

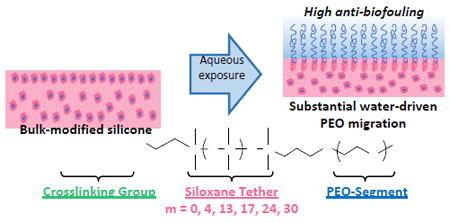
Abstract
Silicones with improved water-driven surface hydrophilicity and anti-biofouling behavior were achieved when bulk-modified with poly(ethylene oxide) (PEO) –silane amphiphiles of varying siloxane tether length: α-(EtO)3Si-(CH2)2-oligodimethylsiloxanem-block-poly(ethylene oxide)8-OCH3 (m = 0, 4, 13, 17, 24, and 30). A PEO8-silane [α-(EtO)3Si-(CH2)3-PEO8-OCH3] served as a conventional PEO-silane control. To examine anti-biofouling behavior in the absence versus presence of water-driven surface restructuring, the amphiphiles and control were surface-grafted onto silicon wafers and used to bulk-modify a medical-grade silicone, respectively. While the surface-grafted PEO-control exhibited superior protein resistance, it failed to appreciably restructure to the surface-water interface of bulk-modified silicone and thus led to poor protein resistance. In contrast, the PEO-silane amphiphiles, while less protein-resistant when surface-grafted onto silicon wafers, rapidly and substantially restructured in bulk-modified silicone, exhibiting superior hydrophilicity and protein resistance. A reduction of biofilm for several strains of bacteria and a fungus was observed for silicones modified with PEO-silane amphiphiles. Longer siloxane tethers maintained surface restructuring and protein resistance while displaying the added benefit of increased transparency.
Table of Contents
Silicones bulk-modified with various PEO-silaneamphiphiles were demonstrated to be resistant to plasma proteins, several bacteria, and a fungus.
Introduction
Silicones such as crosslinked polydimethylsiloxane (PDMS) are widely used in biomedical applications due to their unique properties including thermal and oxidative stability, gas permeability, flexibility, and ease of processing.1-4 Silicone-based medical devices include hemodialysis catheters, cardiac pacing leads, catheter balloons, coated metal stents, extracorporeal device tubing, ophthalmic devices and optical sensors. Unfortunately, due to their hydrophobic nature, silicones lack resistance to protein adsorption.5, 6 Thus, upon implantation of silicone medical devices, plasma proteins are rapidly and substantially adsorbed, leading to subsequent platelet adhesion, activation of coagulation pathways, and eventual thrombosis.5, 6 Bacteria also adhere to device surfaces and form biofilms which are difficult to control with antibiotics.7, 8 Protein adsorption is also considered to facilitate subsequent bacteria biofilm formation.9 To improve the efficacy and safety of silicone-based devices, reduction of both protein and bacterial adsorption and accumulation is essential.
Towards reducing biological adhesion, hydrophilization of silicones with poly(ethylene oxide) (PEO; or poly(ethylene glycol) (PEG)) could be effective given its exceptional protein resistance. This behavior is attributed to hydrophilicity and hydration as well as its configurational mobility which leads to a large excluded volume, steric repulsion, blockage of underlying adsorption sites, and an entropic penalty associated with protein adsorption.10-12 PEO’s biocompatibility13 and recently noted in vivo oxidative stability14 contributes to its widespread use in biomaterials. However, the protein resistance of PEO has largely been demonstrated for chains surface-grafted onto physically stable, model substrates such as gold,4, 15, 16 silicon wafer,17-19 and glass.20, 21 Such “model PEO surfaces” maintain the grafted PEO chains at the surface whether exposed to air or to an aqueous environment (Fig. 1a). This is in contrast to bulk-modified silicones in which PEO chains may undergo surface-reorganization following exposure to different environments. Thus, since protein and bacteria adsorption occurs in an aqueous environment, it is critical that PEO chains migrate to the surface-water interface to create a PEO-enriched silicone surface (Fig. 1b, c). Surface-restructuring of silicones has largely been studied in the case of hydrophobic recovery when exposed to air, a phenomena notably demonstrated by plasma treated silicones.22 This recovery is a result of the low surface energy23 and high chain flexibility of silicones.24 PEO-modified silicones likewise display hydrophobic recovery. For instance, silicones prepared by bulk crosslinking with triethoxysilylpropyl PEO monomethyl ether [(EtO)3Si(CH2)3-(OCH2CH2)n-OCH3]25, 26 as well as allyl PEO mono-methyl ether [CH2=CHCH2-(OCH2CH2)n-OCH3]27 hydrophobically recover. This is also observed for surface-grafted PEO chains such as those prepared with allyl PEO monomethyl ether.27, 28
Fig. 1.
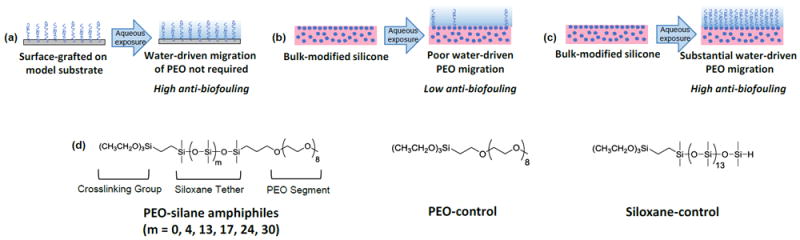
(a-c) The efficacy of PEO to resist biofouling relies on its enrichment at the surface-water interface where biofouling occurs. (d) Herein, silicones were bulk-modified with PEO-silane amphiphiles of variable siloxane tether length (m) and their anti-biofouling behavior compared to each other as well as to silicones bulk-modified with a PEO-control (i.e. a conventional PEO-silane lacking a siloxane tether). Results were compared to surface-grafted analogues that also included a siloxane-control (i.e. no PEO-segment).
Much less studied is the water-driven surface-restructuring of PEO-modified silicones, which is of critical importance to achieve resistance to proteins and bacteria in vivo or in other aqueous environments. In fact, recent reports highlight the poor efficacy of PEO-modified silicones and other polymer matrices to prevent thrombosis that may indicate poor mobilization or retention of the PEO at the surface-water interface.29, 30 Alternatively, silicone modification with an amphiphilic PEO, as reported herein, aims to increase mobilization and water-driven surface-restructuring of the PEO segments.
Previously, we reported the bulk-modification of silicone with three different PEO-silane amphiphiles comprised of a short siloxane tether of three different lengths [α-(EtO)3Si(CH2)2-oligodimethylsiloxanem-block-(OCH2CH2)8-OCH3; m = 0, 4 and 13] (Fig. 1d).31-33 In this way, a hydrophobic, flexible siloxane tether separated the PEO segment and crosslinkable end group. This is in contrast to conventional PEO-silanes that contain a short alkane spacer.25-28 Water-driven surface hydrophilicity as well as protein resistance was improved for silicones bulk-modified with PEO-silane amphiphiles versus the PEO-control (i.e. conventional PEO-silane; [α-(EtO)3Si-(CH2)3-PEO8-OCH3]). Additionally, water-driven surface hydrophilicity increased and protein adsorption decreased with increased siloxane tether length. Bulk-modification of a silica-reinforced silicone with PEO-silane amphiphile (m = 13) likewise demonstrated water-driven surface-restructuring and protein resistance.32 Atomic force microscopy (AFM) analysis verified that PEO segments of the PEO-silane amphiphile (m = 13) were driven to the surface-water interface of the silicone.33 In this way, PEO-silane amphiphiles functioned as effective “surface-modifying additives” (SMAs)34 for silicones. Versus a conventional PEO-silane, the enhanced potential of PEO-silane amphiphiles to migrate to the surface-water interface and reduce protein adsorption was attributed to the siloxane tethers’ molecular flexibility as well as similar hydrophobicity to the silicone matrix that permitted movement of the tether and attached PEO segment through the silicone network. This effect was evidently maximized for the PEO-silane amphiphile with the longest tether (m = 13). We furthermore demonstrated that for PEO-silane amphiphiles (m = 13), water-driven surface hydrophilicity and protein resistance of bulk-modified silicones was superior for a PEO segment length of n = 8 or n = 16 versus n = 3.35
Herein, we sought to determine if PEO-silane amphiphiles with even longer siloxane tethers (m = 17, 24 and 30) would enhance the protein and bacterial biofilm resistance of bulk-modified silicones. Thus, a series of PEO-silane amphiphiles were prepared with an expanded range of siloxane tether lengths [m = 0 (m = 0), Mn = 749 g/mol; m = 4 (m = 4), Mn = 1044 g/mol; m = 13 (m = 13), Mn = 1710 g/mol; m = 17 (m = 17), Mn = 2006 g/mol; m = 24 (m = 24), Mn = 2524 g/mol; and m = 30 (m = 30), Mn = 2968 g/mol] (Fig. 1d, Scheme 1). PEO-silane amphiphiles were used to bulk-modify a medical grade, silica-reinforced room temperature vulcanizing (RTV) silicone and surface-grafted onto a model substrate. Grafting onto silicon wafers permitted the evaluation of surface hydrophilicity as well as protein resistance in the absence of water-driven surface restructuring effects. Given its role in surface-induced thrombosis,36 the adsorption of fibrinogen protein was evaluated. The accumulation of several types of bacteria including Staphylococcus epidermidis (Gram-positive), Staphylococcus aureus (Gram-positive), Pseudomonas aeruginosa (Gram-negative), and Escherichia coli (Gram-negative), as well as the fungus Candida albicans, were also tested due to their prevalence in medical device-related infections.7, 8 A PEO-control (i.e. conventional PEO-silane with no siloxane tether) was utilized for both bulk-modification and surface-grafting to distinguish its behavior versus PEO-silane amphiphiles (Fig. 1d). A siloxane-control (i.e. no PEO segment, m = 13) was used as a hydrophobic control for surface-grafted films (Fig. 1d). Finally, given the necessity for transparency for certain silicone-based devices such as intraocular lenses and optical sensor,37, 38 the transparency of the bulk-modified silicones was also measured.
Scheme 1.
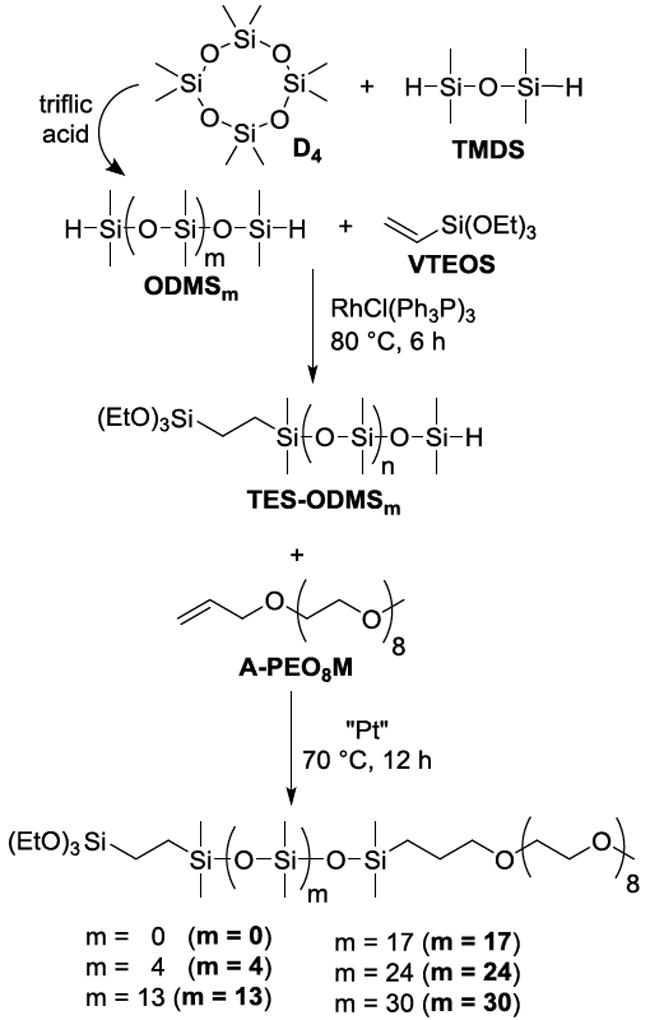
Synthesis of PEO-silane amphiphiles with a siloxane tether of varying length (m) (m = 0, m = 4, m = 13, m = 17, m = 24 and m = 30) and corresponding intermediates (ODMSm and TES-ODMSm).
Results and discussion
Synthesis of PEO-silane amphiphiles (m = 0, m = 4, and m = 13)
PEO-silane amphiphiles with shorter tether (m = 0, m = 4, and m = 13) were prepared as previously reported (Scheme 1).31 Similar protocols were utilized to prepare PEO-silane amphiphiles with longer siloxane tethers as described below.
Synthesis of ODMSm
Triflic acid-catalyzed ring-opening reaction of variable molar ratios of D4 with TMDS was used to produce ODMS17, ODMS24, and ODMS30 in good yields (≥60%) (Scheme 1). The Mn’s of ODMSm were confirmed by 1H NMR end-group analysis.
Synthesis of TES-ODMSm
The Rh-catalyzed regioselective hydrosilylation reaction of equimolar amounts of ODMS17, ODMS24, and ODMS30 each with VTEOS (1:1 molar ratio) effectively produced TES-ODMS17, TES-ODMS24, and TES-ODMS30, respectively, in good yields (≥96%). 1H NMR spectra of TES-ODMS17, TES-ODMS24, and TES-ODMS30 showed a decrease in the Si–H peak integration value by one-half versus the starting material.
The synthesis of TES-ODMSm relies on the Rh-catalyzed regioselective hydrosilylation of the designated ODMSm (Scheme 1). Notably, an increased distance between terminal Si–H groups has been suggested to decrease reactivity towards vinyl-containing compounds.39 In early reports, regioselective hydrosilylation of α,ω-bis(Si-H)oligodimethylsiloxanes was limited to those with 2-4 silicon atoms.40-43 However, we have since reported the successful regioselective hydrosilylation of ODMS0, ODMS4, and ODMS13 with VTEOS.31 Regioselectivity of the hydrosilylation leading exclusively to the “monosubstituted” product was verified with GPC experiments since 1H NMR spectra represent only the average composition of each product. In other words, a pure, monosubstituted product would have the same spectrum as the mixture of the three products obtained from the corresponding nonregioselective hydrosilylation (i.e. monosubstituted as well as disubstituted and nonsubstituted products where the ratio of di- and nonsubstituted would be equal).
Herein, we likewise sought to confirm the regioselectivity of Rh-catalyzed hydrosilylation of ODMS17, ODMS24, and ODMS30 each with VTEOS (1:1 molar ratio). Following hydrosilylation, the products (designated as monosubstituted TES-ODMS17, TES-ODMS24, and TES-ODMS30) were each reacted with CH2=CH–PDMS-n-Bu (Mn = 60,000 g/mol; PDI = 1.38) by Pt-catalyzed hydrosilylation to produce the corresponding materials, designated as mono-17, mono-24, and mono-30, respectively (Scheme S1). If regioselective, the products of the Rh-catalyzed hydrosilylation reaction between each ODMSm and VTEOS would be exclusively mono-17, mono-24, or mono-30. However, if nonregioselective, the reaction between each ODMSm and VTEOS would result in a mixture of three products (i.e. monosubstituted as well as disubstituted and nonsubstituted) which would subsequently react with CH2=CH–PDMS-n-Bu to yield the following mixture: (i) mono-17, mono-24, and mono-30: the products of monosubstituted TES-ODMS17, TES-ODMS24, and TES-ODMS30 each with CH2=CH–PDMS-n-Bu (Mn = 61,582; 62,100; 62,544 g/mol [theoretical values]); (ii) di-17, di-24, and di-30: unreacted α,ω-triethoxysilylethyl-disubstituted products (Mn = 1,772; 2,290; 2,734 g/mol [theoretical values]); and (iii) non-17, non-24, and non-30: the products of ODMS17, ODMS24, and ODMS30 each with CH2=CH–PDMS-n-Bu (1:2 molar ratio) (Mn = 121,334; 121,852; 122,296 g/mol [theoretical values]) (Scheme S1). Due to the differences in Mn, the presence or absence of disubstituted and nonsubstituted products in the GPC chromatographs of mono-17, mono-24, and mono-30 may be used to assess the regioselectivity of the Rh-catalyzed hydrosilylation of ODMSm and VTEOS (designated as TES-ODMS17, TES-ODMS24, and TES-ODMS30).
So that their GPC elution peaks could be identified, products di-17, di-24, and di-30 were individually synthesized in their isolated form by Pt-catalyzed hydrosilylation of ODMS17, ODMS24, and ODMS30, respectively, with VTEOS (1:2 molar ratio) (Scheme S1 and Supporting Information). Products non-17, non-24, and non-30 were synthesized by Pt-catalyzed hydrosilylation of ODMS17, ODMS24, and ODMS30, respectively, with CH2=CH–PDMS-n-Bu (1:2 molar ratio).
In the GPC chromatographs of mono-17, mono-24, and mono-30, the elution peaks of di-17, di-24, and di-30, respectively, are absent (Fig. S1). The elution peaks of non-17, non-24, and non-30 overlap with the elution peaks of mono-17, mono-24, and mono-30 but must be absent as well since di-17, di-24, and di-30 and non-17, non-24, and non-30 would be present in equal amounts, respectively. Thus, the compositions of mono-17, mono-24, and mono-30 may be identified as the product of monosubstituted TES-ODMS17, TES-ODMS24, and TES-ODMS30 (i.e. regioselective products) each with CH2=CH–PDMS-n-Bu. These results confirm the regioselectivity of the Rh-catalyzed hydrosilylation reactions of ODMS17, ODMS24, and ODMS30 each with VTEOS to produce only monosubstituted TES-ODMS17, TES-ODMS24, and TES-ODMS30, respectively.
Synthesis of PEO-silane amphiphiles (m = 17, m = 24, and m = 30)
The Pt-catalyzed hydrosilylation reaction of equimolar amounts of TES-ODMS17, TES-ODMS24, and TES-ODMS30 each with A-PEO8M produced m = 17, m = 24, and m = 30, respectively, in good yields (≥78%). Completion of the reaction was confirmed by IR analysis of m = 17, m = 24, and m = 30, which showed no absorbance at ~2125 cm-1 which corresponds to unreacted Si–H bonds. The Si–H peak (~4.7 ppm) of the 1H NMR spectra of m = 17, m = 24, and m = 30 was also absent.
Characterization of surface-grafted films
XPS
XPS was used to confirm the grafting of PEO-silane amphiphiles (m = 17, m = 24, and m = 30) onto silicon wafers. The elemental surface compositions of these surfaces are reported in Table S1. For the oxidized wafer, the O 1s and Si 2p peaks correspond to the wafer whereas the carbon (C 1s) is attributed to adsorbed contaminants.44, 45 Following grafting, the Si 2p content decreased and the C 1s content increased. The C 1s peak was deconvoluted into two peaks centered at 284.5 eV (C–C and C–Si) and 286.4 eV (C–O), with this latter peak unique to PEO28 (Fig. S2). As expected, versus the PEO-control, the C–C/C–Si content increased and the C–O content generally decreased as the siloxane tether length (m) of the PEO-silane amphiphiles was increased.
Ellipsometry
Protein resistance of surface-grafted PEO chains is influenced by chain spacing (i.e. graft distance, D) and associated conformation.46, 47 Thus, grafted silicon surfaces were confirmed to have similar chain densities. The dry graft layer thickness (h) of the grafted PEO-control, siloxane-control, and PEO-silane amphiphiles were measured, and chain density (σ) values were calculated48-50 (Table 1):
| (1) |
where ρ is the density of the dry grafted layer, NA is Avogadro’s number, and Mn is the number average molecular weight of the chain. Chain “spacing” (D) was also calculated50 (Table 1):
| (2) |
All grafted layers were determined to have similar values of D (1.05-1.49 nm). In order to achieve an extended conformation (brush regime), D must be less than twice the Flory radius (RF).47 Thus, RF was calculated using the length of one monomer unit (a) and the degree of polymerization (N) as follows: (1) for the siloxane-control in a poor solvent (i.e. water), RF = aN1/3, where a = 0.5 nm51 and N = 13 and (2) for the PEO-control in a good solvent (i.e. water), RF = aN3/5, where a = 0.35 nm52 and N = 8.53 For these controls, D < 2 RF. Calculation of RF values for the PEO-silane amphiphiles is complicated by the differing solubility of the PEO segment and siloxane tether in water. Thus, the RF values were determined individually for the PEO segment and the siloxane tether (Table 1). For grafted PEO-silane amphiphiles, D < 2 RF, including for the lower of the two calculated RF values. Thus, all grafted surfaces were confirmed to be in a brush regime.
Table 1.
Ellipsometry data for silicon wafers grafted with PEO-silane amphiphiles, the PEO-control and the siloxane control.
| Grafted Surface | Mn (g/mol) | Density ρ (g/mL) | Thickness h (nm) | Chain Density σ = (hρNA)/Mn (chains/nm2) | Graft Distance D = (4/πσ)1/2 (nm) | PEO Flory Spacing 2RF = 2aN3/5(nm) | Siloxane Flory Spacing 2RF = 2aN1/3 (nm) |
|---|---|---|---|---|---|---|---|
| PEO-control | 588 | 1.13 | 2.3 ± 0.2 | 2.60 | 0.70 | 2.4 | - |
| m = 0 | 749 | 1.02 | 1.4 ± 0.3 | 1.15 | 1.05 | 2.4 | - |
| m = 4 | 1044 | 1.00 | 1.6 ± 0.5 | 0.92 | 1.17 | 2.4 | 1.6 |
| m = 13 | 1710 | 0.98 | 1.5 ± 0.2 | 0.52 | 1.57 | 2.4 | 2.4 |
| m = 17 | 2006 | 0.99 | 2.0 ± 0.2 | 0.59 | 1.46 | 2.4 | 2.5 |
| m = 24 | 2524 | 0.98 | 2.8 ± 0.1 | 0.65 | 1.39 | 2.4 | 2.9 |
| m = 30 | 2968 | 0.98 | 2.9 ± 0.1 | 0.58 | 1.49 | 2.4 | 3.1 |
| Siloxane-control | 1286 | 1.01 | 2.5 ± 0.9 | 1.20 | 1.10 | - | 2.4 |
Contact angle analysis
As physically stable surfaces, silicon wafers maintain grafted chains at the surface whether exposed to an air or aqueous environment. The impact of siloxane tether length (m) on the hydrophilicity of grafted PEO-silane amphiphiles was assessed by measuring υstatic of water droplets. Surfaces grafted with the siloxane-control and PEO-control conveniently serve as a hydrophobic (υstatic > 90°)54 and hydrophilic control, respectively. Measurements were made immediately following droplet placement (0 sec) and at 2 min (Fig. 2, Table S2). Due to the lack of surface restructuring for such stable substrates, υstatic (0 sec) and υstatic (2 min) were very similar, decreasing slightly due to hydration. For the grafted PEO-silane amphiphiles, as the hydrophobic siloxane tether length increased, υstatic values increased as expected.
Fig. 2.
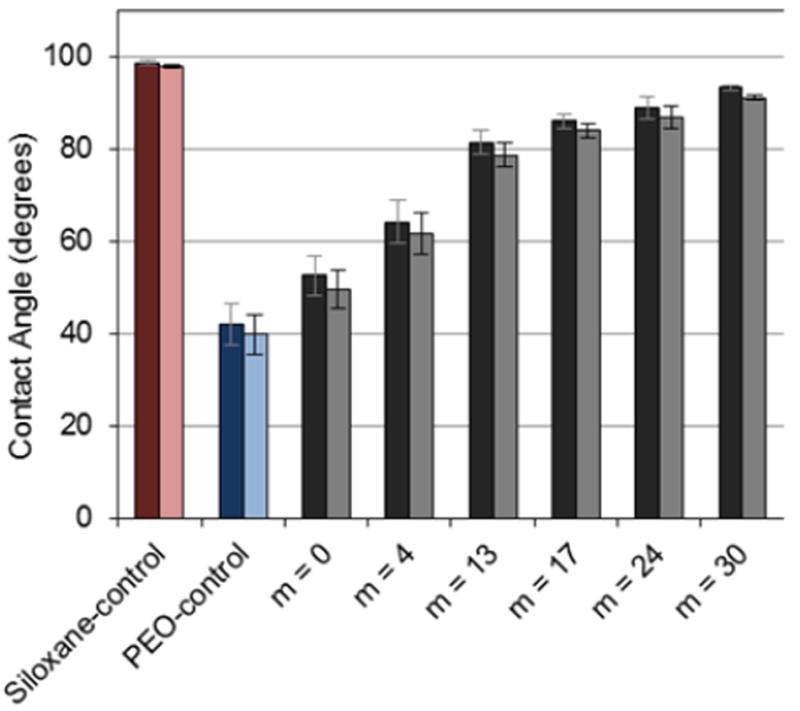
Static contact angles (θstatic) of silicon wafers grafted with PEO-silane amphiphiles, the PEO-control, and the siloxane-control at 0 sec (dark) and 2 min (light) following water droplet placement. Each bar represents the average and standard deviation of three measurements taken from three different areas of the same sample. Using single factor Anova, statistically significant differences in (θstatic (2 min) (p < 0.05) among all specimens were noted.
Protein adsorption via QCM-D
PEO-silane amphiphiles grafted onto silicon wafer permitted the evaluation of protein resistance in the absence of surface restructuring effects. Thus, using QCM-D, adsorption of human fibrinogen (HF) onto these surface-grafted films was evaluated. QCM-D is known to be an effective method to measure protein adsorption onto low-fouling thin films.55, 56 Due to the low dissipation of adsorbed HF,57 the adsorbed mass was calculated using the Sauerbrey approximation and the seventh frequency overtone (Fig. 3).
Fig. 3.
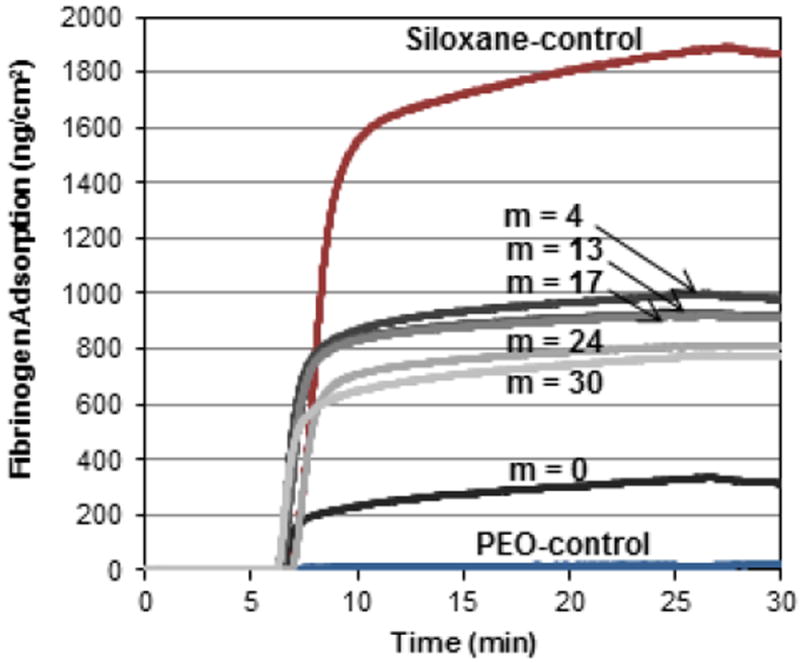
QCM-D-measured adsorption of HF onto silica sensors grafted with PEO-silane amphiphiles, the PEO-control and the siloxane-control. After equilibration for 5 min with PBS, the sensors were exposed to HF for 20 min and then to PBS for 5 min.
It is well established that proteins adsorb more substantially to hydrophobic rather than hydrophilic surfaces.58, 59 Thus, as expected, it was observed that an increase in HF adsorption coincided with the observed relative increase in surface hydrophobicity (Fig 2). For instance, the grafted siloxane-control adsorbed the highest amount of HF whereas the grafted PEO-control was the most resistant. The exceptional protein resistance of the surface-grafted PEO-control is consistent with that observed for PEO chains grafted to stable surfaces.15-21 For grafted PEO-silane amphiphiles, HF adsorption increased substantially as the length of the siloxane tether increased beyond m = 0 with relatively small differences in HF adsorption among those with longer tethers.
Characterization of bulk-modified silicone films
Film preparation, transparency and modulus
The ability of PEO-silane amphiphiles to undergo water-driven surface reorganization to form a PEO-enriched surface and improve protein resistance was examined by bulk-modification of a medical-grade RTV silicone. PEO-silane amphiphiles as well as the PEO-control were each introduced at 50 μmol per gram of silicone and solvent cast onto glass microscope slides (Fig. 4). Film thickness (via electronic calipers) was 0.17 ± 0.02 mm.
Fig. 4.

Photographs of unmodified silicone and silicones bulk-modified with PEO-silane amphiphiles and the PEO-control.
The transparency of the bulk-modified silicones was visually observed to decrease with incorporation the PEO-control as well as PEO-silane amphiphiles with shorter tethers. Transparency of bulk-modified films was also quantified by measuring percent transparency at 500 nm (Fig. S3). First, incorporation of the PEO-control resulted in a substantial reduction in film transparency (70%) versus the unmodified silicone (89%). When modified with PEO-silane amphiphiles, percent transparency improved with increased siloxane tether length (m). Notably, bulk-modified silicones containing m = 0 (62%) and m = 4 (81%) remained relatively nontransparent, whereas those containing m = 13 (88%), m = 17 (92%), m = 24 (87%), and m = 30 (91%) were similar versus the unmodified silicone (89%). This may be attributed to the similar solubility of the siloxane tethers and the silicone matrix, leading to reduced phase separation, particularly as the siloxane tether length was increased.
The characteristic low tensile modulus of unmodified silicone (0.33 ± 0.04 MPa) was maintained upon modification with the PEO-control (0.35 ± 0.03 MPa) and PEO-silane amphiphiles (0.42 ± 0.04 MPa, m = 0; 0.31 ± 0.04 MPa, m = 4; 0.32 ± 0.05 MPa, m = 13; 0.39 ± 0.03 MPa, m = 17; 0.37 ± 0.06 MPa, m = 24; 0.28 ± 0.04 MPa, m = 30).
Contact angle analysis
As noted, we previously used AFM to verify the water-driven formation of a PEO-enriched surface of silicone bulk-modified with PEO-silane amphiphile m = 13.33 This surface restructuring was accompanied by a reduction of the contact angle of a water droplet over time. Likewise, herein, temporal contact angle measurements (0 sec - 2 min) were used to assess the ability of silicones bulk-modified with PEO-silane amphiphiles of varying siloxane tether length (m) to undergo water-driven surface reorganization to rapidly yield a hydrophilic surface (Fig. 5, Table S3).
Fig. 5.
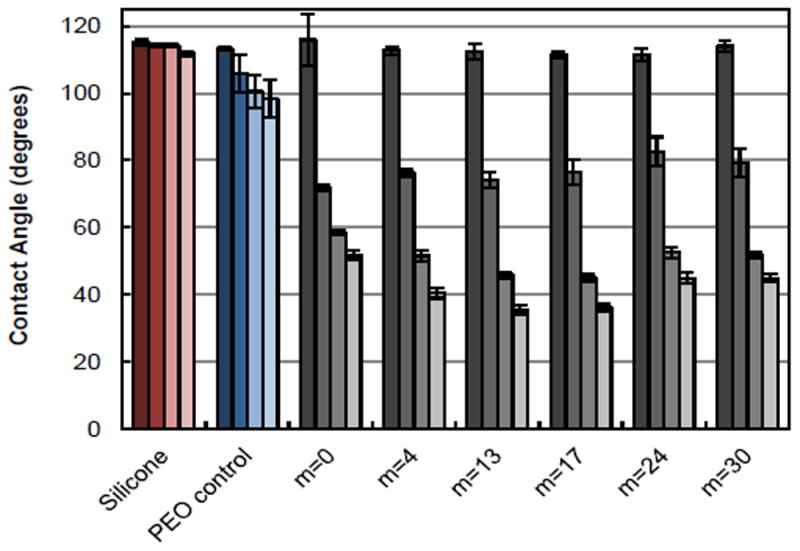
Static contact angles (θstatic) of unmodified silicone and silicones bulk-modified with PEO-silane amphiphiles and the PEO-control. Bars are organized as the time after water droplet placement from dark color to light as follows: 0 sec, 15 sec, 1 min, and 2 min. Each bar represents the average and standard deviation of three measurements on three different areas of the same sample. Using single factor Anova, statistically significant differences in θstatic (2 min) (p < 0.05) among all specimens were noted.
As expected, the unmodified silicone was hydrophobic (υstatic > 90°) with minimal restructuring (i.e. negligible decrease in θstatic; Δ). Notably, when modified with the PEO-control, the silicone was similarly hydrophobic and underwent limited restructuring (Δ = 15°). Thus, the conventional PEO-control was ineffective in its ability to migrate to the surface-water interface. In contrast, all silicones bulk-modified with PEO-silane amphiphiles underwent rapid and substantial water-driven surface reorganization characterized by large Δ values. Initially (θstatic = 0 sec), all silicones modified with PEO-silane amphiphiles were similarly hydrophobic. However, by 15 sec, substantial restructuring had already occurred. While by 2 min, all of these bulk-modified silicones were extremely hydrophilic (i.e. low θstatic), statistically significance differences in θstatic (2 min) (p < 0.05) was determined by single factor Anova. Values of θstatic (2 min) reached to a minimum for silicones modified with PEO-silane amphiphiles having an intermediate siloxane tether length: m = 0 (~52 °) > m = 4 (~41 °) > m = 13 ≈ m = 17 (~36 °) < m = 24 ≈ m = 30 (~45 °). While a longer siloxane tether may improve PEO-silane amphiphile solubility in the silicone, we hypothesize that beyond an intermediate siloxane tether length (i.e. m = 13 or 17), PEO migration potential is slightly diminished by the greater steric barriers.
Protein adsorption via ELISA
The amount of HF adsorbed onto silicones was measured using a modified immunosorbent assay. The amount of HF adsorbed onto silicone films was measured with ELISA in terms of absorbance and quantified via comparison to a HF standard curve (Fig. 6, Table S4).
Fig. 6.
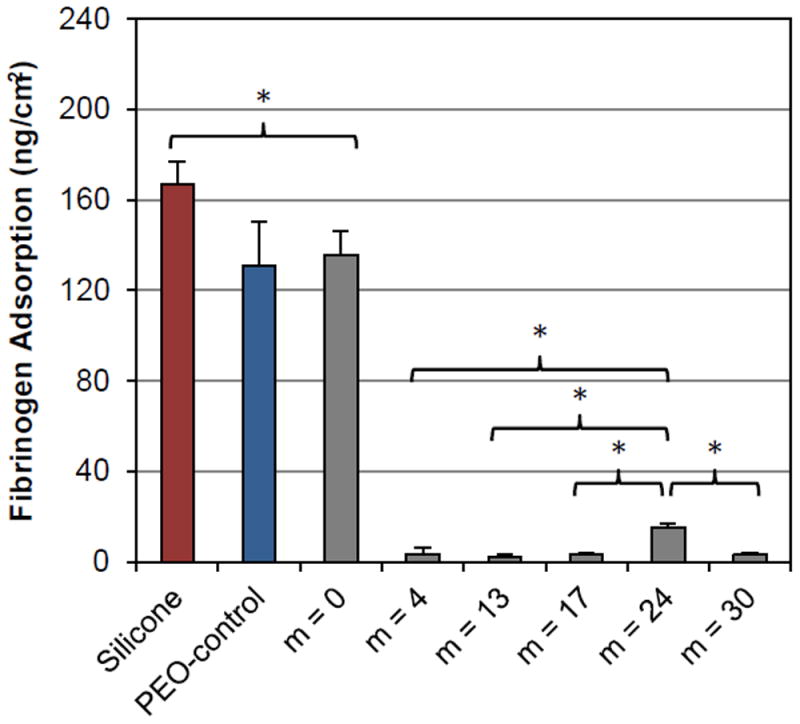
Fibrinogen adsorption on of unmodified silicone and silicones bulk-modified with PEO-silane amphiphiles and the PEO-control. Each bar represents the average and one standard deviation of three replicates. Statistical significance was determined by single factor Anova (where * indicates p < 0.05). For low fouling surfaces, differences in HF adsorption were compared to silicones modified with m = 24.
As expected, the unmodified silicone adsorbed high levels of HF due to its extreme hydrophobicity (Fig. 5). The silicone bulk-modified with the PEO-control also adsorbed high HF levels as a result of its high surface hydrophobicity due to the failure of PEO to successfully migrate to the surface-water interface. For the silicone bulk-modified with the PEO-silane amphiphile with the shortest siloxane tether (m = 0), HF adsorption was similar to that modified with the PEO-control. However, HF adsorption was substantially reduced for all silicones bulk-modified with the PEO-silane amphiphiles having longer siloxane tethers (m = 4, m = 13, m = 17, m = 24 and m = 30). This result coincides with their superior ability to undergo water-driven surface reconstruction, leading to a PEO-enriched, hydrophilic surface (Fig. 5). While very still very low, HF adsorption onto silicone bulk-modified with m = 24 was statistically higher versus for the other modified silicones. Thus, while the PEO-silane amphiphiles exhibited reduced protein resistance versus the PEO-control when surface-grafted onto silicon wafers (Fig. 3), when used to bulk-modify silicones they are superior and highly effective in improving protein resistance when their siloxane tether is of sufficient length (m ≥ 4).
Biofilm adhesion
Analysis of biofilm growth for several microorganisms pertinent to medical device-related infections revealed a difference in behavior among different bacteria and fungus (Fig. 7, Table S5). In the case of S. epidermis, S. aureus, E. coli, and C. albicans, biofilm growth was markedly more inhibited on silicones bulk-modified with most of the PEO-silane amphiphiles versus unmodified silicone and silicone modified with the PEO-control. For these, generally only minor differences in biofilm growth was observed as a function of the siloxane tether length (m) of the PEO-silane amphiphile. In the case of S. aureus, even m = 0 was effective at reducing biofilm levels versus other PEO-silane amphiphiles. For S. epidermis and, to a lesser extent, for E. coli and C. albicans, m = 0 was not as effective in reducing biofilm growth versus m = 4, m = 13, m = 17, m = 24 and m = 30. For E. coli, a slight increase in biofilm growth was observed for m = 24 and m = 30 but still remained substantially lower versus unmodified silicone and the silicone modified with the PEO-control. Only for P. aeruginosa was biofilm accumulation greater for the silicones modified with PEO-silane amphiphiles having longer siloxane tethers (m = 13, m = 17, m = 24 and m = 30) with m = 0 and m = 4 showing the lowest amounts of biofilm. Only silicone modified with m = 0 exhibited statistically significantly lower P. aeruginosa biofilms level versus unmodified silicone. A possible contribution to this result may be related to the unique physiology of this particular bacterial strain. As reported by Roosjen et al.,60 the ability of a given P. aeruginosa strain to adhere to PEO brushes (grafted onto glass or silica) was influenced substantially by cell surface hydrophobicity and the production of biosurfactants of a given strain. Strains possessing both a highly hydrophobic cell surface (contact angle > 130°) and the ability to produce a surfactant-like compound (e.g. rhamnolipids) were shown to adhere in higher numbers to PEO brushes when compared to those isolates possessing an inherently hydrophilic cell surface (contact angle ~ 60°) and incapable of producing a biosurfactant. Thus, it is possible that the P. aeruginosa strain used in this study may have expressed a similar or corresponding ‘adherent’ phenotype, allowing it to overcome the putative interfacial repulsive forces or surface tension imparted by the silicones modified with PEO-silane amphiphiles of longer tether length.
Fig. 7.
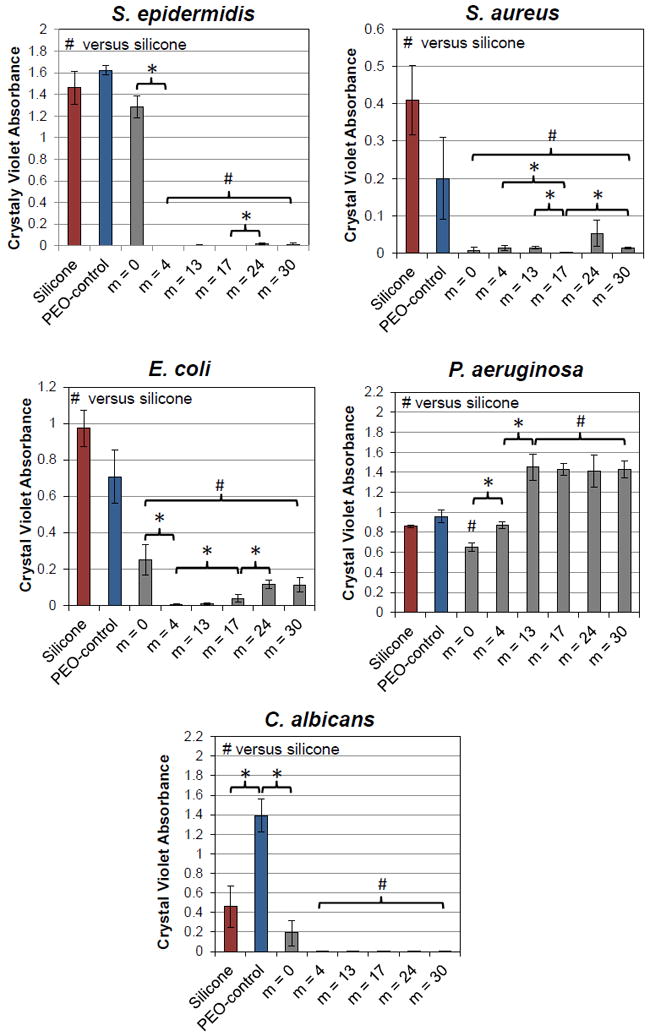
Bacterial and fungal biofilm formation on unmodified silicone and silicones bulk-modified with PEO-silane amphiphiles and the PEO-control. Each bar represents the average and one standard deviation of three replicates. Statistical significance was determined by single factor Anova (where * and # indicates p < 0.05 for “pairs” or to unmodified silicone, respectively.)
Experimental part
Materials
Triflic acid, RhCl(Ph3P)3 (Wilkinson’s catalyst), solvents, sulfuric acid (H2SO4), hexamethyldisilazane (HMDS), and fibrinogen from human plasma (HF; Mw = 340 kDa; lyophilized powder; ≥90% clottable protein) were obtained from Sigma-Aldrich (St. Louis, MO). Solvents were dried over 4 Å molecular sieves prior to use in hydrosilylation reactions. Octamethylcyclotetrasiloxane (D4; MW = 296 g/mol), Pt–divinyltetramethyldisiloxane complex (Karstedt’s catalyst), vinyl-triethoxysilane (VTEOS; MW = 190 g/mol), α,ω-bis-(Si–OH)oligodimethylsiloxanes {ODMS0 or tetramethyldisiloxane (TMDS) [Mn = 118 g/mol per manufacturer’s specifications; Mn = 134 g/mol per 1H NMR end group analysis; 1H NMR (δ, ppm): 0.17–0.21 (d, J = 2.7 Hz, 12H, OSi[CH3]2H) and 4.66–4.72 (m, 2H, SiH)]; ODMS4 [Mn = 400-500 g/mol per manufacturer’s specifications; Mn = 430 g/mol per 1H NMR end group analysis; 1H NMR (δ, ppm): 0.07–0.09 (m, 24H, SiCH3), 0.18–0.19 (d, J = 2.7 Hz, 12H, OSi[CH3]2H), and 4.67–4.73 (m, 2H, SiH)]; ODMS13 [Mn = 1000-1100 g/mol per manufacturer’s specifications; Mn = 1096 g/mol per 1H NMR end group analysis; 1H NMR (δ, ppm): 0.05–0.10 (m, 78H, SiCH3), 0.19 (d, J = 2.7 Hz, 12H, OSi[CH3]2H), and 4.67–4.73 (m, 2H, SiH)]}60, and monovinyl-terminated PDMS (CH2=CH–PDMS-n-Bu) [Mn = 62,700 g/mol, essentially 100% monovinyl-terminated with the nonfunctional end n-butyl-terminated per manufacturer’s specifications] were obtained from Gelest. PEO allyl methyl ether (Polyglycol AM-450; A-PEO8M) [Mn = 292 – 644 g/mol per manufacturer’s specifications; Mn = 424 g/mol per 1H NMR end group analysis; 1H NMR (δ, ppm): 3.35 (s, 3H, OCH3), 3.51–3.66 (m, 32H, OCH2CH2), 4.00 (d, J = 5.4 Hz, 2H, CH2=CHCH2O), 5.13–5.28 (m, 2H, CH2=CHCH2O), and 5.82–5.96 (m, 1H, CH2=CHCH2O)] was obtained from Clariant and was dried overnight under high vacuum prior to use. Silicon wafers (111) were obtained from University Wafers, Inc. (Boston, MA). Silica-coated QCM-D sensors (QSX-303) were obtained from Q-Sense. Hydrogen peroxide (H2O2), glass microscope slides (3″ × 1″), and phosphate buffered saline (PBS, without calcium and magnesium, pH = 7.4) were obtained from Fisher Scientific. Medical-grade silicone (MED-1137) was obtained from NuSil Technology (Carpinteria, CA). Per manufacturer’s specifications, MED-1137 is comprised of α,ω-bis(Si–OH)PDMS, silica (11-21%), methyltriacetoxysilane (<5%), ethyltriacetoxysilane (<5%), and trace amounts of acetic acid. The Alexa Fluor 546 HF conjugate (Mw = 340 kDa; lyophilized powder; 95% clottable protein) was purchased from Invitrogen (Carlsbad, CA). Silicone isolator wells for protein adsorption studies were prepared from silicone sheets (2 mm thick; McMaster Carr) with a die punch (18 mm diameter). The PEO-silane amphiphiles [m = 0, m = 4, and m = 13] and the PEO-control31 as well as the siloxane-control35 were synthesized as previously reported. Tryptic Soy broth (TSB), Luria-Bertani broth (LB), minimal medium M63, yeast nitrogen broth (YNB), RPMI 1640 medium, crystal violet powder (CV), 33% glacial acetic acid, 10X phosphate buffered saline (PBS), Na2HPO4, KH2PO4, KCl, (NH4)2SO4, MgSO4, dextrose, thiamine and biotin were purchased from VWR (Chicago, IL). Bacterial and fungal strains were purchased from the American Type Culture Collection (ATCC) (Manassas, VA): Staphylococcus epidermidis (ATCC 35984), Staphylococcus aureus (ATCC 25923), Escherichia coli (ATTC 12435), Pseudomonas aeruginosa (ATCC 15442), and Candida albicans (ATCC 10231).
Polymer Characterization
1H spectra were obtained on a Mercury 300 MHz spectrometer operating in the Fourier transform mode. Five percent (w/v) CDCl3 (dried over 4 Å molecular sieves) solutions were used to obtain spectra. Residual CDCl3 was used as an internal standard set to 7.26 ppm. IR spectra of neat liquids on NaCl plates were recorded using a Bruker TENSOR 27 Fourier transform infrared spectrometer. GPC analysis was performed on a Tosoh Corporation (Tokyo, Japan) model HLC-8320 EcoSEC system with a two-column set of TOSOH Bioscience TSKgel columns (Super HM-M 6.0 mm ID × 15 cm columns) and a guard column (Super H-H 4 μm). The system was equilibrated at 40 °C in chloroform, which served as the polymer solvent and eluent (flow rate set to 0.6 mL/min). The differential refractometer was calibrated with Polymer Laboratories, Inc. polystyrene standards (580 to 370,000 Da).
General Synthetic Approach
All reactions were run under a nitrogen (N2) atmosphere with a Teflon-covered stir bar to agitate the reaction mixture. ODMSm (ODMS17, ODMS24, and ODMS30) were prepared by a triflic acid-catalyzed ring-opening reaction of D4 with TMDS (Scheme 1).61 D4 and TMDS (4:1, 5:1, and 6:1 molar ratios, respectively) were combined with triflic acid in a 100 mL round bottom flask equipped with a rubber septum at room temperature (RT). After 2.5 h, HMDS was added to the reaction to neutralize the acid. The reaction was then filtered to remove salts, and volatiles were removed under reduced pressure.
α-Triethoxysilylethyl-ω-silane-oligodimethylsiloxanem (TES-ODMS17, TES-ODMS24, and TES-ODMS30) and triethoxysilylethyl-oligodimethylsiloxanem-block-poly(ethylene oxide)8 (m = 17, m = 24, and m = 30) were prepared using a previously reported strategy.31 Briefly, TES-ODMS17, TES-ODMS24, and TES-ODMS30 were synthesized by the Rh-catalyzed regioselective hydrosilylation of equimolar amounts of VTEOS with ODMS17, ODMS24, and ODMS30, respectively (Scheme 1). An equimolar ratio of ODMSm and VTEOS were combined with Wilkinson’s catalyst and toluene and then heated to 80 °C. After 12 h, toluene was removed under reduced pressure, the product was purified by flash column chromatography on silica gel with hexanes/ethyl acetate (2:1 v/v), and volatiles were removed under reduced pressure.
PEO-silane amphiphiles m = 17, m = 24, and m = 30 were synthesized by the Pt-catalyzed hydrosilylation of equimolar amounts of A-PEO8M with TES-ODMS17, TES-ODMS24, and TES-ODMS30, respectively (Scheme 1). TES-ODMSm and A-PEO8M were combined with Karstedt’s catalyst and toluene and then heated to 70 °C. After 12 h, the progress of the reaction was confirmed by the disappearance of the Si–H (~2125 cm-1) absorbance via IR spectroscopy. The catalyst was removed by refluxing the reaction mixture with activated charcoal for 2 h at 80 °C. After filtration, the volatiles were removed under reduced pressure so that m = 17, m = 24, and m = 30 were isolated as colorless liquids.
Synthesis of ODMS17
D4 (20.0 g, 67.6 mmol), TMDS (2.28 g, 17.0 mmol), and triflic acid (40 μL) were reacted as above and quenched with the final addition of HMDS (94 μL). In this way, ODMS17 (13.3 g, 60% yield) was obtained. 1H NMR (δ, ppm): 0.05–0.11 (m, 102H, SiCH3), 0.18–0.19 (d, J = 2.7 Hz, 12H, OSi[CH3]2H), and 4.69–4.72 (m, 2H, SiH).
Synthesis of ODMS24
D4 (20.1 g, 67.9 mmol), TMDS (1.85 g, 13.8 mmol), and triflic acid (40 μL) were reacted as above and quenched with the final addition of HMDS (94 μL). In this way, ODMS24 (18.2 g, 83% yield) was obtained. 1H NMR (δ, ppm): 0.05–0.11 (m, 144H, SiCH3), 0.18–0.20 (d, J = 2.7 Hz, 12H, OSi[CH3]2H), and 4.68–4.73 (m, 2H, SiH).
Synthesis of ODMS30
D4 (20.1 g, 67.9 mmol), TMDS (1.53 g, 11.4 mmol), and triflic acid (40 μL) were reacted as above and quenched with the final addition of HMDS (94 μL). In this way, ODMS30 (16.5 g, 76% yield) was obtained. 1H NMR (δ, ppm): 0.04–0.10 (m, 180H, SiCH3), 0.18–0.20 (d, J = 2.7 Hz, 12H, OSi[CH3]2H), and 4.68–4.73 (m, 2H, SiH).
Synthesis of TES-ODMS17
ODMS17 (7.12 g, 5.11 mmol), VTEOS (0.976 g, 5.14 mmol), and Wilkinson’s catalyst (10 mg) in toluene (50 mL) were reacted as above. In this way, TES-ODMS17 (7.77 g, 96% yield) was obtained. 1H NMR (δ, ppm): 0.05–0.09 (m, 108H, SiCH3), 0.19 (d, J = 2.7 Hz, 6H, OSi[CH3]2H), 0.56 (s, 3H, SiCH2CH2), 1.09 (d, J = 7.5 Hz, 1H, SiCH2CH2), 1.19–1.25 (m, 9H, SiOCH2CH3), 3.82 (q, J = 7.0 Hz, 6H, SiOCH2CH3), and 4.68–4.72 (m, 1H, SiH).
Synthesis of TES-ODMS24
ODMS24 (13.0 g, 6.81 mmol), VTEOS (1.30 g, 6.84 mmol), and Wilkinson’s catalyst (10 mg) in toluene (50 mL) were reacted as above. In this way, TES-ODMS24 (13.8 g, 96% yield) was obtained. 1H NMR (δ, ppm): 0.04–0.11 (m, 150H, SiCH3), 0.18 (d, J = 2.7 Hz, 6H, OSi[CH3]2H), 0.56 (s, 3H, SiCH2CH2), 1.09 (d, J = 7.5 Hz, 1H, SiCH2CH2), 1.19–1.25 (m, 9H, SiOCH2CH3), 3.82 (q, J = 7.0 Hz, 6H, SiOCH2CH3), and 4.68–4.73 (m, 1H, SiH).
Synthesis of TES-ODMS30
ODMS30 (11.9 g, 5.06 mmol), VTEOS (0.966 g, 5.08 mmol), and Wilkinson’s catalyst (10 mg) in toluene (50 mL) were reacted as above. In this way, TES-ODMS30 (12.7 g, 98% yield) was obtained. 1H NMR (δ, ppm): 0.04–0.09 (m, 186H, SiCH3), 0.18 (d, J = 2.7 Hz, 6H, OSi[CH3]2H), 0.56 (s, 3H, SiCH2CH2), 1.09 (d, J = 7.5 Hz, 1H, SiCH2CH2), 1.19–1.25 (m, 9H, SiOCH2CH3), 3.82 (q, J = 7.0 Hz, 6H, SiOCH2CH3), and 4.68–4.72 (m, 1H, SiH).
Synthesis of m = 17
TES-ODMS17 (7.85 g, 4.96 mmol), A-PEO8M (2.10 g, 4.95 mmol), and Karstedt’s catalyst (50 μL) in toluene (100 mL) were reacted as above. In this way, m = 17 (7.76 g, 78% yield) was obtained. 1H NMR (δ, ppm): 0.03–0.09 (m, 114H, SiCH3), 0.48-0.53 (m, 2H, SiCH2CH2CH2), 0.55 (s, 3H, SiCH2CH2), 1.08 (d, J = 7.5 Hz, 1H, SiCH2CH2), 1.18–1.25 (m, 9H, SiOCH2CH3), 1.52–1.65 (m, 2H, SiCH2CH2CH2), 3.37 (s, 3H, OCH3), 3.41 (t, J = 7.2 Hz, 2H, SiCH2CH2CH2), 3.61–3.69 (m, 32H, OCH2CH2), and 3.74 (q, J = 7.0 Hz, 6H, SiOCH2CH3). IR (ν): no Si–H band.
Synthesis of m = 24
TES-ODMS24 (13.4 g, 6.38 mmol), A-PEO8M (2.70 g, 6.37 mmol), and Karstedt’s catalyst (50 μL) in toluene (80 mL) were reacted as above. In this way, m = 24 (14.3 g, 89% yield) was obtained. 1H NMR (δ, ppm): 0.04–0.09 (m, 156H, SiCH3), 0.48–0.54 (m, 2H, SiCH2CH2CH2), 0.56 (s, 3H, SiCH2CH2), 1.09 (d, J = 7.5 Hz, 1H, SiCH2CH2), 1.19–1.25 (m, 9H, SiOCH2CH3), 1.54–1.66 (m, 2H, SiCH2CH2CH2), 3.38 (s, 3H, OCH3), 3.41 (t, J = 7.1 Hz, 2H, SiCH2CH2CH2), 3.62–3.68 (m, 32H, OCH2CH2), and 3.82 (q, J = 7.1 Hz, 6H, SiOCH2CH3). IR (ν): no Si–H band.
Synthesis of m = 30
TES-ODMS30 (12.3 g, 4.83 mmol), A-PEO8M (2.05 g, 4.83 mmol), and Karstedt’s catalyst (50 μL) in toluene (100 mL) were reacted as above. In this way, m = 30 (12.6 g, 88% yield) was obtained. 1H NMR (δ, ppm): 0.04–0.12 (m, 192H, SiCH3), 0.48–0.54 (m, 2H, SiCH2CH2CH2), 0.55 (s, 3H, SiCH2CH2), 1.09 (d, J = 7.2 Hz, 1H, SiCH2CH2), 1.18–1.25 (m, 9H, SiOCH2CH3), 1.53–1.68 (m, 2H, SiCH2CH2CH2), 3.38 (s, 3H, OCH3), 3.41 (t, J = 7.2 Hz, 2H, SiCH2CH2CH2), 3.62–3.70 (m, 32H, OCH2CH2), and 3.82 (q, J = 7.0 Hz, 6H, SiOCH2CH3). IR (ν): no Si–H band.
Preparation of Surface-Grafted Films
Silicon wafers (1″ × 1″) were ultrasonically cleaned in acetone (10 min) followed by rinsing with acetone, repeating with DI water, and then drying in a 120 °C oven overnight. Next, wafers were placed in a 7:3 (v/v) concentrated H2SO4/30% H2O2 (Piranha) solution for 30 min (warning: Piranha must be handled with extreme caution), thoroughly washed with DI water, and dried under a stream of air. The resulting oxidized wafers were then each placed in a sealed jar containing the grafting solution comprised of the designated PEO-silane amphiphile, the PEO-control, or the siloxane-control in HPLC-grade toluene (0.048 M) and placed on a shaker table for 12 h. The grafted wafers were subsequently removed from the grafting solution, dried with a gentle stream of air, and annealed in a vacuum oven (36 mm Hg) at 150 °C for 12 h. To remove unbound chains, the wafers were subjected to sequential soaking (1 h) and sonication (3 min) with ethanol, the sequence repeated with DI water, and lastly dried under a stream of air. Grafted silicon wafers were analyzed by x-ray photoelectron spectroscopy (XPS), ellipsometry, and contact angle analysis. Grafted, silica-coated QCM-D sensors used for protein adsorption measurements were prepared as above following oxidation via exposure to oxygen plasma for 2 min (Harrick Plasma, PDC-00).
Preparation of bulk-modified silicone films
Microscope slides were sequentially washed with acetone, dichloromethane, and acetone and dried in a 100 °C oven for at least 2 h prior to use.
In a scintillation vial, MED-1137 was combined with hexane (1:3, wt:wt) and each PEO-silane amphiphile or the PEO-control at 50 μmol per gram of MED-1137 for a total of 7 solutions. Likewise, unmodified silicone was prepared without the addition of a PEO-silane amphiphile. The sealed vials were placed on a shaker table for 4 h to achieve homogeneous solutions.
Solutions were solvent-cast onto leveled glass microscope slides (1.5 mL per slide) and a polystyrene Petri dish cover placed on top of each, or into 24-well plates (0.25 mL per well) and the lid placed on top for protein and bacteria testing. In this way, solvent evaporation was slowed which prevented the formation of gas bubbles in the resulting cured coatings. Films were allowed to cure at RT. After five days, coated slides were immediately used for contact angle analysis. After seven days, coated slides were immediately used for transparency measurements, and coated well plates were used for fibrinogen and bacteria studies.
Characterization of surface-grafted films and silicone films
X-ray photoelectron spectroscopy (XPS)
Surface composition analysis of surface-grafted films was performed using a Kratos AXIS Ultra Imaging X-ray photoelectron spectrometer with a monochromatised Mg Kα source and operating at a base pressure of ~2% × 10-9 mbar. All analyses were performed over 7 × 3 mm. Survey spectra were obtained from 0 to 1100 eV to detect elements present at the surface of each silicon wafer. High resolution (HR) analyses with pass energy of 40 eV were performed at a take-off angle of 90° to determine elemental atomic percent composition. HR scans (180 s sweeps) were performed at 526 to 536 eV for O 1s, 280 to 295 eV for C 1s, and 96 to 106 eV for Si 2p. The raw data was quantified and analyzed using XPS Peak Processing software.
Ellipsometry
Ellipsometry measurements of surface-grafted films were performed using an Alpha-SE ellipsometer (J.A. Woollam Co., Inc.) with an incident angle of 70° in the spectral range of 380-900 nm and in the high-precision mode (30 sec data acquisition time). Using a standard two-layer (silica-silicon) optical model included in the manufacturer’s software, the average thickness of the silicon wafer oxide layer was determined at three different regions of five individual wafers. The obtained average oxide layer thickness of 2.01 nm is in agreement with literature values.62-64 To measure the thickness of the grafted chains, the oxide layer thickness was utilized in a second optical model that included the third “Cauchy layer” (polymer-silica-silicon). The index of refraction (n) of the PEO-silane amphiphiles and the PEO-control was set to that of crystalline PEO (n = 1.450).47, 65 Each reported thickness value (h) was based on three wafers, each measured at three different regions.
Tensile tests
Tensile properties of the silicone films were performed at RT on a tensile tester (Instron 3345). Rectangular specimens (~37.5 mm × ~5.5 mm × ~0.12 mm) were tested with a gauge length of 5.5 mm and at a crosshead speed of 500 mm/min. From the resulting stress versus strain curves, tensile modulus was determined as the slope from 20-100% strain. Measurements were completed in triplicate.
Transparency measurements
The transparency of silicone films was measured with an ultraviolet-visible (UV-Vis) spectrophotometer (Ocean Optics, Dunedin, FL) in transmittance mode from 390 to 700 nm. Percent transmittance was converted to absorbance, normalized to unmodified silicone based on film thickness, and then converted back to transmittance.
Contact angle measurements
For surface-grafted and silicone films, static (υstatic) contact angles of DI water at the surface-air interface were measured at RT with a CAM200 (KSV Instruments) goniometer equipped with an autodispenser, video camera, and drop-shape analysis software. υstatic of a sessile drop of water (5 μL) was measured at 0 sec, 15 sec, 1 min, and 2 min after deposition onto the coating surface for silicone films and at 0 sec and 2 min for surface-grafted films. The reported υstatic values are an average of three measurements taken from three different areas of the same sample.
Protein adsorption
Surface-grafted films on silica-coated sensors (50 nm thickness; Q-sense) were used for QCM-D measurements. QCM-D was performed with the following sequence: (1) 150 μL/min flow of PBS until the frequency and dissipation values remained constant for >5 min, (2) 150 μL/min flow of 100 μg/mL HF in PBS for 20 min, and (3) 150 μL/min flow of PBS for 5 min to remove loosely bound protein. The raw data was analyzed using Q-Sense software to determine the adsorbed mass of HF.
HF adsorption onto silicone films was measured using a modified immunosorbent assay. Three replicate coated wells of each composition were exposed to 0.15 mL of HF solution prepared in phosphate buffered saline (PBS) (3.0 mg/mL) and statically incubated for 1 h at 37 °C. The protein solution was removed and each well was rinsed three times with PBS before the addition of TBS-T20 (0.50 mL), which was incubated for 30 min at 37 °C. Wells were then rinsed three times with TBS-T20. Next, to each well was added 0.5 mL goat anti-fibrinogen (HRP)-conjugated polyclonal detection antibody (1:50,000 dilution in TBS-T20) and statically incubated for 1 h at 37 °C. Wells were then rinsed three times with TBS-T20. TMB di-HCl substrate solution (0.5 mL) was added and allowed to incubate for 30 min at 37 °C. To stop the reaction, 2 M H2SO4 was added to each well and plates were shaken on an orbital shaker at RT for 15 min. To quantify the amount of adsorbed HF on each surface, 0.15 mL of each resulting solution was transferred to a 96-well plate, absorbance was measured at 450 nm using a spectrophotometer (Tecan Safire2), and the value was compared to a HF standard curve (0.01 to 10,000 ng/mL). An unmodified silicone-coated slide served as a hydrophobic control with well-known high protein adsorption.
Biofilm growth and retention
Biofilm growth and retention on silicone films was characterized for bacteria S. epidermidis, S. aureus, E. coli, and P. aeruginosa as well as for the fungal pathogen C. albicans using a semi-automated, multi-well plate screening methodology.66-68 Overnight cultures of S. epidermidis and S. aureus in Tryptic Soy Broth (TSB) were re-suspended in 1X PBS to 0.6 OD600 and used to prepare 107-108 cells/mL suspensions in deionized (DI) water supplemented with 10.22 g/L Na2HPO4, 3.81 g/L KH2PO4, 1.01 g/L KCl, 0.793 g/L (NH4)2SO4, 0.06 g/L MgSO4, 0.5 g/L dextrose, 1.0 μg/L thiamine and 0.5/L μg biotin. For E. coli and P. aeruginosa, overnight cultures were prepared in LB broth, re-suspended in 1X PBS to 0.4 OD600 and used to prepare 107-108 cells/mL suspensions in minimal medium M63 + 2 g/L dextrose (E. coli) and 0.6 g/L TSB in deionized water (P. aeruginosa). An overnight culture of C. albicans in 1X YNB + 100 mM dextrose was re-suspended in 1X PBS to 0.6 OD600 and used to prepare a 107-108 cells/mL suspension in 1:10 RPMI 1640 medium/deionized water.
One milliliter of the final bacterial and fungal cell suspensions was added to three replicate films prepared 24-well plates66 and incubated statically at 37°C for 72 hr for S. aureus and S. epidermidis and 24 hr for the remaining microorganisms to promote cell attachment and biofilm growth. The films were rinsed 3x with 1X PBS, dried at ambient laboratory conditions for 1 hr and stained with 0.5 mL of a CV dye solution (0.35% in DI water) for 15 min. Excess CV solution was removed and films were rinsed 3x with DI water, dried for 1 hr at ambient laboratory conditions and extracted in 0.5 mL of 33% glacial acetic acid for 15 min to solubilize CV dye bound to retained biofilm. A 0.15 mL aliquot of acetic acid extract from each silicone film was transferred to 96-well plate and measured for absorbance at 600 nm using a multi-well plate spectrophotometer. Three replicates for each film composition were normalized to an assay control (i.e. growth media without microorganism) and the average value was reported for each microorganism.
CONCLUSIONS
Silicones having broad spectrum anti-biofouling behavior towards fibrinogen as well as to bacteria and fungus are expected to improve efficacy and safety when used for implanted medical devices. Herein, we demonstrate the comprehensive anti-biofouling behavior of PEO-silane amphiphiles [α-(EtO)3Si(CH2)2-oligo-dimethylsiloxanem-block-(OCH2CH2)8-OCH3] when used to bulk-modify silicone. While previously prepared with shorter siloxane tether lengths (m = 0, 4, 13), PEO-silane amphiphile SMAs were successfully prepared with an extended siloxane tether length range (m = 17, 24, and 30) using a regioselective hydrosilylation strategy. Additionally, these syntheses were carried out at a multi-gram scale, demonstrating the robustness of the technique. While the protein resistance of PEO grafted to model substrates is well documented, if fails to reveal the ability of PEO to undergo the necessary water-driven surface migration when blended into silicones. Indeed, when PEO-silane amphiphiles were grafted onto silicon wafer, their resulting increased surface hydrophobicity contributed to greater adsorption of HF protein versus grafted PEO-silane (i.e. PEO-control) surfaces. In contrast, silicones bulk-modified with PEO-silane amphiphiles underwent rapid and substantial water-driven surface restructuring, resulting in very hydrophilic surfaces indicative of PEO-enrichment. The PEO-control failed to produce this effect. The enhanced surface-restructuring potential is hypothesized to stem for the molecular mobility and similar solubility of the siloxane tethers and the silicone matrix. Surface hydrophilicity was somewhat maximized for silicones modified with PEO-silane amphiphiles of intermediate tether lengths (m = 13 and 17), perhaps due to steric limitations of longer tethers during restructuring. A topic of future studies for this series, a preliminary report demonstrated the maintenance of surface hydrophilicity and protein resistance for silicones modified with m = 13 and m = 30 after first conditioning in water for 2 weeks.69 While contact angle analysis was utilized to observe water-driven surface restructuring, other factors can contribute to the biological adhesiveness. Thus, we did not predict that contact angle and biological accumulation would correlate in a scalable fashion. Still, a lack of significant surface hydrophilicity nearly always coincided with poor fouling resistance. For example, due to a lack of surface-restructuring, silicones bulk-modified with the PEO-control remained quite hydrophobic and also exhibited poor resistance to HF adsorption and microbial biofilm growth. Silicones modified with m = 0 also failed to achieve a high resistance to HF adsorption as well as to S. epidermis biofilm growth relative to unmodified silicone. Against, E. coli and C. albicans, this modified silicone was less effective at controlling fouling levels versus those modified with PEO-silane amphiphiles of longer tether lengths. Interestingly, for P. aeruginosa, only silicone modified with m = 0 exhibited statistically significantly lower biofilm level versus unmodified silicone. This may be attributed the high adherence of some P. aeruginosa strains to hydrophilic surfaces, like PEO brushes. However, overall, PEO-silane amphiphiles with a siloxane tether length of m ≥ 4 were highly effective in reducing HF adsorption and microbial biofilm growth. Optical transparency, advantageous for ocular and sensing type implanted devices, was best for silicones when modified with PEO-silane amphiphiles whose tether lengths were m ≥ 13. In this way, PEO-silane amphiphiles with moderate to longer tether lengths are effective SMAs for silicones and may improve safety and efficacy when used to form implanted devices.
Supplementary Material
Acknowledgments
We thank Prof. Karen Wooley (Texas A&M University, Department of Chemistry) for GPC and QCM-D use and Prof. Jaime Grunlan (Texas A&M University, Department of Mechanical Engineering) for UV-Vis spectrophotometer use.
The authors thank the Texas A&M Engineering and Experiment Station (TEES) for financial support of this research. M.L. Hawkins thanks the Texas A&M University (TAMU) Diversity Fellowship. M.A. Rufin gratefully acknowledges the NIH (3R01DK95101-02S1). S.J. Stafslien gratefully acknowledges the Office of Naval Research (N00014-17-1-2153).
Footnotes
Electronic Supplementary Information (ESI) available: Additional figures and tables, experimental details, GPC chromatographs, % atomic composition by XPS, HR C 1s XPS spectra, contact angle values for surface grafted films, UV-VIS transmission spectra of silicone films, contact angle values for silicone films, protein adsorption values for silicone films, crystal violet absorbance values of stained biofilms for silicone films. This material is available free of charge via the Internet at http://pubs.acs.org.]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Curtis J, Colas A. In: Biomaterials Science: An Introduction to Materials in Medicine. 2. Ratner BD, Hoffman AS, Schoen FJ, Lemons JE, editors. Elsevier Academic Press; San Diego, CA: 2004. pp. 697–707. [Google Scholar]
- 2.VanDyke ME, Clarson SJ, Arshady R. In: An Introduction to Polymeric Biomaterials. Arshady R, editor. Vol. 1. Citrus Books; London: 2003. pp. 109–135. [Google Scholar]
- 3.El-Zaim HS, Heggers JP. In: Polymeric Biomaterials. 2. Dumitriu S, editor. Marcel Dekker, Inc.; New York, NY: 2002. pp. 79–90. [Google Scholar]
- 4.Feldman K, Hähner G, Spencer ND, Harder P, Grunze M. J Amer Chem Soc. 1999;121:10134–10141. [Google Scholar]
- 5.Anderson JM, Ziats NP, Azeez A, Brunstedt MR, Stack S, Bonfield TL. J Biomater Sci Polym Ed. 1995;7:159–169. doi: 10.1163/156856295x00670. [DOI] [PubMed] [Google Scholar]
- 6.Brash JL. Annals New York Acad Sci. 1977;283:356–371. [Google Scholar]
- 7.Wu H, Moser C, Wang H-Z, Hoiby N, Song Z-J. Int J Oral Sci. 2014;7:1–7. doi: 10.1038/ijos.2014.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Percival SL, Suleman L, Vuotto C, Donelli G. J Med Microbio. 2015;64:323–334. doi: 10.1099/jmm.0.000032. [DOI] [PubMed] [Google Scholar]
- 9.Raad I, Hanna H, Maki D. The Lancet Infect Dis. 2007;7:645–657. doi: 10.1016/S1473-3099(07)70235-9. [DOI] [PubMed] [Google Scholar]
- 10.Lee JH, Lee HB, Andrade JD. Prog Polym Sci. 1995;20:1043–1079. [Google Scholar]
- 11.Jeon SI, Lee JH, Andrade JD, DeGennes PG. J Colloid Interf Sci. 1991;142:149–158. [Google Scholar]
- 12.Knoll D, Hermans J. J Biol Chem. 1983;258:5710–5715. [PubMed] [Google Scholar]
- 13.Poly(ethylene glycol) Chemistry: Biotechnical and Biomedical Applications. Plenum Press; New York: 1992. [Google Scholar]
- 14.Browning MB, Cereceres SN, Luong PT, Cosgriff-Hernandez EM. J Biomed Mater Res A. 2014;102:4244–4251. doi: 10.1002/jbm.a.35096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prime KL, Whitesides GM. J Amer Chem Soc. 1993;115:10714–10721. [Google Scholar]
- 16.Pale-Grosdemange C, Simon ES, Prime KL, Whitesides GM. J Amer Chem Soc. 1991;113:12–20. [Google Scholar]
- 17.Zhang M, Ferrari M. Biomed Microdev. 1998;1:81–89. [Google Scholar]
- 18.Zhang M, Desai T, Ferrari M. Biomaterials. 1998;19:953–960. doi: 10.1016/s0142-9612(98)00026-x. [DOI] [PubMed] [Google Scholar]
- 19.Papra A, Gadegaard N, Larsen NB. Langmuir. 2001;17:1457–1460. [Google Scholar]
- 20.Lee S-W, Laibinis PE. Biomaterials. 1998;19:1669–1675. doi: 10.1016/s0142-9612(98)00044-1. [DOI] [PubMed] [Google Scholar]
- 21.Jo S, Park K. Biomaterials. 2000;21:605–616. doi: 10.1016/s0142-9612(99)00224-0. [DOI] [PubMed] [Google Scholar]
- 22.Owen MJ, Smith PJ. J Adhes Sci Technol. 1994;8:1063–1075. [Google Scholar]
- 23.Owen MJ. In: Siloxane Polymers. Clarson SJ, Semlyen JA, editors. Prentice Hall; Englewood Cliffs: 1993. p. 309. [Google Scholar]
- 24.Lane TH, Burns SA. In: Immunology of silicones. Potter M, Rose NR, editors. Ch. 7 Springer; Berlin: 1996. [Google Scholar]
- 25.Chen H, Brook MA, Sheardown H. Biomaterials. 2004;25:2273–2282. doi: 10.1016/j.biomaterials.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 26.Chen H, Brook MA, Chen Y, Sheardown H. J Biomater Sci Polym Ed. 2005;16:531–548. doi: 10.1163/1568562053700183. [DOI] [PubMed] [Google Scholar]
- 27.Thompson DB, Fawcett AS, Brook MA. In: Silicon Based Polymers. Ganachaud F, Boileau S, Boury B, editors. Springer; 2008. pp. 29–38. [Google Scholar]
- 28.Chen H, Zhang Z, Chen Y, Brook MA, Sheardown H. Biomaterials. 2005;26:2391–2399. doi: 10.1016/j.biomaterials.2004.07.068. [DOI] [PubMed] [Google Scholar]
- 29.Lee JH, Ju YM, Lee WK, Park KD, Kim YH. J Biomed Mater Res Part A. 1998;40:314–323. doi: 10.1002/(sici)1097-4636(199805)40:2<314::aid-jbm17>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 30.Park K, Shim HS, Dewanjee MK, Eigler NL. J Biomater Sci Polym Ed. 2000;11:1121–1134. doi: 10.1163/156856200744228. [DOI] [PubMed] [Google Scholar]
- 31.Murthy R, Cox CD, Hahn MS, Grunlan MA. Biomacromolecules. 2007;8:3244–3252. doi: 10.1021/bm700543c. [DOI] [PubMed] [Google Scholar]
- 32.Hawkins ML, Grunlan MA. J Mater Chem. 2012;22:19540–19546. [Google Scholar]
- 33.Hawkins ML, Rufin MA, Raymond JE, Grunlan MA. J Mater Chem B. 2014;2:5689–5697. doi: 10.1039/c4tb01008f. [DOI] [PubMed] [Google Scholar]
- 34.Tanzi MC. Expert Rev Med Devices. 2005;2:473–492. doi: 10.1586/17434440.2.4.473. [DOI] [PubMed] [Google Scholar]
- 35.Rufin MA, Gruetzner JA, Hurley MJ, Hawkins ML, Raymond ES, Raymond JE, Grunlan MA. J Mater Chem B. 2015;3:2816–2825. doi: 10.1039/C4TB02042A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsai W-B, Grunkemeier JM, Horbett TA. J Biomed Mater Res. 1999;44:130–139. doi: 10.1002/(sici)1097-4636(199902)44:2<130::aid-jbm2>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 37.Bozukova D, Pagnoulle C, De Pauw-Gillet M-C, Desbief S, Lazzaroni R, Ruth N, Jérôme R, Jérôme C. Biomacromolecules. 2007;8:2379–2387. doi: 10.1021/bm0701649. [DOI] [PubMed] [Google Scholar]
- 38.Cao B, Li L, Wu H, Tang Q, Sun B, Dong H, Zhe J, Cheng G. Chem Commun. 2014;50:3234–3237. doi: 10.1039/c3cc48878k. [DOI] [PubMed] [Google Scholar]
- 39.Nagashima H, Tatebe K, Ishibashi T, Sakakibara J, Itoh K. Organometallics. 1989;8:2495–2496. [Google Scholar]
- 40.Crivello JV, Bi D. J Polym Sci Part A. 1993;31:2563–2572. [Google Scholar]
- 41.Crivello JV, Bi D. J Polym Sci Part A. 1993;31:2729–2737. [Google Scholar]
- 42.Crivello JV, Bi D. J Polym Sci Part A. 1993;31:3109–3119. [Google Scholar]
- 43.Crivello JV, Bi D. J Polym Sci Part A. 1993;31:3121–3132. [Google Scholar]
- 44.Moreau O, Portella C, Massicot F, Herry JM, Riquet AM. Surf Coat Tech. 2007;201:5994–6004. [Google Scholar]
- 45.Cole MA, Thissen H, Losic D, Voelcker NH. Surf Sci. 2007;601:1716–1725. [Google Scholar]
- 46.Sofia SJ, Premnath V, Merrill EW. Macromolecules. 1998;31:5059–5070. doi: 10.1021/ma971016l. [DOI] [PubMed] [Google Scholar]
- 47.Unsworth LD, Tun Z, Sheardown H, Brash JL. J Colloid Interf Sci. 2005;281:112–121. doi: 10.1016/j.jcis.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 48.Feng W, Brash JL, Zhu S. Biomaterials. 2006;27:847–855. doi: 10.1016/j.biomaterials.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 49.Sharma S, Johnson RW, Desai TA. Appl Surf Sci. 2003;206:218–229. [Google Scholar]
- 50.Zdyrko B, Klep V, Luzinov I. Langmuir. 2003;19:10179–10187. [Google Scholar]
- 51.Zengin A, Caykara T. Thin Solid Films. 2011;519:3135–3140. [Google Scholar]
- 52.Wagner ML, Tamm LK. Biophys J. 2000;79:1400–1414. doi: 10.1016/S0006-3495(00)76392-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Allen C, Dos Santos N, Gallagher R, Chiu GNC, Shu Y, Li WM, Johnstone SA, Janoff AS, Mayer LD, Webb MS, Bally MB. Biosci Rep. 2002;22:225–250. doi: 10.1023/a:1020186505848. [DOI] [PubMed] [Google Scholar]
- 54.Sangermano M, Bongiovanni R, Malucelli G, Priola A, Pollicino A, Recca A. J Appl Polym Sci. 2003;89:1524–1529. [Google Scholar]
- 55.Lord MS, Stenzel MH, Simmons A, Milthorpe BK. Biomaterials. 2006;27:1341–1345. doi: 10.1016/j.biomaterials.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 56.Cooper MA, Singleton VT. J Mol Recognit. 2007;20:154–184. doi: 10.1002/jmr.826. [DOI] [PubMed] [Google Scholar]
- 57.Hemmersam AG, Foss M, Chevallier J, Besenbacher F. Colloid Surf B. 2005;43:208–215. doi: 10.1016/j.colsurfb.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 58.Geelhood SJ, Horbett TA, Ward WK, Wood MD, Quinn MJ. J Biomed Mater Res Part B Appl Biomater. 2007;81B:251–260. doi: 10.1002/jbm.b.30660. [DOI] [PubMed] [Google Scholar]
- 59.Lu DR, Park K. J Colloid Interf Sci. 1991;144:271–281. [Google Scholar]
- 60.Roosjen A, Busscher HJ, Norde W, Mei HCvd. Microbiol. 2006;152:2673–2682. doi: 10.1099/mic.0.29005-0. [DOI] [PubMed] [Google Scholar]
- 61.Grunlan MA, Mabry JM, Weber WP. Polymer. 2003;44:981–987. [Google Scholar]
- 62.Kohli P, Blanchard GJ. Langmuir. 2000;16:4655–4661. [Google Scholar]
- 63.Fang SJ, Chen W, Yamanaka T, Helms CR. J Electrochem Soc. 1997;144:L231–L233. [Google Scholar]
- 64.Guhathakurta S, Subramanian A. J Electrochem Soc. 2007;154:P136–P146. [Google Scholar]
- 65.Murthy R, Shell CE, Grunlan MA. Biomaterials. 2009;30:2433–2439. doi: 10.1016/j.biomaterials.2009.01.051. [DOI] [PubMed] [Google Scholar]
- 66.Stafslien SJ, Bahr JA, Feser JM, Weisz JC, Chisholm BJ, Ready TE, Boudjouck P. J Comb Chem. 2006;8:156–162. doi: 10.1021/cc050047m. [DOI] [PubMed] [Google Scholar]
- 67.Kugel AJ, Jarabek LE, Daniels JW, VanderWal LJ, Ebert SM, Jepperson MJ, Stafslien SJ, Pieper RJ, Webster DC, Bahr J, Chisholm BJ. J Coat Technol Re. 2009;6:107–121. [Google Scholar]
- 68.Majumdar P, Lee E, Gubbins N, Christianson DA, Stafslien SJ, Daniels J, VanderWal L, Bahr J, Chisholm BJ. J Comb Chem. 2009;11:1115–1127. doi: 10.1021/cc900114e. [DOI] [PubMed] [Google Scholar]
- 69.Rufin MA, Ngo BKD, Barry ME, Page VM, Hawkins ML, Stafslien SJ, Grunlan MA. Green Materials. 2017;5:1–10. doi: 10.1680/jgrma.16.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.