivation predisposes female lymphocytes for increased expression from the inactive X. Proc Natl Acad Sci U S A. 2016;113:E2029–2038. doi: 10.1073/pnas.1520113113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jager N, et al. Hypermutation of the inactive X chromosome is a frequent event in cancer. Cell. 2013;155:567–581. doi: 10.1016/j.cell.2013.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tower J. Sex-specific regulation of aging and apoptosis. Mech Ageing Dev. 2006;127:705–718. doi: 10.1016/j.mad.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 16.Camus MF, et al. Mitochondria, maternal inheritance, and male aging. Curr Biol. 2012;22:1717–1721. doi: 10.1016/j.cub.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 17.Tower J. Programmed cell death in aging. Ageing Res Rev. 2015;23:90–100. doi: 10.1016/j.arr.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pomatto LC, et al. Sexual Dimorphism and Aging Differentially Regulate Adaptive Homeostasis. J Gerontol A Biol Sci Med Sci. 2017;00:109. doi: 10.1093/gerona/glx083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang J, et al. Synchronized age-related gene expression changes across multiple tissues in human and the link to complex diseases. Scientific reports. 2015;5:15145. doi: 10.1038/srep15145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Landis G, et al. Gene expression changes in response to aging compared to heat stress, oxidative stress and ionizing radiation in Drosophila melanogaster. Aging (Albany NY) 2012;4:768–789. doi: 10.18632/aging.100499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Magalhaes JP, et al. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 2009;25:875–881. doi: 10.1093/bioinformatics/btp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tower J. Heat shock proteins and Drosophila aging. Exp Gerontol. 2011;46:355–362. doi: 10.1016/j.exger.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morrow G, Tanguay RM. Drosophila melanogaster Hsp22: a mitochondrial small heat shock protein influencing the aging process. Frontiers in genetics. 2015;6:1026. doi: 10.3389/fgene.2015.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwekel JC, et al. Age and sex dependent changes in liver gene expression during the life cycle of the rat. BMC Genomics. 2010;11:675. doi: 10.1186/1471-2164-11-675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JS, et al. Meta-analysis of gene expression in the mouse liver reveals biomarkers associated with inflammation increased early during aging. Mech Ageing Dev. 2012;133:467–478. doi: 10.1016/j.mad.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 26.Amador-Noguez D, et al. Alterations in xenobiotic metabolism in the long-lived Little mice. Aging Cell. 2007;6:453–470. doi: 10.1111/j.1474-9726.2007.00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu X, et al. Gene expression atlas of the mouse central nervous system: impact and interactions of age, energy intake and gender. Genome Biol. 2007;8:R234. doi: 10.1186/gb-2007-8-11-r234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berchtold NC, et al. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc Natl Acad Sci U S A. 2008;105:15605–15610. doi: 10.1073/pnas.0806883105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan L, et al. Gender-specific proteomic alterations in glycolytic and mitochondrial pathways in aging monkey hearts. J Mol Cell Cardiol. 2004;37:921–929. doi: 10.1016/j.yjmcc.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 30.Vijay V, et al. Sexual dimorphism in the expression of mitochondria-related genes in rat heart at different ages. PLoS One. 2015;10:e0117047. doi: 10.1371/journal.pone.0117047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Isensee J, et al. Sexually dimorphic gene expression in the heart of mice and men. J Mol Med (Berl) 2008;86:61–74. doi: 10.1007/s00109-007-0240-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kapahi P, et al. Dietary restriction and lifespan: Lessons from invertebrate models. Ageing Res Rev. 2016 doi: 10.1016/j.arr.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hur JH, et al. Complex-I-ty in aging. Journal of bioenergetics and biomembranes. 2014;46:329–335. doi: 10.1007/s10863-014-9553-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Munkacsy E, Rea SL. The paradox of mitochondrial dysfunction and extended longevity. Exp Gerontol. 2014;56:221–233. doi: 10.1016/j.exger.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palikaras K, et al. Mitophagy: In sickness and in health. Mol Cell Oncol. 2016;3:e1056332. doi: 10.1080/23723556.2015.1056332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Austad SN, Bartke A. Sex Differences in Longevity and in Responses to Anti-Aging Interventions: A Mini-Review. Gerontology. 2015;62:40–46. doi: 10.1159/000381472. [DOI] [PubMed] [Google Scholar]
- 37.Lapierre LR, et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nature communications. 2013;4:2267. doi: 10.1038/ncomms3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toth ML, et al. Longevity Pathways Converge on Autophagy Genes to Regulate Life Span in Caenorhabditis elegans. Autophagy. 2008:4. doi: 10.4161/auto.5618. [DOI] [PubMed] [Google Scholar]
- 39.Sun N, et al. The Mitochondrial Basis of Aging. Mol Cell. 2016;61:654–666. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swindell WR. Dietary restriction in rats and mice: a meta-analysis and review of the evidence for genotype-dependent effects on lifespan. Ageing Res Rev. 2012;11:254–270. doi: 10.1016/j.arr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Magwere T, et al. Sex differences in the effect of dietary restriction on life span and mortality rates in female and male Drosophila melanogaster. J Gerontol A Biol Sci Med Sci. 2004;59:3–9. doi: 10.1093/gerona/59.1.b3. [DOI] [PubMed] [Google Scholar]
- 42.Clancy DJ, et al. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- 43.Miller RA, et al. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell. 2014;13:468–477. doi: 10.1111/acel.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bjedov I, et al. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010;11:35–46. doi: 10.1016/j.cmet.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang HM, et al. Moderate lifelong overexpression of tuberous sclerosis complex 1 (TSC1) improves health and survival in mice. Scientific reports. 2017;7:834. doi: 10.1038/s41598-017-00970-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khazaeli AA, et al. Heat-induced longevity extension in Drosophila. I. Heat treatment, mortality, and thermotolerance. J Gerontol A Biol Sci Med Sci. 1997;52:B48–52. doi: 10.1093/gerona/52a.1.b48. [DOI] [PubMed] [Google Scholar]
- 47.Lin R, et al. Genetic analysis of dTSPO, an outer mitochondrial membrane protein, reveals its functions in apoptosis, longevity, and Ab42-induced neurodegeneration. Aging Cell. 2014;13:507–518. doi: 10.1111/acel.12200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kanfi Y, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–221. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- 49.Enns LC, et al. Disruption of protein kinase A in mice enhances healthy aging. PLoS One. 2009;4:e5963. doi: 10.1371/journal.pone.0005963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harrison DE, et al. Acarbose, 17-alpha-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell. 2014;13:273–282. doi: 10.1111/acel.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Danilov A, et al. Influence of non-steroidal anti-inflammatory drugs on Drosophila melanogaster longevity. Oncotarget. 2015;6:19428–19444. doi: 10.18632/oncotarget.5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi C, Murphy CT. Mating induces shrinking and death in Caenorhabditis mothers. Science. 2014;343:536–540. doi: 10.1126/science.1242958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maures TJ, et al. Males shorten the life span of C. elegans hermaphrodites via secreted compounds. Science. 2014;343:541–544. doi: 10.1126/science.1244160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tower J, et al. Mifepristone/RU486 acts in Drosophila melanogaster females to counteract the life span-shortening and pro-inflammatory effects of male Sex Peptide. Biogerontology. 2017 doi: 10.1007/s10522-017-9703-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gendron CM, et al. Drosophila life span and physiology are modulated by sexual perception and reward. Science. 2014;343:544–548. doi: 10.1126/science.1243339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bartke A, et al. Somatotropic signaling: trade-offs between growth, reproductive development, and longevity. Physiol Rev. 2013;93:571–598. doi: 10.1152/physrev.00006.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clark RI, et al. Distinct Shifts in Microbiota Composition during Drosophila Aging Impair Intestinal Function and Drive Mortality. Cell reports. 2015;12:1656–1667. doi: 10.1016/j.celrep.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reiff T, et al. Endocrine remodelling of the adult intestine sustains reproduction in Drosophila. eLife. 2015;4:e06930. doi: 10.7554/eLife.06930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ren C, et al. Increased internal and external bacterial load during Drosophila aging without life-span trade-off. Cell Metab. 2007;6:144–152. doi: 10.1016/j.cmet.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 60.Rera M, et al. Organ-specific mediation of lifespan extension: more than a gut feeling? Ageing Res Rev. 2013;12:436–444. doi: 10.1016/j.arr.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gelino S, et al. Intestinal Autophagy Improves Healthspan and Longevity in C. elegans during Dietary Restriction. PLoS Genet. 2016;12:e1006135. doi: 10.1371/journal.pgen.1006135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Portal-Celhay C, et al. Control of intestinal bacterial proliferation in regulation of lifespan in Caenorhabditis elegans. BMC Microbiol. 2012;12:49. doi: 10.1186/1471-2180-12-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dominianni C, et al. Sex, body mass index, and dietary fiber intake influence the human gut microbiome. PLoS One. 2015;10:e0124599. doi: 10.1371/journal.pone.0124599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Prusator DK, Chang L. Sex-Related Differences in GI Disorders. Handbook of experimental pharmacology. 2017;239:177–192. doi: 10.1007/164_2016_121. [DOI] [PubMed] [Google Scholar]
- 65.Shen J, et al. Multiple Metazoan Life-span Interventions Exhibit a Sex-specific Strehler-Mildvan Inverse Relationship Between Initial Mortality Rate and Age-dependent Mortality Rate Acceleration. J Gerontol A Biol Sci Med Sci. 2017;72:44–53. doi: 10.1093/gerona/glw005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Strehler BL, Mildvan AS. General theory of mortality and aging. Science. 1960;132:14–21. doi: 10.1126/science.132.3418.14. [DOI] [PubMed] [Google Scholar]
- 67.Gavrilov LA, Gavrilova NS. The Biology of Life Span: A Quantitative Approach. Harwood Academic Publisher; 1991. [Google Scholar]
- 68.Tarkhov AE, et al. Strehler-Mildvan correlation is a degenerate manifold of Gompertz fit. Journal of theoretical biology. 2017;416:180–189. doi: 10.1016/j.jtbi.2017.01.017. [DOI] [PubMed] [Google Scholar]
- 69.Nakagawa S, et al. Comparative and meta-analytic insights into life extension via dietary restriction. Aging Cell. 2012;11:401–409. doi: 10.1111/j.1474-9726.2012.00798.x. [DOI] [PubMed] [Google Scholar]
- 70.Yen K, et al. Validated analysis of mortality rates demonstrates distinct genetic mechanisms that influence lifespan. Exp Gerontol. 2008;43:1044–1051. doi: 10.1016/j.exger.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 71.Simons MJ, et al. Dietary restriction of rodents decreases aging rate without affecting initial mortality rate -- a meta-analysis. Aging Cell. 2013;12:410–414. doi: 10.1111/acel.12061. [DOI] [PubMed] [Google Scholar]
- 72.Hughes BG, Hekimi S. Different Mechanisms of Longevity in Long-Lived Mouse and Caenorhabditis elegans Mutants Revealed by Statistical Analysis of Mortality Rates. Genetics. 2016;204:905–920. doi: 10.1534/genetics.116.192369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liao CY, et al. Genetic variation in the murine lifespan response to dietary restriction: from life extension to life shortening. Aging Cell. 2010;9:92–95. doi: 10.1111/j.1474-9726.2009.00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu J, et al. Longevity effect of IGF-1R(+/−) mutation depends on genetic background-specific receptor activation. Aging Cell. 2014;13:19–28. doi: 10.1111/acel.12145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hughes BG, Hekimi S. A mild impairment of mitochondrial electron transport has sex-specific effects on lifespan and aging in mice. PLoS One. 2011;6:e26116. doi: 10.1371/journal.pone.0026116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liggett MR, et al. High-glucose diets have sex-specific effects on aging in C. elegans: toxic to hermaphrodites but beneficial to males. Aging (Albany NY) 2015;7:383–388. doi: 10.18632/aging.100759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morsci NS, et al. Age-Related Phasic Patterns of Mitochondrial Maintenance in Adult Caenorhabditis elegans Neurons. J Neurosci. 2016;36:1373–1385. doi: 10.1523/JNEUROSCI.2799-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tower J. Stress and stem cells. Wiley interdisciplinary reviews. Developmental biology. 2012;1:789–802. doi: 10.1002/wdev.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Margineantu DH, Hockenbery DM. Mitochondrial functions in stem cells. Curr Opin Genet Dev. 2016;38:110–117. doi: 10.1016/j.gde.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 80.Truong TH, Carroll KS. Redox regulation of protein kinases. Crit Rev Biochem Mol Biol. 2013;48:332–356. doi: 10.3109/10409238.2013.790873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Palikaras K, et al. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature. 2015 doi: 10.1038/nature14300. [DOI] [PubMed] [Google Scholar]
- 82.Tawo R, et al. The Ubiquitin Ligase CHIP Integrates Proteostasis and Aging by Regulation of Insulin Receptor Turnover. Cell. 2017;169:470–482. e413. doi: 10.1016/j.cell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McCullough LD, et al. Stroke sensitivity in the aged: sex chromosome complement vs. gonadal hormones. Aging (Albany NY) 2016;8:1432–1441. doi: 10.18632/aging.100997. [DOI] [PMC free article] [PubMed] [Google Scholar]