

Abstract
An effective, nontoxic, tumor-specific immunotherapy is the ultimate goal in the battle against cancer, especially the metastatic disease. Checkpoint blockade-based immunotherapies have been shown to be extraordinarily effective but benefit only the minority of patients whose tumors have been pre-infiltrated by T cells. Here, we show that Zn-pyrophosphate (ZnP) nanoparticles loaded with the photosensitizer pyrolipid (ZnP@pyro) can kill tumor cells upon irradiation with light directly by inducing apoptosis and/or necrosis and indirectly by disrupting tumor vasculature and increasing tumor immunogenicity. Furthermore, immunogenic ZnP@pyro photodynamic therapy (PDT) treatment sensitizes tumors to checkpoint inhibition mediated by a PD-L1 antibody, not only eradicating the primary 4T1 breast tumor but also significantly preventing metastasis to the lung. The abscopal effects on both 4T1 and TUBO bilateral syngeneic mouse models further demonstrate that ZnP@pyro PDT treatment combined with anti-PD-L1 results in the eradication of light-irradiated primary tumors and the complete inhibition of untreated distant tumors by generating a systemic tumor-specific cytotoxic T cell response. These findings indicate that nanoparticle-mediated PDT can potentiate the systemic efficacy of checkpoint blockade immunotherapies by activating the innate and adaptive immune systems in tumor microenvironment.
Graphical Abstract
INTRODUCTION
Breast cancer is the most common cancer for females in the United States and the second most common cause of cancer-related death in women.1 In particular, metastatic triple-negative breast cancer (mTNBC) is associated with a poor prognosis and has no effective targeted therapy available, making this breast cancer subtype almost fatal.2 The relative ineffectiveness of surgical interventions, radiation, and cytotoxic chemotherapies has driven interest in immunotherapy as a primary treatment modality.3 Tumor immunotherapy operates on the premise that cancer cells can be eliminated by host cytotoxic CD8+ T cells,4 although these cells themselves can be subjected to various suppressive mechanisms including inhibition by regulatory T (Treg) cells,5 myeloid derived suppressor cells,6 and induced expression of programmed death-1 (PD-1) and other inhibitory checkpoint receptors,7 all limiting the antitumor functions of cytotoxic lymphocytes.
Targeting T cell inhibitory checkpoint signaling pathways overexpressed in tumors with antibodies has provided a promising strategy for tumor-specific immunotherapy.8 The unusually high density of transmembrane protein PD-L1 expressed on tumors presents the PD-1/PD-L1 pathway as a valuable target:9 two PD-1 targeted antibodies, nivolumab and pembrolizumab, and one PD-L1 targeted antibody, atezolizu-mab, have already been approved by the Food and Drug Administration for the treatments of advanced melanoma, non-small cell lung cancer, and bladder cancer, respectively.10 However, only a small minority of cancer patients respond to checkpoint inhibition due to its reliance on high expression of PD-L1 on tumors and/or pre-existing tumor-infiltrating CD8+ T cells expressing PD-1.7a,11 This evidence indicates that strategies that can induce immunogenic tumor microenvironments to enhance T cell infiltration might sensitize tumors to checkpoint therapy and improve response rates.4d,12
Photodynamic therapy (PDT) is a clinically used, minimally invasive therapeutic procedure that has also been shown to induce antitumor immunity.13 In PDT, a photosensitizer (PS) accumulated in tumors is activated with a specific wavelength of light in the presence of oxygen to generate reactive oxygen species (ROS), predominantly the singlet oxygen (1O2), which kills tumor cells directly by inducing necrosis and/or apoptosis and indirectly by disrupting tumor vasculature and producing tumor-specific immunity.14 The precise mechanisms involved in PDT-mediated induction of antitumor immunity are not yet fully understood. Potential contributing factors include alterations in the tumor microenvironment via stimulation of proinflammatory cytokines and direct effects of PDT on the tumor that increase immunogenicity.15 We hypothesize that highly effective PDT can sensitize tumors to checkpoint blockade therapy by inducing acute inflammation and increasing tumor immunogenicity to broaden the use of checkpoint blockade immunotherapies in metastatic cancers.
Selective accumulation of PSs in tumors is critical for effective PDT by minimizing collateral damage to surrounding healthy tissues. However, typically PSs are hydrophobic and aggregate in aqueous media, which deleteriously affects their photophysical (decreased 1O2 formation), chemical (decreased solubility) and biological (insufficient tumor localization) properties, thereby diminishing the PDT efficacy.16 Nano-particles can increase the solubility of hydrophobic therapeutic or PDT agents and offer proper size and surface properties to prolong blood circulation, allowing for their selective accumulation in tumors via the enhanced permeability and retention (EPR) effect.17 Tumor accumulation may be further improved by modifying the particle surface with cancer targeting ligands.18 Indeed, a number of nanoparticles have been explored as promising delivery vehicles for molecule- or material-based PDT alone or combined with chemotherapeutic agents to cancers in order to enhance the phototreatment efficiency, and in some cases, encouraging preclinical and clinical data are emerging.19
Here we report the design of nontoxic core–shell nano-particles (ZnP@pyro) with a coordination polymer of Zn and pyrophosphate (ZnP) in the core and the photosensitizer pyrolipid (a lipid conjugate of pyropheophorbide-a) in the shell for highly effective PDT. ZnP@pyro is optimally biocompatible as both Zn and pyrophosphate are endogenously found in blood plasma and pyrolipid is nontoxic without light activation.20 The particles showed minimal uptake by the mononuclear phagocyte system (MPS), prolonged blood circulation, and preferential accumulation in the tumor after systemic injection, due to the EPR effect. The dual selectivity of tumor-targeted nanomedicine and the spatially controlled light irradiation minimizes damage to normal tissues to reduce systemic toxicity associated with classical PDT. This novel nanomedicine harnessed the power of PDT for direct cell killing and stimulation of systemic immune response for cancer treatment. We demonstrated that ZnP@pyro PDT treatment could sensitize tumors to checkpoint blockade therapy (Figure 1): the combination of ZnP@pyro PDT treatment with PD-L1 checkpoint blockade therapy not only eradicated the primary tumors, but also significantly prevented lung metastases in a 4T1 mTNBC murine model. In addition, the combination therapy produced an efficient abscopal effect on two bilateral syngeneic mouse models, 4T1 and TUBO, leading to the complete inhibition of the non-irradiated pre-existing distant tumors. These findings indicate that the proportion of cancers responding to checkpoint therapy can be substantially increased by combining checkpoint blockade with immunogenic conventional therapies such as PDT.
Figure 1.
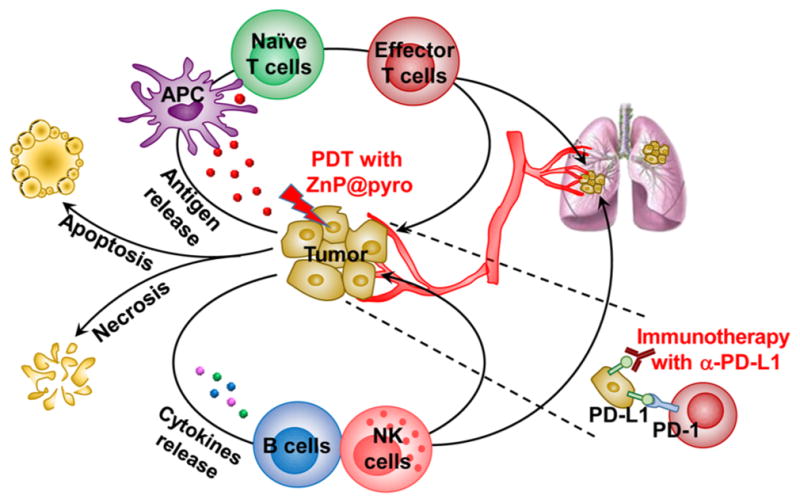
Immunogenic ZnP@pyro PDT sensitizes tumors to PD-L1 blockade immunotherapy for the treatment of metastatic tumors. ZnP@pyro PDT induces immunogenic cell death (ICD) and releases tumor-associated antigens, which are presented to naïve T cells to stimulate the production and proliferation of tumor-specific effector T cells. ZnP@pyro PDT also elicits an inflammatory environment to enhance the infiltration of effector T cells and other immune cells, such as B cells and NK cells, into both primary and metastatic tumors. When combined with PD-L1 checkpoint blockade, ZnP@pyro PDT not only eradicates the primary tumors, but also rejects the metastatic tumors by a systemic antitumor immune response.
RESULTS AND DISCUSSION
Preparation and Characterization of ZnP@pyro
ZnP nanoparticles were first synthesized by the polymerization between Zn2+ ions and pyrophosphate in the presence of 1,2-dioleoyl-sn-glycero-3-phosphate sodium salt (DOPA, Figure S1). The coordination polymerization between Zn2+ ions and pyrophosphate linkers was confirmed by extended X-ray absorption fine structure (EXAFS) studies (Figure S1 and Table S1). ZnP particles are capped with a DOPA monolayer via the interactions between phosphate groups of DOPA and free Zn coordination sites on ZnP which are reinforced by hydrophobic–hydrophobic interactions between DOPA molecules. The DOPA coating not only controls the particle size but also makes the nanoparticles dispersible in organic solvents, facilitating pyrolipid loading into the shell. ZnP exhibited a number-average diameter of 25.1 ± 0.7 nm and a polydispersity index (PDI) of 0.13 ± 0.01, as determined by dynamic light scattering (DLS, Figure S2). The transmission electron microscopy (TEM) image showed that ZnP was generally spherical in shape with good monodispersity (Figure S3).
ZnP was further coated with a mixture of lipids containing 1,2-dioleyl-sn-glycero-3-phosphocholine (DOPC), cholesterol, pyrolipid, and 1,2-diastearoyl-sn-glycero-3-phosphoethanol-amine-N-[amino(polyethylene glycol)2000] (DSPE-PEG2k) in a 2:1:1:1 molar ratio to afford the core–shell nanoparticle ZnP@pyro. The self-assembled asymmetric lipid bilayer contained pyrolipid as a PS for PDT, DOPC as a lipid component to form a lipid bilayer, cholesterol as a lipid excipient to order, condense, and stabilize the lipid bilayer structure, and DSPE-PEG2k to endow “stealth” and long circulation properties (Figure 2A). Zn@pyro was observed by TEM to be well-dispersed, uniformly spherical nanoparticles (Figure 2B). DLS measurements gave a number-average diameter, PDI, and zeta potential of 45.4 ± 2.8 nm, 0.13 ± 0.01, and –1.5 ± 0.3 mV, respectively (Figure 2C). ZnP@pyro also exhibited favorable structural stability in a physiological environment, as evidenced by consistent size and PDI when incubated in phosphate buffer solution (PBS) containing 5 mg/mL bovine serum albumin (BSA) for up to 24 h (Figure S4). We hypothesize that the small size, near neutral surface charge, and high stability of ZnP@pyro should endow the particle with low MPS uptake, prolonged blood circulation, and improved tumor uptake, making ZnP@pyro ideal for in vivo therapeutic applications.21
Figure 2.

Preparation and characterization of ZnP@pyro. (A) Scheme showing the Zn-pyrophosphate core and the asymmetric lipid bilayer shell of ZnP@pyro. (B) TEM image showing the spherical and nearly monodispersed morphology of ZnP@pyro (scale bar = 200 nm). (C) Number-average diameter of ZnP@pyro in PBS, measured by DLS. (D) Blood concentration of pyrolipid over time after i.v. injection of ZnP@pyro at a pyrolipid dose of 6 mg/kg. (E) Biodistribution and tumor uptake of ZnP@pyro in 4T1 tumor-bearing mice. Data are expressed as means ± s.d. (n = 3).
Pyrolipid was incorporated into lipid bilayer at the very high loading of 10.6 ± 0.5 wt%, as determined by UV–vis at 669 nm. Due to the high loading, > 97% of the pyrolipid fluorescence was self-quenched when the lipid layer was intact. After the addition of Triton X-100 to disrupt the lipid bilayer, pyrolipid was freed from the ordered structure of ZnP@pyro and regained its fluorescence (Figure S5). The 1O2 generation efficiency of ZnP@pyro was determined in the presence of singlet oxygen sensor green (SOSG) regent. When the lipid bilayer was intact, ZnP@pyro generated very little singlet oxygen, likely due to the quenching of pyrolipid excited states before it can transfer energy to triplet oxygen. After addition of Triton X-100 to ZnP@pyro, 1O2 generation upon light irradiation was restored to a similar efficiency to that of free pyrolipid at the same concentration (Figure S6).
ZnP@pyro Shows Long Circulation and High Tumor Accumulation
The pharmacokinetics (PK) and biodistribution studies on orthotopic 4T1 tumor-bearing BALB/c mice showed that ZnP@pyro exhibited a prolonged blood circulation half-life of 14.5 ± 2.2 h after intravenous (i.v.) injection (Figure 2D). ZnP@pyro also showed low distribution in the liver (<5 ID%/g), spleen (<8 ID%/g), and kidneys (<6 ID%/g), suggesting that ZnP@pyro can avoid MPS uptake. Slow blood clearance and low MPS uptake led to high tumor accumulation, with the highest tumor uptake measured to be 15.6 ± 2.5 ID%/g at 24 h post i.v. administration (Figure 2E). The confocal laser scanning microscope (CLSM) imaging confirmed the high distribution of ZnP@pyro in the tumor at 24 h after i.v. injection (Figure S7). By contrast, free pyrolipid showed a low tumor accumulation of 3.2 ± 1.7 ID%/g and very high accumulation in liver, heart, and spleen at 24 h post i.v. administration (Figure S8).
ZnP@pyro PDT Induces Cell Apoptosis and/or Necrosis in Vitro and in Vivo
ZnP@pyro was rapidly internalized by 4T1 tumor cells, with most uptake occurring within 1 h followed by stable amounts measured over 24 h (Figure S9). High cellular uptake and negligible eflux (<2%) (Figure S10) ensured the high cellular accumulation of ZnP@pyro. Confocal images showed that the fluorescence of ZnP@pyro was quite dim in the first 2 h incubation, but became much brighter after 2 h incubation (Figure S11). The initial dim signal indicates fluorescence quenching and suggests that ZnP@pyro maintains its structural integrity in cells for the first 2 h, followed by the lipid layer dissociation and pyrolipid release. After release, pyrolipid can absorb light to generate cytotoxic ROS, killing tumor cells by inducing apoptosis and/or necrosis.
ZnP@pyro induced no cytotoxicity in cells without irradiation (IC50 > 5 μM), but exhibited very high cytotoxicity after irradiation at a light dose of 54 J/cm2, given at 60 mW/cm2 for 15 min, as shown by a significant decrease in the IC50 value (0.42 ± 0.02 μM) (Figure S12 and Table S2), confirming that ZnP@pyro is nontoxic without light activation, and the local application of light can specifically control the cytotoxic effect. Flow cytometry assay showed that ZnP@pyro at a concentration of 0.2 μM failed to induce apoptosis and/or necrosis without irradiation, but evoked high levels of apoptosis and/or necrosis consistent with free pyrolipid under irradiation (Figure 3A and Figure S13), which was further confirmed by confocal imaging. Cells treated by ZnP@pyro with irradiation could be stained by both Annexin V and propidium iodide (PI), showing that cells were in late apoptosis and/or necrosis (Figure 3B and Figure S14).
Figure 3.

ZnP@pyro PDT induces cell apoptosis and/or necrosis in vitro and in vivo. (A) Apoptosis and/or necrosis of 4T1 tumor cells treated with ZnP@pyro plus light irradiation (54 J/cm2 given at 60 mW/cm2 for 15 min) by flow cytometry analysis. (B) Confocal images showing apoptosis and/or necrosis of 4T1 tumor cells induced by ZnP@pyro PDT treatment in vitro (scale bar = 50 μm). (c) Apoptosis and/or necrosis of 4T1 tumor induced by ZnP@pyro PDT in vivo, as shown by H&E staining (top, scale bar = 100 μm) and TUNEL assay (bottom, scale bar = 50 μm). Syngeneic 4T1 tumor-bearing mice were i.v. injected with free pyrolipid or ZnP@pyro at an equivalent dose of 6 mg/kg, followed by light irradiation at a dose of 180 J/cm2 (670 nm, 100 mW/cm2 for 30 min). Tumors were collected, sectioned, and subjected to H&E staining and TUNEL assay. “(+)” in the figure legends refers to treatment with irradiation.
We also investigated the ability of ZnP@pyro and light irradiation to induce apoptosis and/or necrosis in vivo. Orthotopic 4T1 tumor-bearing mice were i.v. injected with free pyrolipid or ZnP@pyro at the same dose of 6 mg/kg. Twenty-four hours after injection, tumors were irradiated with a 670 nm light-emitting diode (LED) at a light dose of 180 J/cm2, given at 100 mW/cm2 for 30 min. After treatment, tumors were collected, sectioned, and subjected to hematoxylin and eosin (H&E) staining and TdT-mediated dUTP nick end labeling (TUNEL) assay. Histological analysis of tumors treated with free pyrolipid showed compact tumor cells with intact structure, compared to the sparse and separated tumor cells in tumors treated with ZnP@pyro PDT (Figure 3C). The presence of green fluorescence from DNA fragmentation by the TUNEL assay and the high percentage of TUNEL-positive cells (around 80%) further confirmed the ability of ZnP@pyro PDT to induce apoptosis and/or necrosis in vivo (Figure 3C and Figure S15). The drastic difference in the ability of free pyrolipid and ZnP@pyro to induce apoptosis and/or necrosis in vivo is likely due to the low distribution of free pyrolipid in tumor tissues at the time of light irradiation, highlighting the need for ZnP@pyro nanoparticles to take advantage of the EPR effect for high tumor accumulation.
ZnP@pyro PDT Induces 4T1 Tumor Cell Immune Phenotypes in Vitro and in Vivo
Calreticulin (CRT) is a chaperone protein abundant in the endoplasmic reticulum (ER) that is transported to the cell surface in response to ER stress as an indicator of immunogenic cell death (ICD).22 PDT has been reported to cause CRT exposure through a distinct and rapid pathway that relies on protein kinase RNA-like endoplasmic reticulum kinase (PERK).23 Once on the cell surface, CRT serves as an “eat-me” signal, stimulating the engulfment of dying tumor cells and their apoptotic debris by macrophages and immature dendritic cells (DCs).24 We tested the ability of ZnP@pyro under irradiation to induce immunogenic phenotypes on 4T1 tumor cells by determining the CRT exposure. After incubation with ZnP@pyro and irradiation at a light dose of 54 J/cm2, cells were stained with Alexa Fluor 488-CRT antibody and analyzed by flow cytometry. ZnP@pyro with irradiation induced CRT exposure comparable to free pyrolipid, in terms of CRT fluorescence intensity and CRT-positive cells. As shown in Figure 4A,B, ZnP@pyro and free pyrolipid with irradiation induced CRT exposure on ~87% and 90% of cells, respectively. The CLSM images confirmed the flow cytometry results that ZnP@pyro only induced CRT exposure under irradiation (Figure 4C and Figure S16), suggesting that PDT but not pyrolipid itself induces 4T1 tumor cell immunogenic properties.
Figure 4.

ZnP@pyro PDT induces tumor cell immune phenotypes and acute inflammation. (A,B) Quantification of CRT exposure on the surface of 4T1 cells after treatment with free pyrolipid or ZnP@pyro plus light irradiation (54 J/cm2) by flow cytometry analysis. (C,D) Confocal images showing the CRT exposure on 4T1 tumor cells in vitro (C) and in vivo (D) after treatment with free pyrolipid or ZnP@pyro plus light irradiation (scale bar = 50 μm). (E) Pro-inflammatory cytokine levels in the sera of mice treated with PDT of ZnP@pyro from day 0 to day 3. Arrows represent the time of nanoparticle administration (black) and irradiation (red). “(+)” in the figure legends refers to treatment with irradiation. Data are expressed as means ± s.d. (n = 3).
Building on these observations, we tested the CRT exposure on 4T1 tumors treated with ZnP@pyro plus irradiation. As shown in Figure 4D, ZnP@pyro PDT treatment significantly increased CRT staining within 4T1 tumor nodules, a result that mirrors our in vitro findings. However, free pyrolipid PDT treatment induced minimal CRT exposure in vivo due to inefficient tumor accumulation at the time of light irradiation. These data demonstrate that ZnP@pyro PDT can induce immunogenic phenotypes in 4T1 tumor cells both in vitro and in vivo.
ZnP@pyro PDT Induces Acute Inflammation in Vivo
To evaluate the level and duration of inflammation evoked by PDT of ZnP@pyro, blood was collected daily from orthotopic 4T1 tumor-bearing mice from day 0 when the mice received the ZnP@pyro injection to day 3. The serum was separated and analyzed by enzyme-linked immunosorbent assay (ELISA) to determine the production of TNF-α, IL-6, and IFN-γ cytokines. Release of such cytokines indicates acute inflammation, an important mechanism in inducing antitumor immunity by PDT.13c,25 High levels of TNF-α, IL-6, and IFN-γ were observed on day 2 post light irradiation in mice treated by ZnP@pyro PDT, indicating that PDT can successfully cause inflammation (Figure 4E). However, 2 days after PDT treatment, all three proinflammatory cytokine levels rapidly dropped to baseline levels, suggesting that inflammation caused by ZnP@pyro PDT was only an acute response.
ZnP@pyro PDT Combined with PD-L1 Blockade Eradicates Primary 4T1 Tumor and Prevents Lung Metastasis
In order to determine whether antitumor immunity triggered by ZnP@pyro PDT could be harnessed for sensitizing tumors to checkpoint blockade therapy, we investigated the antitumor activity and antimetastatic effect of ZnP@pyro PDT combined with anti-PD-L1 (α-PD-L1, Clone: 10F.9G2, Catalog No. BE0101, BioXCell) on 4T1 tumors. Orthotopic 4T1 tumors in the mammary fat pads of mice produce spontaneous metastases to the lung, making it a suitable experimental animal model for stage IV human breast cancer.26 4T1 tumor-bearing mice were i.v. injected with ZnP@pyro at a pyrolipid dose of 6 mg/kg every 2 days for a total of three treatments. Twenty-four hours post injection, tumors were irradiated with a 670 nm LED at an irradiance of 100 mW/cm2 for 30 min. After irradiation, mice were intra-peritoneally (i.p.) injected with anti-PD-L1 antibody at a dose of 75 μg/mouse. As indicated in Figure 5A–C, anti-PD-L1 itself failed to delay 4T1 tumor progression. In contrast, ZnP@pyro PDT treatment significantly inhibited 4T1 tumor growth with a 68% reduction in tumor volume and a 75% reduction in tumor weight compared to the PBS control group. Notably, ZnP@pyro PDT combined with anti-PD-L1 treatment completely eradicated the primary 4T1 tumor, indicating that the combination treatment was markedly better than either ZnP@pyro PDT or anti-PD-L1 alone. In addition, no weight loss was observed in ZnP@pyro PDT plus anti-PD-L1 treated group, indicating the absence of severe systemic toxicity (Figure S17).
Figure 5.

ZnP@pyro PDT combined with PD-L1 blockade eradicates primary 4T1 tumors and prevents lung metastasis. PBS or ZnP@pyro was i.v. injected into an orthotopic 4T1 mouse model at a pyrolipid dose of 6 mg/kg, then tumors were irradiated (670 nm, 100 mW/cm2) for 30 min at 24 h after each injection. (A) Tumor growth curves. Arrows represent the time of nanoparticle administration (black) and irradiation (red). (B) Tumor weights at the end point. (C) Photographs of excised tumors at the end point. From top to bottom: PBS, α-PD-L1, ZnP@pyro (+), and ZnP@pyro (+) + α-PD-L1. Rectangle indicates tumors disappeared in the ZnP@pyro (+) + α-PD-L1 group. (D) Representative pictures showing the gross appearance of tumor nodules in the lungs. (E) The numbers of tumor nodules present in the lungs. (F) Representative lung sections stained with H&E. (G) Percentage of lung in metastasis. (H) Representative pictures showing the colonies formed after culturing in the presence of 6-thiogunine for 10 days. (I) Normalized absorbance of crystal violet in different treatment groups. “(+)” in the figure legends refers to treatment with irradiation. * P < 0.05, **P < 0.01, ***P < 0.001.
At the end of the study, 23 days after tumor inoculation, mice were sacrificed and assessed for the extent of metastasis to the lungs by gross examination of tissue for tumor nodules. Compared to the PBS control, ZnP@pyro PDT or anti-PD-L1 alone showed little effect on preventing lung metastasis, while the combination treatment significantly reduced tumor nodules: only one or two tumor nodules were found on the lungs of those receiving combination treatment, compared to 31 ± 6 tumor nodules observed in the PBS control group (Figure 5D,E). Lungs were further sectioned and stained with H&E to quantify the proportion of the metastasis area to the whole lung. As shown in Figure 5F,G, about 37%, 30%, and 26% of lungs were occupied by tumors in PBS-, ZnP@pyro PDT-, and anti-PD-L1-treated groups, respectively. Combination treatment significantly decreased the percentage of metastasis in the lung to only 0.4%, indicating that the combination treatment was much more effective in preventing lung metastasis than either ZnP@pyro PDT or anti-PD-L1 alone. Lungs were also digested, and the cells were cultured in the presence of 60 μM 6-thioguanine for 10 days. After fixation with menthol, colonies formed by clonogenic metastatic cancer cells were stained with 0.1% crystal violet. Because 4T1 tumor cells are resistant to 6-thioguanine, only metastasized tumor cells can proliferate and form colony.27 As shown in Figure 5H, the combination treatment significantly reduced the colonies number to only 6 ± 3, compared to PBS-, ZnP@pyro PDT-, and anti-PD-L1-treated groups, which all formed numerous colonies. The quantitative results showed that the absorbance of the combination treatment group was only 4.5 ± 1.5% of the PBS control group (Figure 5I), which indicates that there were much less clonogenic metastatic cancer cells in the lungs treated by ZnP@pyro PDT plus anti-PD-L1 than that treated by PBS.
Our findings were consistent with previous reports that 4T1 tumor showed no response to anti-PD-L1, possibly due to their low expression of PD-L1.28 However, literature reports indicate that checkpoint blockade immunotherapy can be enhanced by combining with other immunogenic therapies. For example, ibrutinib was able to convert a weak antitumor T-cell immune response induced by anti-PD-L1 antibody into a powerful one, although it did not affect the PD-L1 expression level in tumors.28 In another example, the combination of oxaliplatin with cyclophosphamide was shown to induce tumor cell immune phenotypes, trigger adaptive and innate immunity, and sensitize tumors to checkpoint blockade therapy.29 We demonstrated that PDT treatment can also increase tumor immunogenicity and induce acute inflammation, thereby producing tumor-specific immunity. The tumor-specific immunity evoked by PDT enhanced the effect of immune checkpoint therapy, resulting in the eradication of primary tumor and the prevention of lung metastasis.
ZnP@pyro PDT Combined with PD-L1 Blockade Not Only Prevents Metastasis but Also Completely Inhibits Larger, Pre-existing Metastatic Tumors
A bilateral subcutaneous 4T1 model was used to determine whether the induced antitumor immune response by ZnP@pyro PDT plus anti-PD-L1 antibody could be effective against larger, pre-existing metastatic tumors. ZnP@pyro was systemically injected but only the right (primary) tumors were irradiated. As indicated in Figure 6A–C, anti-PD-L1 alone exhibited very little effect on the inhibition of either the primary or the distant tumors. ZnP@pyro with irradiation effectively controlled primary tumor growth but did not significantly inhibit the distant tumors, compared to the anti-PD-L1 group. However, the combination of ZnP@pyro with irradiation and PD-L1 blockade induced complete eradication of the irradiated primary tumors (synergistic effect) and effective control of the nonirradiated distant tumors (abscopal effect), eliciting a 92% reduction in tumor size compared to the PBS control group. These results indicate that tumors can be sensitized to PD-L1 blockade immunotherapy by ZnP@pyro PDT-mediated tumor-specific immune responses, and the combination of ZnP@pyro PDT and PD-L1 blockade has the potential to become a potent immunotherapy strategy in the management of patients with metastatic cancer.
Figure 6.

ZnP@pyro PDT sensitizes tumors to immune checkpoint therapy. Bilateral syngeneic tumor models of 4T1 and TUBO were developed by subcutaneously injecting cancer cells into both the right and left flank regions of each animal. The right tumors were designated as primary tumors for light irradiation, and the left tumors were designated as distant tumors and not subjected to light irradiation. ZnP@pyro was i.v. injected into mice, followed by light irradiation at a dose of 180 J/cm2 (670 nm, 100 mW/cm2) at 24 h after nanoparticle injection and i.p. injection of anti-PD-L1 at a dose of 75 μg/mouse. The treatment was carried out every other day for a total of three treatments. Primary and distant tumor growth curves in 4T1 (A,B) and TUBO (D,E) models. The arrows represent the time of nanoparticle administration (black) and irradiation (red). (C,F) Weight of 4T1 (C) and TUBO (F) tumors at the end point of the experiment. “(+)” in the figure legends refers to treatment with irradiation. *P < 0.05, ** P < 0.01, *** P < 0.001.
ZnP@pyro PDT Sensitizes Other Tumors to Immune Checkpoint Therapy
Finally, we tested whether the antitumor immunity of ZnP@pyro PDT could also sensitize other tumors to immune checkpoint therapy. We explored another syngeneic murine breast cancer model, TUBO, and similarly found that TUBO-bearing mice also failed to respond to anti-PD-L1 antibody. However, when combined with ZnP@pyro PDT, anti-PD-L1-mediated checkpoint blockade therapy completely eradicated primary tumors and significantly inhibited the growth of distant tumors (Figure 6D–F). These results demonstrate that our findings in the 4T1 mouse model could be extended to other tumor types. To further validate earlier findings that our nanoparticle formulation of pyrolipid is necessary to observe the in vivo effects, we also determined the efficacy of free pyrolipid PDT plus anti-PD-L1, which showed no significant difference from anti-PD-L1 alone (Figure 6D–F). This result was consistent with the earlier results that free pyrolipid PDT induced low apoptosis/necrosis, low CRT exposure, and low cytokines level in vivo, compared to ZnP@pyro PDT, due to its inefficient accumulation in tumor tissues.
ZnP@pyro PDT Combined with PD-L1 Blockade Activates Systematic Antitumor Immune Response
The inhibition of distant tumors in 4T1- and TUBO-bearing mice treated with ZnP@pyro PDT plus anti-PD-L1 implied an effective induction of systemic antitumor immune response. We first validated this hypothesis in TUBO model with an IFN-γ ELISPOT assay to determine the presence or absence of tumor-specific cytotoxic T cells. Splenocytes were harvested from TUBO-bearing mice at day 12 after the first treatment and stimulated with antigen-presenting 3T3/NKB cells, which express TUBO-derived antigen neu, and the IFN-γ spot-forming cells (SFC) were determined.30 We found that the numbers of IFN-γ SFC significantly increased in both PD-L1 blockade (IFN-γ SFC/104 cells = 2.20 ± 1.54) and ZnP@pyro PDT plus anti-PD-L1 groups (IFN-γ SFC/104 cells = 2.66 ± 1.95), compared to the PBS control group (IFN-γ SFC/104 cells = 0.29 ± 0.31) (Figure 7A).
Figure 7.

ZnP@pyro PDT plus immune checkpoint therapy activates systematic antitumor immune response. Bilateral syngeneic tumor models of TUBO were developed by subcutaneously injecting cancer cells into both the right and left flank regions of each animal. The right tumors were designated as primary tumors for light irradiation, and the left tumors were designated as distant tumors and not subjected to light irradiation. ZnP@pyro was i.v. injected into mice, followed by light irradiation at a dose of 180 J/cm2 (670 nm, 100 mW/cm2) at 24 h after nanoparticle injection and i.p. injection of anti-PD-L1 at a dose of 75 μg/mouse. The treatment was carried out every other day for a total of three treatments. On day 22 (12 days post initial treatment), splenocytes from different treatment groups were harvested and stimulated with 3T3/NKB cells or control 3T3/KB cells for 48 h, the IFN-γ SFC were determined by ELISPOT (A). The distant tumors were harvested for flow cytometry, the percentages of CD45+ leukocytes (CD45+PI–) (B), NK cells (CD45+CD3e–NKp46+PI–) (C), CD8+ T cells (CD45+CD3e+CD8+PI–) (D), CD4+ T cells (CD45+CD3e+CD4+PI–) (E) and B cells (CD45+CD3e–B220+PI–) (F) in total tumor cells were determined. The distant tumors were also sectioned and subjected to immunofluorescence staining. (G) Representative CLSM images of tumors after immunofluorescence staining (scale bar = 200 μm).
After initial indication of systemic immune response, we further profiled infiltrating leukocytes in the distant tumors. The percentage of CD45+ leukocytes in the total tumor cells significantly increased by about 45% in the ZnP@pyro PDT plus anti-PD-L1 treatment group (25.4 ± 6.01%), compared to the PBS control group (17.5 ± 1.45%) (Figure 7B). Specifically, the percentages of NK cells (17.2 ± 3.89%), CD8+ T cells (1.45 ± 0.65%) and CD4+ T cells (0.97 ± 0.16%) all significantly increased in the anti-PD-L1 treated group compared to the PBS control group (NK cells, 9.28 ± 3.07%; CD8+ T cells, 0.63 ± 0.20%; CD4+ T cells, 0.33 ± 0.18%) (Figure 7C–E), while the percentage of B cells significantly increased in ZnP@pyro PDT treated group (6.65 ± 3.64%) compared to the PBS control group (3.01 ± 2.06%) (Figure 7F). These results suggest that PD-L1 checkpoint blockade plays an important role in promoting the dramatically increased NK cell infiltration and accumulation in the distant tumor sites and activating tumor-specific T cells responses to control the distant tumors, while ZnP@pyro PDT evokes B cells infiltration to the distant tumors, which can potentially induce antitumor humoral immune responses. PDT and checkpoint blockade therapy each initiate unique forms of immune response, which were both found in ZnP@pyro PDT plus anti-PD-L1 treatment (NK cells, 17.6 ± 6.17%; CD8+ T cells, 1.72 ± 0.66%; CD4+ T cells, 1.04 ± 0.32%; B cells, 8.28 ± 3.25%). The increase in CD8+ T cell infiltration and activity may have been influenced by the PD-L1 blockade, as PD-L1 has been reported to negatively regulate T cells.31 There was also a slight, though not statistically significant, decrease in the percentage of regulatory T cells in tumors treated with ZnP@pyro PDT combined with PD-L1 blockade (Figure S18), which may have contributed to the increased CD8+ T cells activity. A combination of these immune responses were likely required for the eradication of the primary tumor and inhibition of distant tumor, supporting the increased efficacy of ZnP@pyro PDT combined with immune checkpoint blockade therapy.
The antitumor immune response elicited by ZnP@pyro PDT in combination with anti-PD-L1 was further confirmed by immunofluorescence assay. We found that ZnP@pyro with irradiation plus anti-PD-L1 treatment instigated CD3+ T cell infiltration within the distant tumor tissues, whereas no tumor-infiltrating CD3+ T cells were observed in the PBS control group. In addition, a large fraction of the tumor-infiltrating CD3+ T cells were CD8+ (Figure 7G), indicating the ability of ZnP@pyro PDT plus anti-PD-L1 to promote CD8+ T-cell infiltration into tumors.
CONCLUSION
We have developed nontoxic and immunogenic ZnP@pyro nanoparticles for the effective treatment of metastatic breast cancer by combining PDT and checkpoint blockade immunotherapy. ZnP@pyro showed prolonged blood circulation and enhanced tumor accumulation after systemic administration, thereby effectively inhibiting tumor growth upon light irradiation. More importantly, ZnP@pyro-mediated PDT induced an immunogenic environment in tumors and sensitized tumors to PD-L1 checkpoint blockade therapy. As a result, ZnP@pyro PDT combined with anti-PD-L1 not only eradicated the primary tumors, but also prevented the lung metastasis and inhibited the pre-existing metastatic tumors by generating systemic antitumor immunity. Our results indicate that immunogenic therapies may provide immediate clinical benefit by expanding the small proportion of cancer patients who respond to current immune checkpoint treatments.
Supplementary Material
Acknowledgments
We thank the National Cancer Institute (U01-CA198989), the University of Chicago Medicine Comprehensive Cancer Center (NIH CCSG: P30 CA014599), the CBI Training Grant (NIH 5T32GM008720-15), the Cancer Research Foundation, and the Ludwig Institute for Metastasis Research for funding support. We thank Mr. Zekai Lin for help with EXAFS experiments.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b09538.
Experimental details, supporting Figures S1–S18 and Tables S1 and S2 (PDF)
References
- 1.DeSantis C, Ma J, Bryan L, Jemal A. Ca-Cancer J Clin. 2014;64:52. doi: 10.3322/caac.21203. [DOI] [PubMed] [Google Scholar]
- 2.(a) Schmadeka R, Harmon BE, Singh M. Am J Clin Pathol. 2014;141:462. doi: 10.1309/AJCPQN8GZ8SILKGN. [DOI] [PubMed] [Google Scholar]; (b) Bayraktar S, Glück S. Breast Cancer Res Treat. 2013;138:21. doi: 10.1007/s10549-013-2421-5. [DOI] [PubMed] [Google Scholar]
- 3.(a) Vanneman M, Dranoff G. Nat Rev Cancer. 2012;12:237. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Couzin-Frankel J. Science. 2013;342:1432. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 4.(a) Restifo NP, Dudley ME, Rosenberg SA. Nat Rev Immunol. 2012;12:269. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Cell. 2015;160:48. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schumacher TN, Schreiber RD. Science. 2015;348:69. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]; (d) Gajewski TF, Schreiber H, Fu YX. Nat Immunol. 2013;14:1014. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishikawa H, Sakaguchi S. Curr Opin Immunol. 2014;27:1. doi: 10.1016/j.coi.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 6.Talmadge JE, Gabrilovich DI. Nat Rev Cancer. 2013;13:739. doi: 10.1038/nrc3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Sharma P, Allison JP. Science. 2015;348:56. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]; (b) Topalian SL, Drake CG, Pardoll DM. Cancer Cell. 2015;27:450. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Pardoll DM. Nat Rev Cancer. 2012;12:252. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, et al. Nature. 2014;515:577. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Topalian SL, Taube JM, Anders RA, Pardoll DM. Nat Rev Cancer. 2016;16:275. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Meng X, Huang Z, Teng F, Xing L, Yu J. Cancer Treat Rev. 2015;41:868. doi: 10.1016/j.ctrv.2015.11.001. [DOI] [PubMed] [Google Scholar]; (b) Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Proc Natl Acad Sci U S A. 2002;99:12293. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Blank C, Gajewski TF, Mackensen A. Cancer Immunol Immunother. 2005;54:307. doi: 10.1007/s00262-004-0593-x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Topalian SL, Drake CG, Pardoll DM. Curr Opin Immunol. 2012;24:207. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Webster RM. Nat Rev Drug Discovery. 2014;13:883. doi: 10.1038/nrd4476. [DOI] [PubMed] [Google Scholar]; (b) Leventakos K, Mansfield AS. BioDrugs. 2016;30:397. doi: 10.1007/s40259-016-0187-0. [DOI] [PubMed] [Google Scholar]
- 11.(a) Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. Nature. 2014;515:568. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Science. 2015;348:124. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, Spranger S. Curr Opin Immunol. 2013;25:268. doi: 10.1016/j.coi.2013.02.009. [DOI] [PubMed] [Google Scholar]; (b) Joyce JA, Fearon DT. Science. 2015;348:74. doi: 10.1126/science.aaa6204. [DOI] [PubMed] [Google Scholar]; (c) Shahabi V, Postow MA, Tuck D, Wolchok JD. Am J Clin Oncol. 2015;38:90. doi: 10.1097/COC.0b013e3182868ec8. [DOI] [PubMed] [Google Scholar]
- 13.(a) Shams M, Owczarczak B, Manderscheid-Kern P, Bellnier DA, Gollnick SO. Cancer Immunol Immunother. 2015;64:287. doi: 10.1007/s00262-014-1633-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kwitniewski M, Juzeniene A, Glosnicka R, Moan J. Photochem Photobiol Sci. 2008;7:1011. doi: 10.1039/b806710d. [DOI] [PubMed] [Google Scholar]; (c) Castano AP, Mroz P, Hamblin MR. Nat Rev Cancer. 2006;6:535. doi: 10.1038/nrc1894. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) He C, Duan X, Guo N, Chan C, Poon C, Weichselbaum RR, Lin W. Nat Commun. 2016;7:12499. doi: 10.1038/ncomms12499. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lu K, He C, Guo N, Chan C, Ni K, Weichselbaum RR, Lin W. J Am Chem Soc. 2016;138:12502. doi: 10.1021/jacs.6b06663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Garg AD, Agostinis P. Photochem Photobiol Sci. 2014;13:474. doi: 10.1039/c3pp50333j. [DOI] [PubMed] [Google Scholar]; (b) Dolmans DE, Fukumura D, Jain RK. Nat Rev Cancer. 2003;3:380. doi: 10.1038/nrc1071. [DOI] [PubMed] [Google Scholar]; (c) Garg AD, Nowis D, Golab J, Agostinis P. Apoptosis. 2010;15:1050. doi: 10.1007/s10495-010-0479-7. [DOI] [PubMed] [Google Scholar]; (d) Gollnick SO, Brackett CM. Immunol Res. 2010;46:216. doi: 10.1007/s12026-009-8119-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Mroz P, Hashmi JT, Huang YY, Lange N, Hamblin MR. Expert Rev Clin Immunol. 2011;7:75. doi: 10.1586/eci.10.81. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Mijan MC, Longo JPF, de Melo LND, Simioni AR, Tedesco AC, Azevedo RB. J Nanomed Nanotechnol. 2014;5:218. [Google Scholar]
- 15.(a) Sanabria LM, Rodríguez ME, Cogno IS, Vittar NBR, Pansa MF, Lamberti MJ, Rivarola VA. Biochim Biophys Acta, Rev Cancer. 2013;1835:36. doi: 10.1016/j.bbcan.2012.10.001. [DOI] [PubMed] [Google Scholar]; (b) Gollnick S, Evans S, Baumann H, Owczarczak B, Maier P, Vaughan L, Wang W, Unger E, Henderson B. Br J Cancer. 2003;88:1772. doi: 10.1038/sj.bjc.6600864. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Galluzzi L, Kepp O, Kroemer G. EMBO J. 2012;31:1055. doi: 10.1038/emboj.2012.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Celli JP, Spring BQ, Rizvi I, Evans CL, Samkoe KS, Verma S, Pogue BW, Hasan T. Chem Rev. 2010;110:2795. doi: 10.1021/cr900300p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chatterjee DK, Fong LS, Zhang Y. Adv Drug Delivery Rev. 2008;60:1627. doi: 10.1016/j.addr.2008.08.003. [DOI] [PubMed] [Google Scholar]; (c) Paszko E, Ehrhardt C, Senge MO, Kelleher DP, Reynolds JV. Photodiagn Photodyn Ther. 2011;8:14. doi: 10.1016/j.pdpdt.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 17.(a) Wang AZ, Langer R, Farokhzad OC. Annu Rev Med. 2012;63:185. doi: 10.1146/annurev-med-040210-162544. [DOI] [PubMed] [Google Scholar]; (b) Maeda H, Nakamura H, Fang J. Adv Drug Delivery Rev. 2013;65:71. doi: 10.1016/j.addr.2012.10.002. [DOI] [PubMed] [Google Scholar]; (c) Stylianopoulos T. Ther Delivery. 2013;4:421. doi: 10.4155/tde.13.8. [DOI] [PubMed] [Google Scholar]; (d) Allison R, Mota H, Bagnato VS, Sibata C. Photodiagn Photodyn Ther. 2008;5:19. doi: 10.1016/j.pdpdt.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 18.(a) Friedman A, Claypool S, Liu R. Curr Pharm Des. 2013;19:6315. doi: 10.2174/13816128113199990375. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Florence AT. J Controlled Release. 2012;164:115. doi: 10.1016/j.jconrel.2012.03.022. [DOI] [PubMed] [Google Scholar]; (c) Subramani K, Hosseinkhani H, Khraisat A, Hosseinkhani M, Pathak Y. Curr Nanosci. 2009;5:135. [Google Scholar]; (d) Stuchinskaya T, Moreno M, Cook MJ, Edwards DR, Russell DA. Photochem Photobiol Sci. 2011;10:822. doi: 10.1039/c1pp05014a. [DOI] [PubMed] [Google Scholar]
- 19.(a) Lovell JF, Jin CS, Huynh E, Jin H, Kim C, Rubinstein JL, Chan WC, Cao W, Wang LV, Zheng G. Nat Mater. 2011;10:324. doi: 10.1038/nmat2986. [DOI] [PubMed] [Google Scholar]; (b) Jin CS, Cui L, Wang F, Chen J, Zheng G. Adv Healthcare Mater. 2014;3:1240. doi: 10.1002/adhm.201300651. [DOI] [PubMed] [Google Scholar]; (c) Samia AC, Chen X, Burda C. J Am Chem Soc. 2003;125:15736. doi: 10.1021/ja0386905. [DOI] [PubMed] [Google Scholar]; (d) Cheng Y, Doane TL, Chuang CH, Ziady A, Burda C. Small. 2014;10:1799. doi: 10.1002/smll.201303329. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chen J, Wang D, Xi J, Au L, Siekkinen A, Warsen A, Li ZY, Zhang H, Xia Y, Li X. Nano Lett. 2007;7:1318. doi: 10.1021/nl070345g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Lal S, Clare SE, Halas NJ. Acc Chem Res. 2008;41:1842. doi: 10.1021/ar800150g. [DOI] [PubMed] [Google Scholar]; (g) Idris NM, Gnanasammandhan MK, Zhang J, Ho PC, Mahendran R, Zhang Y. Nat Med. 2012;18:1580. doi: 10.1038/nm.2933. [DOI] [PubMed] [Google Scholar]; (h) Tian J, Ding L, Xu HJ, Shen Z, Ju H, Jia L, Bao L, Yu JS. J Am Chem Soc. 2013;135:18850. doi: 10.1021/ja408286k. [DOI] [PubMed] [Google Scholar]; (i) Luo D, Carter KA, Miranda D, Lovell JF. Adv Sci. 2016 doi: 10.1002/advs.201600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Sian L, Mingyan X, Miller LV, Tong L, Krebs NF, Hambidge KM. Am J Clin Nutr. 1996;63:348. doi: 10.1093/ajcn/63.3.348. [DOI] [PubMed] [Google Scholar]; (b) Lomashvili KA, Narisawa S, Millan JL, O'Neill WC. Kidney Int. 2014;85:1351. doi: 10.1038/ki.2013.521. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang M, Zhang Z, Blessington D, Li H, Busch TM, Madrak V, Miles J, Chance B, Glickson JD, Zheng G. Bioconjugate Chem. 2003;14:709. doi: 10.1021/bc034038n. [DOI] [PubMed] [Google Scholar]
- 21.(a) Duan X, Li Y. Small. 2013;9:1521. doi: 10.1002/smll.201201390. [DOI] [PubMed] [Google Scholar]; (b) Hirsjärvi S, Dufort S, Gravier J, Texier I, Yan Q, Bibette J, Sancey L, Josserand V, Passirani C, Benoit JP, Coll JL. Nanomedicine. 2013;9:375. doi: 10.1016/j.nano.2012.08.005. [DOI] [PubMed] [Google Scholar]; (c) Kulkarni SA, Feng SS. Pharm Res. 2013;30:2512. doi: 10.1007/s11095-012-0958-3. [DOI] [PubMed] [Google Scholar]
- 22.(a) Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, et al. Nat Med. 2007;13:54. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]; (b) Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Annu Rev Immunol. 2013;31:51. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 23.Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, Rubio N, Firczuk M, Mathieu C, Roebroek AJ, et al. EMBO J. 2012;31:1062. doi: 10.1038/emboj.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, Catani JPP, Hannani D, Duret H, Steegh K, et al. Immunity. 2013;38:729. doi: 10.1016/j.immuni.2013.03.003. [DOI] [PubMed] [Google Scholar]; (b) Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Immunity. 2013;39:74. doi: 10.1016/j.immuni.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 25.(a) Yom SS, Busch TM, Friedberg JS, Wileyto EP, Smith D, Glatstein E, Hahn SM. Photochem Photobiol. 2003;78:75. doi: 10.1562/0031-8655(2003)078<0075:esclim>2.0.co;2. [DOI] [PubMed] [Google Scholar]; (b) Ziółkowski P, Symonowicz K, Milach J, Zawirska B, Szkudlarek T. Neoplasma. 1996;44:192. [PubMed] [Google Scholar]
- 26.(a) Pulaski BA, Ostrand-Rosenberg S. Curr Protoc Immunol. 2001;39:20.2.1. doi: 10.1002/0471142735.im2002s39. [DOI] [PubMed] [Google Scholar]; (b) Tao K, Fang M, Alroy J, Sahagian GG. BMC Cancer. 2008;8:228. doi: 10.1186/1471-2407-8-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shan D, Chen L, Njardarson JT, Gaul C, Ma X, Danishefsky SJ, Huang XY. Proc Natl Acad Sci U S A. 2005;102:3772. doi: 10.1073/pnas.0500658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sagiv-Barfi I, Kohrt HEK, Czerwinski DK, Ng PP, Chang BY, Levy R. Proc Natl Acad Sci U S A. 2015;112:E966. doi: 10.1073/pnas.1500712112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, Lin YJ, Wojtkiewicz G, Iwamoto Y, Mino-Kenudson M, Huynh TG, Hynes RO, Freeman GJ, Kroemer G, Zitvogel L, Weissleder R, Pittet MJ. Immunity. 2016;44:343. doi: 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park S, Jiang Z, Mortenson ED, Deng L, Radkevich-Brown O, Yang X, Sattar H, Wang Y, Brown NK, Greene M, Liu Y, Tang J, Wang S, Fu YX. Cancer Cell. 2010;18:160. doi: 10.1016/j.ccr.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.(a) Blank C, Gajewski TF, Mackensen A. Cancer Immunol Immunother. 2005;54:307. doi: 10.1007/s00262-004-0593-x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, Kuchroo VK, Freeman GJ, Sharpe AH. Proc Natl Acad Sci U S A. 2004;101:10691. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.