

Abstract
Plasticity of the cell state has been proposed to drive resistance to multiple classes of cancer therapies, thereby limiting their effectiveness1–4. A high-mesenchymal cell state observed in human tumours and cancer cell lines has been associated with resistance to multiple treatment modalities across diverse cancer lineages, but the mechanistic underpinning for this state has remained incompletely understood1–6. Here we molecularly characterize this therapy-resistant high-mesenchymal cell state in human cancer cell lines and organoids and show that it depends on a druggable lipid-peroxidase pathway that protects against ferroptosis, a nonapoptotic form of cell death induced by the build-up of toxic lipid peroxides7,8. We show that this cell state is characterized by activity of enzymes that promote the synthesis of polyunsaturated lipids. These lipids are the substrates for lipid peroxidation by lipoxygenase enzymes8,9. This lipid metabolism creates a dependency on pathways converging on the phospholipid glutathione peroxidase (GPX4), a selenocysteine-containing enzyme that dissipates lipid peroxides and thereby prevents the iron-mediated reactions of peroxides that induce ferroptotic cell death8. Dependency on GPX4 was found to exist across diverse therapy-resistant states characterized by high expression of ZEB1, including epithelial-mesenchymal transition in epithelial-derived carcinomas, TGFβ-mediated therapy- resistance in melanoma, treatment-induced neuroendocrine transdifferentiation in prostate cancer, and sarcomas, which are fixed in a mesenchymal state owing to their cells of origin. We identify vulnerability to ferroptic cell death induced by inhibition of a lipid peroxidase pathway as a feature of therapy-resistant cancer cells across diverse mesenchymal cell-state contexts.
We hypothesized that the existence of distinct cell states in cultured cells1,2,4 could be exploited to discover vulnerabilities of therapy-resistant cell states by mining cancer cell-line sensitivity data for compounds whose activities correlate with bulk cell-line expression of resistance markers. To explore this, we selected three cellular and patient-derived signatures of high mesenchymal state proposed to yield a therapy-resistant state5,6,10. We first computed a mesenchymal score for 516 epithelium-derived cancer cell lines (carcinomas) used to generate small-molecule sensitivity measurements available in the Cancer Therapeutics Response Portal (CTRP at http://portals.broadinstitute.org/ctrp.v2.2/) (Supplementary Table 1). Geneexpression data were obtained from the CCLE portal (https://portals.broadinstitute.org/ccle/home)11. We performed single-sample gene set enrichment analysis12 using gene-expression signatures of mesenchymal states associated with therapy resistance5,6,10. Correlating these cell-line mesenchymal scores with an area-under-the-curve (AUC) metric of cell-line sensitivity (from 16 concentrations measured in duplicate) to each of 481 performance-diverse compounds yielded a range of compound sensitivity-mesenchymal score correlation coefficients13 (Fig. 1a and Supplementary Table 2). Tyrosine kinase inhibitors known to inhibit epithelial cancer cell targets (for example, epidermal growth factor receptor) were among the compounds most correlated with epithelial cell-state sensitivity, increasing our confidence in this approach (Fig. 1a). Also, ML239, a compound that was initially identified for its ability to preferentially kill breast cancer cells that had been induced to undergo EMT14, and has recently been shown to increase levels of unsaturated lipids in cancer cells13, correlated with mesenchymal state sensitivity, further validating this strategy (Fig. 1a and Supplementary Table 2).
Figure 1. Gene-signature, proteomic and lineage-based correlation analyses identify mesenchymal state-targeting compounds.
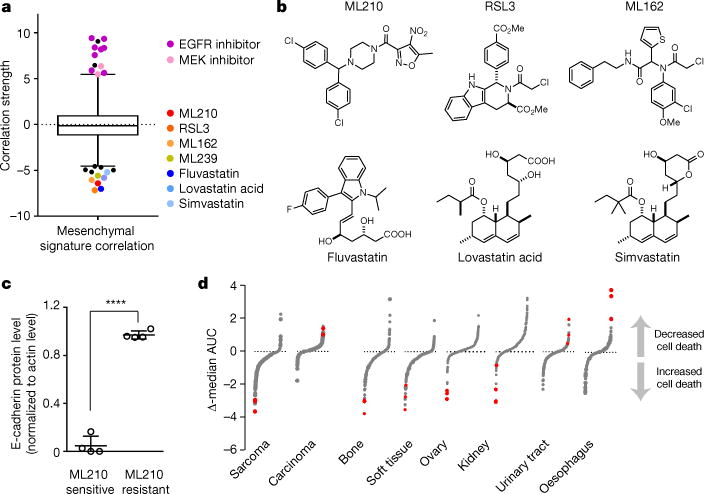
a, Box-and-whisker plots show the extent of correlation between cytotoxic effects of each compound (black and coloured dots) and cell-line mesenchymal score (516 carcinoma cell lines). Plotted values are z scored Pearson’s correlation coefficients (see Methods); line, median; box, 25th–75th percentile; whiskers, 2.5th and 97.5th percentile expansion; dotted line marks 0.0; P < 0.005. b, Structures of mesenchymal statetargeting compounds. c, Column scatterplot with mean ± s.d. showing the relationship between cell-line levels of E-cadherin protein and sensitivity to a mesenchymal state-targeting compound (ML210) across a panel of pancreatic and gastric cancer-derived cell lines (P < 0.0001). d, Relative activity of each of 481 compounds in the indicated lineage (see Methods). Ferroptosisinducing subset of mesenchymal state-correlated compounds (red dots) are preferentially active in sarcoma (mesenchymal-derived) compared to carcinoma (epithelial-derived), as well as among cancer cell lines derived from individual mesenchymal-origin versus epithelial lineages.
In addition to the lipid-modifying compound ML239 (ref. 13), two distinct classes of compounds strongly correlated with selective cell death induction of epithelial cancer-derived cell lines with highmesenchymal state gene-expression scores (Fig. 1a). The first class comprises RSL3, ML210 and ML162, compounds known to induce ferroptosis, a non-apoptotic, oxidative form of regulated cell death involving lipid hydroperoxides7,8 (Fig. 1b). The second class comprises fluvastatin, lovastatin acid and simvastatin, compounds (statins) that inhibit 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR) (Fig. 1b). We refer to these seven compounds collectively as mesenchymal state-targeting compounds.
The correlation of low protein levels of E-cadherin, a canonical epithelial state marker, and high protein levels of vimentin, a marker of mesenchymal states, with cell-line sensitivity to mesenchymal statetargeting compounds across a panel of eight pancreatic and gastric cancer cell lines provided further validation of the mesenchymal statetargeting effects of these compounds (Fig. 1c and Extended Data Figs 1, 2). In addition to exhibiting selectivity for epithelial cancer-derived cell lines with high expression of mesenchymal state genes, the ferroptosis-inducing subset of mesenchymal state-targeting compounds were the most selective compounds for cancer cell lines of mesenchymal origin (sarcomas) (Fig. 1d). Cell lines derived from ovarian and kidney cancers also demonstrated selective targeting by ferroptosis-inducing mesenchymal state-targeting compounds (Fig. 1d). We hypothesize that this stems from the mesenchymal mesonephric embryonal origin of the ovaries and kidneys, with retention of a mesenchymal state-vulnerability in ovarian and kidney cancers arising from these tissues15,16.
Figure 2. Mesenchymal state-targeting compounds act on a lipidperoxidase pathway converging on GPX4.
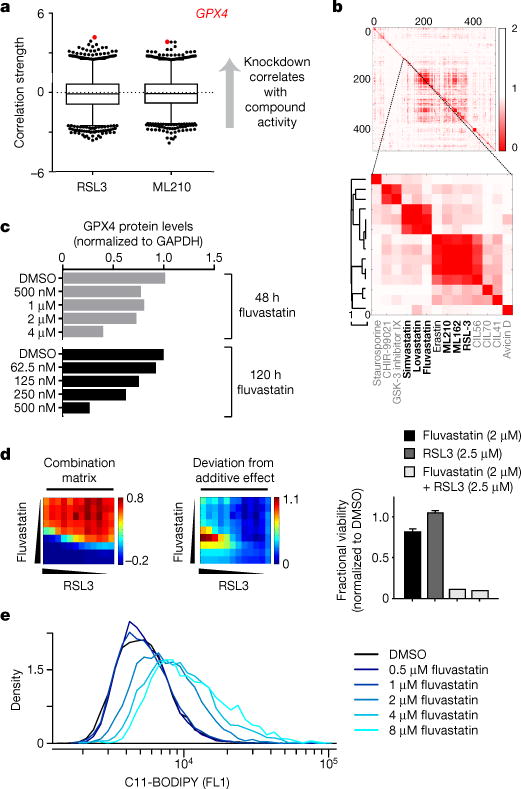
a, Box-and-whisker plots show the extent of correlation between the indicated compound’s cytotoxic effects and sensitivity to knockdown of genes (black dots) across 136 (RSL3) and 132 (ML210) cancer cell lines (excluding haematopoietic cell lines). Plotted values are z scored Pearson’s correlation coefficients (see Methods), line, median; box, 25th–75th percentile; whiskers, 1st and 99th percentile expansions; dotted line marks 0.0; P < 0.0002 for coloured dots. b, Hierarchical clustering of a pairwise similarity matrix for 481 compounds, where each matrix element represents the strength of correlation between the sensitivity patterns of two compounds in up to 664 cell lines11. Black compounds are those that fall into the two clusters of interest. The grey compounds fall outside these two clusters. c–e, Fluvastatin treatment of HT-1080 cells decreases GPX4 protein levels in a time-and concentration-dependent manner (c) synergizes with a direct GPX4 inhibitor (RSL3) (d) and leads to accumulation of lipid hydroperoxides (e). Bar graphs for fluvastatin + RSL3 in d are two technical replicates (fractional viability normalized to DMSO); other bar graphs shown mean ± s.d. of four technical replicates. Data in c–e are representative of two independent biological experiments.
Using publically available genome-wide short hairpin RNA (shRNA) sensitivity data for 349 cancer cell lines (http://portals.broadinstitute.org/achilles), we found that cell-line sensitivity to knockdown of GPX4 was the top correlate of sensitivity to the ferroptosis-inducing subset of compounds (Fig. 2a and Extended Data Fig. 3). This suggested that the distinguishing feature of these compounds lies in their unique ability to inhibit GPX4, a hypothesis that was particularly attractive in light of the identification of GPX4 as the relevant direct target of RSL3 (ref. 8). GPX4, one of 25 selenocysteine-containing proteins encoded in the human genome17, uses glutathione as a cofactor to reduce allylic lipid hydroperoxides to their corresponding alcohols8. Using a lysate-based assay that reports on the ability of cellular GPX4 to reduce exogenous phosphatidylcholine hydroperoxide, we were able to confirm for RSL3 (ref. 8) and to demonstrate for ML210 and ML162, that these mesenchymal state-targeting compounds inhibit GPX4 activity (Extended Data Fig. 4).
Statins, which are small-molecule inhibitors of HMGCR, may mediate their selective cell death induction of high-mesenchymal state cancer cells (Fig. 1a) through their effects on isopentenylation of the selenocysteine-charged transfer RNA (tRNA) as part of selenocysteine-tRNA maturation18. By inhibiting the production of isopentenyl pyrophosphate through the mevalonate pathway, statins are known to inhibit selenoprotein biosynthesis18. The adjacent clustering of statins and direct GPX4 inhibitors in a hierarchical clustering of the CTRP AUC-similarity matrix11 (Fig. 2b), which aims to identify compounds that have similar patterns of sensitivity across hundreds of cancer cell lines, supported a mechanism of action for statins that is closely related to, albeit distinct from, that of direct GPX4 inhibitors. Treatment of cells with fluvastatin led to decreased expression of GPX4 in a time-and concentration-dependent manner (Fig. 2c and Supplementary Fig. 1) and synergized with co-treatment with the direct GPX4 inhibitor RSL3 (Fig. 2d). Unlike the effects of direct GPX4 inhibitors, statin treatment could not be rescued by lipophilic antioxidants (Extended Data Fig. 5). However, statin treatment led to increased levels of cellular lipid peroxidation (Fig. 2e), consistent with inhibition of GPX4 protein expression8.
The existence of not just one, but multiple high-mesenchymal cancer cell states derived from different inputs has become apparent in recent years19. To gain a deeper insight into the relationship between these states and GPX4 dependency, we analysed several isogenic cell-line systems that model cancer-relevant mesenchymal states. High-mesenchymal state cancer cells derived from artificially enforced expression of the EMT transcription factors SNAIL1 (also known as SNAI1) or TWIST1 (refs 20, 21) did not show consistent sensitization to GPX4 inhibition (Extended Data Fig. 6). By contrast, we found that the EMT regulator and lipogenic factor ZEB1 (refs 22, 23) was strongly correlated with mesenchymal state sensitivity to GPX4 inhibition (Fig. 3a). More importantly, ZEB1 deletion abolished sensitivity to GPX4 inhibition (Fig. 3b, Extended Data Fig. 7 and Supplementary Fig. 1), showing that ZEB1 is required for the dependency of high-mesenchymal state cells on GPX4. High-mesenchymal state populations arising in response to targeted therapy represent an additional isogenic model of therapy-resistant mesenchymal cancer cell states24. We found that HCC4006 non-small-cell lung cancer cells that have acquired a high-mesenchymal state together with resistance to gefinitib24 were preferentially sensitive to GPX4 inhibition compared with parental HCC4006 cells (Fig. 3c and Extended Data Fig. 8). Two patient-derived pancreatic cancer cell lines were found to have GPX4 inhibitor sensitivity that correlated positively with levels of mesenchymal state protein markers and inversely with sensitivity to erlotinib (Extended Data Fig. 1). Neuroendocrine transdifferentiation in the setting of prostate cancer has been shown to yield therapy-resistant cells with increased expression of mesenchymal state genes25,26. Across a panel of patient-derived prostate cancer organoids and benign prostate cells exhibiting a range of epithelial versus mesenchymal characteristics25, we found therapy-induced, high ZEB1 neuroendocrine transition to be associated selectively with GPX4 dependency (Fig. 3d and Extended Data Fig. 8).
Figure 3. Validation of dependency of therapy-resistance-associated high-mesenchymal state cancer cells on GPX4.
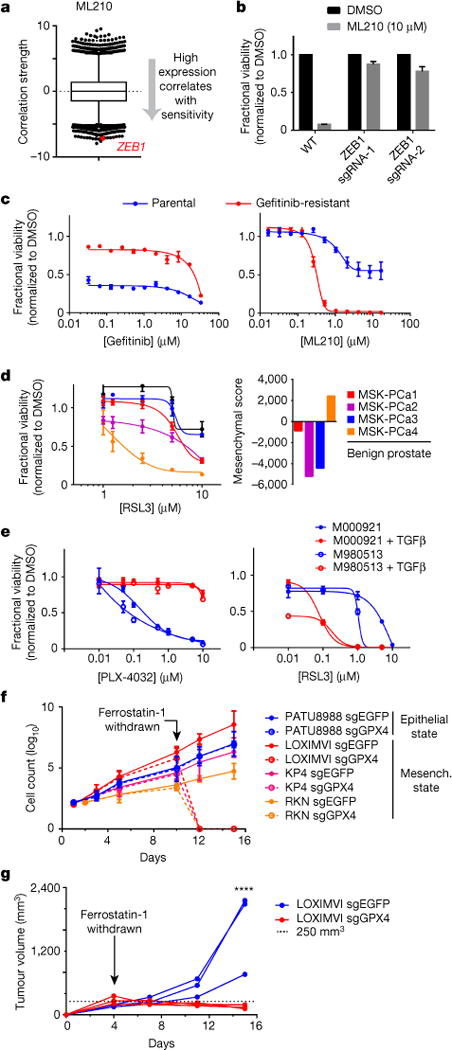
a, High baseline ZEB1 transcripts levels (red dot) are strongly correlated with sensitivity to a GPX4 inhibitor (ML210) across 610 cancer cell lines (excludes nonadherent cell lines). Plotted values are z scored Pearson’s correlation coefficients (see Methods); line, median; box, 25th–75th percentile; whiskers, 1st and 99th percentile expansion; dotted line marks 0.0. b, Knockout of ZEB1 prevents cell death induced by GPX4 inhibition in KP4 high-mesenchymal state pancreatic cancer cells. c, HCC4006 lung cancer cells that have acquired a high-mesenchymal state concomitant with becoming resistant to gefitinib (red curve) exhibit increased sensitivity to GPX4 inhibition. d, A patient-derived prostate cancer organoid exhibiting high mesenchymal and ZEB1 gene expression related to therapy-induced neuroendocrine transition (MSK-PCa4)25 is selectively sensitive to GPX4 inhibition (three technical replicates representative of two separate experiments). e, Patient-derived melanoma lines induced into a therapy-resistant mesenchymal state with TGFβ treatment acquire resistance to BRAF inhibition and sensitivity to GPX4 inhibition. f, In vitro growth curves of GPX4 wild-type (sgEGFP) and GPX4 knockout (sgGPX4) cancer cells in the presence or absence of a lipophilic antioxidant (ferrostatin-1). g, In vivo growth of GPX4-wild-type (sgEGFP; blue curves) and GPX4-knockout (sgGPX4; red curves) melanoma xenografts in mice dosed with ferrostatin-1 and upon cessation of dosing with ferrostatin-1. Individual curves represent individual mice with one xenograft each. ****P < 0.0001 at day 15. Data reflect a single cohort experiment. Data plotted in b–f are mean ± s.d. of three (b, d–f) or four (c) technical replicates and are representative of two separate biological experiments.
In the context of BRAF-mutant melanoma, a non-mutational cell state observed in human tumours has been implicated in resistance to targeted therapy and immunotherapy1–4. This melanoma therapy-resistant state resembles that of activated fibroblasts (for example, myofibroblasts) and is characterized by increased expression of mesenchymal markers including AXL, decreased expression of the melanocyte lineage-specific transcription factor MITF and can be induced in therapy-sensitive cells by treatment with TGFβ1. We found that a gene-expression signature2 of this melanoma therapy-resistant state correlated strongly with sensitivity to GPX4 inhibitors within the CTRP (Extended Data Fig. 8) and that treatment of two therapy-sensitive patient-derived melanoma lines27 with TGFβ caused resistance to BRAF inhibition and induced GPX4 dependency (Fig. 3e).
While attempting to demonstrate that GPX4 remains intact as a mesenchymal state dependency in vivo, we were confronted by pharmacokinetic liabilities of existing GPX4 inhibitors that preclude their use in animals8. To circumvent this challenge, we used a genetic approach that exploits the ability of lipophilic antioxidants (for example, vitamin E (ref. 8), ferrostatin-1 (ref. 7)) to rescue the effects of GPX4 loss. We used CRISPR-Cas9 technology to knockout GPX4 across a panel of cancer cell lines (Extended Data Fig. 7 and Supplementary Fig. 1) with a range of mesenchymal scores. This was achieved in the presence of ferrostatin-1, allowing the survival of GPX4-knockout cells that are dependent on GPX4. Both epithelial and mesenchymal state GPX4-knockout cancer cells proliferated at rates comparable to their wild-type (GPX4-wild-type) counterparts when in the presence of ferrostatin-1 (Fig. 3f). Withdrawal of ferro-statin-1 uncovered the loss of GPX4 function, inducing ferroptosis in mesenchymal state GPX4-knockout cells, whereas epithelial state GPX4-knockout cells remained unaffected (Fig. 3f).
We adapted this approach to an in vivo context by optimizing a ferrostatin-1 dosing strategy for mice that enabled the survival of GPX4-dependent GPX4-knockout cancer cells injected subcutaneously. We generated xenografts of both GPX4-wild-type and GPX4-knockout clones of the high-mesenchymal state, therapy-resistant melanoma cell line LOXIMVI (Fig. 3g). Cessation of ferrostatin-1 dosing led to tumour regression selectively in the GPX4-knockout xenograft, whereas the GPX4-wild-type xenograft continued to grow rapidly (Fig. 3g). These results demonstrate in a cell-autonomous context that mesenchymal state dependency on GPX4 remains intact in vivo, despite potential differences between in vitro and in vivo metabolic contexts.
GPX4 dependency was more pronounced in cancer cells adopting a therapy-resistant mesenchymal state than across a panel of non-transformed mesenchymal cell lines (Extended Data Fig. 9). We have discovered that this differential sensitivity can be further expanded by pre-treatment of cells with lipophilic antioxidants such as vitamin E (Extended Data Fig. 9; see also Fig. 4), which indirectly complement the actions of GPX4 (ref. 8). For example, the protective effects of vitamin E persisted longer in non-transformed mesenchymal cells compared to therapy-resistant cancer cells (Extended Data Fig. 9), reminiscent of the differential effects of folinic acid between transformed and non-transformed cells in the context of methotrexate-based therapy28.
Figure 4. Schematic of a pathway that characterizes a therapy-resistant mesenchymal cancer cell state and its dependency on lipid peroxide dissipation.
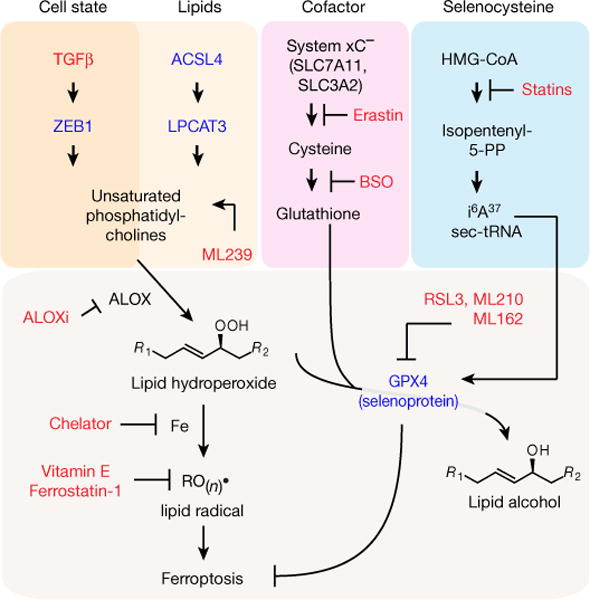
This pathway centres on the ZEB1-dependent synthesis, storage and use of long-chain PUFAs, which are susceptible to becoming reactive lipid peroxides through the action of lipoxygenase enzymes. GPX4 dissipates these reactive peroxides and prevents their downstream iron-dependent reactions that ultimately result in ferroptotic cell death. Red text denotes chemical perturbations used to probe and uncover this circuitry; blue text denotes genetic perturbations that have been carried out. ACSL4, acyl-CoA synthetase long-chain family member 4; ALOX, arachidonate lipoxygenase; ALOXi, ALOX inhibitor; BSO, buthionine sulfoximine; GPX4, glutathione peroxidase 4; LPCAT3, lysophosphatidylcholine acyltransferase 3; sec-tRNA, selenocyteine tRNA; TGFβ, transforming growth factor-β; ZEB1, zinc finger E-box binding homeobox 1.
We have defined, through perturbations and functional characterization, a pathway comprising thirteen distinct molecular nodes that underlies a broadly relevant mesenchymal therapy-resistant state and its dependency on a lipid-peroxidase pathway (Fig. 4). This pathway centres on the synthesis, storage and use of long-chain polyunsaturated fatty acids (PUFAs) (for example, arachidonic acid), which are susceptible to becoming reactive lipid peroxides through the action of lipoxygenase enzymes8. GPX4 dissipates these reactive peroxides and thus protects against ferroptotic cell death8. Consistent with this model, we find that knockout of upstream regulators of PUFA metabolism (ACSL4, LPCAT3)9 and inhibition of lipoxygenases prevent cell death due to inhibition of GPX4 (Extended Data Fig. 10). A convergence on this theme is also consistent with the mesenchymal state-targeting compound ML239 (Fig. 1a) acting on fatty acid desaturase-2 (FADS2) to promote the generation of PUFAs that are substrates for lipid peroxidation13 (Fig. 4). By contrast, direct treatment of cells with a lipid peroxide does not show selectivity for cells in a mesenchymal state (Extended Data Fig. 10). This result supports that the GPX4-dependent feature of the therapy-resistant mesenchymal state we describe is not sensitivity to exogenous lipophilic peroxides themselves, but rather the propensity of specific cellular lipids to be substrates for lipid-oxidizing enzymes (Extended Data Fig. 10).
ZEB1, which we linked causally to GPX4 dependency (Fig. 3b), provides a bridge between mesenchymal gene expression and lipidperoxide vulnerability23. ZEB1 has been shown to play an essential role in cellular lipid metabolism, in part through direct transcriptional regulation of PPARγ, a master regulator of lipid biology22. Growing evidence shows that ZEB1 regulates the uptake, accumulation and mobilization of lipids, and affects EMT-associated remodelling of the plasma membrane29, which is where lipoxygenase-mediated oxidation of PUFAs occurs. The resulting dependency of ZEB1-high cells on lipid-peroxidase activity is most effectively exploited by direct inhibition of GPX4, but can also be targeted through the suppression of GPX4 biosynthesis (statins) and distal perturbations of the metabolic pathways regulating the levels of the GPX4 cofactor glutathione (erastin, buthionine sulfoximine)7,8 (Extended Data Fig. 8).
Dysregulation of the apoptotic cascade is characteristic of cancer cells in a therapy-resistance state and may explain their broad resistance to diverse therapeutic modalities30. Our results suggest that these same cells have an enhanced ability to undergo ferroptosis, a non-apoptotic form of cell death. More broadly, the results presented here illustrate an approach to understand and overcome cancer therapy resistance that transcends traditional approaches based on driver oncogenes and resistance mutations.
METHODS
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Omic datasets
Gene-expression data (RMA-processed, quantile normalized) were obtained from the CCLE portal (https://portals.broadinstitute.org/ccle/home). shRNA data and ATARiS gene solutions were obtained from the Project Achilles portal (http://portals.broadinstitute.org/achilles).
Computation of mesenchymal score
Mesenchymal scores were computed by single-sample gene set enrichment analysis (ssGSEA)12, which estimates the degree of coordinate up- or downregulation of members of a given gene set within a transcriptionally profiled sample. Published EMT gene signatures5,6,10 and Axl-positive melanoma gene signatures2 were analysed using this method, yielding a single ‘mesenchymal score’ per signature for each cell line.
Category-specific Δ-median AUC calculations (Fig. 1d)
For each class assignment (for example, bone lineage), a median AUC was computed for each compound across all cell lines belonging to the class. The median AUC for each compound across all cell lines not belonging to that class was then subtracted from the within-class median AUC, to yield a Δ-median AUC value. Non-adherent cell lines were excluded from this analysis. Significance testing was applied using a Student’s t-test assuming unequal variance. Tests to evaluate AUC distributions for each compound within a given class assignment (for example, bone lineage) were regarded as a family of hypotheses for the purpose of multiple hypothesistest correction. Compounds within each class are plotted in rank order of their Δ-median AUC (smallest to largest). Compounds that meet a P value cut-off of 0.05 after correction for multiple hypothesis testing are plotted as larger dots than those compounds that do not meet the P value cut-off of 0.05.
Correlation analyses
AUC values, as a metric of cell-line sensitivity to compound treatment11, were correlated with cellular features (gene-expression, geneknockdown sensitivity)13, ssGSEA-derived cell-line mesenchymal scores and with one another (across compounds). The number of cell lines used in individual correlation analyses, as well as specific cell-line exclusions (for example, exclusion of haematopoietic cell lines) are noted in the main text and figure legends. Correlation data are plotted as z scores computed analytically using Fisher’s z transformation31 to account for individual compounds having been exposed to different numbers of cell lines. Box-and-whisker plots and scatter plots were produced in GraphPad Prism.
Clustering analysis
CTRP cluster (Fig. 2b). Pairwise distances between compounds were computed using Pearson’s correlation distances normalized by Fisher’s z transformation to account for pairs of compounds exposed to different numbers of cell lines in common31. Normalized correlation z scores were further transformed using a monotonic double-sigmoid function to emphasize variation in the most relevant part of the resulting dendrograms11,31. Non-adherent cell lines were excluded from the clustering application11.
Cell lines
HT-1080 fibrosarcoma cells, LOXIMVI melanoma cells and WI38 lung fibroblasts were purchased from ATCC. RKN leiomyosarcoma were procured from HSRRB and KP4 pancreatic cancer cells from JCRB (Japan). Mesenchymal stem cells (MSCs), human umbilical vein endothelial cells (HUVECs) and haematopoietic progenitor (CD34+) cells were obtained from Lonza. BJeH foreskin fibroblasts were procured from the Weinberg laboratory (Whitehead Institute), MCF-7 ER-Snail1 cells20 were obtained from the Haber laboratory (Massachusetts General Hospital), H358 ER-Twist1 cells21 were obtained from the McCormick laboratory (UCSF) and gefitinib-sensitive and -resistant HCC4006 lung cancer cells24 were obtained from the Engelman laboratory (Massachusetts General Hospital). M000921 and M980513 cells27 were obtained from the Levesque laboratory (University of Zurich Hospital). AA01 and AA02 patient-derived cell lines were developed from ascites (AA01) or pleural fluid (AA02) of patients with metastatic pancreatic ductal adenocarcinoma using IRB-approved protocols at the Dana-Farber Cancer Institute and the Brigham and Women’s Hospital. Cells were cultured by the Cell Line Factory at the Broad Institute (https://portals.broadinstitute.org/cellfactory) using conditional reprogramming with a ROCK inhibitor and feeder cells as previously described32. In brief, each patient-derived sample was placed into culture on a layer of irradiated 3T3-J2 feeder cells in F-medium containing ROCK inhibitor. At passage number five, cell pellets were collected and KRAS mutation status was confirmed by targeted PCR-Sanger sequencing using isolated genomic DNA. Exome sequencing later confirmed the purity of the cell lines (99% tumour) and provided detailed mutational and copy-number characterization data. Cells were effectively grown in DMEM-based medium without ROCK inhibitor beyond passage number five.
HT-1080, WI38, MCF-7 ER-Snail1, AA01 and AA02 cells were maintained in DMEM, supplemented with 10% FBS and 1% penicillin-streptomycin (all medium components from Sigma-Aldrich). BJeH cells were maintained in 65% DMEM, 20% Medium 199 (Sigma-Aldrich) and 15% FBS supplemented with 1% penicillinstreptomycin. KP4 cells were cultured in a 1:1 ratio of DMEM:Ham’s-F12 medium and RKN cells in Ham’s-F12 medium, all supplemented with 10% FBS and 1% penicillin-streptomycin (Sigma-Aldrich). M000921 and M980513 cells27 were maintained in RPMI supplemented with 5 mM glutamine, 1 mM sodium pyruvate and 10% heat-inactivated fetal calf serum (Invitrogen). HCC4006 gefitinib-resistant cells were maintained in 10 μM gefitinib (SelleckChem), which was replaced every three days. MSCs were grown in MSC growth medium (Lonza), HUVECs in EGM-2 medium (Lonza) and CD34 cells in serum-free HPGM medium (Lonza) supplemented with 25 ng ml−1 stem cell factor, 50 ng ml−1 thrombopoietin and 50 ng ml−1 FLT-3 ligand (all cytokines from Peprotech).
Cell lines included in CTRP were recently validated with fingerprinting analysis. Selected cell lines were tested and confirmed as not having mycoplasma contamination using the commercially available MycoAlert kit.
Compounds
1S,3R-RSL3 was synthesized according to previously published protocols8. All other compounds were obtained from Broad Institute Compound Management Platform or purchased from SelleckChem or Sigma-Aldrich.
Compound treatment
CTRP viability experiments11,13 were performed in 384-well format using opaque white plates (Corning). Twenty-four hours after seeding at optimized densities, cells were exposed to compounds at indicated concentrations for 72 h. Cellular ATP levels (as a surrogate for viability) were measured using CellTiter-Glo (Promega). For co-treatment experiments, spent medium was removed 24 h after cell seeding and replaced with medium containing a single concentration of the modulator of interest (for example, antioxidant). Compound treatment began one hour later. HCC4006 gefitinib-resistant cells24 were exposed to compounds in the absence of gefitinib. For experiments characterizing GPX4 protein levels in fluvastatin-treated HT-1080 cells (Fig. 2c), spent medium was removed every 48 h and replaced with fresh medium containing the indicated concentration of compound.
Compound treatment of inducible mesenchymal state models
MCF-7 ER- Snail1 cells20 and H358 ER-Twist1 cells21 were induced to undergo EMT with 1 μM and 100 nM 4-hydroxytamoxifen (4-OHT, Sigma-Aldrich), respectively, which was replaced every 72 h, for the number of indicated days. Induced cells were washed thoroughly to remove 4-OHT and were then exposed to compounds for 72 h (in the absence of 4-OHT). The upregulation of EMT-related genes in 4-OHT-induced cells under these conditions was confirmed using a Fluidigm Real-Time PCR system (Supplementary Table 3).
M000921 and M980513 melanoma cells27 were treated with 5 ng ml−1 human recombinant TGFβ-1 (R&D Systems) for a minimum of 14 days, with TGFβ replaced every three days. Cells cultured in regular growth medium or supplemented with TGFβ were seeded in 96-well plates and treated with the indicated concentrations of PLX-4032 or RSL3 (Selleckchem). After 72 h, cell viability was measured by incubating cells for 2 h with a Resazurin solution (0.15 mg ml−1 in PBS) (Sigma-Aldrich) diluted 1:10 in regular growth medium. Fluorescence was read using a standard plate reader (Tecan) with excitation/emission of 535/595 nm.
RSL3-fluvastatin synergy analysis (Fig. 2d)
HT-1080 cells were exposed to the indicated concentrations of fluvastatin for 48 h. At the end of this period, fluvastatin-containing medium was removed and replaced with medium containing the indicated concentrations of RSL3 and fluvastatin. Deviation from expected response was calculated assuming a Loewe additivity model for compound interactions33.
Lipophilic antioxidant rescue of transformed versus non-transformed cells (Extended Data Fig. 9)
Cells that had been allowed to adhere overnight in 24-well dishes were exposed for 18 h to medium containing the indicated lipophilic antioxidant (2.5 μM ferrostatin-1 (ref. 7) or 200 μM α-tocopherol acetate). Following this exposure, cells were washed three times with 1 ml medium and three times with 1 ml PBS. Cells were then cultured continually in medium containing 0.5 μM RSL3 for the duration of the experiment. Cells that became confluent during the course of the experiment were split directly into compound-containing medium. Cellular viability was assessed using CellTiter-Glo (Promega).
Western blot
Cells were seeded in six-well dishes and cultured to 70% confluence. RIPA buffer (Thermo Scientific) supplemented with phosphatase inhibitor (Thermo Scientific) and protease inhibitor cocktails (Roche) was used to lyse cells. Lysate was centrifuged at 12,000 r.p.m. for 10 min at 4 °C and the protein concentration in the supernatant was quantified using a Bradford reagent (Bio-rad). Approximately 40 μg of protein was mixed with sample buffer (Bio-rad) and boiled at 95 °C for 10 min, followed by incubation on ice for 5 min. Samples were separated by SDS-PAGE (Novex 4–12% Bis-Tris gel, Life Technologies) and transferred to a nitrocellulose membrane using an iBlot system (Life Technologies). Membranes were blocked with Odyssey Blocking Buffer (LI-COR) and incubated with primary antibodies overnight at 4 °C and LI-COR secondary antibodies for 1 h at room temperature. Membranes were imaged using the LI-COR Odyssey Imaging System and protein levels were quantified using ImageJ software. Antibodies used: anti-E-cadherin (BD Biosciences, BD610181, 1:1,000), anti-vimentin (Cell Signaling, 5741, 1:1,000), anti-β-actin (Santa Cruz, sc-47778, 1:200), anti-GPX4 (Abcam, ab41787, 1:1,000) and ZEB1 (CST, D80D3, 1:1,000).
GPX4 activity assay
GPX4 activity was assayed as previously described8. In brief, 20 million HT-1080 cells were exposed to vehicle (0.1% DMSO) or 10 μM RSL3, ML210 or ML162 for 1.5 h. Sonication was used to lyse cells and the resulting solution was centrifuged at 14,000 r.p.m. for 10 min. Subsequently, 200 μg of cleared lysate was incubated with phosphatidylcholine hydroperoxide and reduced glutathione for 30 min at 37 °C. Lipids were extracted from this mixture using a chloroform:methanol (2:1) solution and evaporated using a Rotavap. The extract was reconstituted in 100% ethanol and injected into an Ultimate 3000 Rapid Separation Liquid Chromatography system (Thermo Scientific) coupled to an amaZon SL ion trap mass spectrometer (Bruker).
Lipid-peroxidation assay
Measurement of cellular lipid peroxidation was performed as previously described8. HT-1080 cells were seeded in six-well plates at the following densities: 50,000 cells per well for vehicle (0.05% DMSO) and 0.5 μM fluvastatin, 150,000 cells per well for 1 μM fluvastatin, 250,000 cells for 2 μM and 4 μM fluvastatin and 350,000 cells for 8 μM fluvastatin. Following 96 h of compound exposure, cells were collected, resuspended in 500 μl of Hank’s Balanced Salt Solution (HBSS; Gibco) and incubated with 2 μM BODIPY 581/591 C11 (Life Technologies) for 10 min at 37 °C. Cells were then washed twice in HBSS and analysed using a C6 flow cytometry system (BD Accuri cytometer).
Generation of knockout cells
Single-guide RNAs (sgRNAs) targeting GPX4 were cloned into the lentiviral vector pLentiCRISPRv2 as previously described34 using the following guide sequence: GPX4-GPP1: CACGCCCGATACGCTGAGTG. Lentiviral particles expressing sgRNAs were produced by co-transfection of pLentiCRISPRv2 plasmids with the packaging and envelope plasmids psPAX2 (Addgene plasmid 12260) and pMD2.G (Addgene plasmid 12259) into HEK293 cells. Target cells were transduced with lentivirus in the presence of 2.5 μM ferrostatin-1 and transduced cells were selected using puromycin at a concentration of 2 μg ml−1. Single-cell clones were derived from this population using limiting dilution and then assayed for knockout of GPX4 by western blotting and functional validation by withdrawal of ferrostatin-1. ACSL4, LPCAT3 and ZEB1 knockout cells were generated by transfecting cells with a ribonucleoprotein complex containing Cas9 protein and target-specific sgRNAs as previously described35. Targeting sequences used were TCTGTGAGACCAGGGCCGAA, GGTCTCACAGAAGATGGCAA, CTAGCTGTAATAGACATCCC, TGATGC ATCATCACTCCCTT for ACSL4; GCTGTTGACTACTTTGACGG for LPCAT3; and GAGCACTTAAGAATTCACAG, GCTTCACCCATACAACAAGG for ZEB1.
In vivo xenograft experiment (Fig. 3g)
The University of Arizona Animal Care and Use Committee (Tuscon, AZ) approved the following xenograft studies (Protocol 14-533), permitting a maximum tumour volume of < 10% of the animal’s original body weight or up to 4,000 mm3. In none of the experiments, these limits were exceeded. Xenografts for LOXIMVI sgEGFP (WT) and LOXIMVI sgGPX4 (KO) cells were established by injecting 10 million cells in a 1:1 PBS:Matrigel mixture containing 2.5 μM ferrostatin-1 into the flanks of athymic mice (NRC Nude, Taconic). Animals were dosed daily with 2 mg kg−1 ferrostatin-1 by intraperitoneal injections. Tumour volume was measured twice a week. Ferrostatin-1 treatment was withdrawn once the tumour volume reached 250 mm3 and mice were euthanized once tumour volume exceeded 2,000 mm3.
Data availability
All data are available from the authors upon reasonable request. This manuscript is accompanied by three Supplementary Tables and data are deposited in the NCBI BioProject database with identifier PRJNA381497.
Extended Data
Extended Data Figure 1. Correlation of E-cadherin and vimentin protein levels of cell lines with sensitivity to mesenchymal statetargeting compounds.
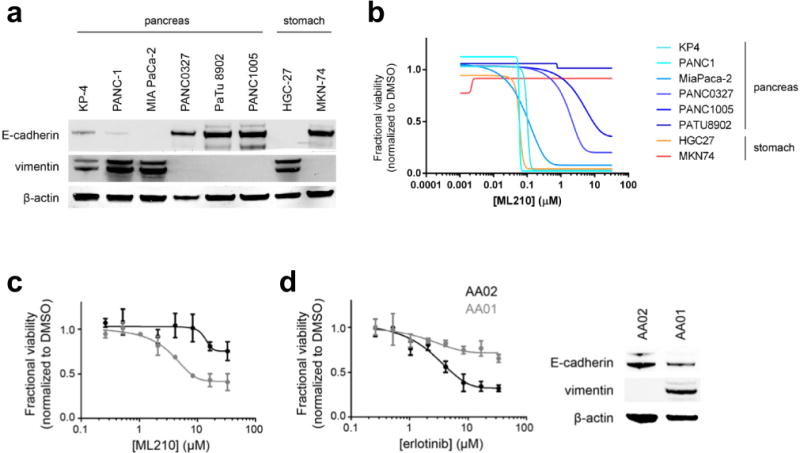
a, b, Pancreatic and gastric cancer cell lines with low E-cadherin protein levels have high levels of vimentin (a) and are preferentially sensitive to ML210, a mesenchymal state-targeting compound (b). Concentration-response curves are from CTRP c, d, Two patient-derived pancreatic cancer cell lines with differing sensitivity to erlotinib (d), show GPX4-inhibitor sensitivity and levels of epithelial and mesenchymal protein markers correlating in the predicted direction with erlotinib sensitivity. Data plotted in c and d are mean ± s.d. of four technical replicates and are representative of two biological replicates.
Extended Data Figure 2. Lineage-specific AUC-mesenchymal score correlations.
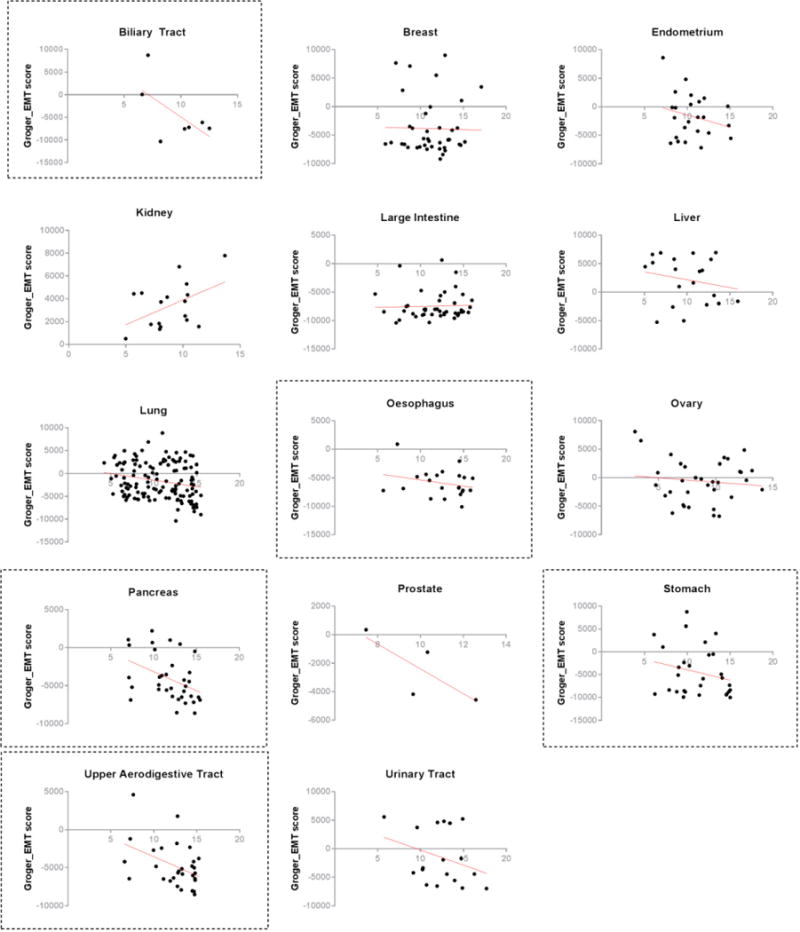
Scatter plots with linear regression line (red) show the relationship between cancer cell-line mesenchymal score and cell-line sensitivity to ML210 (a ferroptosis-inducing, mesenchymal state-targeting compound) within different epithelium-derived cancer lineages. Gastrointestinal cancer lineages showing stronger correlations are demarcated with dashed boxes.
Extended Data Figure 3. Correlation of individual cell-line features with mesenchymal state-targeting compounds.
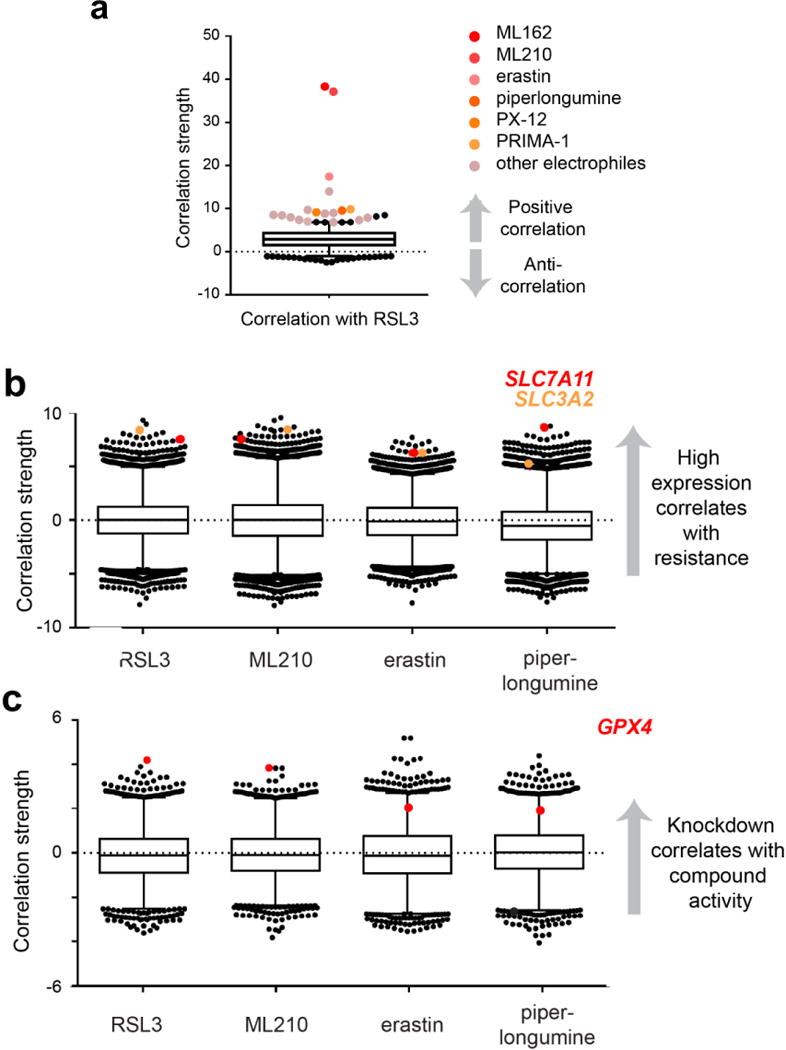
a, Box-and-whisker plot shows the coefficient of correlation between the cytotoxic effects of the GPX4 inhibitor RSL3 and cytotoxic effects of 481 other compounds (black dots; inducers of electrophilic stress in shades of orange) across 656 cancer cell lines (excludes suspension cell lines). Plotted values are absolute correlation coefficients z scored using Fisher’s z transformation to account for individual compounds having been exposed to different numbers of cell lines; line, median; box, 25th–75th percentile; whiskers, expansion of the 2.5th and 97.5th percentile outlier compounds (black and coloured dots); dotted line marks 0.0. Data for compounds indicated to the right of the plot are significant with a P value of less than 0.005. b, c, Box-and-whisker plots show the extent of correlation between baseline expression of gene-expression transcripts (b) or sensitivity to gene knockdown (c) and cytotoxic effects of compounds with strong (RSL3, ML210), intermediate (erastin) and weak (piperlongumine) mesenchymal state-targeting properties despite otherwise sharing similar cell death-inducing profiles (shown in a). Plotted values in b and c are z scored Pearson’s correlation coefficients (see Methods); line, median; box, 25th-75th percentile; whiskers, expansion of 1st and 99th percentile outlier correlates; dotted line marks 0.0. Data highlighted with coloured dots are significant with a P value of less than 0.0002. Plots in b are derived from 610–631 cancer cell lines (excludes non-adherent cell lines). SLC7A11 (red dots) and SLC3A2 (orange dots) are shared gene-expression outlier correlates among the shown electrophilic stress-inducing compounds. Plots in c are derived from 132–136 cancer cell lines (excluding haematopoietic cell lines). Sensitivity to GPX4 knockdown (red dots) is uniquely correlated with sensitivity to mesenchymal state-targeting electrophilic compounds.
Extended Data Figure 4. Effect of RSL3, ML162 and ML210 on GPX4 activity in cellular lysates.
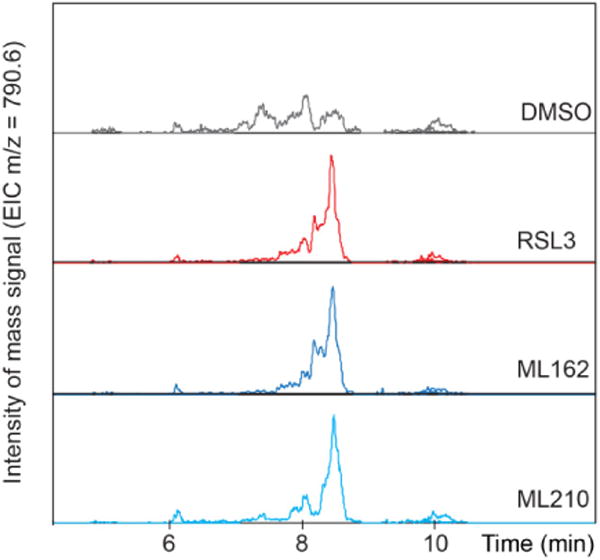
Treatment of cells with RSL3, ML162 or ML210 inhibits the ability of cellular lysates to reduce exogenous phosphatidylcholine hydroperoxide (m/z of 790.6). Data reflect the results of single biological experiment.
Extended Data Figure 5. Modulation of statins by mevalonate pathway intermediates and antioxidants.
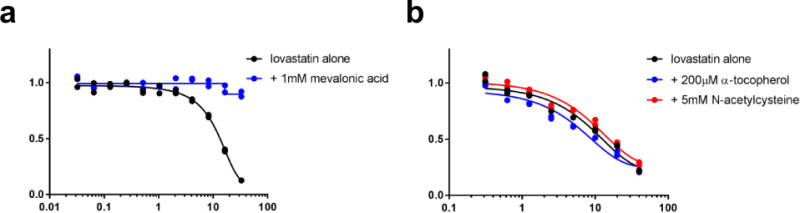
a, b, The effect of statins on HT-1080 fibrosarcoma-derived cells is rescued by co-treatment with mevalonic acid (a), but not by co-treatment with a lipophilic antioxidant (b). Data for two technical replicates are plotted; data represent two separate biological experiments.
Extended Data Figure 6. Relative GPX4 inhibitor sensitivity of cell lines modelling EMT driven by inducible expression of EMT transcription factors.
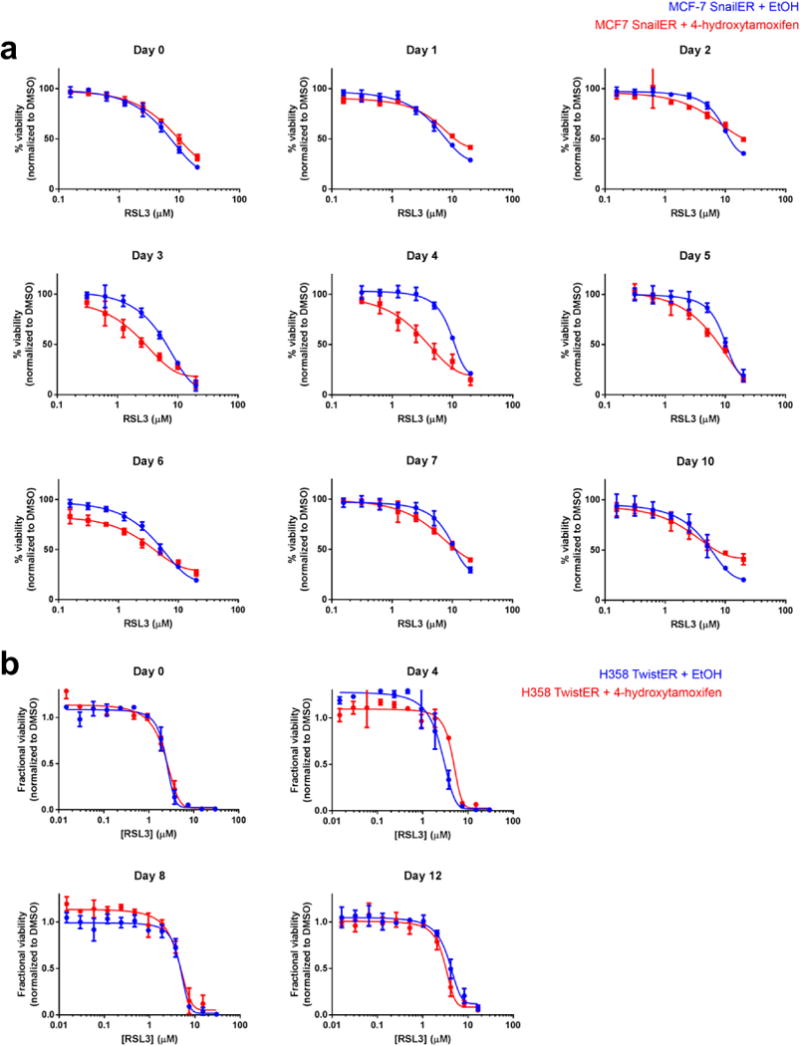
a, Engineered MCF-7 breast cancer cells induced with a small molecule (4-hydroxytamoxifen; 4-OHT) to express high levels of SNAIL1 and undergo EMT (red curve). b, Engineered H358 lung cancer cells induced with 4-OHT to express high levels of TWIST1 and undergo EMT (red curve). Data plotted are mean ± s.d. of four technical replicates and are representative of two biological replicates.
Extended Data Figure 7. Protein-level validation of successful gene knockout in CRISPR-Cas9-engineered cells.
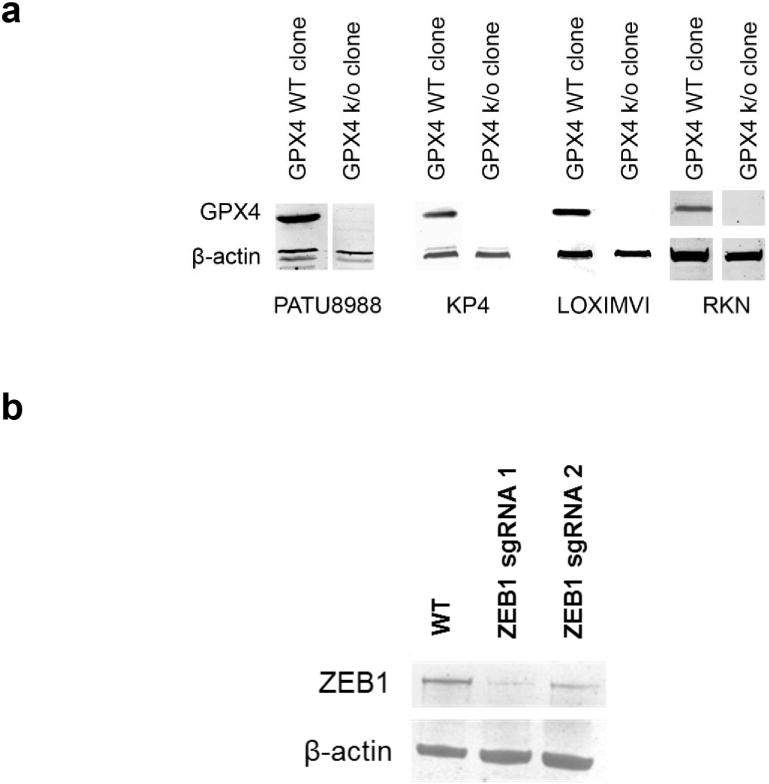
a, GPX4 protein levels in GPX4-wild-type (WT) and GPX4-knockout (k/o) clones generated using CRISPR-Cas9 technology. b, ZEB1 protein levels in KP4 pancreatic cancer cells exposed to ZEB1-targeting CRISPR-Cas9 technology.
Extended Data Figure 8. Relative compound sensitivity of epithelial versus mesenchymal state cancer models.
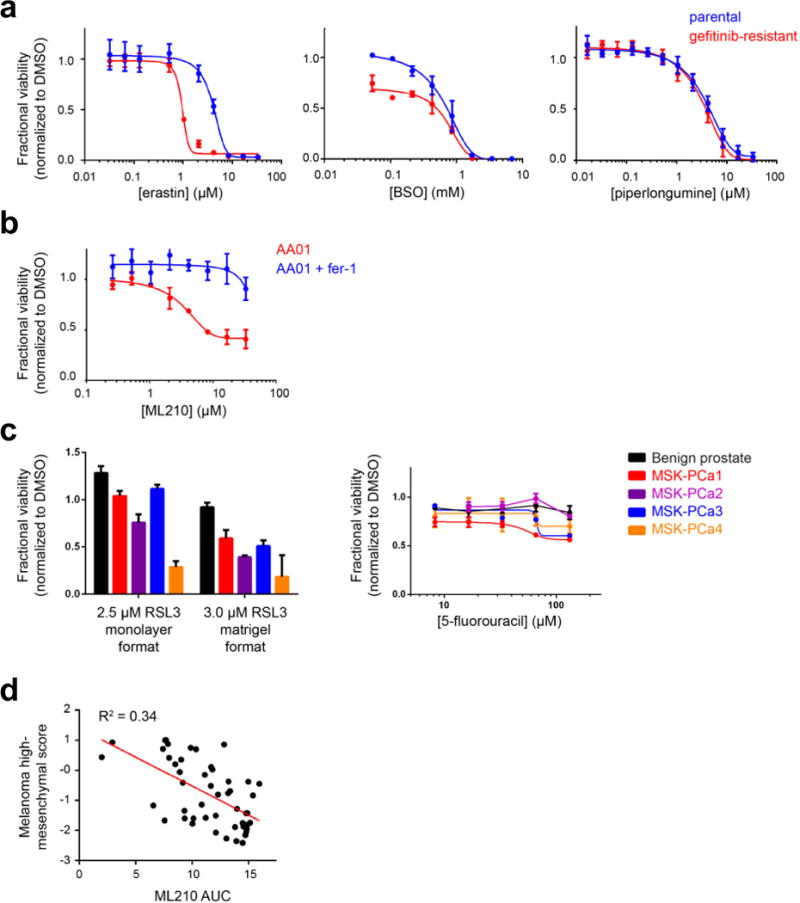
a, HCC4006 lung cancer cells that have undergone EMT as a mechanism of resistance to gefitinib (red curve). Erastin and buthionine sulfoximine (BSO) are ferroptosis inducers, while piperlongumine is an electrophile that induces a non-ferroptotic form of oxidative cell death. b, Mesenchymal state patient-derived pancreatic cancer cells (AA01) undergo ferroptosis in response to GPX4 inhibition as evidenced by the ability of ferrostatin-1 to rescue loss of viability due to GPX4 inhibition. c, Patient-derived prostate cancer organoid lines show sensitivity to a GPX4 inhibitor (RSL3) in a manner correlated with mesenchymal gene-expression score (Fig. 3d), in both collagen-based and Matrigel-based culture conditions. This correlation with mesenchymal score is not seen for a control lethal agent (5-fluorouracil). d, Scatter plot with linear regression line (red) showing the correlation between a melanoma-specific high mesenchymal state score and sensitivity to a GPX4 inhibitor (ML210) across 49 melanoma-derived cell lines from CTRP Data plotted in a–c are mean ± s.d. of four technical replicates (a and b) and three technical replicates (c) and are representative of two biological replicates.
Extended Data Figure 9. Effect of lipophilic antioxidants on rescuing GPX4 inhibitor-mediated cell death in transformed versus non-transformed high-mesenchymal state cells.
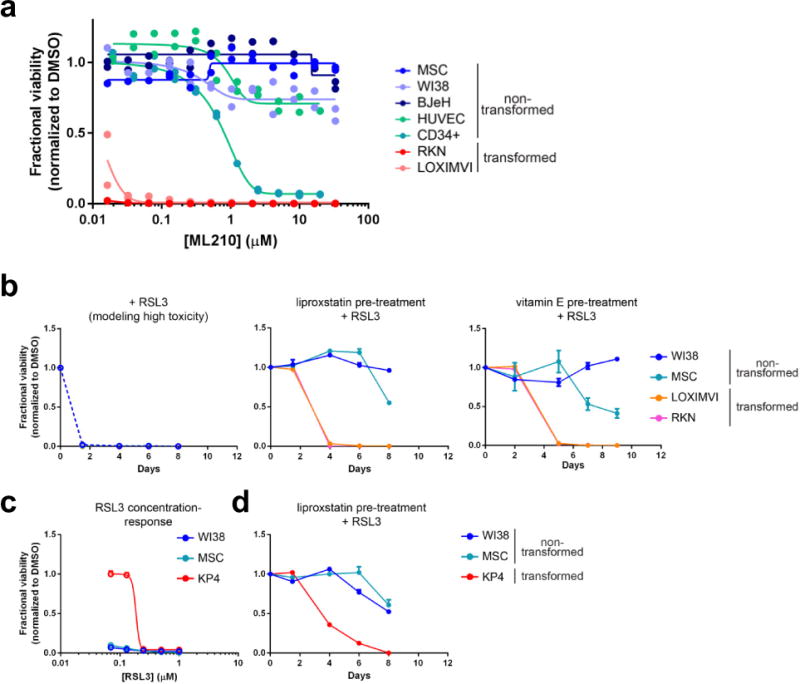
a, Relative sensitivity of transformed and non-transformed high-mesenchymal state cell lines to GPX4 inhibition. Data for two technical replicates are plotted and represent two separate biological experiments. Concentration-response curves collected over a period of several months are plotted on a single set of axes to aid comparison of cell-line sensitivities. BJeH, foreskin fibroblasts; CD34+ cells, haematopoietic progenitor cells; HUVEC, human umbilical vein endothelial cells; LOXIMVI, melanoma-derived cells; MSC, mesenchymal stem cells; RKN, leiomyosarcoma-derived cells; WI38, lung fibroblasts. b, A single pre-treatment of cells with a lipophilic antioxidant (liproxstatin-1 or vitamin E) protects non-transformed mesenchymal state cells for a longer period of time than transformed high-mesenchymal state cells, from prolonged treatment with a high concentration of a GPX4 inhibitor (RSL3). c, d, Transformed high-mesenchymal state cells that are less sensitive to GPX4 inhibition (KP4) than non-transformed mesenchymal state cells (WI38, MSC) can be killed preferentially by pretreating cells with a lipophilic antioxidant before initiating treatment with a GPX4 inhibitor. Data plotted in b–d are mean ± s.d. of four technical replicates and are representative of two biological replicates.
Extended Data Figure 10. Relationship of GPX4 dependence and modulation of cellular lipid peroxides.
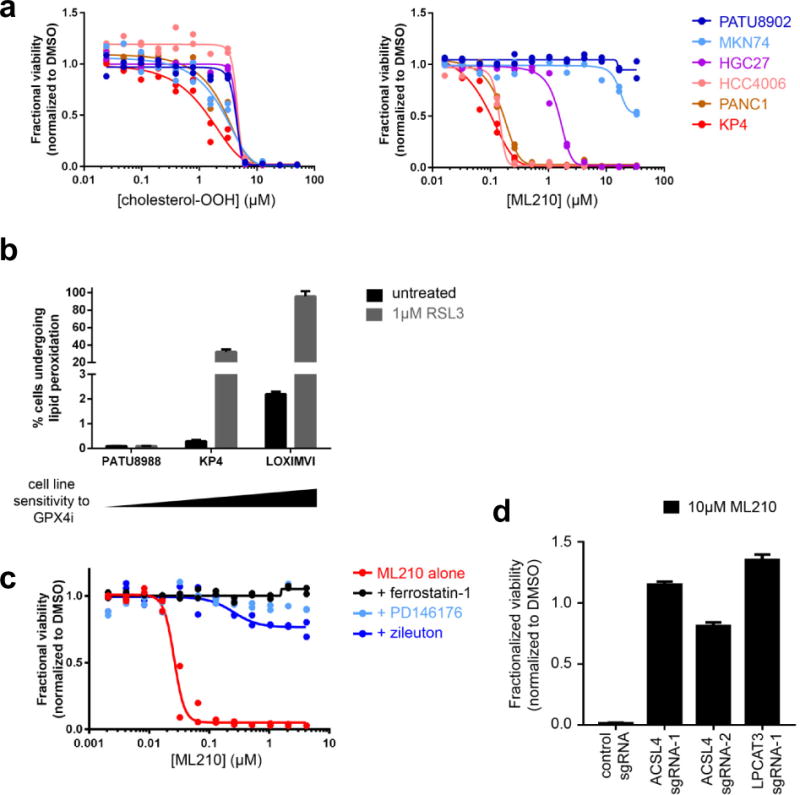
a, Cell-line sensitivity to exogenous lipid peroxides (for example, cholesterol peroxide) does not correlate with differential cell-line sensitivity to a GPX4 inhibitor (ML210). b, Cell-line sensitivity to GPX4 inhibition correlates positively with induction of lipid peroxidation upon GPX4 inhibition (GPX4i). c, d, Small-molecule inhibitors of arachidonic acid lipoxygenases (PD146176, zileuton) (c) and genetic knockout of two upstream regulators of arachidonic acid metabolism (ACSL4, LPCAT3) (d) prevent cell death induced by a GPX4 inhibitor (ML210) in KP4 cells. Data in a and c are two technical replicates whereas data in b and d are mean ± s.d. of three technical replicates. All panels represent two separate biological experiments.
Supplementary Material
Acknowledgments
We thank M. J. Hangauer, M. T McManus, F. McCormick, K. Dutton-Regester, L. V. Kemeny, D. J. Adams and Y. Drier for valuable discussions and L. Hartman for execution of in vivo studies. This project has been supported by grants from the National Cancer Institute (Cancer Target Discovery and Development Network grant U01CA176152 to S.L.S., U01CA168397 to M.E.B., 5R01CA097061 and R01CA161061 to B.R.S., NCI-CA129933 to D.A.H., P30CA008748 to Y.C.), the National Institutes of Health (R01GM038627 to S.L.S., 5R01GM085081 to B.R.S.), the Swiss National Fund (310030_149946, to M.P.L.) and Howard Hughes Medical Institute (D.A.H., S.L.S.).
Footnotes
Online Content Methods, along with any additional Extended Data display items and Source Data, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Supplementary Information is available in the online version of the paper.
Author Contributions S.L.S. directed the project; S.L.S. and V.S.V. wrote the manuscript; V.S.V. and M.J.R. performed research; H.D.D. performed in vivo experiments; S.G. performed CRISPR experiments; O.M.E. performed TGFβ treatment of melanoma cell lines; S.R.V. and S.Che. generated CRISPR reagents; S.D.K. performed organoid experiments; B.S.-L., A.J.A., M.G.R. and P.T. generated mesenchymal scores; K.S. performed the lipid peroxidation assay; W.S.Y performed the GPX4 activity assay; Z.V.B. and J.K.E synthesized compounds; S.Cha. and C.H. contributed to profiling of non-transformed cell lines; J.M.C. and B.M.W. collected patient samples; A.J.A., X.W. and Y.-Y.T generated patient-derived pancreatic cancer cell lines; J.PM., D.A.H., J.A.E., J.S.B., D.G., S.J., Y.C., W.C.H., M.PL., J.G.D., E.M.R and M.E.B. contributed reagents; J.D.K., C.S.H., B.R.S. and A.F.S. provided project support; P.A.C. oversaw data analysis; V.S.V. and PA.C. performed large-scale data analysis.
The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewer Information Nature thanks N. Chandel, T Sato and the other anonymous reviewer(s) for their contribution to the peer review of this work.
References
- 1.Hoek KS, et al. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008;68:650–656. doi: 10.1158/0008-5472.CAN-07-2491. [DOI] [PubMed] [Google Scholar]
- 2.Tirosh I, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bu X, Mahoney KM, Freeman GJ. Learning from PD-1 resistance: new combination strategies. Trends Mol Med. 2016;22:448–451. doi: 10.1016/j.molmed.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaffer SM, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546:431–435. doi: 10.1038/nature22794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gröger CJ, Grubinger M, Waldhör T, Vierlinger K, Mikulits W. Metaanalysis of gene expression signatures defining the epithelial to mesenchymal transition during cancer progression. PLoS ONE. 2012;7:e51136. doi: 10.1371/journal.pone.0051136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byers LA, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19:279–290. doi: 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dixon SJ, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang WS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dixon SJ, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10:1604–1609. doi: 10.1021/acschembio.5b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taube JH, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci USA. 2010;107:15449–15454. doi: 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seashore-Ludlow B, et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 2015;5:1210–1223. doi: 10.1158/2159-8290.CD-15-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barbie DA, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rees MG, et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat Chem Biol. 2016;12:109–116. doi: 10.1038/nchembio.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Germain AR, et al. Identification of a selective small molecule inhibitor of breast cancer stem cells. Bioorg Med Chem Lett. 2012;22:3571–3574. doi: 10.1016/j.bmcl.2012.01.035. [DOI] [PubMed] [Google Scholar]
- 15.Chen VW, et al. Pathology and classification of ovarian tumors. Cancer. 2003;97(Suppl):2631–2642. doi: 10.1002/cncr.11345. [DOI] [PubMed] [Google Scholar]
- 16.Tun HW, et al. Pathway signature and cellular differentiation in clear cell renal cell carcinoma. PLoS ONE. 2010;5:e10696. doi: 10.1371/journal.pone.0010696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kryukov GV, et al. Characterization of mammalian selenoproteomes. Science. 2003;300:1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 18.Warner GJ, et al. Inhibition of selenoprotein synthesis by selenocysteine tRNA[Ser]Sec lacking isopentenyladenosine. J Biol Chem. 2000;275:28110–28119. doi: 10.1074/jbc.M001280200. [DOI] [PubMed] [Google Scholar]
- 19.Nieto MA, Huang RYJ, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 20.Javaid S, et al. Dynamic chromatin modification sustains epithelialmesenchymal transition following inducible expression of Snail-1. Cell Rep. 2013;5:1679–1689. doi: 10.1016/j.celrep.2013.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salt MB, Bandyopadhyay S, McCormick F. Epithelial-to-mesenchymal transition rewires the molecular path to PI3K-dependent proliferation. Cancer Discov. 2014;4:186–199. doi: 10.1158/2159-8290.CD-13-0520. [DOI] [PubMed] [Google Scholar]
- 22.Gubelmann C, et al. Identification of the transcription factor ZEB1 as a central component of the adipogenic gene regulatory network. eLife. 2014;3:e03346. doi: 10.7554/eLife.03346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang P, Sun Y, Ma L. ZEB1: at the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle. 2015;14:481–487. doi: 10.1080/15384101.2015.1006048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ware KE, et al. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogenesis. 2013;2:e39. doi: 10.1038/oncsis.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao D, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159:176–187. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKeithen D, Graham T, Chung LWK, Odero-Marah V. Snail transcription factor regulates neuroendocrine differentiation in LNCaP prostate cancer cells. Prostate. 2010;70:982–992. doi: 10.1002/pros.21132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng PF, et al. Methylation-dependent SOX9 expression mediates invasion in human melanoma cells and is a negative prognostic factor in advanced melanoma. Genome Biol. 2015;16:42. doi: 10.1186/s13059-015-0594-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen IJ, Wolff JE. How long can folinic acid rescue be delayed after high-dose methotrexate without toxicity? Pediatr Blood Cancer. 2014;61:7–10. doi: 10.1002/pbc.24770. [DOI] [PubMed] [Google Scholar]
- 29.Mathow D, et al. Zeb1 affects epithelial cell adhesion by diverting glycosphingolipid metabolism. EMBO Rep. 2015;16:321–331. doi: 10.15252/embr.201439333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song J. EMT or apoptosis: a decision for TGFβ. Cell Res. 2007;17:289–290. doi: 10.1038/cr.2007.25. [DOI] [PubMed] [Google Scholar]
- 31.Dančík V, et al. Connecting small molecules with similar assay performance profiles leads to new biological hypotheses. J Biomol Screen. 2014;19:771–781. doi: 10.1177/1087057113520226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X, et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol. 2012;180:599–607. doi: 10.1016/j.ajpath.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fitzgerald JB, Schoeberl B, Nielsen UB, Sorger PK. Systems biology and combination therapy in the quest for clinical efficacy. Nat Chem Biol. 2006;2:458–466. doi: 10.1038/nchembio817. [DOI] [PubMed] [Google Scholar]
- 34.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuris JA, et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol. 2015;33:73–80. doi: 10.1038/nbt.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the authors upon reasonable request. This manuscript is accompanied by three Supplementary Tables and data are deposited in the NCBI BioProject database with identifier PRJNA381497.